

Abstract
Glial cell line-derived neurotrophic factor (GDNF), a recently described and cloned member of the transforming growth factor (TGF)-β superfamily, has been shown to have marked trophic activity on several populations of central neurons. Survival-promoting and injury protectant activity in vitro and in vivo, using several paradigms, has been demonstrated for ventral mesencephalic dopaminergic neurons and spinal cord motoneurons. In view of a proposed commonality of mechanisms, involving intracellular free radical generation, depolarization-induced Ca2+ influx, and mitochondrial respiratory enzyme injury, between such GDNF-responsive paradigms and those of ischemia-induced injury, we tested the effects of GDNF on the extent of neural degeneration induced by transient middle cerebral artery (MCA) occlusion. We now report that intracerebroventricular and intraparenchymal administration of GDNF potently protects the cerebral hemispheres from damage induced by MCA occlusion. In addition, the increase in nitric oxide that accompanies MCA occlusion and subsequent reperfusion is blocked almost completely by GDNF. Thus, this protein may play an important role in the treatment of cerebrovascular occlusive disease.
Keywords: nitric oxide, cerebral ischemia, MCA ligation, GDNF, neuroprotection, TGF-β superfamily
Ischemia-induced brain injury is a major cause of mortality worldwide (Brown et al., 1996; Menotti et al., 1996;Rastenyte et al., 1996) for which there are no effective pharmacotherapies to date. Cell injury or death after cerebral vascular occlusion has been postulated to result from a number of interacting pathophysiological factors, including depolarization-induced calcium entry via NMDA receptors, intracellular free radical generation, damage to mitochondrial respiratory enzymes, and induction of programmed cell death, to name but a few proposed mechanisms (Hajimohammadreza et al., 1995; Vornov, 1995; Dalkara et al., 1996; Strijbos et al., 1996).
Glial cell line-derived neurotrophic factor (GDNF), a recently identified and cloned transforming growth factor (TGF)-β superfamily trophic factor for central dopaminergic neurons (Lin et al., 1993;Hoffer et al., 1994; Beck et al., 1995; Hudson et al., 1995), spinal cord motorneurons (Henderson et al., 1994; Yan et al., 1995; Trok et al., 1996), and kidney (Pichel et al., 1996), has been shown to protect dopaminergic neurons from damage induced by neurotoxins that elevate intracellular free radicals and produce damage to mitochondrial respiratory enzymes (Bowenkamp et al., 1995; Tomac et al., 1995; Gash et al., 1996). This molecule has additional nondopaminergic neuroprotective activity (Williams et al., 1996a,b). It has been found that the expression of TGF-β1 mRNA is increased during regeneration of renal tubules after acute ischemic injury (Basile et al., 1996). Similarly, TGF-β1 transcript expression is enhanced in the hippocampus after transient forebrain ischemia (Knuckey et al., 1996). These data suggest that TGF-β superfamily molecules are actively released after acute ischemia.
Extracellular nitric oxide (NO) is released during ischemia and reperfusion (Malinski et al., 1993; Kumura et al., 1994; Sato et al., 1994). Inhibition of NO synthesis reduces hypoxia or ischemia-mediated tissue damage (Hamada et al., 1994; Kozniewska et al., 1995; Zhang et al., 1996). Previous studies indicate that NMDA receptors are activated during ischemia or hypoxia (Simon et al., 1984; Benveniste et al., 1988), and NO is believed to be an important mediator of glutamatergic neurotoxicity during brain ischemia (Strosznajder et al., 1994). Administration of the NMDA antagonists ketamine or MK801 reduces NOS activity (Lin et al., 1996) and the amount of infarction (Caldwell et al., 1994; Earley et al., 1996) induced by cerebral ischemia. Recent studies have also indicated that TGF-β1 inhibits NO synthase (NOS) mRNA and NO production in vascular smooth muscle during septic shock (Perrella et al., 1996). Thus, it is possible that TGF-β1 superfamily molecules can also attenuate cerebral NOS activity and the size of infarction during ischemia.
In the present experiments, we studied the effects of GDNF on a model of cerebral ischemia, induced by occlusion of the middle cerebral artery (MCA). Previous experiments have demonstrated that ligation of MCA activates NOS (Kader et al., 1993; Lin et al., 1996) and induces cortical infarction (Menzies et al., 1992; Du et al., 1996). We show that GDNF potently protects the cerebral cortex from ischemia-induced injury and blocks the elevation of NO that accompanies MCA occlusion and reperfusion.
MATERIALS AND METHODS
Animals. A total of 59 adult Sprague Dawley rats were used. Animals were divided into two groups: an aged group (weight 584.8 ± 16.4 gm; n = 46) and a relatively younger group (weight 392.2 ± 11.4 gm; n = 13). Of the aged group, 22 were used for electrochemical studies of extracellular NO levels (10 controls and 12 pretreated with GDNF) and 24 were used for histological experiments (11 controls without PBS pretreatment, 6 controls with PBS pretreatment, and 7 with GDNF in PBS). All animals in the younger group (n = 13) were used for histological studies (seven controls with PBS pretreatment and six with GDNF in PBS). The rats were anesthetized using either chloral hydrate (400 mg/kg, i.p., for histological studies) or urethane (1.25 g/kg, i.p., for electrochemical studies). Two sets of control animals were used. One set received intraparenchymal and intraventricular injections of PBS alone at the same volumes, sites, and time intervals used for the GDNF injections before arterial ligation. The other set received no GDNF or PBS injections but only arterial occlusion.
