

Abstract
A nonconservative leucine to proline mutation in peripheral myelin protein 22 (PMP22) causes the Trembler-J (TrJ) neuropathy in mice and humans. The expression levels and localization of the PMP22 protein in the TrJ mouse have not been previously determined. The aim of our studies was to reevaluate the extent of myelin deficit in genotyped heterozygous and homozygous animals and to examine how the TrJ mutation alters the normal in vivo post-translational processing of PMP22. Morphological studies show evidence for primary dysmyelination and myelin instability in affected animals. As expected, Western blot analysis indicates that in adult heterozygousTrJ animals, the level of PMP22 is markedly decreased, similar to myelin basic protein and protein zero, whereas myelin-associated glycoprotein is largely unaffected. The decrease in myelin protein expression is associated with an increase in lysosomal biogenesis, suggestive of augmented endocytosis or autophagy. Double-immunolabeling experiments show the accumulation of PMP22 in endosomal/lysosomal structures of TrJSchwann cells, and chloroquine treatment of nerve segments indicates that the degradation of protein zero, PMP22, and myelin basic protein is augmented in TrJ nerves. These studies suggest that the TrJmutation alters myelin stability and that the mutant protein is likely degraded via the lysosomal pathway.
Keywords: peripheral myelin protein 22, Schwann cells, peripheral nervous system, myelin, neuropathy, Trembler-J, endosomal-lysosomal pathway, protein processing
The Trembler (Tr) andTrembler-J (TrJ) mice are animal models for the human hereditary neuropathy Charcot-Marie-Tooth (CMT) disease, a group of common (1/2500) (Skre, 1974) heterogeneous peripheral neuropathies. In mice, the semidominant and dominantly inherited mutations in the pmp22 gene result in the nonconservative leucine-to-proline (L16P) or glycine-to-aspartic acid (G160D) replacements in the PMP22 protein in theTrJ or Tr mouse, respectively (Suter et al., 1992a,b). These mutations are believed to be responsible for the PNS deficits. The majority of human patients with CMT, however, do not have point mutations in the PMP22gene, but instead harbor a submicroscopic chromosomal duplication on human chromosome 17p11.2–12, which encompasses the PMP22gene (for review, see Suter and Snipes, 1995a). The similarity of the disease phenotypes, including disease progression, as well as the finding of the identical TrJ single point mutation in a severely affected CMT disease type 1A (CMT1A) family (Valentijn et al., 1992) substantiates the use of these mice as models of human disease and provide a system for elucidating the biology of the PMP22 protein.
The ways in which pmp22 mutations perturb myelination are complex, particularly because the function of the PMP22 protein is unknown (for review, see Suter and Snipes, 1995b). Previous morphological studies of the TrJ mouse used the close linkage of the TrJphenotype to the vestigial tail marker on mouse chromosome 11 to predict the genotype of the progeny of heterozygous (TrJ/+) crosses, although the homozygous (TrJ/TrJ) genotype remained ambiguous. From these studies,TrJ/+ mice were shown to live a normal life span, and only in adulthood did they show clinical signs of myelin deficit (Henry and Sidman, 1983). In contrast, putativeTrJ/TrJanimals were severely affected early in development and did not live beyond postnatal day 16 (P16) to P18 (Henry and Sidman, 1983). That at least part of the TrJ phenotype is unique to this mutation and is not simply a result of general myelin loss is suggested by comparison to homozygous Tr mice. In contrast toTrJ/TrJ,Tr/Tr mice live a normal life span even with a virtual absence of peripheral myelin (Henry and Sidman, 1988). InTr/+ mice, the progression of the disease is more rapid than in TrJ/+ mice, leading to prominent histological abnormalities (e.g., “onion bulb” formation) in aged animals (Henry et al., 1983). Although “onion bulbs,” which consist of redundant Schwann cell processes and connective tissue surrounding axons, are not common in TrJ nerves, both Tr and TrJ animals show severe PNS hypomyelination and overproduction of Schwann cells (Henry et al., 1983). Thus, there are significant differences in the phenotypes of Tr and TrJanimals that may reflect the unique effects of individualpmp22 mutations.
The hypomyelination evident in adult Tr nerves is reflected in the severe reduction in the levels of mRNAs and proteins for the structural components of compact myelin, protein zero (P0), and myelin basic proteins (MBP) (Jacque et al., 1983; Bascles et al., 1992; Garbay and Bonnet, 1992), whereas myelin associated glycoprotein (MAG) levels are unchanged (Inuzuka et al., 1985). These results agree with morphological studies in Tr nerves demonstrating the presence of loose, uncompacted myelin (Henry et al., 1983) which are immunoreactive for MAG (Trapp and Quarles, 1982). In adultTrJ/+ sciatic nerves, ultrastructural immunolocalization studies showed the presence of MAG-positive mesaxon-like membranes and P0-containing Golgi-associated vesicles in some of the Schwann cells (Heath et al., 1991). Thus, there is growing evidence that post-transcriptional intracellular events such as protein synthesis, transport, and processing are critical determinants of successful myelination and are important variables in our understanding of the effects of pmp22 mutations on Schwann cell biology.
Despite the rapid advances in elucidating the genetics of theTr and TrJ mutations, to date the level and localization of the PMP22 protein in trembler animals have not been examined. The aim of our studies was to determine the effects of the pmp22 mutation inTrJ mice and to understand how alterations in PMP22 expression lead to the demyelinating phenotype with particular emphasis on post-transcriptional events. In these studies, we found low levels of complex glycosylated PMP22 and an abundance of lysosomal constituents, containing PMP22 immunoreactivity, in TrJ Schwann cells. Our results indicate that PMP22 is transported beyond the endoplasmic reticulum (ER) and transverses the Golgi apparatus, followed by accumulation in lysosomes.
MATERIALS AND METHODS
Heterozygous TrJ animals in the C57BL/6J background were obtained from Jackson Laboratories (Bar Harbor, ME) and were used to establish a breeding colony at Stanford University.
Genotyping. Genomic DNA was isolated from the tails of newborn mouse pups, and a 103 bp fragment of the pmp22 gene containing the mutated region was amplified using PCR (forward primer 5′-GATCCCGAGCCCAACTC, reverse primer 5′-CTGACGATGGTGGAGAC). Reaction conditions were as follows: initial denaturation at 97°C for 1 min, followed by three cycles of 65°C for 1 min, 72°C for 40 sec, and 97°C for 30 sec. During a 4 min time delay period at 72°C, an additional 2.5 U of AmpliTaq DNA polymerase (Perkin-Elmer, Norwalk, CT) was added to each sample. Next, 30 cycles at 94°C for 30 sec, 60°C for 1 min, and 72°C for 40 sec, followed by a final extension at 72°C for 10 min, were completed in a Perkin-Elmer Cetus DNA Thermal Cycler. An aliquot of each PCR product was incubated withBanI restriction enzyme (New England Biolabs, Beverly, MA) for 90 min at 37°C. Control and enzyme-treated samples were analyzed on 6% acrylamide gels using a standard Tris-borate-EDTA buffer system, and DNA bands were visualized by ethidium bromide staining.
Morphological studies. For the morphological studies, all reagents were obtained from Electron Microscopy Sciences (Fort Washington, PA). Sciatic nerves were collected from P10 and adult genotyped animals. Samples were fixed by immersion in ice-cold 2% glutaraldehyde/2% paraformaldehyde in 0.1 m sodium cacodylate buffer, pH 7.4, overnight at 4°C. After five 10 min rinses in PBS, nerves were infiltrated with 80 mm sucrose containing PBS and processed for standard Epon embedding. Plastic sections (0.5 mm) were stained with toluidine blue enhanced with silver. Measurements were performed on images captured with a Cohu CCD camera (Model 4915-4000) digitized through a Scion VG5 frame grabber onto a Macintosh computer running the public domain National Institutes of Health Image program (v1.59, available through the Internet athttp://rsb.info.nih.gov/nih-image/, developed at NIH). Statistics were performed by two-tailed ANOVA using the SPSS software package.
