

Abstract
Moderate hypothermia significantly diminishes consequences of spinal and cerebral anoxia. One component of this neuroprotection has been hypothesized to be suppression of excitotoxic transmitter release. Whether this suppression is attributable to reduced hypoxic injury that induces release or an alteration of the release process itself is unclear. We sought to characterize the temperature sensitivity (Q10) of basal and evoked calcitonin gene-related peptide (CGRP) and amino acid release from dorsal horn slices of rat spinal cord over a range of temperatures from 40 to 8°C. At 40°C, potassium (60 mm) and capsaicin (10 μm) evoked a 21- and 32-fold increase in basal CGRP concentrations, respectively. Capsaicin had no effect on glutamate release, but potassium evoked a 2.7-fold increase. Release evoked by either potassium or capsaicin was reduced in a biphasic fashion with declining temperature. Over the range of 40 to 34°C, theQ10 values for evoked release for CGRP were 11.3 (potassium) and 39.7 (capsaicin) and for glutamate, 5.5 (potassium). Over the range of 34 to 8°C,Q10 values were near unity for all evoked release (0.8 and 1.3 for CGRP and 1.2 for glutamate). Although serine, glycine, glutamine, taurine, and citrulline showed no evoked release, basal levels were reduced at temperatures below 34°C. The pronounced temperature dependency of evoked transmitter release between 40 and 34°C is consistent with the profound cerebral protection observed with mild hypothermia in which metabolic activity is only slightly depressed.
Keywords: hypothermia, hyperthermia, glutamate, CGRP, spinal cord superfusion, evoked transmitter release, dorsal horn, capsaicin
Reduction of brain or spinal temperature by 3–5°C has been shown in preclinical and clinical models to attenuate neuronal damage and dysfunction after ischemia (Vacanti and Ames, 1984;Busto et al., 1989). Several mechanisms have been proposed to account for the protection produced by such hypothermia. The first assumes that ischemic injury occurs secondary to depletion of metabolic stores and that hypothermia preserves these stores by reducing cerebral metabolic rate of oxygen consumption (CMRO2). In animal models of cerebral ischemia, CMRO2 declines monotonically by ∼4.5% per degree over the range of 37–38°C (normothermia) to 15–18°C (profound hypothermia) (Michenfelder and Milde, 1992). These findings are clinically relevant, because recovery of normal function in humans can be observed after 60 min of circulatory arrest when the arrest occurs at 15–18°C (Tharion et al., 1982). Although such profound hypothermia has been widely acknowledged, considerable data suggest that a surprisingly moderate hypothermia (32–34°C) can provide neurological protection in the face of cerebral and spinal ischemia. Such a modest reduction in tissue temperature has little effect on CMRO2 and does not preserve metabolic stores (Hagerdal et al., 1975; Sano et al., 1992).
A second mechanism that may underlie hypothermic neuroprotection involves excitotoxicity. Activation of NMDA and non-NMDA receptors has been shown to induce neurodegeneration (Olney and de Gubareff, 1978;Choi, 1988). Glutamate receptor antagonists reduce neurological and histopathological indices of ischemic injury in brain (Meldrum et al., 1987; Newell et al., 1995) and spinal cord (Faden et al., 1990), a finding consistent with the observation that ischemia/anoxia increases excitatory amino acid release from brain (Busto et al., 1989) and spinal cord (Marsala et al., 1994a). Recent work has shown that this glutamate release after ischemia is exquisitely sensitive to modest reductions in local brain (Illievich et al., 1994) or spinal temperature (Marsala et al., 1994b).
These data suggest a covariance between the effects of hypothermia on ischemia-induced injury and transmitter release (vs an effect mediated solely by suppression of CMRO2). Whether the effect on release is attributable to a reduction of the hypoxic injury that otherwise induces the release or in fact alters the release process itself is unclear. Consideration of the literature surprisingly revealed no systematic analysis describing the temperature coefficients for either basal or evoked transmitter release from neuronal systems. The lack of information led us to consider the overall role of temperature by examining the in vitro release of calcitonin gene-related peptide (CGRP) and glutamate from rat lumbar dorsal horn over a range of temperatures from 40 to 8°C.
MATERIALS AND METHODS
Animal care and preparation. Male Sprague Dawley rats (325–400 gm; Harlan Industries, Indianapolis, IN) were housed pairwise in cages and maintained on a 12 hr light/dark cycle with access to food and water at all times. Under terminal halothane anesthesia (4%), rats were decapitated, and the spinal cords were hydraulically extruded. The spinal cords were placed in ice-cold isotonic buffer and then dissected on a filter paper-covered glass plate placed on crushed ice. A 2 cm segment of the lumbar enlargement was isolated and hemisected longitudinally into lateral halves. These halves were hemisected again, and the dorsal quadrants were retained. These dorsal segments were sliced cross-sectionally into 2 mm prisms. Prisms were dispersed on Millipore filters (13 mm diameter, 5 μm pore size) that were placed inside perfusion chambers (modified Millipore filter units; Millipore, Bedford, MA).
In vitro perfusion. The prisms of one lumbar enlargement were used in each perfusion study and dispersed at random to three or four perfusion chambers (five prisms per chamber). Perfusion chambers were immersed in a water bath maintained at 37°C and perfused with artificial CFS (ACSF) at a rate of 0.2 ml/min via peristaltic pump. The ACSF reservoir for each study was placed in the same water bath as the perfusion chambers, and pH was adjusted by bubbling with 5% CO2/95% O2 for 30 min before and throughout the study. After an initial washout period of 45 min to allow tissue stabilization (see Fig. 1), an additional 20 min washout was initiated to allow for temperature changes.
Fig. 1.

Basal glutamate outflow. Resting release of glutamate is described over the total time course of the experiment at 37 (•), 34 (▪), and 25°C (▴) to demonstrate that a stable baseline is present during aliquot collection time points. Time 0 of the tracings represents the beginning of the washout period after tissue harvest, with baseline collections taken atT = 65 min.