MCA ligation. The ligation of the right MCA and bilateral common carotids (CCAs) was performed using methods suggested by Chen et al. (1986). The bilateral CCAs were identified and isolated through a ventral midline cervical incision. The CCAs were ligated with nontraumatic arterial clips. A craniotomy of ∼2 × 2 mm2 was made in the right squamosal bone. The right MCA was ligated with a 10-O suture. The craniotomy was then covered with gelfoam. Two paradigms (40 and 90 min ligation) were used to study the protective effects of GDNF in the aged and young rats, respectively. As reported previously, the 40 min ligation did not cause brain infarction in the young rats. Ninety minute ligations, however, induced maximal infarctions in the young rats (Du et al., 1996). Our initial experiments demonstrated that 40 min ligations were enough to produce maximal infarction in the cortex in the aged rats. Twenty-four hours after reperfusion, animals were killed and perfused intracardially with saline. The brain tissue was then removed, immersed in cold saline for 5 min, and sliced into 2.0 mm sections. The brain slices were incubated in 2% triphenyltetrazolium chloride (TTC) dissolved in PBS for 30 min at 37°C and then transferred to 5% formaldehyde solution for fixation (Chen et al., 1986). The volume of infarction was measured in each slice and summed using computerized planimetry (In Situ, LSR Ltd.)
In vivo NO measurement.In vivochronoamperometric measurements of extracellular NO concentration were performed with a microcomputer-controlled apparatus (IVEC-10, Medical Systems, Green-vale, NY) as described previously (Lin et al., 1996). The recordings were taken within the cerebral cortex (2.2 mm posterior to the bregma, 5.0–5.5 mm lateral to the midline, 0.9 mm below the cortical surface) 30 min before MCA ligation and lasted for 1–2 hr, depending on the duration of ligation (40 or 90 min ligations, respectively). Remote from this site, miniature Ag/AgCl reference electrodes were inserted into the brain and cemented in place with dental acrylic. The working electrodes were made of two carbon fiber filaments (30 μm in diameter; Textron, Lowell, MA). The sensor was first coated with Nafion (5% solution; Aldrich Chemical, Milwaukee, WI) at 65°C to decrease any interference from extracellular ascorbic acid (Gerhardt et al., 1984). The electrodes were then coated with 2 mm Ni Mesotetra (N-methyl-4- pyridyl) porphine tetratosylate (TMPP-Ni) in 0.1 m NaOH, at 0.9 V for 30 min. Each electrode was tested for sensitivity and selectivity to NOin vitro. Calibration of NO was made using 10 μmS-nitroso-N-acetyl-dl-penicillamine (SNAP) in 0.1 mm phosphate buffer, pH 7.4 (Feelisch, 1991). Only electrodes showing selectivity for NO, compared with ascorbate, of >100,000 in vitro were used in the in vivorecordings. The NO current generated by application of an oxidation potential of +0.9 V, relative to a Ag/AgCl reference electrode, was recorded in vivo continuously at a rate of 1 Hz. Allin vivo signals were expressed as nanomolar changes in NO using the in vitro calibration factors.
Drug administration. Human recombinant GDNF, obtained from Synergen, was used in this study. GDNF, dissolved in PBS, was applied locally through a Hamilton syringe for histological experiments. GDNF was injected initially into the left lateral cerebral ventricle at a dose of 4 μg (0.4 μg/μl × 10 μl). Thirty minutes after intracerebroventricular GDNF injection, the squamosal bone (about 2 × 2 mm2) overlying the right frontal and temporal cortex was removed. Three injections of GDNF (0 4 μg/μl × 5 μl × 3 sites) were made directly into the cortex through a Hamilton syringe adjacent to the MCA. Five minutes after intracerebral GDNF injection, the left MCA and bilateral CCAs were ligated for 40 or 90 min in the aged and young animals, respectively. The animals were killed 24 hr later for TTC staining.
GDNF, dissolved in PBS, was also applied locally through micropipettes for electrochemical studies. The NO sensor and the micropipette were mounted together with sticky wax (Kerr Inc.); tips were separated by 100–150 μm. Local application of GDNF (0.4 μg/μl × 5 μl) from the micropipettes was performed by pressure ejection using a pneumatic pump (PPM-2, Medical Systems). The ejection volume was monitored by recording the change in the fluid meniscus in the pipette, before and after ejection, with a dissection microscope. GDNF (0.4 μg/μl × 10 μl) was also injected intracerebroventricularly into the left ventricle in these experiments.