Immunocytochemical studies. Sciatic nerves were dissected out from genotyped animals at P10, P18, and adult. Samples were frozen by immersion in freezing N-methyl butane (isopentane). Frozen sections (10 μm thick) were dried for 1 hr on Superfrost/Plus microslides (Fisher, Pittsburgh, PA) followed by fixation with 1% paraformaldehyde in 90% ethanol for 2 min at room temperature. Samples were blocked by incubation in PBS containing 20% normal goat serum for at least 30 min. Primary antibodies (see below) were added in the same blocking solution overnight at 4°C. After four 10 min rinses in PBS, the sections were incubated with anti-rat IgG FITC conjugate (Boehringer Mannheim, Indianapolis, IN) or anti-rabbit IgG Texas red conjugate (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1–2 hr. Hoechst dye #33258 (Polysciences, Warrington, PA) was included in the secondary antibody solution at 0.5 μg/ml. Samples were mounted with Citifluor (University of Kent, Canterbury, UK). Slides were examined and photographed on a Nikon Microphot FXA microscope.
Primary antibodies. Rabbit polyclonal anti-PMP22 at 1:250, anti-P0 (the gift of Dr. M. T. Filbin, Hunter College, New York) at 1:2000, anti-MAG (the gift of Dr. M. B.Tropak, S. Lunenfeld Research Institute, Toronto, Canada) at 1:1000, and anti-MBP (the gift of Dr. A. Campagnoni, University of California Los Angeles) at 1:2000; and rat monoclonal anti-ER (clone H-69, Developmental Studies Hybridoma Bank, Iowa City, IA) at 12 μg/ml and anti-lysosome associated membrane protein (LAMP1) (clone 1D4B, Developmental Studies Hybridoma Bank, Iowa City, IA) at 13 μg/ml were used for the immunocytochemical studies.
Deglycosylation and Western analysis. Sciatic nerves were collected from genotyped 18-d-old and adult animals and were frozen immediately in liquid nitrogen. Nerves pooled from two to four animals were pulverized under liquid nitrogen, and total tissue homogenates were prepared in SDS gel sample buffer (62.5 mmTris, pH 6.8, 10% glycerol, 3% SDS) or in denaturation buffer (0.5% SDS, 1% β-mercaptoethanol), supplied with the endoglycosidases, for the deglycosylation studies. Protein concentrations were determined using BCA reagents (Pierce Chemicals, Rockford, IL) or the Lowry method. Endoglycosidase digestions with N-glycosidase F (PNGase F) and endoglycosidase H (endoH), both from New England Biolabs, were performed according to the manufacturer’s suggestions, for 16 hr at 37°C. Protein samples were separated on 12.5% SDS gels under reducing conditions, and gels were transferred to nitrocellulose membranes. The same polyclonal antibodies as above, anti-PMP22 at 1:1000, anti-MBP at 1:4000, anti-P0 at 1:4000, anti-MAG at 1:1000, and anti-cathepsin D (CatD), purified rabbit IgG (Cortex Biochem, San Leandro, CA) at 1:100, and rat monoclonal anti-LAMP1 undiluted supernatant were used. Bound antibodies were detected using the ECL chemiluminescent detection method (Amersham Corporation, Arlington Heights, IL).
Chloroquine treatment. To prevent lysosomal degradation, excised normal and TrJ sciatic nerves were incubated in DMEM, 10% fetal calf serum, and 100 ng/ml nerve growth factor, with and without the addition of 100 μmchloroquine (Calbiochem, La Jolla, CA), overnight at 37°C. Samples were quickly frozen and processed for immunocytochemical studies and Western analysis as described above.
RESULTS
Myelinating homozygous and heterozygousTrJ nerves show evidence for primary dysmyelination
Now that the gene defect in theTrJ neuropathy has been identified as a mutation in pmp22, a peripheral nerve myelin protein, we reanalyzed the morphology of the peripheral nerves inTrJ mice for clues as to whether the mutation primarily affects general Schwann cell function, myelin formation, or myelin stability. At the DNA level, the thymidine-to-cytosine transition mutation at position +90 in theTrJ mouse pmp22 sequence introduces a novel BanI restriction site (Fig.1A). We genotyped all of the mice used in this study by PCR to amplify a 103 bp region of the mousepmp22 gene containing the mutated site, followed byBanI restriction digestion and electrophoresis (Fig.1B). Previous studies ofTrJ/+ and putativeTrJ/TrJsciatic nerves showed increased amounts of endoneurial tissue, abnormally thin myelin, increased number of Schwann cell nuclei, and irregular axonal contours in affected animals (Henry et al., 1983). We have reconfirmed many of these findings on genotyped animals. Epon-embedded sciatic nerve samples were stained with toluidine blue enhanced with silver to evaluate the extent of myelination (Fig.2). In agreement with the findings of Henry et al. (1983), we found differences in the abundance and thickness of myelin, the diameter of the axons, increased amount of extracellular matrix, and increased number of Schwann cells per unit area of nerve. Furthermore, we also found a low frequency of abnormally thick myelin (tomacula) and myelin fragmentation, the latter indicating active myelin breakdown, in sciatic nerves of 10-d-oldTrJ/+ mice (Fig.2F).
Fig. 1.

Identification of theTrJ genotype. TheTrJ mutation introduces a novelBanI restriction site in the pmp22sequence (A). A 103 bp fragment of thepmp22 gene, including the mutated site, was amplified by PCR. BanI restriction enzyme untreated (−) and treated (+) samples were analyzed on an ethidium bromide-stained 6% acrylamide gel (B). Homozygous (TrJ/TrJ) and heterozygous (TrJ/+)TrJ animals are identified by the cleavage of the 103 bp PCR-amplified fragment into two smaller pieces (70 and 33 bp). The numbers on the left of the gel indicate DNA fragment size in base pairs.
Fig. 2.
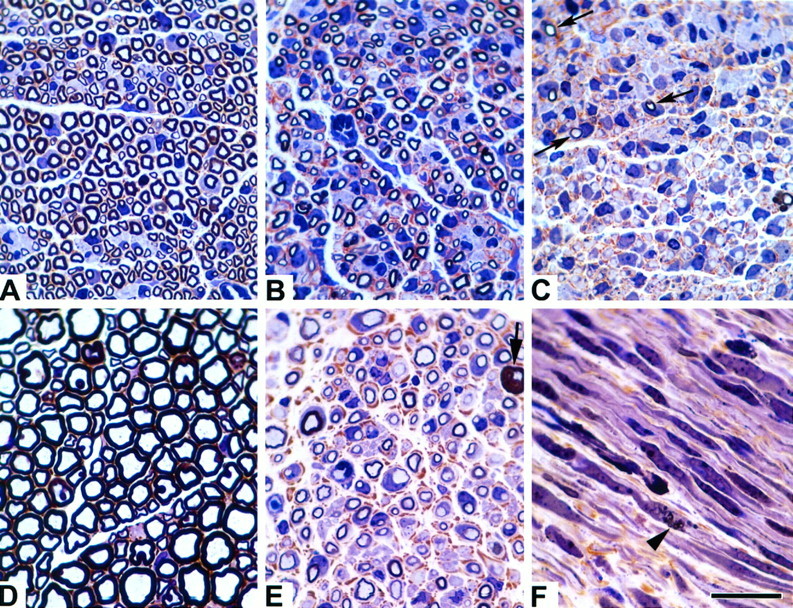
Morphological studies of normal andTrJ sciatic nerves. Microscopic sections at equivalent magnifications from normal (A,D), heterozygous TrJ(B, E, F), and homozygous TrJ(C) sciatic nerves from 10-d-old (A–C, F) and adult (D, E) mice are shown. Note the marked increase in axon caliber and myelin thickness during nerve development between normal (A, D) andTrJ/+ mice (B,E). Also note the increasing severity of dysmyelination, even at 10 d of age between the +/+,TrJ/+, andTrJ/TrJnerves (A–C, respectively). Homozygous TrJ have only small amounts of myelin (C, arrows). AdultTrJ/+ animals have increased endoneurial connective tissue (E, brown material) and occasional tomaculae are observed (bold arrow). Myelinic debris (arrowhead) can be found in longitudinal sections from 10-d-old TrJ/+ animals (F). Scale bar, 20 μm.