Perfusion and stimulation solutions. Standard ACSF consisted of the following (in mm): 21 NaHCO3, 2.5 Na2HPO4/7 H2O, 125 NaCl, 2.6 KCl, 0.9 MgCl2/6 H2O, 1.3 CaCl2, 3.9d-glucose. When a high potassium solution was used as a stimulus, KCl was increased to 60 mm and NaCl reduced to 67.6 mm to maintain osmolarity. Capsaicin was dissolved in 25% β-propyl cyclodextrin in normal saline to a concentration of 1 mg/ml and diluted to 10 μm with ACSF for all capsaicin studies. All ACSF solutions described were oxygenated for 30 min before and throughout use and used at 37°C unless indicated otherwise. Calcium-free perfusions were conducted using ACSF in which CaCl2 (1.3 mm) was replaced with 1.5 mm EGTA in both washout and stimulation solutions.
Temperature conditions. As stated above, the 45 min washout period was followed by a second period (20 min). This second period served as an interval for affecting temperature changes while still allowing the tissue to stabilize at a physiological temperature. Hypothermic conditions (8, 18, 25, and 34°C) were generated by simultaneously siphoning out a portion of the water bath and replacing the volume with chipped ice. Hyperthermic (40°C) conditions were created by increasing the set point of the water heater. Perfusion chambers remained immersed in the water bath, and the entire system could be cooled/heated to the target temperature. Target temperature was monitored throughout the experiment using a thermocouple (36Ga, Type T, Omega Instruments) permanently mounted inside one of the perfusion chambers.
CGRP and glutamate studies. All perfusate samples were collected in chilled polystyrene tubes on ice. After completion of both washout periods, a 10 min perfusate fraction was collected (∼2 ml) to assess basal release levels. The tissue was then stimulated with either capsaicin (10 μm) or potassium (60 mm) in ACSF. A 2 min collection period was discarded after perfusion was initiated with the stimulation solution to allow for tubing volume, after which a 10 min stimulation fraction was collected. A 100 μl aliquot from all samples collected was pipetted to polycarbonate tubes and frozen (−70°C) for HPLC assay of amino acids. The remaining perfusate sample was frozen at −70°C, lyophilized, and analyzed for CGRP immunoreactivity using a polyclonal competitive radioimmunoassay (RIA) as described below.
Amino acid analysis. Perfusate samples were analyzed for glutamate, aspartate, serine, glycine, glutamine, taurine, and citrulline using the phenyl isothiocyanate derivatization procedure (Bennett and Solomon, 1986), a Waters (Milford, MA) HPLC with reverse-phase C18 column (3.9 × 300 mm, 4 μm particle), and a UV detector. Samples were delivered by refrigerated autoinjector, and detector data were collected by an IBM PC interface. Methionine sulfone was added to each amino acid sample as an internal standard. Sensitivity was 5–10 pmol/sample with a coefficient of variation typically in the 10% range. In cases where amino acids were below detection limits, content was assumed to be 5 pmol/sample. Amino acid peak heights were initially normalized to the methionine sulfone peak and then quantified based on a linear relationship between peak height and amounts of corresponding external standards, which were run daily.
CGRP antisera development. The CGRP polyclonal antisera G2027 was obtained from an immunized New Zealand white rabbit (male, 3 kg). Rat CGRP-α (0.5 mg) was coupled to ovalbumin (6.0 mg) using EDAC carbodimide (30 mg) as a coupling agent in PBS buffer, pH 6.0, at 4°C for 18 hr. The peptide conjugate was administrated intradermally with Freund’s complete adjuvant in the first injection. The dose administered corresponded to 20 μg of unconjugated peptide. Booster injections of CGRP-α conjugates in Freund’s incomplete adjuvant were administered subcutaneously once a month, 30 d after the first immunization. The animal was bled at 4 week intervals starting 2 weeks after the booster injection. The antisera G2027 was taken from the ninth bleeding, and the working dilution was 1:21,000. The cross-reactivity of the antibody was examined by competitive RIA in the presence of varying concentrations of human CGRP I and II, rat CGRP α and β, rat CGRP 29–37 fragment, substance P, neurokinin A, cholycystokinin B, calcitonin, and amylin.
CGRP RIA. CGRP iodination was accomplished using the chloramine-T reaction. TyrOCGRP-α (5.0 μg) in 50 μl of 0.5 m NaPO4 buffer was coupled with 10 μl (1 mCi) NaI125. Lyophilized samples or rat CGRP-α (as standard) were reconstituted in assay buffer (0.04 mKH2PO4, 0.01 m EDTA, 0.02% thimersol, 0.25% trasylol, 1% BSA, 4% polyethylene glycol 8000, pH 6.5) and incubated with 0.1 ml G2027 antisera (1:3000) overnight at 4°C. 125I-TryOCGRP-α was then incubated for an additional 24 hr at 4°C. After precipitation of the bound complex using goat anti-rabbit serum (1:300) and centrifugation, the pellet was counted in a gamma counter. Assays were carried out in duplicate with nonspecific binding and blanks, and minimum assay sensitivity was 2.8 fmol/assay tube.
CGRP immunoreactivity characterization. The CGRP immunoreactivity of G2027 was characterized by reverse-phase HPLC (C-18 Bondupack, Waters). The elution gradient was 0–100% acetonitrile in 0.1% trifluoroacetic acid (1–5 min, 0%; 6–35 min, 0–100%; 36–40 min, 100%). The flow rate was 1 ml/min, and 1 min aliquots were collected for RIA with G2027. The elution pattern of CGRP immunoreactivity with synthetic rat CGRP-α and β (American Peptide, Sunnyvale, CA) was compared with that of the standard CGRP-α (Peninsula, Belmont, CA) used for RIAs. To assess G2027 immunoreactivity to CGRP within spinal tissue and whether short-term temperature changes affected tissue CGRP immunoreactivity profile, spinal tissue was harvested and perfused as above for 45 min, after which chamber temperature was maintained at 40, 37, or 34°C for 20 min. Spinal tissue was then removed from the chambers, frozen at −70°C, extracted, and assayed for CGRP content by HPLC-RIA.
Data analysis and statistics. Basal amino acid and CGRP levels were compared across temperature using one-way ANOVA with Scheffé’s S correction for multiple comparisons. Scheffé’s S test was selected for both its allowance of unequal number of subjects per category as well as the fact that all pairwise comparisons are made. The time course of glutamate outflow was compared at three different temperatures using repeated measures ANOVA (one between, one within). Basal glutamate and CGRP release were not significantly different across temperature, so basal levels were pooled and used for comparison to evoked release. Evoked release of glutamate and CGRP was compared across temperature using one-way ANOVA with Dunnett’s two-tailed post hoc procedure for comparing all means to control. Calcium-free trials were compared in the same fashion.