RESULTS
Previous experiments have demonstrated that ligation of MCA induces cortical infarction in rats (Chen et al., 1986; Du et al., 1996), similar to the level of infarction we saw in the control group (see below). We found that pretreatment with GDNF diminished the volume of cortical infarction in aged rats, induced by 40 min of MCA ligation, as measured by TTC staining (Figs. 1, 2). In all the control animals studied (n = 11), MCA ligation and reperfusion resulted in clear-cut infarction of the cortex. Both the incidence and volume of infarction were not altered by PBS pretreatment as compared with the noninjected animals (Fig.2A,B). In contrast, only two of seven rats that received GDNF pretreatment (doses given as one intracerebroventricular and three local injections) showed mild infarction after MCA ligation. The incidence (p< 0.05; χ2 test) and the volume (120.8 ± 28.8 mm3 vs 26.5 ± 18.5 mm3;p < 0.05; one-way ANOVA + Dunn’s test) of infarctions were reduced significantly by GDNF pretreatment (Fig. 2). Furthermore, the number of infarcted slices in each rat was reduced significantly (Fig. 2C) from 5.4 ± 0.7 slice/rat in non-PBS treated rats or 5.3 ± 0.8 slice/rat in PBS-treated rats to 1.1 ± 0.7 slice/rat in the GDNF-treated rats (p < 0.05; one-way ANOVA + Dunn’s test). These data suggest that the GDNF diminished not only the volume but also the extent of infarction in the ischemic brain.
Fig. 1.
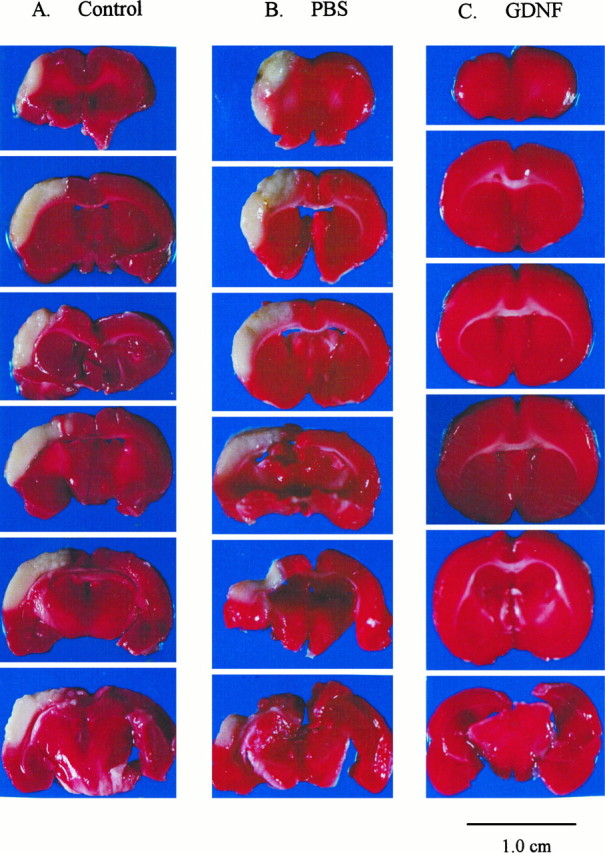
Pretreatment with GDNF markedly reduces cortical infarction induced by middle cerebral arterial ligation in aged rats.A, The right MCA was ligated for 40 min after bilateral common carotid arterial ligation in a control rat without PBS pretreatment. The brain tissue was removed 24 hr later and sliced coronally at 2 mm thickness for TTC staining. In this animal, an infarcted (white) zone in the right cerebral cortex was found in almost all the slices. B, A similar extent of cortical infarction was seen in another animal receiving MCA ligation and PBS pretreatment (10 μl, i.c.v.) and three local injections (5 μl × 3 sites). C, In a third, similarly anesthetized rat, GDNF was injected initially into the left lateral cerebral ventricle at a dose of 4 μg (0.4 μg/μl × 10 μl). An additional three injections of GDNF (0.4 μg/μl × 5 μl × 3 sites) were made directly into the cortex close to the MCA 30 min after removal of the squamosal bone. The TTC staining showed almost no cortical infarction in this animal after 40 min MCA ligation and 24 hr reperfusion.
Fig. 2.

Pretreatment with GDNF attenuates cortical infarction induced by MCA ligation for 40 min in aged rats.A, Bar graphs illustrate the incidence of infarction (number of animals with infarction/number of animals studied) in aged rats after 40 min MCA ligation and 24 hr reperfusion. The area of infarction was calculated after TTC staining. Cortical infarction was found in 11 of 11 control rats without PBS injection (−PBS) and six of six control rats with PBS pretreatment (+PBS). In contrast, only two of seven rats pretreated with GDNF (hatched bar) developed infarction. (*p < 0.05; χ2 test).B, Volume of infarction was reduced significantly by pretreatment with GDNF as compared with controls (p < 0.05; one-way ANOVA + Dunn’s test). The volume of infarction = 2 mm (thickness of the slice) × [sum of the infarction area in all brain slices (mm2)]. The infarction volumes for the GDNF-treated animals were averaged from all the GDNF-treated rats in this and the succeeding figure.C, The number of infarcted slices/rat was attenuated by GDNF (p < 0.05; one-way ANOVA + Dunn’s test). D, The area of the largest infarction in a slice from a given rat was not altered significantly by GDNF (p > 0.05; one-way ANOVA).
Similarly, we found that GDNF diminished the incidence of infarction in the young rats (393.2 ± 11.4 gm; n = 13) after a 90 min MCA ligation and 24 hr reperfusion (Fig. 3). In all the PBS-pretreated rats, MCA ligation induced brain infarction (n = 7). On the other hand, only two of the six rats pretreated with GDNF developed mild infarction (Fig. 3A) (p < 0.05; Fisher’s Exact test). In these young rats, GDNF significantly diminished the volume (Fig.3B) of cortical infarction (95.5 ± 17.5 mm3 vs 34.0 ± 26.3 mm3; p< 0.05; Mann–Whitney Rank Sum test) and the area of infarction (Fig.3D) in the most severely infarcted slice (14.1 ± 1.4 mm2 vs 3.7 ± 2.4 mm2; p< 0.05; t test).