These morphological studies showing only minor differences between 10-d-old +/+ and TrJ/+ nerves, and marked differences in older animals, suggest that the heterozygousTrJ neuropathy is progressive. In agreement with the histological findings, +/+ andTrJ/+ animals are behaviorally indistinguishable at young age (until 3–4 weeks of age), whereasTrJ/+ animals display noticeable tremors in adulthood. The neuropathy often progresses to complete paralysis of the hind legs in older animals. In comparison,TrJ/TrJpups display tremors, problems with movements soon after birth (P5–P7), remain smaller in size, and generally die at ∼P18–P22 (weaning age), possibly because of inability to obtain proper nutritional support.
Although the 10-d-old +/+ and TrJ/+ nerves appear similar, there are significant differences between the ratios of the fiber (axon and myelin together) to axon diameters (referred as g ratios: +/+, 0.47 ± 0.09;TrJ/+, 0.55 ± 0.11;p < 0.001, ratios calculated from all myelinated axons in representative fields on at least 50 fibers) (compare Fig. 2,A and B). Because the overall axon diameters are similar between +/+ and TrJ/+ animals at this age (average axon diameters: +/+, 1.41 ± 0.49 μm;TrJ/+, 1.39 ± 0.37 μm), the differences in the g ratios indicate a significant decrease in the thickness of myelin in TrJ/+ animals, compared with normal littermates as early as P10. In agreement with previous investigators, the increased amount of extracellular connective tissue (stained brown; compare Fig. 2, A andB) and increased number of nuclei per cross-section are already evident in affected nerves of 10-d-old mice (TrJ/+ andTrJ/TrJhave approximately twofold more nuclei per cross-section than littermate controls). Adult TrJ/+ animals show severe signs of demyelination (Fig. 2E), and when compared with the normal adult nerve, only a few of the axons are surrounded by thick myelin (compare Figs. 2, D andE, and 3). Taken together, these data suggest that the heterozygous TrJ phenotype is expressed during early myelin formation and appears more severe in older animals because of an arrest in the size of the axon–Schwann cell units. However, even when normalized for axon size, scatterplots demonstrate that axons from TrJ/+ mice have less myelin than littermate controls, at least for axon diameters >1 mm (Fig. 3).
Fig. 3.

Scatterplot analysis of 10-d-old and adult nerves. Plots of fiber diameter (including axon and myelin) versus axon diameter for 10-d-old (A) and adult (B) wild-type (+) and TrJ/+ (open circles) nerves reveal a marked tendency for theTrJ/+ myelinated fibers to have thinner myelin sheaths (fiber diameter) for a given axon size. The best-fit linear regression lines are shown for comparison purposes and do not necessarily imply a linear relationship between the two parameters.
PMP22 protein levels are reduced out of proportion to other myelin proteins in the TrJneuropathy
The abnormally thin myelin observed in younger animals and the nonconservative nature of the TrJmutation are consistent with the notion that the mutated allele either interferes with myelin formation or enhances myelin degradation through an intracellular mechanism. As a first step in evaluating the intracellular processing of PMP22, we performed immunoblots to compare the levels of PMP22 protein expression with those of other known peripheral myelin protein constituents. Accordingly, total sciatic nerve lysates of P18 and adult mice were analyzed for the expression of MAG, P0, MBP, and PMP22, as shown in Figure 4. As expected from the pathology, severely affectedTrJ/TrJnerves contained nearly undetectable amounts of P0 and MBP and essentially no anti-PMP22 immunoreactive protein even when up to 100 μg of nerve lysate protein was analyzed. In adult heterozygous animals, the levels of P0 and MBP, the major structural proteins of PNS myelin, were severely reduced. We found PMP22 protein levels similarly decreased. In comparison, we did not detect a significant alteration in the amount of MAG in TrJ/+ mice between P18 and adulthood. Similar to the TrJ/+ condition, MAG levels inTrJ/TrJanimals remained approximately normal; however, a low molecular weight form of the molecule, possibly resulting from altered glycosylation (Johnson et al., 1989; our unpublished observations), was also detected. We found no evidence of a higher molecular weight form of MAG in any of the nerve lysates, as has been described previously inTr animals (Inuzuka et al., 1985). The relative differences in P0 and MBP protein levels between the +/+ andTrJ/+ animals are much less pronounced at P18. In contrast, PMP22 protein expression is apparently reduced in P18 heterozygotes compared with normals.
Fig. 4.

PMP22, P0, and MBP protein levels are reduced significantly in TrJ nerves, whereas MAG remains normal. Total sciatic nerve homogenates of 18-d-old (P 18) wild-type (+/+), heterozygous (−/+), homozygous (−/−), and adult wild-type (+/+) and heterozygous (−/+) mice were analyzed for the expression of four different myelin proteins, MAG, P0, PMP22, and MBP. All lanes contain 25 μg of total protein. Molecular weights are indicated in kilodaltons.
Most of the steady-state PMP22 protein undergoes complex glycosylation in heterozygous TrJnerves
We investigated the acquisition of PMP22 glycosylation inTrJ/+ nerves to determine whether we could detect differences in the intracellular processing of the PMP22 protein in affected animals. Previous pulse-chase studies have demonstrated that PMP22 is synthesized as a core 18 kDa protein and then progressively modified by glycosylation to a 22 kDa high-mannose form (endoH-sensitive) and finally to a 22 kDa complex form (endoH-resistant) (Pareek et al., 1993). Thus, we compared the glycosylation profile of the protein in 18-d-old and adult normal andTrJ/+ mice (Fig. 5) as a marker for the extent of its intracellular processing through the Golgi network. As expected, PNGase F digestion revealed that the size of the core peptide is approximately the same in all samples (18 kDa). During normal peripheral nerve development, a greater fraction of the total PMP22 becomes endoH-resistant; however, a significant endoH-sensitive pool remains, even in normal adult nerves, probably representing an ER and/or Golgi pool of newly synthesized PMP22. At P18, the ratio of endoH-sensitive to endoH-resistant protein is similar in +/+ andTrJ/+ nerves. In adultTrJ/+ nerves, however, PMP22 almost exclusively contains complex-type sugars. As a control, anti-P0 immunoblots of identical samples revealed a similar but less pronounced shift of the P0 protein toward complex glycosylation in adultTrJ/+ nerves (Fig. 5). This result is in agreement with the accumulation of P0 in Golgi-associated vesicles in TrJ/+ nerves (Heath et al., 1991). Because of the low levels of PMP22 inTrJ/TrJanimals, similar Western blot studies on the carbohydrate content of the protein could not be performed. The lack of detectable levels of endoH sensitive PMP22 in adult TrJ/+ nerves suggests that at steady-state, most of PMP22 is transported and processed through the trans-Golgi network inTrJ/+ nerves without accumulating in the ER or the early Golgi network. However, these data alone do not rule out the possibility that the endoH-resistant PMP22 represents only the product of the normal allele.
Fig. 5.

Increased proportion of PMP22 and P0 is endoH-resistant in TrJ/+ nerves. Sciatic nerve lysates of 18-d-old and adult, wild-type (+/+), and heterozygous TrJ (−/+) mice were treated with PNGase F (N) or endoH (H) and immunoblotted with anti-PMP22 and anti-P0 antibodies. Samples incubated without the addition of enzyme are analyzed in lanes C. Top open arrowsindicate endoH-resistant proteins, and bottom arrowsshow the migration position of the deglycosylated protein. Lanes for the wild-type samples (+/+) contain 25 μg of total protein, and lanes for the heterozygous (−/+) samples contain 75 μg of total protein. Molecular weights are indicated in kilodaltons.
