

Abstract
Inducible nitric oxide synthase (iNOS), an enzyme that produces toxic amounts of nitric oxide, is expressed in a number of brain pathologies, including cerebral ischemia. We used mice with a null mutation of the iNOS gene to study the role of iNOS in ischemic brain damage. Focal cerebral ischemia was produced by occlusion of the middle cerebral artery (MCA). In wild-type mice, iNOS mRNA expression in the post-ischemic brain begun between 24 and 48 hr peaked at 96 hr and subsided 7 d after MCA occlusion. iNOS mRNA induction was associated with expression of iNOS protein and enzymatic activity. In contrast, mice lacking the iNOS gene did not express iNOS message or protein after MCA occlusion. The infarct and the motor deficits produced by MCA occlusion were smaller in iNOS knockouts than in wild-type mice (p < 0.05). Such reduction in ischemic damage and neurological deficits was observed 96 hr after ischemia but not at 24 hr, when iNOS is not yet expressed in wild-type mice. The decreased susceptibility to cerebral ischemia in iNOS knockouts could not be attributed to differences in the degree of ischemia or vascular reactivity between wild-type and knockout mice. These findings indicate that iNOS expression is one of the factors contributing to the expansion of the brain damage that occurs in the post-ischemic period. iNOS inhibition may provide a novel therapeutic strategy targeted specifically at the secondary progression of ischemic brain injury.
Keywords: cerebral ischemia, knockout mice, gene expression, neuroprotection, stroke, reverse-transcription PCR
Inducible nitric oxide synthase (iNOS) is an enzyme that produces toxic levels of nitric oxide (NO) (for review, see Gross and Wolin, 1995). NO produced by iNOS is responsible for the toxicity of activated microglia–macrophages and aggravates glutamate-mediated neuronal damage in vitro (Chao et al., 1992; Dawson et al., 1994; Hewett et al., 1994). In brain, iNOS is expressed in several pathological states, including tumors, trauma, demyelination, AIDS dementia, Alzheimer’s disease, and cerebral ischemia (Endoh et al., 1994; Bagasra et al., 1995; Iadecola et al., 1995b; Adamson et al., 1996; Clark et al., 1996; Hara et al., 1996;Vodovotz et al., 1996). However, the role that NO produced by iNOS plays in the mechanisms of these brain diseases has not been defined.
In cerebral ischemia produced by occlusion of the rat middle cerebral artery (MCA), iNOS expression begins 12 hr after induction of ischemia, peaks at 48 hr, and subsides at 7 d (Iadecola et al., 1995b). Investigations using the iNOS inhibitor aminoguanidine have suggested that iNOS expression may contribute to ischemic brain injury (for review, see Iadecola, 1997). Administration of aminoguanidine 24 hr after MCA occlusion reduces the volume of the resulting infarct by ∼30% (Iadecola et al., 1995a). These data raise the possibility that NO produced by iNOS contributes to the late stages of ischemic brain damage. However, this evidence is far from conclusive because aminoguanidine also has neuroprotective effects unrelated to iNOS inhibition. These include inhibition of advanced glycation endproducts formation (Zimmerman et al., 1995) and inhibition of diamino oxidase, an enzyme that produces toxic aldehydes from polyamines oxidation (Brunton et al., 1994; Sessa and Perin, 1994). Therefore, the role of iNOS expression in the mechanisms of cerebral ischemia remains to be firmly established.
Mice with a null mutation of the iNOS gene have recently been developed using homologous recombination (MacMicking et al., 1995; Wei et al., 1995). These mice are relatively protected from hypotension and death resulting from septic shock and from the deleterious effects of carrageenin-induced local inflammation (MacMicking et al., 1995; Wei et al., 1995). In the present study, therefore, we used iNOS null mice (MacMicking et al., 1995) to investigate the role of iNOS expression in ischemic brain damage. We found that the knockouts do not express iNOS after focal cerebral ischemia, develop smaller infarcts, and have less pronounced neurological deficits than wild-type controls. The reduction in infarct volume and neurological deficits is observed at 96 hr but not at 24 hr after induction of ischemia. The data provide nonpharmacological evidence that NO produced by iNOS plays an important role in the delayed progression of ischemic brain damage and provide the rational basis for using inhibition of iNOS as a treatment for stroke.
MATERIALS AND METHODS
Animals
All animal procedures were approved by the Animal Care Committee of the University of Minnesota. C57BL/6 and SV129 mice were obtained from Jackson Laboratories (Bar Harbor, ME). Breeding pairs of iNOS knockouts were kindly provided by Drs. C. Nathan (Cornell University Medical College) and J. Mudgett (Merck Research Laboratories) (MacMicking et al., 1995). Mice were bred to obtain a colony of homozygous iNOS knockouts. After each experiment, knockouts were genotyped to verify the lack of the iNOS gene and the presence of the targeting vector.
Induction of focal cerebral ischemia
Focal cerebral ischemia was produced by occlusion of the MCA (Zhang and Iadecola, 1992). Mice were anesthetized with 2% halothane in 100% oxygen. Body temperature was maintained at 37 ± 0.5°C by a thermostatically controlled infrared lamp. A 2 mm hole was drilled in the inferior portion of the temporal bone to expose the left MCA. The MCA was elevated and cauterized distal to the origin of the lenticulostriate branches (Zhang and Iadecola, 1992). Wounds were sutured, and mice were allowed to recover and then returned to their cages. Rectal temperature was controlled until mice regained full consciousness. Mice were killed at different time points after MCA occlusion for determination of mRNA, for the iNOS assay, for immunocytochemistry, or measurement of infarct size (see below).