Fig. 3.

Pretreatment with GDNF attenuates cortical infarction induced by 90 min of MCA occlusion in young rats.A, The incidence of infarction (number of animals with infarction/number of animals studied) in young rats after 90 min MCA ligation and 24 hr reperfusion was reduced significantly by GDNF pretreatment (hatched bar). Cortical infarction was found in seven of seven control rats pretreated with PBS. In contrast, only two of six rats pretreated with GDNF developed infarction (*p < 0.05; Fisher’s Exact test).B, Volume of infarction was reduced significantly by pretreatment with GDNF as compared with the controls (p < 0.05; Mann–Whitney Rank Sum test).C, The number of infarcted slices/rat was not altered by GDNF (p > 0.05; t test).D, The area of the largest infarction in a slice from a given rat was attenuated significantly by GDNF (p < 0.05; t test).
We have demonstrated previously that MCA ligation can induce cortical NO release (Lin et al., 1996). In the present study, we found that NO can be released from the cortex during both the ischemic and reperfusion periods. MCA ligation for 40 min gradually increased extracellular NO concentration to 20 nm, which then declined (Fig. 4). After removal of the arterial ligature, NO concentration again increased to 2 nm (Fig. 4). Intraparenchymal application of GDNF alone (0.4 μg/μl × 5 μl), or in conjunction with intraventricular administration of GDNF (0.4 μg/μl × 10 μl), significantly reduced the peak, rise time, and decay of NO production during ischemia and reperfusion (Fig. 4; Tables 1 and 2;p < 0.05; t test), suggesting that the production of NO was attenuated by GDNF pretreatment.
Table 1.
NO release during MCA ligation
| Control | GDNF (local) | GDNF (local + intracerebroventricular) | |
|---|---|---|---|
| NO (nm) | 17.7 ± 2.4 | 4.6 ± 2.1* | 1.4 ± 0.7* |
| Rise time (sec) | 397.1 ± 125.1 | 418.6 ± 240.3 | 115.4 ± 58.5* |
| T1/2(sec) | 784.7 ± 167.0 | 574.3 ± 332.9 | 232.4 ± 112.9* |
| N | 10 | 7 | 5 |
Significantly different from control, one-way ANOVA, and Newman-Keuls test (p < 0.05). Data are expressed as mean ± SEM. N, Number of animals.
Table 2.
NO release during reperfusion
| Control | GDNF (local, or local + intracerebroventricular) | |
|---|---|---|
| NO (nm) | 6.3 ± 1.4 | 0.5 ± 0.2* |
| Rise time (sec) | 537.3 ± 150.1 | 45.1 ± 24.1* |
| T1/2(sec) | 655.9 ± 158.5 | 66.6 ± 35.0* |
| N | 7 | 7 |
Significantly different from control, Student’s ttest (p < 0.05). Data are expressed as mean ± SEM.N, Number of animals.
Fig. 4.

GDNF markedly reduces NO release from the cortex during cerebral ischemia and reperfusion in aged rats.A1, Ischemia was induced by MCA ligation (arrow) for 40 min in this urethane-anesthetized rat. Extracellular NO concentration gradually increased to 20 nmand then declined. A2, Forty minutes after the cerebral ischemia, the arterial ligature was removed (arrow, reperfusion). NO concentration increased to 2.1 nm in this animal. B1, The production of NO during ischemia was reduced by GDNF (0.4 μg/μl × 5 μl) locally applied by microejection 5 min before MCA ligation. B2, NO increases, induced by reperfusion, were also reduced by the local application of GDNF. C1, Intraventricular injection of GDNF (0.4 μg/μl × 10 μl, 30 min before the ligation) and local application (0.4 μg/μl × 5 μl, 5 min before the ligation) attenuated the NO elevation during ischemia and (C2) during reperfusion. D, Bar graphs showing the peak NO concentration during MCA ligation. In the control animals (clear bar), ischemia elicited an average NO release of 17.7 ± 2.4 nm. Local application of GDNF significantly attenuated NO release (hatched bar; one-way ANOVA + Newman–Keuls test). Local plus intracerebroventricular injection of GDNF further diminished NO release (solid bar; p < 0.05). E, Bar graphs showing reperfusion-induced NO release in control (clear bar) and GDNF-pretreated rats (hatched bar). NO release was again significantly diminished by the GDNF pretreatment (p < 0.05).
DISCUSSION
It has been shown that no infarction is present 1 d after short-term MCA occlusion (e.g., 30 min) in Long–Evans rats. On the other hand, maximal infarction (∼150 mm3) is found 1 d after 90 min of MCA occlusion in young (300–350 gm) rats in this strain (Du et al., 1996). Previous experiments have demonstrated that body weight and/or age may play an important role in the degree of ischemia-induced infarction (Menzies et al., 1992). In the present study, we found that 40 min of ischemia is sufficient to induce maximal infarction (∼140 mm3) in relatively old Sprague Dawley rats (584.8 ± 16.4 gm), but 90 min is required in younger animals. Our results thus also suggest that weight (age) and the strain of the rat may interact with duration of ischemia to determine the degree of infarction.