Immunoreactive PMP22 protein accumulates in the cytoplasm ofTrJ/+ Schwann cells where it co-localizes, in part, with lysosomal markers
To follow further the processing of PMP22 inTrJ/+ animals, we used immunostaining of frozen nerve sections with polyclonal anti-PMP22 antibodies. In the normal peripheral nerve, PMP22 is found distributed uniformly in compact myelin and is absent from the nodes of Ranvier (Fig.6A) (see also Snipes and Suter, 1995). In contrast, nerve sections of adultTrJ/+ mice show low levels of myelin-like anti-PMP22 immunoreactivity, whereas a significant proportion of the PMP22 protein appears to accumulate in some of the Schwann cell perikarya (Fig. 6D,arrowheads). We interpret the myelin-like immunoreactivity as representing both myelin and cytoplasmic staining of PMP22. In longitudinal sections, the Schwann cells with cytoplasmic immunoreactive PMP22 from the TrJ/+ nerves can be distinguished morphologically by their rounded cell bodies and nuclei (Fig. 6F) from the Schwann cells of normal mice, which have elongated cell bodies and nuclei (Fig.6C). Comparison of the nerve sections stained with the nuclear Hoechst dye also reveals the increased density of nuclei inTrJ/+ (Fig.6F), compared with +/+ mice (Fig.6C) (see above). In some nerve sections of 10- and 18-d-oldTrJ/+ mice, we were able to detect PMP22-like immunoreactivity in myelin sheaths resembling normal nerve (Fig. 6G). Nonetheless, the staining intensity was usually reduced, compared with wild type, probably reflecting the thin myelin and decreased density of myelinated fibers as noted in the resin sections. Similar, but less abundant, myelin-like profiles were observed in adult TrJ/+ nerves (Fig.2). InTrJ/TrJnerves, we could not detect anti-PMP22 immunoreactive myelin (data not shown).
Fig. 6.
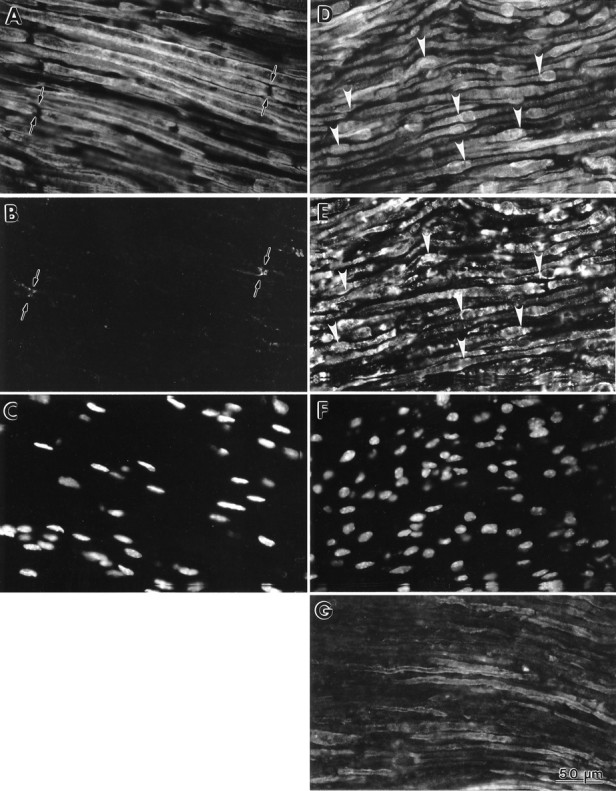
Co-localization of PMP22 and LAMP1 inTrJ Schwann cells. Fresh-frozen sections of adult normal (A–C) andTrJ/+ (D–F) sciatic nerves were double-immunolabeled with polyclonal anti-PMP22 (A,D) and monoclonal LAMP1 (B,E) antibodies. Nuclei were stained with Hoechst dye (C, F). In the normal nerve, PMP22 is found in myelin (A) and is absent from the nodes of Ranvier (A, arrows). In comparison, PMP22 accumulates in the cell bodies of some of the Schwann cells inTrJ/+ nerve (D,arrowheads). A low level of LAMP1-like immunoreactivity is found in the normal nerve (B, arrows), which is markedly increased in theTrJ/+ nerve (E). Several Schwann cells are identified in theTrJ/+ sample and show co-localization of PMP22 and LAMP1 (arrowheads in D andE, respectively). In 10-d-old heterozygousTrJ nerve, PMP22-like immunoreactivity can be detected in the Schwann cell membrane (G). Scale bar (shown in G): 50 μm.
Intracellular accumulation of PMP22 inTrJ/+ Schwann cells is suggestive of altered protein processing, transport, and possibly, degradation. Therefore, we performed double-immunolabeling with rat monoclonal organelle markers for ER and lysosomes. These studies revealed co-localization of PMP22 with LAMP1 (Fig. 6E), but not with the ER marker (clone H-69) (see Materials and Methods) (data not shown). There is a dramatic increase in the abundance of LAMP1 immunoreactive lysosomal structures inTrJ/+ nerves (Fig.6E), compared with normals (Fig.6B). In the normal nerve, very low levels of LAMP1-like immunoreactivity were detected, usually concentrated at paranodal regions (Fig. 6B). Paranodal distribution of lysosomal constituents in normal myelinated nerves has been described previously (Hildebrand et al., 1992). Inhibition of lysosomal degradation by chloroquine treatment before immunostaining augmented the intracellular staining of PMP22 inTrJ/+ nerves (data not shown) (see also Fig. 9).
Fig. 9.

Chloroquine treatment enhances PMP22 protein levels in TrJ/+ nerves. Chloroquine-untreated (−) and chloroquine-treated (+) sciatic nerve explants of adult wild-type (+/+) andTrJ/+ mice were analyzed for the levels of MAG, P0, PMP22, and MBP (A). All lanes contain 25 μg of total protein. Molecular weights are indicated in kilodaltons.
We also examined the anatomical distribution of three other myelin constituents, MAG, P0 and MBP, to determine whether their expression pattern was altered. High levels of mouse Igs inTrJ nerves (our unpublished observations) prevented double-immunolabeling studies with polyclonal anti-PMP22 antiserum and mouse origin monoclonal anti-myelin protein antibodies. In adjacent sections, however, using polyclonal anti-myelin protein antibodies, we found that the normal distribution of MAG is highly altered in TrJ/+ andTrJ/TrJnerves. In the normal nerve, MAG is found in the periaxonal region of the myelin sheath and in areas of uncompacted myelin, such as Schmidt–Lanterman incisures and the paranodes (Fig.7A) (see also Martini and Schachner, 1986, and references therein). As with PMP22, significant levels of MAG (Fig.7B) and LAMP1 immunoreactivity (Fig. 7C) co-localize to intracellular Schwann cell compartments inTrJ/+ (Fig.7B,C, arrowheads) andTrJ/TrJnerves (data not shown). In 10-d-oldTrJ/+ mice, some punctate staining of paranodal structures was identified by their MAG immunoreactivity, which was absent from 10-d-old homozygous animals (data not shown). Compared with the normal nerve (Fig. 7D), the abundance of P0 (Fig. 7E) and MBP (data not shown) reactive myelin segments was severely reduced in adultTrJ/+ nerves. Nevertheless, we were able to detect both P0 and MBP apparently at or near the surface of Schwann cells. Low levels of intracellular P0-like immunoreactivity were also found in some of the TrJ/+ Schwann cells of the adult nerve (Fig. 7E), in agreement with the findings of Heath et al. (1991). In contrast to P0, MAG, and PMP22, we did not detect intracellular accumulation of MBP (data not shown).
Fig. 7.