Temperature-dependent effects were also expressed as theQ10 of transmitter release evoked by capsaicin (10 μm in ACSF) or potassium (60 mm in ACSF) of glutamate and CGRP. The Q10 was calculated as described below, where X2 equals the experimental value at the higher absolute temperature (T2) and X1 equals the experimental value at the lower absolute temperature (T1): Q10 = (X2/X1)[10/(T2− T1)] (Kimura and Meves, 1979).
RESULTS
Basal release of amino acids
Immediately after placement in the perfusion chamber, glutamate levels were high, as would be expected immediately after tissue harvest, and they displayed a subsequent exponential decay that reached asymptote after 45 min (Fig. 1). The mean basal glutamate release rate was shown to be 163 ± 8 pmol/min. There was no significant difference in basal glutamate release as a function of chamber temperature (p = 0.18).
In contrast to glutamate, the amino acids aspartate, serine, glycine, and glutamine demonstrated a decrease in basal perfusate release rates with hypothermia, and a clear inflection point below 37°C was observed (p < 0.001) (Table 1). Basal citrulline was depressed significantly at 25 and 8°C (P < 0.05), and taurine demonstrated a similar decreasing trend that did not reach statistical significance.
Table 1.
Basal amino acid perfusate content expressed as pmol/min ± SEM was compared across temperature
| 40°C (n = 12) | 37°C (n = 22) | 34°C (n = 17) | 25°C (n = 17) | 18°C (n = 12) | 8°C (n = 15) | |
|---|---|---|---|---|---|---|
| Aspartate | 74.5 ± 6.4 | 114.4 ± 9.8 | 66.5 ± 8.5† | 40.4 ± 6.1‡ | 64.6 ± 11.7† | 59.0 ± 5.0† |
| Glutamate | 177.0 ± 13.0 | 169.9 ± 16.9 | 168.9 ± 12.5 | 128.6 ± 17.0 | 184.6 ± 42.2 | 133.4 ± 16.5 |
| Serine | 348.9 ± 16.3 | 390.7 ± 34.2 | 142.3 ± 18.3‡ | 86.2 ± 11.6‡ | 99.7 ± 9.5‡ | 114.0 ± 22.0‡ |
| Glycine | 360.9 ± 15.7 | 378.3 ± 27.3 | 149.3 ± 15.2‡ | 64.0 ± 9.1‡ | 100.6 ± 15.6‡ | 100.2 ± 15.7‡ |
| Glutamine | 167.0 ± 8.9 | 177.6 ± 14.0 | 49.7 ± 10.1‡ | 36.0 ± 6.0‡ | 52.2 ± 6.1‡ | 36.0 ± 2.6‡ |
| Taurine | 41.3 ± 7.4 | 34.2 ± 5.6 | 27.4 ± 4.2 | 31.8 ± 11.2 | 29.3 ± 7.8 | 14.1 ± 2.7 |
| Citrulline | 40.0 ± 12.0 | 43.6 ± 7.7 | 24.7 ± 4.6 | 15.8 ± 2.9* | 20.4 ± 4.2 | 11.8 ± 0.5* |
Significant differences from resting normothermic release for each amino acid (ANOVA/Scheffé’s) are indicated by
p< 0.05;
p < 0.01; or
p < 0.0001.
Basal release of CGRP
After the initial washout period, mean basal CGRP release was 75 ± 6 fmol/min. Basal CGRP levels did not differ across temperatures (p = 0.26).
Evoked amino acid release
Glutamate perfusate levels were significantly elevated by potassium (60 mm in ACSF), with the magnitude of release varying directly with temperature. As shown in Figure 2, release rates were enhanced in the presence of potassium at 40°C as compared with 37°C and depressed in studies conducted at or below 34°C. Thus, at and below 34°C, there was no significant increase in glutamate release with potassium stimulation. Evoked release at 37°C was significantly greater (p < 0.002) than under hypothermic conditions, and hyperthermic conditions (40°C) produced evoked release that was significantly greater than at 37°C (p < 0.02).
Fig. 2.

Temperature-sensitive evoked glutamate release. The top panel describes the basal outflow of glutamate over all temperatures investigated (8, 18, 25, 34, 37, and 40°C). Temperature dependency of potassium-evoked glutamate release is shown in the bottom panel as a plot of temperature (°C) versus percentage change from baseline. Statistically significant increases from baseline are indicated by ** (p < 0.01). Hyperthermia produced significant increases in evoked release relative to normothermia, as indicated by † (p < 0.02).
Capsaicin did not alter basal glutamate release rates at any temperature examined. Neither capsaicin nor potassium significantly altered release rates of aspartate, serine, glycine, glutamine, citrulline, or taurine from basal at any temperature (data not shown;n = 4–17 at each temperature).
Evoked CGRP release
CGRP release rates were significantly increased from baseline by both potassium and capsaicin. As shown in Figure 3, release rates were enhanced at 40°C as compared with 37°C in the presence of potassium (p < 0.02) and capsaicin (p < 0.0001), and normothermic and hyperthermic release rates were significantly enhanced (p < 0.01) relative to hypothermic conditions. For both potassium and capsaicin, release rates were significantly diminished but not completely eliminated in studies conducted at or below 34°C. Thus, smaller but still significant increases (p < 0.05) from basal CGRP levels were evoked by potassium at 25°C and capsaicin at 8°C.
Fig. 3.

Temperature-sensitive evoked CGRP release. The top panel describes the basal outflow of CGRP over all temperatures investigated (8, 18, 25, 34, 37, and 40°C). Temperature dependency of potassium (•)- and capsaicin (○)-evoked CGRP release is shown in the bottom panelas a plot of temperature (°C) versus percentage change from basal release. Statistically significant increases from baseline are indicated by * (p < 0.05) or ** (p < 0.01). Hyperthermia produced significant increases in evoked release relative to normothermia, as indicated by † (p < 0.02) and ‡ (p < 0.0001).
Temperature dependency of release
Given the changes in release with temperature for glutamate and CGRP, Q10 values were calculated as described byKimura and Meves (1979). As shown in Table2, the Q10 of potassium-evoked glutamate release for 40 to 34°C and 34 to 8°C were 5.5 and 1.0, respectively. For CGRP release, potassium producedQ10 values of 11.3 and 1.2, respectively, and capsaicin evoked CGRP Q10 values of 39.7 and 0.8, respectively.