In this study, we injected GDNF locally and intraventricularly shortly before MCA ligation. The MCA supplies blood to a wide cortical area. Local GDNF (5 μl × 3 sites) may protect a limited area in the cortex. The combination of local and intracerebroventricular injection of GDNF could increase GDNF concentrations locally but also over a wider area more congruent with the area of MCA perfusion. We have reported previously that NO is released shortly after ligation of the MCA (Lin et al., 1996). The time interval in this study was chosen to match the onset and duration of NO release that we found previously during ischemia.
Our data show a profound protective action of GDNF on ischemic-induced cerebral cortical injury. In this respect, it is of interest that fetal and neonatal brain tissue, which is more resistant to ischemia than adult CNS (Olson et al., 1984; Bickler, 1996), manifest much higher levels of GDNF mRNA expression (Stromberg et al., 1993; Springer et al., 1994). Moreover, the expression of GDNF mRNA in adult telencephalic structures can be upregulated by strong depolarizing inputs that activate receptors for excitatory amino acids. Recent studies have indicated that neuronal excitation may induce the expression of GDNF mRNA (Ho et al., 1995). The mRNA for GDNF in hippocampus or dentate can be upregulated in the adult rats after systemic injections of kainate (Humpel et al., 1994) or pilocarpine (Schmidt-Kastner et al., 1994). Pretreatment with high doses of dizocilpine maleate (MK801) blocks the kainate-induced seizures and the expression of GDNF mRNA (Humpel et al., 1994), suggesting that excitatory amino acids may be involved in this modulatory response. Taken together, these considerations suggest that GDNF may be part of an endogenous neuroprotective mechanism that could limit the extent of infarction after ischemia.
We and others have reported previously that porphyrine-coated electrodes have a high sensitivity for exogenously applied NO donors, such as SNAP or nitroprosside in vivo and in vitro (Malinski et al., 1993; Lin et al., 1996). We have also reported that the release of NO, as measured by chronoamperometric procedures at 0.9 V, during MCA ligation can be antagonized by the NOS inhibitor L-NAME (Lin et al., 1996). It has been found that catecholamines can be detected by nonporphyrine electrodes at 0.55 V (Gerhardt et al., 1984) after local injections of low doses of KCl in catecholamine-enriched areas, such as striatum, substantia nigra, or locus coeruleus. Application of KCl to the cortex did not induce electrochemical signals in this area. Taken together, these data indicate that the levels of dopamine or norepinephrine in the cortex are too low for chronoamperometric detection. In this study, the NO electrode was placed in the cortex (0.9 mm below the pial surface), which therefore would sense little or no signals from the catecholamines.
We found that NO release peaks at 397.1 ± 125.1 sec after MCA ligation and lasts for 20–30 min. This time course is consistent with the activity of NOS, which is increased sharply from baseline 10 min after MCA occlusion and then declines in 50 min (Kader et al., 1993).
Three NOS isoforms have been cloned and sequenced: two calcium-dependent constitutive isoforms, which are present in the endothelium and neurons, and one calcium-independent inducible isoform found mainly in activated immune cells and vascular smooth muscle. Animals treated with 7-nitroindazole, a selective inhibitor of neuronal NOS, showed a significant reduction in focal infarction after MCA occlusion (Yoshida et al., 1994). Infarct volumes of mice deficient in neuronal NOS activity were decreased significantly compared with those in normal mice after MCA occlusion. After inhibition of endothelial NO synthesis, however, the infarct size in the mutants became larger (Huang et al., 1994). Taken together, these data suggest that neuronal NO production may exacerbate acute ischemic injury, whereas endothelial NO may protect nerve cells after MCA occlusion.
It has been reported that NO generated in response to activation of NMDA receptors in vivo is neuronally derived and not caused by vascular production (Faraci and Breese, 1993). We found recently that local application of NMDA induces NO release in neuronal-enriched cortical cultures but not in the glial-enriched cultures. Furthermore, inhibition of synthesis of the R1 subunit of the NMDA receptors by treatment with antisense oligonucleotides prevents the neurotoxicity elicited by NMDA and reduces the volume of focal ischemic infarction produced by occlusion of the MCA (Wahlestedt et al., 1993). These data suggest that blocking NMDA receptors may limit injury during or after ischemic insults possibly via inhibition of neuronal NO production. Because NO release during the acute ischemic phase of MCA ligation is sensitive to NMDA antagonists (Lin et al., 1996) and GDNF, it is possible that GDNF inhibits NO release and infarction through inhibition of neuronal NOS activity during ischemia.
In conclusion, our data indicate that GDNF is a neuroprotectant not only for ventral mesencephalic DA neurons and for spinal motorneurons but also for cerebral cortex after ischemia. These findings open up a new pharmacotherapeutic approach to the treatment of stroke and other disorders of the cerebral vasculature.
Footnotes
This study was supported by the National Sciences Council of Taiwan, Republic of China, and the United States Public Health Service. We thank Synergen, Inc., for supplying the GDNF used in this study.
Correspondence should be addressed to Dr. Barry Hoffer, National Institute on Drug Abuse, 5500 Nathan Shock Drive, Baltimore, MD 21224.