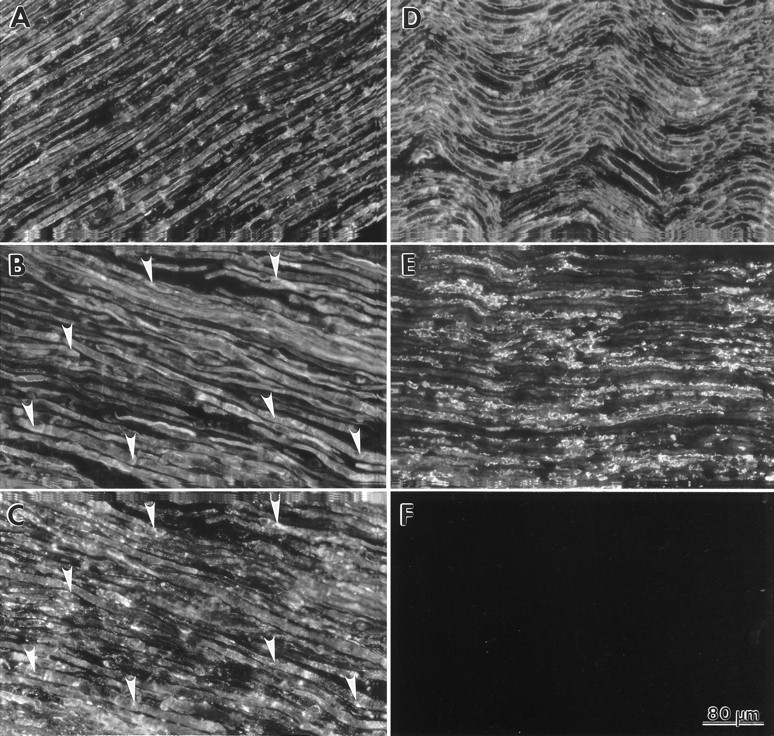
The TrJ mutation alters the normal distribution pattern of MAG and P0. Fresh-frozen sections of adult normal (A, D) andTrJ/+ (B,C, E) sciatic nerves were examined for the distribution of MAG (A, B) and P0 (D, E). The normal localization pattern of MAG is shown in A. Intracellular MAG-like immunoreactivity (B, arrowheads) is co-localized with LAMP1 (C, arrowheads) in TrJ/+ nerves. In the normal nerve, P0 is found in compact myelin (D). Reduced levels of myelin-like P0 immunoreactivity are present inTrJ/+ nerves (E). A nerve sample processed without primary antibody incubation is shown inF. Scale bar (shown in F): 80 μm.
Components of the endosomal-lysosomal pathway are upregulated in heterozygous TrJ/+ nerves
The co-localization of PMP22 with LAMP1 prompted us to investigate whether the lysosomal pathway is activated inTrJ/+ Schwann cells. We used Western blots to examine nerve lysates (aliquots of the lysates that had been probed for myelin proteins in Fig. 4) for the abundance of LAMP1 and CatD (Fig. 8). LAMP1 is a structural component of lysosomes that has been used as a marker for lysosomal vesicles (Chen et al., 1985), whereas CatD levels are taken as indicators of lysosomal enzyme activity (Cataldo et al., 1995). Abnormally high levels of LAMP1 and CatD are present in TrJ/+ nerves (Fig. 8), suggesting a generalized increase in protein degradation. Particularly striking is the marked increase in LAMP1 levels inTrJ/+, compared with adult normal nerves. As our morphological studies suggest (Fig.2F), there is an active myelin breakdown inTrJ/+ nerves. The parallel decrease in structural myelin components (P0, MBP, and PMP22) and increase in lysosomal constituents were reconfirmed by reprobing the same blots with all six different antibodies (MAG, P0, PMP22, MBP, LAMP, and CatD).
Fig. 8.

Upregulation of the lysosomal pathway inTrJ/+ mice. Total sciatic nerve homogenates of 18-d-old (P 18) wild-type (+/+), heterozygous (+/−), homozygous (−/−), and adult wild-type (+/+) and heterozygous (+/−) mice were analyzed for the expression of LAMP1 and protein hydrolase CatD. Arrows point to LAMP1 and the 48 kDa precursor and 34 kDa mature forms of CatD. The open arrow indicates a nonspecific band that is immunoreactive against the polyclonal anti-CatD antibody. All lanes contain 25 μg of total protein. Molecular weights are indicated in kilodaltons.
To correlate the intracellular localization of PMP22, MAG, and P0 with the enhanced lysosomal activity and apparent myelin instability inTrJ/+ nerves, chloroquine-treatedex vivo nerve samples were analyzed (Fig. 9). Chloroquine, a weak base, is thought to inhibit lysosomal enzyme activity by increasing the pH of the acidic vesicles leading to the accumulation of proteins that are destined for degradation (Brown et al., 1984). After a 16 hr chloroquine treatment, we found a notable accumulation of PMP22, P0, and MBP inTrJ/+ nerves (Fig. 9A). Compared with these compact myelin proteins, MAG levels were less affected. We detected only a small increase in the levels of the same proteins in chloroquine-treated normal nerves, likely reflecting general protein degradation secondary to early Wallerian degeneration during the overnight incubation period. Chloroquine sensitivity of PMP22, P0, and MBP degradation further supports increased endocytosis/autophagy in TrJ/+ nerves.
DISCUSSION
Recent and emerging studies reveal a variety of pathogenetic mechanisms by which mutations in myelin genes can cause hereditary myelinopathies. In humans, a large proportion of patients with CMT and hereditary neuropathy with liability to pressure palsies are caused by altered PMP22 gene dosage resulting from a duplication or deletion, respectively, of a submicroscopic portion of chromosome 17 containing the human PMP22 gene (see Suter and Snipes, 1995a). Although controversial, the altered PMP22 gene dosage in CMT1A appears to be reflected by increased levels of PMP22 mRNA in Schwann cells (Yoshikawa, 1994) and PMP22 protein in peripheral myelin (Vallat, 1996; see also Hanemann, 1994). In mouse models alone, altered dosage of normal myelin genes causing myelination defects has now been described for the major compact myelin proteins in both the CNS and PNS, namely for MBP (Shine et al., 1992), P0 (Giese et al., 1992;Martini et al., 1995), PLP (Nave, 1994), and PMP22 (Adlkofer et al., 1996; Magyar et al., 1996; Sereda et al., 1996). In addition, apparent gain of function, as well as dominant negative mutations, has been identified among this group of proteins (Warner et al., 1996). It is within this background that we discuss the effects of the L16P mutation in the first putative transmembrane domain of the PMP22 protein in theTrJ mouse.
Since the original description of the Tr andTrJ mice, these animals have been used as models of human hereditary peripheral neuropathies (Nave, 1994). We have begun to address the cellular pathogenesis of the PMP22 myelinopathies by focusing on the TrJneuropathy. Our studies confirmed and extended earlier morphological (Henry et al., 1983) and immunochemical (Heath et al., 1991) findings in TrJ mice. Data indicate an increasing disparity between normal and diseased nerves in theTrJ neuropathy with advancing age, similar to the disease progression in humans with CMT1A and the Tr mouse (Ayers et al., 1976; Low, 1977). We show that theTrJ neuropathy is manifest as a primary dysmyelination early in myelin formation. Studies on children with CMT1A also reveal heterogeneous nonprogressive slowing of nerve conduction from early childhood consistent with a dysmyelinating process with full genetic penetrance (Nicholson, 1991; Kaku et al., 1993a,b). The TrJ and CMT1A clinical phenotypes are similar; namely, both are progressive sensorimotor neuropathies, although their pathologies and axon–myelin thickness relationships differ (Gabreels-Festen, 1995). Thus, although there are many similarities between the TrJneuropathy caused by a point mutation and CMT1A attributable toPMP22 duplication, there are likely to be fundamental pathogenetic similarities and differences that remain to be elucidated.