Table 2.
Q10 values for capsaicin and potassium-evoked transmitter release
| Q10 (40-34°C) | Q10 (34-8°C) | |
|---|---|---|
| CGRP | ||
| Potassium | 11.31 | 1.23 |
| Capsaicin | 39.72 | 0.80 |
| Glutamate | ||
| Potassium | 5.54 | 1.03 |
| Capsaicin | 0.69 | 1.26 |
Calcium dependency of release
Replacement of perfusate calcium with EGTA had no effect on basal release rates but abolished potassium-evoked increases in glutamate release at 37°C (see Fig. 5). Under the same calcium-free conditions, the potassium-evoked increase in CGRP levels observed at 37 and 25°C was abolished (p < 0.01 and 0.05, respectively) (Fig. 4).
Fig. 5.
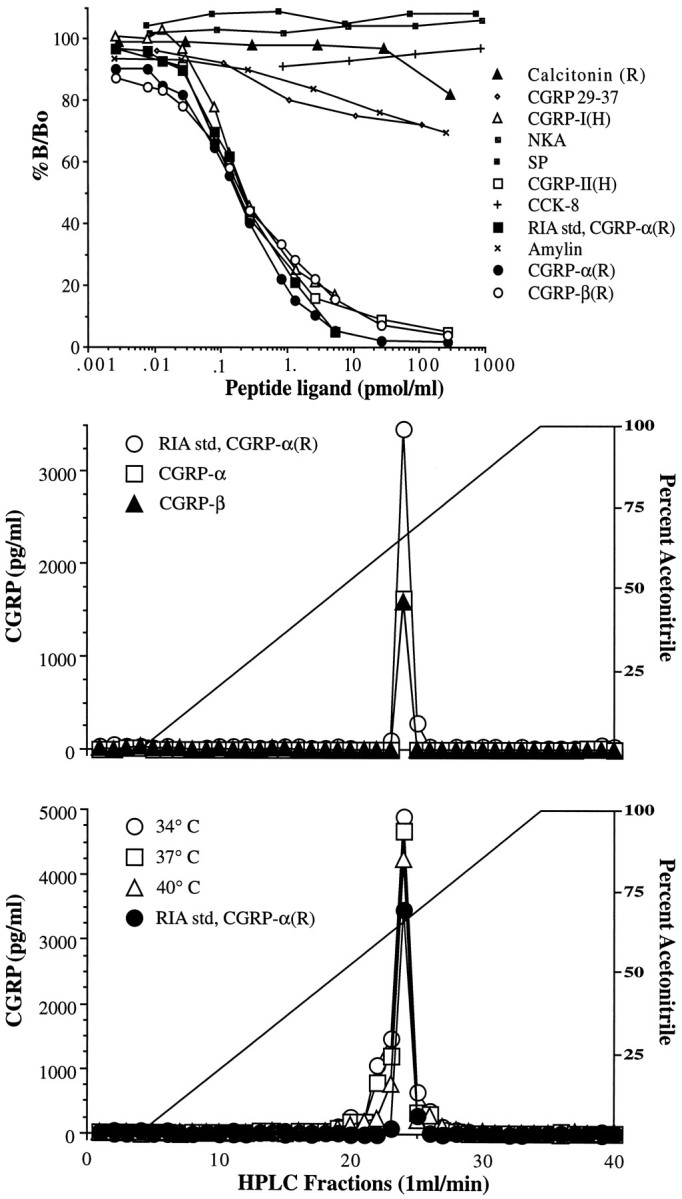
Characterization of G2027. The top panel describes the cross-reactivity of G2027 to various structurally related and unrelated peptides presented as a competition curve of percentage of total binding (%B/Bo) versus the logarithm of competing peptide concentrations. The middle panel presents the elution of standard rat (R) CGRP-α (□) and -β (▴) from the HPLC column as described in Materials and Methods compared with rat CGRP-α RIA standard (○). Peak immunoreactivity was observed during the 24 min fraction in each sample. The bottom panel demonstrates the elution profile of CGRP immunoreactivity from extracted spinal cord slices after perfusion at 34 (○), 37 (□), and 40°C (▵) as compared with standard rat CGRP-α (•).
Fig. 4.
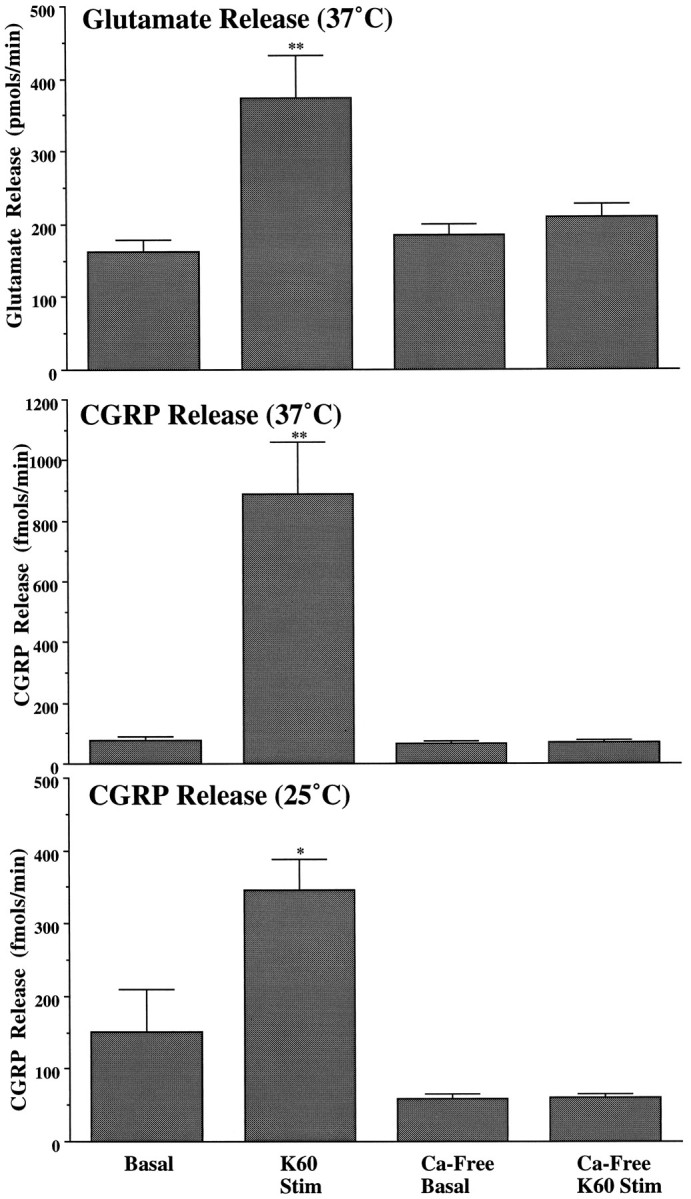
Calcium-dependent evoked transmitter release. In all three panels, basal and potassium-evoked transmitter release are compared under calcium-containing and calcium-free conditions. Calcium-containing examples are redrawn from Figures 2 and 3 for the sake of comparison. The top panel describes the calcium dependency of potassium-evoked glutamate release at 37°C. Themiddle panel describes the calcium dependency of potassium-evoked CGRP release at 37°C, and the bottom panel describes the same potassium-evoked release and calcium dependency for CGRP at 25°C. Statistically significant increases from basal release are indicated by * (p < 0.05) or ** (p < 0.01).