REFERENCES
- 1.Basile DP, Rovak JM, Martin DR, Hammerman MR. Increased transforming growth factor-beta 1 expression in regenerating rat renal tubules following ischemic injury. Am J Physiol Renal. 1996;39:F500–F509. doi: 10.1152/ajprenal.1996.270.3.F500. [DOI] [PubMed] [Google Scholar]
- 2.Beck KD, Valverde J, Alexi T, Poulsen K, Moffat B, Vandlen RA, Rosenthal A, Hefti F. Mesencephalic dopaminergic neurons protected by GDNF from axotomy-induced degeneration in the adult brain. Nature. 1995;373:339–341. doi: 10.1038/373339a0. [DOI] [PubMed] [Google Scholar]
- 3.Benveniste H, Jorgensen MB, Diemer NH, Hansen AJ. Calcium accumulation by glutamate receptor activation is involved in hippocampal cell damage after ischemia. Acta Neurol Scand. 1988;78:529–536. doi: 10.1111/j.1600-0404.1988.tb03697.x. [DOI] [PubMed] [Google Scholar]
- 4.Bickler P. Anoxia-tolerant CA1 neurons from rat hippocampus: anoxia-evoked calcium changes and NMDA receptor inactivation. Soc Neurosci Abstr. 1996;22:3. [Google Scholar]
- 5.Bowenkamp KE, Hoffman AF, Gerhardt GA, Henry MA, Biddle PT, Hoffer BJ, Granholm AE. Glial cell line-derived neurotrophic factor supports survival of injured midbrain dopaminergic neurons. J Comp Neurol. 1995;355:479–489. doi: 10.1002/cne.903550402. [DOI] [PubMed] [Google Scholar]
- 6.Brown RD, Whisnant JP, Sicks JD, O’Fallon WM, Wiebers DO. Stroke incidence, prevalence, and survival: secular trends in Rochester, Minnesota, through 1989. Stroke. 1996;27:373–380. [PubMed] [Google Scholar]
- 7.Caldwell M, O’Neill M, Earley B, Leonard B. NG-Nitro-l-arginine protects against ischaemia-induced increases in nitric oxide and hippocampal neuro-degeneration in the gerbil. Eur J Pharmacol. 1994;260:191–200. doi: 10.1016/0014-2999(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 8.Chen ST, Hsu CY, Hogan EL, Maricq H, Balentine JD. A model of focal ischemic stroke in the rat: reproducible extensive cortical infarction. Stroke. 1986;17:738–743. doi: 10.1161/01.str.17.4.738. [DOI] [PubMed] [Google Scholar]
- 9.Dalkara T, Ayata C, Demirci M, Erdemli G, Onur R. Effects of cerebral ischemia on N-methyl-d-aspartate and dihydropyridine-sensitive calcium currents: an electrophysiological study in the rat hippocampus in situ. Stroke. 1996;27:127–133. doi: 10.1161/01.str.27.1.127. [DOI] [PubMed] [Google Scholar]
- 10.Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after mild focal cerebral ischemia: a role for apoptosis? J Cereb Blood Flow Metab. 1996;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Earley B, Canney M, Clune B, Caldwell M, Leonard BE, Junien JL. The effects of MK-801, ifenprodil, JO 1784, JO 1994 and JO 1997 on PK 11195 receptor binding, nitric oxide synthase (NO synthase) activity and infarct volume in a mouse model of focal cerebral ischaemia. Neurochem Int. 1996;28:509–521. doi: 10.1016/0197-0186(95)00144-1. [DOI] [PubMed] [Google Scholar]
- 12.Faraci FM, Breese KR. Nitric oxide mediates vasodilatation in response to activation of N-methyl-d-aspartate receptors in brain. Circ Res. 1993;72:476–480. doi: 10.1161/01.res.72.2.476. [DOI] [PubMed] [Google Scholar]
- 13.Feelisch M (1991) The biochemical pathways of nitric oxide formation from nitrovasodilators: appropriate choice of exogenous NO donors and aspects of preparation and handling of aqueous NO solutions. J Cardiovasc Pharmacol 17[Suppl 3]:S25–S33.