The major finding in this paper is that the endosomal/lysosomal pathways are activated in the TrJneuropathy and that PMP22 and other myelin proteins are degraded at a high rate in TrJ/+ nerves. There are several cellular mechanisms by which a mutant transmembrane glycoprotein may be degraded. First, if the mutation interferes with the normal folding or oligomerization of the protein, then it is retained and degraded in the ER (Bonifacio and Lippincott-Schwartz, 1991). A second possible pathway involves the direct transport of the glycosylated mutant protein from the trans-Golgi network to endosomal/lysosomal vesicles. This pathway has been described for the transport of lysosomal proteins via specific targeting sequences (Green et al., 1987; Kornfeld and Mellman, 1989), but has not yet been implicated in the elimination of mutant proteins destined for the plasma membrane. Third, the mutant protein could translocate to the plasma membrane, where its insertion may be unstable. Autophagy of the mutant protein (or abnormal membranes) would lead to degradation by the lysosomal pathway.
Based on recent in vitro studies of the Trmutation in pmp22 (Naef et al., 1997) and several proteolipid protein mutations (Gow et al., 1994), we had reason to expect that the nonconservative L16P mutation in PMP22 underlying theTrJ neuropathy would result in its ER retention and degradation. In these mutations, the abnormal protein is apparently recognized as defective and is retained in the ER. Degradation of misfolded or incorrectly oligomerized proteins from the ER is a commonly used pathway (Bonifacio and Lippincott-Schwartz, 1991) for ensuring the integrity of protein synthesis. As will be discussed below, we find no evidence that the effect of theTrJ mutation is manifest at the level of the ER. That not all mutations are detected by ER fail-safe mechanisms is clearly shown by studies of mutations in transmembrane proteins underlying other human diseases (for review, see Amara et al., 1992). For example, depending on the specific mutation in the rhodopsin molecule, the mutant protein may be retained in the ER or may be transported to the plasma membrane (Sung et al., 1991). We hypothesize that the differences observed between Tr andTrJ mice with regards to the intracellular processing of PMP22 reflect a similar pleiotropism. Alternative explanations, however, including species differences attributable to distinct genetic backgrounds and methodological differences, will need to be explored further.
Although the products of the normal and the mutated pmp22alleles were not differentiated in heterozygousTrJ/+ mice, we do not believe that the mutated PMP22 in TrJ/+ animals is selectively eliminated by ER retention and degradation for several reasons. First, our immunohistochemical results co-localizing most of the intracellular PMP22 in TrJ/+ nerves with lysosomal markers, and not ER markers, argue against a mutation-specific ER retention. This histochemical localization is consistent with our inability to detect a pool of ER- and/or Golgi-resident endoH-sensitive PMP22 inTrJ/+ nerves, even though this pool is readily visible in normal nerves. One interpretation of this finding is that newly glycosylated PMP22 is used more rapidly inTrJ/+ nerves than in normal nerves, reflecting increased myelin turnover. Furthermore, simply removing the mutated PMP22 protein from the ER (assuming no other effects of the mutated protein) would lead to a 50% reduction in functional protein, which should result in the phenotype of heterozygous PMP22 knock-out (PMP220/+) mice. PMP220/+ mice display prominent tomacula at a young age (Adlkofer et al., 1995). In contrast, we and others (Henry et al., 1983) find only a few of these structures in TrJ/+ nerves. Moreover, homozygouspmp22 knock-out mice live past 20 weeks of age, whereasTrJ/TrJanimals die at P18, again indicating either a gain of function or a dominant-negative effect for the TrJprotein. The effects of different genetic backgrounds in these examples are unlikely to be significant, because both PMP220/+ andTrJ/+ mice appear to faithfully reflect their human disease counterparts, hereditary neuropathy with liability to pressure palsies (Adlkofer et al., 1995) and variant CMT1A (Valentijn et al., 1992). Finally, ER retention and degradation of mutated PMP22 would not explain the accumulation of PMP22, MBP, and P0 in endosomes/lysosomes after chloroquine treatment.
It is difficult to distinguish from the data presented here whether there is direct transport of mutated PMP22 from the Golgi network to lysosomes, or whether the mutation leads to increased instability of myelin, without determining if the mutated PMP22 protein is incorporated into myelin. A specific shunting of trans-Golgi mutant PMP22 resulting in a 50% reduction of PMP22 is unlikely based on the observed differences between PMP220/+ andTrJ mice. Furthermore, if the mutated PMP22 is shunted directly to endosomes/lysosomes without being incorporated into myelin, it would have to be associated with other myelin proteins, perhaps as “premyelin” vesicles, to account for the increased accumulation with P0 and MBP in endosomes/lysosomes after chloroquine treatment. The possibility of an effect on “premyelin” vesicles seems particularly unlikely for MBP, a cytoplasmic protein synthesized on free polyribosomes in close proximity to myelin assembly (for review, see Campagnoni and Macklin, 1988). Ultimately though, this putative pathway fails to explain why the leftover wild-type PMP22 and the other compact myelin proteins cannot support the formation of normal myelin over a longer period of time.
The possibility that the L16P PMP22 mutation might behave either as a dominant negative mutation by disrupting the interaction of PMP22 with itself or other myelin proteins, or as a gain of function mutation by sequestering interacting proteins or overwhelming degradation pathways during biosynthesis and processing or within the myelin membranes, cannot be readily dismissed. The altered expression pattern and intracellular detection of P0 and MAG inTrJ/+ nerves may indicate an interaction of PMP22 with MAG and/or P0. These transmembrane myelin proteins carry adhesion motifs and are thought to have a role in PNS myelin formation and maintenance (for review, see Snipes and Suter, 1995). In vivo homotypic and/or heterotypic interaction and likely multimerization of myelin proteins have been proposed but has only been documented in vitro (D’Urso et al., 1990; Sinoway et al., 1994; Jung et al., 1995). These protein interactions may take place during intracellular trafficking, such as in the case of PLP and DM20 (Sinoway et al., 1994) and/or in the plasma membrane (D’Urso et al., 1990; Shapiro et al., 1996). Our morphological and biochemical studies suggest that the TrJ mutation in PMP22 leads to altered distribution of both MAG and P0. We and others (Heath et al., 1991) have found abnormal intracellular accumulation of PMP22, P0, and MAG inTrJ/+ Schwann cells either in Golgi-associated vesicles or associated with LAMP1-positive structures, suggesting that if these dominant-negative or gain-of-function mechanisms are operative, they affect post-Golgi compartments.
In conclusion, our results are most consistent with the possibility that the effect of the L16P mutation in PMP22 is to increase myelin instability in TrJ/+ nerves. Augmented myelin breakdown in excess of myelin synthesis would account for the morphological and biochemical hypomyelination observed in theTrJ/+ mouse at all ages. This hypothesis is supported additionally by our observation that PMP22 protein in TrJ/+ Schwann cells is found in abnormally large quantities in the cytoplasm, apparently co-localizing with the lysosomal marker LAMP1. In addition, LAMP1 and CatD protein levels are upregulated, whereas the steady-state levels of PMP22, MBP, and P0 proteins are reduced. We hypothesize that the mutantTrJ PMP22 is normally glycosylated and is incorporated into myelin along with MBP and P0. The myelin containing these three proteins then becomes unstable and undergoes autophagy and/or endocytosis by the Schwann cells that degrade the myelin proteins and recycle the lipids through the endosomal/lysosomal pathway which is upregulated in the TrJneuropathy. The final proof of this hypothesis will await studies that examine directly whether PMP22 carrying the L16P mutation is incorporated into myelin and direct measurements of myelin turnover inTrJ/+ nerves. Although we have outlined significant differences between the TrJneuropathy and CMT1A, it will be of considerable interest to determine whether CMT1A also shows evidence for increased myelin turnover as a final common pathway for one class of myelin disorders.
Footnotes
This study was supported by National Institutes of Health (NIH) Grant NS 09694–01 and National Multiple Sclerosis Society Grant FG 1120-A-1 and the Giannini Foundation (L.N.); NIH Grant NS04270 and American Paralysis Association Grant SBR1–9501 and the Muscular Dystrophy Association (E.M.S.); and the Medical Research Council of Canada (G.J.S.). We wish to thank Dr. Ueli Suter for helpful discussions during the course of these studies and Martha Hernandez for the assistance with the chloroquine experiments.