CGRP antibody and immunoreactivity characterization
The antisera G2027 cross-reacted 100% with rat CGRP-α and -β as well as with human CGRP-I and -II and did not cross-react with substance P, neurokinin A, or cholecystokinin-B (<1%) at 1 nmol/ml. G2027 displayed cross-reactivity with the CGRP fragment 29–37 (32%), amylin (30%), and calcitonin (18%) at 1 nmol/ml. Predictably, the highest levels of cross-reactivity were apparent with those peptides of closely related structure (Fig. 5A). As shown in Figure 5B, rat CGRP-α and -β standard immunoreactivity co-eluted in the same fraction (24 min) as the RIA CGRP-α standard during HPLC-RIA characterization. Because CGRP-α and -β are both found within rat spinal cord (Mulderry et al., 1988), we sought to determine which one was released in the present study; however, we were unable to obtain a reliable separation based on the single amino acid difference that distinguishes the two variants. Accordingly, although we are certain that the identity of the immunoreactivity is a CGRP-like peptide, we cannot determine which variant is released or rule out the presence of immunoreactive CGRP metabolic fragments.
The identity of the immunoreactivity was not altered as a function of temperature. Spinal tissue perfused as above at chamber temperatures of 40, 37, and 34°C was collected. In these samples, tissue CGRP content after perfusion was identical: 29.3, 28.7, and 28.3 pmol/mg wet weight, respectively. As shown in Figure 5C, tissue CGRP immunoreactivity in these samples eluted in the same fraction (24 min) as standard regardless of chamber temperature, demonstrating that the immunoreactivity profile did not vary with the temperature of tissue perfusion. This consistency argues against the presence of immunoreactive metabolites, because products of enzymatic cleavage would be temperature-sensitive, and any change attributable to stoichiometric changes in CGRP and its metabolites would be observed as a change in the immunoreactivity profile across temperature.
DISCUSSION
Normothermic (37°C) superfusion of lumbar dorsal horn tissue reveals an ongoing release of several substances, including glutamate and CGRP. Addition of depolarizing potassium concentrations increases the rate at which glutamate and CGRP but not serine, glycine, and glutamine appear in the perfusate (Donnerer and Amann, 1990; Donnerer, 1991). Addition of capsaicin, which depolarizes small sensory afferent terminals, increases the release rate of CGRP but not glutamate (Donnerer, 1991; but see Ueda et al., 1993).
Release properties
Several arguments may be marshaled suggesting that evoked release from harvested spinal cord reflects normal terminal function. (1) Release increases as a function of stimulating agent concentration (Malmberg and Yaksh, 1994). (2) Evoked release is subject to modulation by receptors on terminals from which the transmitter is released. Thus, CGRP is contained in primary afferent terminals that possess α2 adrenoceptors that negatively modulate evoked release (Takano et al., 1993). (3) Evoked release is dependent on extracellular calcium. Although examples of calcium-independent exocytosis have been reported (Hirsch and Gibson, 1984), calcium dependency is widely considered to reflect terminal release (Smith and Augustine, 1988). Thus, the increased release rate for glutamate and CGRP associated with depolarizing stimuli reflects normal terminal function, whereas basal release reflects either an ongoing leakage from injured terminals or calcium-independent release.
Release versus clearance
Increased CGRP or glutamate perfusate concentrations might represent a reduced clearance and not an increased release rate. The fact that CGRP increases could be induced by two different agents (capsaicin and potassium), however, and glutamate increases were not accompanied by changes in other amino acids not considered to be transmitters, argues that increased concentrations reflect increased release. Accordingly, we believe it justi- fiable to refer to changes in extracellular levels as changes in glutamate and CGRP release rates.
Origin of dorsal horn transmitters
Several cell populations may contribute to CGRP and glutamate release. CGRP is stored in dense-core vesicles and present in Lamina I and II and dorsal root ganglion cells (Carlton et al., 1987; Harmann et al., 1988). Depletion of CGRP by neonatal capsaicin or dorsal rhizotomy (Chung et al., 1988; McNeill et al., 1988a,b) and release by acute capsaicin (Takano et al., 1993) suggest that extracellular CGRP levels originate from small-caliber primary afferent terminals. Such terminals would also be depolarized by excess potassium.
Although glutamate has been identified in small afferent terminals (Miller et al., 1988), failure to reduce basal glutamate levels in neonatal-treated animals (Skilling and Larson, 1993) and the controversy as to whether it is released by capsaicin (Donnerer, 1991;Ueda et al., 1993) suggest that extracellular glutamate does not arise primarily from small afferent terminals. Glutamate, typically observed in small clear-core vesicle, is found in Laminae I–III terminals, which are believed to derive from spinopetal projections and interneurons (Merighi et al., 1991).
Temperature-dependent release
From 40 to 8°C, there was a biphasic relationship between temperature and evoked but not basal release, with an inflection at 34°C. Evoked release between 40 and 34°C displayed a steep temperature dependency with high Q10 values for glutamate and CGRP. From 34 to 8°C, release was characterized byQ10 values near unity. Unlike glutamate, significant increases in CGRP release were noted even with severe hypothermia. These characteristics seem to represent a fundamental property of evoked terminal release and were independent of transmitter or mechanism of terminal activation (potassium vs capsaicin for CGRP). Importantly, between 40 and 8°C there were only modest changes in CGRP or glutamate perfusate concentration in the absence of stimulation.