- 14.Gash DM, Zhang ZM, Ovadia A, Cass WA, Yi A, Simmerman L, Russell D, Martin D, Lapchak PA, Collins F, Hoffer BJ, Gerhardt GA. Functional recovery in parkinsonian monkeys treated with GDNF. Nature. 1996;380:252–255. doi: 10.1038/380252a0. [DOI] [PubMed] [Google Scholar]
- 15.Gerhardt GA, Oke AF, Nagy G, Moghaddam B, Adams RN. Nafion-coated electrodes with high selectivity for CNS electrochemistry. Brain Res. 1984;290:390–395. doi: 10.1016/0006-8993(84)90963-6. [DOI] [PubMed] [Google Scholar]
- 16.Hajimohammadreza I, Probert AW, Coughenour LL, Borosky SA, Marcoux FW, Boxer PA, Wang KK. A specific inhibitor of calcium/calmodulin-dependent protein kinase-II provides neuroprotection against NMDA- and hypoxia/hypoglycemia-induced cell death. J Neurosci. 1995;15:4093–4101. doi: 10.1523/JNEUROSCI.15-05-04093.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamada Y, Hayakawa T, Hattori H, Mikawa H. Inhibitor of nitric oxide synthesis reduces hypoxic-ischemic brain damage in the neonatal rat. Pediatr Res. 1994;35:10–14. doi: 10.1203/00006450-199401000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Henderson CE, Phillips HS, Pollock RA, Davies AM, Lemeulle C, Armanini M, Moffet B, Vandlen RA, Simpson LC, Koliatsos V, Rosenthal A. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 1994;266:1062–1064. doi: 10.1126/science.7973664. [DOI] [PubMed] [Google Scholar]
- 19.Ho A, Gore AC, Weickert CS, Blum M. Glutamate regulation of GDNF gene expression in the striatum and primary striatal astrocytes. NeuroReport. 1995;6:1454–1458. doi: 10.1097/00001756-199507100-00023. [DOI] [PubMed] [Google Scholar]
- 20.Hoffer BJ, Hoffman A, Bowenkamp K, Huettl P, Hudson J, Martin D, Lin LF, Gerhardt GA. Glial cell line-derived neurotrophic factor reverses toxin-induced injury to midbrain dopaminergic neurons in vivo. Neurosci Lett. 1994;182:107–111. doi: 10.1016/0304-3940(94)90218-6. [DOI] [PubMed] [Google Scholar]
- 21.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 22.Hudson J, Granholm AC, Gerhardt GA, Henry MA, Hoffman A, Biddle P, Leela NS, Mackerlova L, Lile JD, Collins F, Hoffer BJ. Glial cell line-derived neurotrophic factor augments midbrain dopaminergic circuits in vivo. Brain Res Bull. 1995;36:425–432. doi: 10.1016/0361-9230(94)00224-o. [DOI] [PubMed] [Google Scholar]
- 23.Humpel C, Hoffer B, Stromberg I, Bektesh S, Collins F, Olson L. Neurons of the hippocampal formation express glial cell line-derived neurotrophic factor messenger RNA in response to kainate-induced excitation. Neuroscience. 1994;59:791–795. doi: 10.1016/0306-4522(94)90284-4. [DOI] [PubMed] [Google Scholar]
- 24.Kader A, Frazzini VI, Solomon RA, Trifiletti RR. Nitric oxide production during focal cerebral ischemia in rats. Stroke. 1993;24:1709–1716. doi: 10.1161/01.str.24.11.1709. [DOI] [PubMed] [Google Scholar]
- 25.Knuckey NW, Finch P, Palm DE, Primiano MJ, Johanson CE, Flanders KC, Thompson NL. Differential neuronal and astrocytic expression of transforming growth factor beta isoforms in rat hippocampus following transient forebrain ischemia. Mol Brain Res. 1996;40:1–14. doi: 10.1016/0169-328x(96)00016-2. [DOI] [PubMed] [Google Scholar]
- 26.Kozniewska E, Roberts TP, Tsuura M, Mintorovitch J, Moseley ME, Kucharczyk J. NG-nitro-l-arginine delays the development of brain injury during focal ischemia in rats. Stroke. 1995;26:282–288. doi: 10.1161/01.str.26.2.282. [DOI] [PubMed] [Google Scholar]
- 27.Kumura E, Kosaka H, Shiga T, Yoshimine T, Hayakawa T. Elevation of plasma nitric oxide end products during focal cerebral ischemia and reperfusion in the rat. J Cereb Blood Flow Metab. 1994;14:487–491. doi: 10.1038/jcbfm.1994.60. [DOI] [PubMed] [Google Scholar]
- 28.Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- 29.Lin SZ, Chiou AL, Wang Y. Ketamine antagonizes nitric oxide release from cerebral cortex after middle cerebral artery ligation in rats. Stroke. 1996;27:747–752. doi: 10.1161/01.str.27.4.747. [DOI] [PubMed] [Google Scholar]
- 30.Malinski T, Bailey F, Zhang ZG, Chopp M. Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1993;13:355–358. doi: 10.1038/jcbfm.1993.48. [DOI] [PubMed] [Google Scholar]
- 31.Menotti A, Jacobs DR, Blackburn H, Kromhout D, Nissinen A, Nedeljkovic S, Buzina R, Mohacek I, Seccareccia F, Giampaoli S, Dontas A, Aravanis C, Toshima H. Twenty-five-year prediction of stroke deaths in the seven countries study: the role of blood pressure and its changes. Stroke. 1996;27:381–387. doi: 10.1161/01.str.27.3.381. [DOI] [PubMed] [Google Scholar]
- 32.Menzies SA, Hoff JT, Betz AL. Middle cerebral artery occlusion in rats: a neurological and pathological evaluation of a reproducible model. Neurosurgery. 1992;31:100–107. doi: 10.1227/00006123-199207000-00014. [DOI] [PubMed] [Google Scholar]
- 33.Olson L, Bjorklund H, Hoffer BJ. Camera bulbi anterior: new vistas on a classical locus for neural tissue transplantation. In: Sladek J, Gash D, editors. Neuronal transplants, development and function. Plenum; New York: 1984. pp. 373–406. [Google Scholar]