Correspondence should be addressed to Dr. Eric M. Shooter, Department of Neurobiology, Stanford University School of Medicine, Stanford, CA 94305.
REFERENCES
- 1.Adlkofer K, Martini R, Aguzzi A, Zielasek J, Toyka KV, Suter U. Hypermyelination and demyelinating peripheral neuropathy in Pmp22-deficient mice. Nat Genet. 1995;11:274–280. doi: 10.1038/ng1195-274. [DOI] [PubMed] [Google Scholar]
- 2.Amara JF, Cheng SH, Smith AE. Intracellular protein trafficking defects in human disease. Trends Cell Biol. 1992;2:145–149. doi: 10.1016/0962-8924(92)90101-r. [DOI] [PubMed] [Google Scholar]
- 3.Ayers MM, Anderson RM. Development of onion bulb neuropathy in the trembler mouse. Acta Neuropathol. 1976;36:137–152. doi: 10.1007/BF00685276. [DOI] [PubMed] [Google Scholar]
- 4.Bascles L, Bonnet J, Garbay B. Expression of the PMP22 gene in trembler mutant mice: comparison with other myelin protein genes. Dev Neurosci. 1992;14:336–341. doi: 10.1159/000111680. [DOI] [PubMed] [Google Scholar]
- 5.Bonifacio JS, Lippincott-Schwartz J. Degradation of proteins within the endoplasmic reticulum. Curr Opin Cell Biol. 1991;3:592–600. doi: 10.1016/0955-0674(91)90028-w. [DOI] [PubMed] [Google Scholar]
- 6.Brown WJ, Constantinescu E, Farquhar MG. Redistribution of mannose-6-phosphate receptors induced by tunicamycin and chloroquine. J Cell Biol. 1984;99:320–326. doi: 10.1083/jcb.99.1.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campagnoni AT, Macklin WB. Cellular and molecular aspects of myelin protein gene expression. Mol Neurobiol. 1988;2:41–89. doi: 10.1007/BF02935632. [DOI] [PubMed] [Google Scholar]
- 8.Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Bursztajn S, Lippa C, Nixon RA. Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron. 1995;14:671–680. doi: 10.1016/0896-6273(95)90324-0. [DOI] [PubMed] [Google Scholar]
- 9.Chen JW, Murphy TL, Willingham MC, Pastan I, August JT. Identification of two lysosomal membrane glycoproteins. J Cell Biol. 1985;101:85–95. doi: 10.1083/jcb.101.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D’Urso D, Brophy PJ, Staugaitis SM, Gillespie CS, Frey AB, Stempak G, Colman DR. Protein zero of peripheral nerve myelin: biosynthesis, membrane insertion, and evidence for homotypic interaction. Neuron. 1990;2:449–460. doi: 10.1016/0896-6273(90)90057-m. [DOI] [PubMed] [Google Scholar]
- 11.Gabreels-Festen AA, Bolhuis PA, Hoogendijk JE, Valentijn LJ, Eshuis EJ, Gabreels FJ. Charcot-Marie-Tooth disease type 1A: morphological phenotype of the 17p duplication versus PMP22 point mutations. Acta Neuropathol. 1995;90:645–649. doi: 10.1007/BF00318579. [DOI] [PubMed] [Google Scholar]
- 12.Garbay B, Bonnet J. P0 protein in normal, trembler heterozygous/homozygous mice during active PNS myelination. NeuroReport. 1992;3:594–596. doi: 10.1097/00001756-199207000-00012. [DOI] [PubMed] [Google Scholar]
- 13.Giese KP, Martini R, Lemke G, Soriano P, Schachner M. Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, and degeneration of myelin and axons. Cell. 1992;71:565–576. doi: 10.1016/0092-8674(92)90591-y. [DOI] [PubMed] [Google Scholar]
- 14.Gow A, Friedrich VL, Jr, Lazzarini RA. Many naturally occurring mutations of myelin proteolipid protein impair its intracellular transport. J Neurosci Res. 1994;37:574–583. doi: 10.1002/jnr.490370504. [DOI] [PubMed] [Google Scholar]
- 15.Green SA, Zimmer K-P, Griffits G, Mellman I. Kinetics of intracellular transport and sorting of lysosomal membrane and plasma membrane proteins. Cell Biol. 1987;105:1227–1240. doi: 10.1083/jcb.105.3.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanemann CO, Stoll G, D’Urso D, Fricke W, Martin JJ, Van Broeckhoven C, Mancardi GL, Bartke I, Muller HW. Peripheral myelin protein-22 expression in Charcot-Marie-Tooth disease type 1a sural nerve biopsies. J Neurosci Res. 1994;37:654–659. doi: 10.1002/jnr.490370513. [DOI] [PubMed] [Google Scholar]
- 17.Heath JW, Inuzuka T, Quarles RH, Trapp BD. Distribution of P0 protein and the myelin-associated glycoprotein in peripheral nerves from Trembler mice. J Neurocytol. 1991;20:439–449. doi: 10.1007/BF01252272. [DOI] [PubMed] [Google Scholar]
- 18.Henry EW, Sidman RL. The murine mutation Trembler-J: proof of semidominant expression by use of the linked vestigial tail marker. J Neurogenet. 1983;1:39–52. doi: 10.3109/01677068309107071. [DOI] [PubMed] [Google Scholar]
- 19.Henry EW, Sidman RL. Long lives for homozygous Trembler mutant mice despite virtual absence of peripheral nerve myelin. Science. 1988;241:344–346. doi: 10.1126/science.3388045. [DOI] [PubMed] [Google Scholar]
- 20.Henry EW, Cowen JS, Sidman RL. Comparison of Trembler and Trembler-J mouse phenotypes: varying severity of peripheral hypomyelination. J Neuropathol Exp Neurol. 1983;42:688–706. doi: 10.1097/00005072-198311000-00008. [DOI] [PubMed] [Google Scholar]
- 21.Hildebrand C, Remahl S, Persson H, Bjartmar C. Myelinated nerve fibers in the CNS. Prog Neurobiol. 1992;40:319–384. doi: 10.1016/0301-0082(93)90015-k. [DOI] [PubMed] [Google Scholar]
- 22.Inuzuka T, Quarles RH, Heath J, Trapp BD. Myelin-associated glycoprotein and other proteins in Trembler mice. J Neurochem. 1985;44:793–797. doi: 10.1111/j.1471-4159.1985.tb12885.x. [DOI] [PubMed] [Google Scholar]
- 23.Jacque C, Delassalla A, Raoul M, Baumann N. Myelin basic protein deposition in the optic and sciatic nerves of dysmyelinating mutants quaking, jimpy, trembler, MLD, and shiverer during development. J Neurochem. 1983;41:1335–1340. doi: 10.1111/j.1471-4159.1983.tb00830.x. [DOI] [PubMed] [Google Scholar]
- 24.Johnson PW, Abramow-Newerly W, Seilheimer B, Sadoul R, Tropak MB, Arquint M, Dunn RJ, Schachner M, Rode JC. Recombinant myelin-associated glycoprotein confers neuronal adhesion and neurite outgrowth function. Neuron. 1989;3:377–385. doi: 10.1016/0896-6273(89)90262-6. [DOI] [PubMed] [Google Scholar]
- 25.Jung M, Schneider A, Nave K-A (1995) Dominant-negative action of mutations in the PLP/DM20 gene and direct interaction of PLP polypeptides in vivo. J Neurochem 64 [Suppl]:S101.