Mechanisms of temperature dependency
Glutamate/CGRP stores
Decreased release observed with mild hypothermia may result from decreased spinal synthesis/storage of transmitters, leading to decreased tissue content. We consider this unlikely for several reasons. (1) The actual cooling occurred only during the brief interval before and during stimulation. It is not reasonable to expect that essentially complete depletion of releasable stores would occur during this brief interval. (2) Although tissue levels were not systematically measured for amino acids, we noted that CGRP content did not differ after incubation at 34, 37, or 40°C, and column elution of tissue extracts revealed no changes in either the recovery of CGRP immunoreactivity or the distribution of that immunoreactivity. (3) Although the cellular origin of the basal CGRP or glutamate is unknown, if tissue depletion exceeded synthesis rate, we would anticipate that basal values should have shown a similar decline. It thus seems unlikely that the marked hypothermic effects on evoked transmitter release can be explained by decreased transmitter synthesis/storage.
Membrane channel function
Calcium entry though voltage-sensitive or ligand-gated ion channels leads to vesicle migration to, and fusion with, the plasma membrane (Miller, 1987; Smith and Augustine, 1988). Nobile and colleagues (1990) demonstrated in chick DRG neurons that L- and N-type calcium current amplitude and channel opening times decreased in a linear fashion with hypothermia (17–37°C). Hippocampal CA1 NMDA and non-NMDA ionotropic receptors demonstrated linear temperature-dependent decreases in calcium current amplitude (14–24°C) (McLarnon and Curry, 1990). These observations suggest that temperature-dependent transmitter release may be a reflection of calcium channel kinetics; however, the linearity of channel function over a broad temperature range is inconsistent with the biphasic nature of observed transmitter release.
Membrane physical properties
Decreases in temperature shift the lipid bilayer from a fluid to a gel phase. Such loss of membrane fluidity would affect not only integral (e.g., ion channel kinetics) and peripheral proteins (e.g., docking proteins) associated with exocytotic release, but would hinder vesicular fusion into membranes. In lipid bilayers, synaptosomal fusion is decreased as a simple linear function of temperature (27–37°C) (Almeida et al., 1994). Such models may in fact reveal that lipid fluidity could be rate-limiting for temperature-dependent exocytosis and release. They cannot define, however, the role of accessory proteins that serve to coordinate vesicle fusion and might be influenced by modest changes in temperature. Such information is not presently available.
Temperature-dependent release and neuroprotection
The potent neuroprotective effect of hypothermia in human and animal models of ischemia is well established. Although this protection may reflect reduced metabolic requirements, at 34°C CMRO2in rats decreases by only 10% (Hagerdal et al., 1975), whereasin vivo models have shown significant neuronal preservation. Additionally, mild hypothermia (32–34°C) in vivo has been shown to significantly reduce glutamate release from brain and spinal cord that is otherwise induced by ischemia (Minamisawa et al., 1990;Marsala et al., 1994b; Patel et al., 1994). This correlation between release and protection as a function of mild hypothermia is consistent with the importance of excitotoxicity associated with ischemia-induced glutamate release. The present study also demonstrated that an increased temperature (40°C) augmented transmitter release. Consistent with this enhancement, hyperthermia increases postischemic deficit and glutamate release in cerebral ischemia (Mitani and Kataoka, 1991).
The unexpectedly pronounced sensitivity of evoked release observed over physiologically accessible temperatures may have an impact on normal spinal physiology. A modest increase to 40°C resulted in a 1.2- and twofold increase from normothermic glutamate and CGRP release, respectively. Conversely, 34°C trials resulted in a 3- and 5- to 10-fold suppression from normothermic release, respectively. Effects on synaptic transmission over this range might be anticipated to exert an important influence on spinal function. First, it is interesting to note that temperature-sensitive neurons are present in the spinal cord, such that heating the spinal cord evokes temperature-regulating behavior in mammals and birds (Thauer, 1968). Spinal recordings describe single-unit activity with heat sensitivity that peaked at 40°C and dropped off sharply with cooling (Wunnenberg and Bruck, 1970; Simon and Iriki, 1971). Similar to transmitter release in the present study, these heat-sensitive units demonstrated an inflection point at 35°C, after which there was a continuing, low level of activity. Thus, one aspect of the temperature sensitivity of release mirrors the activity of heat-sensitive spinofugal fibers believed to be associated with thermoregulation. Second, hypersensitivity is observed during pyrexia (Watkins et al., 1994). Although some of this sensitivity may reflect on peripheral terminal sensitization by prostanoids or cytokines, fever-related hyperthermic augmentation of spinal CGRP and glutamate release itself would lead to a facilitation of afferent input and an enhanced behavioral response to a given stimulus.
In conclusion, the present study demonstrates that glutamate and CGRP release can be evoked under normoxic conditions from dorsal spinal slices in a temperature-dependent manner by potassium, with capsaicin also evoking CGRP, but not glutamate, release. In either case, the release demonstrated a steep temperature dependency with highQ10 values between 40 and 34°C. This relationship between release and a surprisingly narrow temperature range suggests that slight changes in temperature can profoundly affect depolarization-evoked release and, by extrapolation, may play a major role in spinal synaptic transmission and the protective effect associated with mild hypothermia during spinal ischemia.
Footnotes
This work was supported in part by National Institutes of Health Grants GM07552 (D.M.D.), DA05726 (D.M.D.), HL50403 (X.-Y.H.), and DA02110 (T.L.Y.). We thank Allan Moore, Fran Simonet-Magnuson, and Christine Nguyen for their assistance with the RIA and HPLC assays, as well as Dr. Linda Sorkin for many helpful discussions on neuroanatomy.
Correspondence should be addressed to Dr. Tony L. Yaksh, Department of Anesthesiology, University of California, San Diego, 9500 Gilman Drive, Mail Code 0818, La Jolla, CA 92093-0818.