- 34.Perrella MA, Hsieh CM, Lee WS, Shieh S, Tsai JC, Patterson C, Lowenstein CJ, Long NC, Haber E, Shore S, Lee ME. Arrest of endotoxin-induced hypotension by transforming growth factor beta 1. Proc Natl Acad Sci USA. 1996;93:2054–2059. doi: 10.1073/pnas.93.5.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pichel JG, Shen L, Sheng HZ, Granholm A-C, Drago J, Grinberg A, Lee EJ, Huang SP, Saarmas M, Hoffer BJ, Sariola H, Westphal H. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- 36.Rastenyte D, Tuomilehto J, Sarti C, Cepaitis Z, Bluzhas J. Increasing trends in mortality from cerebral infarction and intracerebral hemorrhage in Kaunas, Lithuania. Cerebrovasc Dis. 1996;6:216–221. [Google Scholar]
- 37.Sato S, Tominaga T, Ohnishi T, Ohnishi ST. Electron paramagnetic resonance study on nitric oxide production during brain focal ischemia and reperfusion in the rat. Brain Res. 1994;647:91–96. doi: 10.1016/0006-8993(94)91402-8. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt-Kastner R, Tomac A, Hoffer B, Bektesh S, Rosenzweig B, Olson L. Glial cell-line derived neurotrophic factor (GDNF) mRNA upregulation in striatum and cortical areas after pilocarpine-induced status epilepticus in rats. Brain Res. 1994;26:325–330. doi: 10.1016/0169-328x(94)90106-6. [DOI] [PubMed] [Google Scholar]
- 39.Simon RP, Swan JH, Griffiths T, Meldrum BS. Blockade of N-methyl-d-aspartate receptors may protect against ischemic damage in the brain. Science. 1984;226:850–852. doi: 10.1126/science.6093256. [DOI] [PubMed] [Google Scholar]
- 40.Springer JE, Mu X, Bergmann LW, Trojanowski JQ. Expression of GDNF mRNA in rat and human nervous tissue. Exp Neurol. 1994;127:167–170. doi: 10.1006/exnr.1994.1091. [DOI] [PubMed] [Google Scholar]
- 41.Strijbos PJLM, Leach MJ, Garthwaite J. Vicious cycle involving Na+ channels, glutamate release, and NMDA receptors mediates delayed neurodegeneration through nitric oxide formation. J Neurosci. 1996;16:5004–5013. doi: 10.1523/JNEUROSCI.16-16-05004.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stromberg I, Bjorklund L, Johansson M, Tomac A, Collins F, Olson L, Hoffer B, Humpel C. Glial cell line-derived neurotrophic factor is expressed in the developing but not adult striatum and stimulates developing dopamine neurons in vivo. Exp Neurol. 1993;124:401–412. doi: 10.1006/exnr.1993.1214. [DOI] [PubMed] [Google Scholar]
- 43.Strosznajder J, Chalimoniuk M, Samochocki M, Gadamski R. Nitric oxide: a potent mediator of glutamatergic neurotoxicity in brain ischemia. Ann NY Acad Sci. 1994;723:429–432. [PubMed] [Google Scholar]
- 44.Tomac A, Lindqvist E, Lin LF, Ogren SO, Young D, Hoffer BJ, Olson L. Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature. 1995;373:335–339. doi: 10.1038/373335a0. [DOI] [PubMed] [Google Scholar]
- 45.Trok K, Hoffer B, Olson L. Glial cell line-derived neurotrophic factor enhances survival and growth of prenatal and postnatal spinal cord transplants. Neuroscience. 1996;71:231–241. doi: 10.1016/0306-4522(95)00412-2. [DOI] [PubMed] [Google Scholar]
- 46.Vornov JJ. Toxic NMDA-receptor activation occurs during recovery in a tissue culture model of ischemia. J Neurochem. 1995;65:1681–1691. doi: 10.1046/j.1471-4159.1995.65041681.x. [DOI] [PubMed] [Google Scholar]
- 47.Wahlestedt C, Golanov E, Yamamoto S, Yee F, Ericson H, Yoo H, Inturrisi CE, Reis DJ. Antisense oligodeoxynucleotides to NMDA-R1 receptor channel protect cortical neurons from excitotoxicity and reduce focal ischaemic infarctions. Nature. 1993;363:260–263. doi: 10.1038/363260a0. [DOI] [PubMed] [Google Scholar]
- 48.Williams LR, Inouye G, Cummins V, Pelleymounter MA. Glial cell line-derived neurotrophic factor sustains axotomized basal forebrain cholinergic neurons in vivo: dose-response comparison to nerve growth factor and brain-derived neurotrophic factor. J Pharmacol Exp Ther. 1996a;277:1140–1151. [PubMed] [Google Scholar]
- 49.Williams LR, Inouye G, Cummins V, Du C. ICV pretreatment with GDNF reduces infarct volume in a rat model of transient middle cerebral artery occlusion. Soc Neurosci Abstr. 1996b;22:1667. [Google Scholar]
- 50.Yan Q, Matheson C, Lopez OT. In vivo neurotrophic effects of GDNF on neonatal and adult facial motor neurons. Nature. 1995;373:341–344. doi: 10.1038/373341a0. [DOI] [PubMed] [Google Scholar]
- 51.Yoshida T, Limmroth V, Irikura K, Moskowitz MA. The NOS inhibitor, 7-nitroindazole, decreases focal infarct volume but not the response to topical acetylcholine in pial vessels. J Cereb Blood Flow Metab. 1994;14:924–929. doi: 10.1038/jcbfm.1994.123. [DOI] [PubMed] [Google Scholar]
- 52.Zhang ZG, Reif D, MacDonald J, Tang WX, Kamp DK, Gentile RJ, Shakespeare WC, Murray RJ, Chopp M. ARL 17477, a potent and selective neuronal NOS inhibitor decreases infarct volume after transient middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab. 1996;16:599–604. doi: 10.1097/00004647-199607000-00009. [DOI] [PubMed] [Google Scholar]