- 26.Kaku DA, Parry GJ, Malamut R, Lupski JR, Garcia CA. Nerve conduction studies in Charcot-Marie-Tooth polyneuropathy associated with a segmental duplication of chromosome 17. Neurology. 1993a;43:1806–1808. doi: 10.1212/wnl.43.9.1806. [DOI] [PubMed] [Google Scholar]
- 27.Kaku DA, Parry GJ, Malamut R, Lupski JR, Garcia CA. Uniform slowing of conduction velocities in Charcot-Marie-Tooth polyneuropathy type 1. Neurology. 1993b;43:2664–2667. doi: 10.1212/wnl.43.12.2664. [DOI] [PubMed] [Google Scholar]
- 28.Kornfeld S, Mellman I. The biogenesis of lysosomes. Annu Rev Cell Biol. 1989;5:483–525. doi: 10.1146/annurev.cb.05.110189.002411. [DOI] [PubMed] [Google Scholar]
- 29.Low PA. The evolution of “onion bulbs” in the hereditary hypertrophic neuropathy of the trembler mouse. Neuropathol Appl Neurobiol. 1977;3:81–92. [Google Scholar]
- 30.Magyar JP, Martini R, Ruelicke T, Aguzzi A, Adlkofer K, Dembic Z, Zielasek J, Toyka KV, Suter U. Impaired differentiation of Schwann cells in transgenic mice with increased PMP22 gene dosage. J Neurosci. 1996;16:5351–5360. doi: 10.1523/JNEUROSCI.16-17-05351.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martini R, Schachner M. Immunoelectron microscopic localization of neuronal cell adhesion molecules (L1, N-CAM and MAG) and their shared carbohydrate epitope and myelin basic protein in developing sciatic nerve. J Cell Biol. 1986;103:2439–2448. doi: 10.1083/jcb.103.6.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martini R, Zielasek J, Toyka KV, Gieise KP, Schachner M. Protein zero (P0)-deficient mice show myelin degeneration in peripheral nerves characteristic of inherited human neuropathies. Nat Genet. 1995;11:281–286. doi: 10.1038/ng1195-281. [DOI] [PubMed] [Google Scholar]
- 33.Naef R, Adlkofer K, Lescher B, Suter U (1997) Aberrant protein trafficking in Trembler suggests a disease mechanism for hereditary human peripheral neuropathies. Mol Cell Neurosci, in press. [DOI] [PubMed]
- 34.Nave K-A. Neurological mouse mutants and the genes of myelin. J Neurosci Res. 1994;38:607–612. doi: 10.1002/jnr.490380602. [DOI] [PubMed] [Google Scholar]
- 35.Nicholson G. Penetrance of the hereditary motor and sensory neuropathy Ia mutation: assessment by nerve conduction studies. Neurology. 1991;41:547–552. doi: 10.1212/wnl.41.4.547. [DOI] [PubMed] [Google Scholar]
- 36.Pareek S, Suter U, Snipes GJ, Welcher AA, Shooter EM, Murphy RA. Detection and processing of peripheral myelin protein PMP22 in cultured Schwann cells. J Biol Chem. 1993;268:10372–10379. [PubMed] [Google Scholar]
- 37.Sereda M, Griffits I, Puhlhofer A, Stewart H, Rossner MJ, Zimmermann F, Magyar JP, Schneider A, Hund E, Meinck H-M, Suter U, Nave K-A. A transgenic rat model for Charcot-Marie-Tooth disease. Neuron. 1996;16:1049–1060. doi: 10.1016/s0896-6273(00)80128-2. [DOI] [PubMed] [Google Scholar]
- 38.Shapiro L, Doyle JP, Hensley P, Colman DR, Hendrickson WA. Crystal structure of the extracellular domain from P0, the major structural protein of peripheral myelin. Neuron. 1996;17:435–449. doi: 10.1016/s0896-6273(00)80176-2. [DOI] [PubMed] [Google Scholar]
- 39.Shine HD, Readhead C, Popko B, Hood L, Sidman RL. Morphometric analysis of normal mutant and transgenic CNS: correlation of myelin basic protein expression to myelinogenesis. J Neurochem. 1992;58:342–349. doi: 10.1111/j.1471-4159.1992.tb09316.x. [DOI] [PubMed] [Google Scholar]
- 40.Sinoway MP, Kitagawa K, Timsit S, Hashim GA, Colman DR. Proteolipid protein interactions in transfectants: implications for myelin assembly. J Neurosci Res. 1994;37:551–562. doi: 10.1002/jnr.490370502. [DOI] [PubMed] [Google Scholar]
- 41.Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin Genet. 1974;6:98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x. [DOI] [PubMed] [Google Scholar]
- 42.Snipes GJ, Suter U. Molecular anatomy and genetics of myelin proteins in the peripheral nervous system. J Anat. 1995;186:483–494. [PMC free article] [PubMed] [Google Scholar]
- 43.Sung C, Schneider BG, Agarwal N, Papermaster DS, Nathans J. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88:8840–8844. doi: 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suter U, Snipes GJ. Biology and genetics of hereditary motor and sensory neuropathies. Annu Rev Neurosci. 1995a;18:45–75. doi: 10.1146/annurev.ne.18.030195.000401. [DOI] [PubMed] [Google Scholar]
- 45.Suter U, Snipes GJ. Peripheral myelin protein 22: facts and hypothesis. J Neurosci Res. 1995b;40:145–151. doi: 10.1002/jnr.490400202. [DOI] [PubMed] [Google Scholar]
- 46.Suter U, Moskow JJ, Welcher AA, Snipes GJ, Kosaras B, Sidman RL, Buchberg AM, Shooter EM. A leucine-to-proline mutation in the putative first transmembrane domain of the 22 kDa peripheral myelin protein in trembler-J mouse. Proc Natl Acad Sci USA. 1992a;89:4382–4386. doi: 10.1073/pnas.89.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suter U, Welcher AA, Ozcelik T, Snipes GJ, Kosaras B, Francke U, Billings-Gagliardi S, Sidman RL, Shooter EM. Trembler mouse carries a point mutation in a myelin gene. Nature. 1992b;356:241–244. doi: 10.1038/356241a0. [DOI] [PubMed] [Google Scholar]
- 48.Trapp BD, Quarles RH. Presence of myelin-associated glycoprotein correlates with alterations in the periodicity of peripheral myelin. J Cell Biol. 1982;92:877–882. doi: 10.1083/jcb.92.3.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Valentijn LJ, Baas F, Wolterman RA, Hoogendijk JE, van den Bosh HA, Zorn I, Gabreels-Festen AWM, deWisser M, Bolhuis PA. Identical point mutations of PMP-22 in Trembler-J mouse and Charcot-Marie-Tooth disease type 1A. Nat Genet. 1992;2:288–291. doi: 10.1038/ng1292-288. [DOI] [PubMed] [Google Scholar]
- 50.Vallat JM, Sindau P, Preux PM, Tabaroud F, Milor AM, Couratier P, Leguern E, Brice A. Ultrastructural PMP22 expression in inherited demyelinating neuropathies. Ann Neurol. 1996;39:813–817. doi: 10.1002/ana.410390621. [DOI] [PubMed] [Google Scholar]
- 51.Warner LE, Hilz MJ, Appel SH, Killian JM, Kolodny EH, Karpati G, Carpenter S, Watters GV, Wheeler C, Witt D, Bodell A, Nelis E, VanBroeckhoven C, Lupski JR. Clinical phenotypes of different MPZ (P0) mutations may include Charcot-Marie-Tooth type 1B, Dejerine-Sottas, and congenital hypomyelination. Neuron. 1996;17:451–460. doi: 10.1016/s0896-6273(00)80177-4. [DOI] [PubMed] [Google Scholar]
- 52.Yoshikawa H, Nishimura T, Nakatsuji Y, Fujimura H, Himoro M, Hayasaka K, Sakoda S, Yanagihara T. Elevated expression of messenger RNA for peripheral myelin protein 22 in biopsied peripheral nerves of patients with Charcot-Marie-Tooth disease type 1A. Ann Neurol. 1994;35:445–450. doi: 10.1002/ana.410350412. [DOI] [PubMed] [Google Scholar]