REFERENCES
- 1.Almeida MT, Ramalho-Santos J, Oliveira CR, de Lima MC. Parameters affecting fusion between liposomes and synaptosomes: role of proteins, lipid peroxidation, pH and temperature. J Membr Biol. 1994;142:217–222. doi: 10.1007/BF00234943. [DOI] [PubMed] [Google Scholar]
- 2.Bennett HP, Solomon S. Use of Pico-Tag methodology in the chemical analysis of peptides with carboxyl-terminal amides. J Chromatogr. 1986;359:221–231. doi: 10.1016/0021-9673(86)80076-0. [DOI] [PubMed] [Google Scholar]
- 3.Busto R, Globus MY, Dietrich WD, Martinez E, Valdes I, Ginsberg MD. Effect of mild hypothermia on ischemia-induced release of neurotransmitters and free fatty acids in rat brain. Stroke. 1989;20:904–910. doi: 10.1161/01.str.20.7.904. [DOI] [PubMed] [Google Scholar]
- 4.Carlton SM, McNeill DL, Chung K, Coggeshall RE. A light and electron microscopic level analysis of calcitonin gene-related peptide (CGRP) in the spinal cord of the primate: an immunohistochemical study. Neurosci Lett. 1987;82:145–150. doi: 10.1016/0304-3940(87)90119-4. [DOI] [PubMed] [Google Scholar]
- 5.Choi DW. Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988;11:465–469. doi: 10.1016/0166-2236(88)90200-7. [DOI] [PubMed] [Google Scholar]
- 6.Chung K, Lee WT, Carlton SM. The effects of dorsal rhizotomy and spinal cord isolation on calcitonin gene-related peptide-labeled terminals in the rat lumbar dorsal horn. Neurosci Lett. 1988;90:27–32. doi: 10.1016/0304-3940(88)90781-1. [DOI] [PubMed] [Google Scholar]
- 7.Donnerer J. Depolarization-evoked release of glutamate, aspartate and gamma-aminobutyric acid from rat dorsal spinal cord slices does not originate from capsaicin-sensitive neurons. Brain Res. 1991;555:332–334. doi: 10.1016/0006-8993(91)90360-8. [DOI] [PubMed] [Google Scholar]
- 8.Donnerer J, Amann R. Capsaicin-evoked neuropeptide release is not dependent on membrane potential changes. Neurosci Lett. 1990;117:331–334. doi: 10.1016/0304-3940(90)90686-4. [DOI] [PubMed] [Google Scholar]
- 9.Faden AI, Ellison JA, Noble LJ. Effects of competitive and non-competitive NMDA receptor antagonists in spinal cord injury. Eur J Pharmacol. 1990;175:165–174. doi: 10.1016/0014-2999(90)90227-w. [DOI] [PubMed] [Google Scholar]
- 10.Hagerdal M, Harp J, Nilsson L, Siesjo BK. The effect of induced hypothermia upon oxygen consumption in the rat brain. J Neurochem. 1975;24:311–316. doi: 10.1111/j.1471-4159.1975.tb11881.x. [DOI] [PubMed] [Google Scholar]
- 11.Harmann PA, Chung K, Briner RP, Westlund KN, Carlton SM. Calcitonin gene-related peptide (CGRP) in the human spinal cord: a light and electron microscopic analysis. J Comp Neurol. 1988;269:371–380. doi: 10.1002/cne.902690305. [DOI] [PubMed] [Google Scholar]
- 12.Hirsch JA, Gibson GE. Selective alteration of neurotransmitter release by low oxygen in vitro. Neurochem Res. 1984;9:1039–1049. doi: 10.1007/BF00964800. [DOI] [PubMed] [Google Scholar]
- 13.Illievich UM, Zornow MH, Choi KT, Strnat MA, Scheller MS. Effects of hypothermia or anesthetics on hippocampal glutamate and glycine concentrations after repeated transient global cerebral ischemia. Anesthesiology. 1994;80:177–186. doi: 10.1097/00000542-199401000-00025. [DOI] [PubMed] [Google Scholar]
- 14.Kimura JE, Meves H. The effect of temperature on the asymmetrical charge movement in squid giant axons. J Physiol (Lond) 1979;289:479–500. doi: 10.1113/jphysiol.1979.sp012748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malmberg AB, Yaksh TL. Capsaicin-evoked prostaglandin E2 release in spinal cord slices: relative effect of cyclooxygenase inhibitors. Eur J Pharmacol. 1994;271:293–299. doi: 10.1016/0014-2999(94)90786-2. [DOI] [PubMed] [Google Scholar]
- 16.Marsala M, Sorkin LS, Yaksh TL. Transient spinal ischemia in rat: characterization of spinal cord blood flow, extracellular amino acid release, and concurrent histopathological damage. J Cereb Blood Flow Metab. 1994a;14:604–614. doi: 10.1038/jcbfm.1994.75. [DOI] [PubMed] [Google Scholar]
- 17.Marsala M, Vanicky I, Yaksh TL. Effect of graded hypothermia (27° to 34° C) on behavioral function, histopathology, and spinal blood flow after spinal ischemia in rat. Stroke. 1994b;25:2038–2046. doi: 10.1161/01.str.25.10.2038. [DOI] [PubMed] [Google Scholar]
- 18.McLarnon JG, Curry K. Single channel properties of the N-methyl-d-aspartate receptor channel using NMDA and NMDA agonists: on-cell recordings. Exp Brain Res. 1990;82:82–88. doi: 10.1007/BF00230840. [DOI] [PubMed] [Google Scholar]
- 19.McNeill DL, Chung K, Carlton SM, Coggeshall RE. Calcitonin gene-related peptide immunostained axons provide evidence for fine primary afferent fibers in the dorsal and dorsolateral funiculi of the rat spinal cord. J Comp Neurol. 1988a;272:303–308. doi: 10.1002/cne.902720212. [DOI] [PubMed] [Google Scholar]
- 20.McNeill DL, Coggeshall RE, Carlton SM. A light and electron microscopic study of calcitonin gene-related peptide in the spinal cord of the rat. Exp Neurol. 1988b;99:699–708. doi: 10.1016/0014-4886(88)90186-0. [DOI] [PubMed] [Google Scholar]
- 21.Meldrum BS, Evans MC, Swan JH, Simon RP. Protection against hypoxic/ischaemic brain damage with excitatory amino acid antagonists. Med Biol. 1987;65:153–157. [PubMed] [Google Scholar]
- 22.Merighi A, Polak JM, Theodosis DT. Ultrastructural visualization of glutamate and aspartate immunoreactivities in the rat dorsal horn, with special reference to the co-localization of glutamate, substance P and calcitonin-gene related peptide. Neuroscience. 1991;40:67–80. doi: 10.1016/0306-4522(91)90175-n. [DOI] [PubMed] [Google Scholar]
- 23.Michenfelder JD, Milde JH. The effect of profound levels of hypothermia (below 14° C) on canine cerebral metabolism. J Cereb Blood Flow Metab. 1992;12:877–880. doi: 10.1038/jcbfm.1992.120. [DOI] [PubMed] [Google Scholar]
- 24.Miller KE, Clements JR, Larson AA, Beitz AJ. Organization of glutamate-like immunoreactivity in the rat superficial dorsal horn: light and electron microscopic observations. Synapse. 1988;2:28–36. doi: 10.1002/syn.890020106. [DOI] [PubMed] [Google Scholar]
- 25.Miller RJ. Multiple calcium channels and neuronal function. Science. 1987;235:46–52. doi: 10.1126/science.2432656. [DOI] [PubMed] [Google Scholar]
- 26.Minamisawa H, Nordstrom CH, Smith ML, Siesjo BK. The influence of mild body and brain hypothermia on ischemic brain damage. J Cereb Blood Flow Metab. 1990;10:365–374. doi: 10.1038/jcbfm.1990.66. [DOI] [PubMed] [Google Scholar]
- 27.Mitani A, Kataoka K. Critical levels of extracellular glutamate mediating gerbil hippocampal delayed neuronal death during hypothermia: brain microdialysis study. Neuroscience. 1991;42:661–670. doi: 10.1016/0306-4522(91)90035-m. [DOI] [PubMed] [Google Scholar]
- 28.Mulderry PK, Ghatei MA, Spokes RA, Jones PM, Pierson AM, Hamid QA, Kanse S, Amara SG, Burrin JM, Legon S, Polak JM, Bloom SR. Differential expression of alpha-CGRP and beta-CGRP by primary sensory neurons and enteric autonomic neurons of the rat. Neuroscience. 1988;25:195–205. doi: 10.1016/0306-4522(88)90018-8. [DOI] [PubMed] [Google Scholar]
- 29.Newell DW, Barth A, Malouf AT. Glycine site NMDA receptor antagonists provide protection against ischemia-induced neuronal damage in hippocampal slice cultures. Brain Res. 1995;675:38–44. doi: 10.1016/0006-8993(95)00039-s. [DOI] [PubMed] [Google Scholar]
- 30.Nobile M, Carbone E, Lux HD, Zucker H. Temperature sensitivity of Ca currents in chick sensory neurones. Pflügers Arch. 1990;415:658–663. doi: 10.1007/BF02584002. [DOI] [PubMed] [Google Scholar]
- 31.Olney JW, de Gubareff T. Glutamate neurotoxicity and Huntington’s chorea. Nature. 1978;271:557–559. doi: 10.1038/271557a0. [DOI] [PubMed] [Google Scholar]
- 32.Patel PM, Drummond JC, Cole DJ, Yaksh TL. Differential temperature sensitivity of ischemia-induced glutamate release and eicosanoid production in rats. Brain Res. 1994;650:205–211. doi: 10.1016/0006-8993(94)91783-3. [DOI] [PubMed] [Google Scholar]
- 33.Sano T, Drummond JC, Patel PM, Grafe MR, Watson JC, Cole DJ. A comparison of the cerebral protective effects of isoflurane and mild hypothermia in a model of incomplete forebrain ischemia in the rat. Anesthesiology. 1992;76:221–228. doi: 10.1097/00000542-199202000-00011. [DOI] [PubMed] [Google Scholar]
- 34.Simon E, Iriki M. Sensory transmission of spinal heat and cold sensitivity in ascending spinal neurons. Pflügers Arch. 1971;328:103–120. doi: 10.1007/BF00592439. [DOI] [PubMed] [Google Scholar]
- 35.Skilling SR, Larson AA. Capsaicin inhibits whereas rhizotomy potentiates substance P-induced release of excitatory amino acids in the rat spinal cord in vivo. Neurosci Lett. 1993;150:107–111. doi: 10.1016/0304-3940(93)90119-6. [DOI] [PubMed] [Google Scholar]
- 36.Smith SJ, Augustine GJ. Calcium ions, active zones and synaptic transmitter release. Trends Neurosci. 1988;11:458–464. doi: 10.1016/0166-2236(88)90199-3. [DOI] [PubMed] [Google Scholar]
- 37.Takano M, Takano Y, Yaksh TL. Release of calcitonin gene-related peptide (CGRP), substance P (SP), and vasoactive intestinal polypeptide (VIP) from rat spinal cord: modulation by alpha 2 agonists. Peptides. 1993;14:371–378. doi: 10.1016/0196-9781(93)90055-l. [DOI] [PubMed] [Google Scholar]
- 38.Tharion J, Johnson DC, Celermajer JM, Hawker RM, Cartmill TB, Overton JH. Profound hypothermia with circulatory arrest: nine years’ clinical experience. J Thorac Cardiovasc Surg. 1982;84:66–72. [PubMed] [Google Scholar]
- 39.Thauer R. Thermosensitivity of the spinal cord. In: Hardy JD, Gagge AP, Stolwijk AJ, editors. Physiological and behavioral temperature regulation. Charles C. Thomas; Springfield, IL: 1968. pp. 472–492. [Google Scholar]
- 40.Ueda M, Kuraishi Y, Satoh M. Detection of capsaicin-evoked release of glutamate from spinal dorsal horn slices of rat with on-line monitoring system. Neurosci Lett. 1993;155:179–182. doi: 10.1016/0304-3940(93)90702-m. [DOI] [PubMed] [Google Scholar]
- 41.Vacanti FX, Ames A., III Mild hypothermia and Mg++ protect against irreversible damage during CNS ischemia. Stroke. 1984;15:695–698. doi: 10.1161/01.str.15.4.695. [DOI] [PubMed] [Google Scholar]
- 42.Watkins LR, Wiertelak EP, Goehler LE, Mooney-Heiberger K, Martinez J, Furness L, Smith KP, Maier SF. Neurocircuitry of illness-induced hyperalgesia. Brain Res. 1994;639:283–299. doi: 10.1016/0006-8993(94)91742-6. [DOI] [PubMed] [Google Scholar]
- 43.Wunnenberg W, Bruck K. Studies on the ascending pathways from the thermosensitive region of the spinal cord. Pflügers Arch. 1970;321:233–241. doi: 10.1007/BF00588444. [DOI] [PubMed] [Google Scholar]