Determination of mRNA by reverse transcription (RT)-PCR
iNOS or COX-2 mRNA was detected in the ischemic brain using RT-PCR, as described previously (Iadecola et al., 1996; Ross and Iadecola, 1996; Nogawa et al., 1997). Briefly, a 2-mm-thick coronal brain slice was cut at the level of the optic chiasm, and the infarcted cortex was dissected using the corpus callosum as a ventral landmark. The homotopic region of the contralateral cortex was also sampled. Total RNA was extracted, and the integrity of the RNA was determined on denaturing formaldehyde gels. First-strand cDNA synthesis was then performed using 0.25, 0.5, and 1.0 μg of total RNA, oligo dT primer (BRL, Bethesda, MD) and Moloney Murine leukemia virus reverse transcriptase (New England BioLabs, Beverly, MA). Aliquots (5 μl each) from the RT reaction were then used for PCR amplification with primer pairs for the mRNA of interest and the ubiquitously expressed control sequence porphobilinogen deaminase (PBD). The iNOS primer sequences used were Forward, 5′-ACAACGTGAAGAAAACCCCTTGTG-3′; Reverse, 5′-ACAGT TCCGAGCGTCAAAGACC-3′. The COX-2 primers were Forward, 5′-CCATGTCAAAACCGTGGTGAATG-3′; Reverse, 5′-ATGGG AGTTGGGCAGTCATCAG-3′. The “hot start” method was used (Stratagene, La Jolla, CA) with the following cycle parameters: 94°C, 15 sec; 65°C, 30 sec; 73°C, 25 sec, × 5 cycles; then 94°C, 15 sec; 60°C, 30 sec; 73°C, 25 sec, × 35 cycles. Reaction products were then separated on an 8% acrylamide gel, ethidium-stained, and photographed. The PCR products were sequenced to confirm their identity. Each set of PCR reactions included control samples run without RNA/RT. Relative optical density (OD) of the bands was measured using an image analysis system (M4, Imaging Research Inc.). All measurements were normalized to the relative OD of the PBD band (Iadecola et al., 1995b; Nogawa et al., 1997).
iNOS enzymatic activity and immunocytochemistry
iNOS catalytic activity of the brain samples was determined by the citrulline assay of Bredt and Snyder modified for detection of calcium-independent activity as described previously (Ross and Iadecola, 1996). Immunocytochemical procedures were identical to those described previously (Iadecola et al., 1996; Nogawa et al., 1997). Sections (7 μm) from formalin-fixed, paraffin-embedded brains were incubated overnight (4°C) with an iNOS polyclonal antibody (Upstate Biotechnology, Lake Placid, NY) (dilution 1:200), washed, and incubated with the secondary antibody (Vector, Laboratories, Burlingame, CA) for 30 min. The immunocomplex was visualized using the ABC complex (Vectastain Elite Kit, Vector). Alternate sections were processed for immunocytochemistry for glial fibrillary acidic protein (GFAP) (Boehringer Mannheim, Indianapolis, IN) or myeloperoxidase (Dako, Carpinteria, CA).
Determination of infarct volume
Mice were killed 24 or 96 hr after MCA occlusion. Brains were removed and frozen in cooled isopentane (−30°C). Coronal forebrain sections (thickness 30 μm) were serially cut in a cryostat, collected at 150 μm intervals, and stained with thionin for determination of infarct volume by an image analyzer (MCID, Imaging Research) (Zhang and Iadecola, 1992). To factor out the contribution of ischemic edema to the total volume of the lesion, infarct volume in cerebral cortex was corrected for swelling as described previously (Zhang and Iadecola, 1994; Iadecola et al., 1995a). The correction method is based on the determination of ischemic swelling by comparing the volume of ischemic and nonischemic hemispheres (Lin et al., 1993).
Determination of neurological deficits
Neurological deficits were assessed by a neurological scoring system widely used in mice (Huang et al., 1994; Yang et al., 1994). Because distal MCA occlusion produced no neurological deficits measurable by this scale, in these experiments the MCA was occluded at a more proximal site, a procedure that results in a larger infarct area and measurable neurological deficits. The examiner was not aware of the identity of the mice. The neurological scores were as follows: 0, normal motor function; 1, flexion of torso and contralateral forelimb when mouse was lifted by the tail; 2, circling to the contralateral side when mouse was held by the tail on a flat surface, but normal posture at rest; 3, leaning to the contralateral side at rest; 4, no spontaneous motor activity (Huang et al., 1994; Yang et al., 1994). Mice were evaluated before MCA occlusion and at 24 hr intervals up to 96 hr after MCA occlusion.
Determination of cerebral blood flow (CBF)
Techniques used for studying the cerebral circulation in mice were similar to those described previously (Dalkara et al., 1995; Sobey and Faraci, 1997). Mice were anesthetized with halothane (maintenance 1%), and the femoral artery and trachea were cannulated. Mice were artificially ventilated with an oxygen–nitrogen mixture by a mechanical ventilator (SAR-830, CWI Inc., Ardmore, PA). The inspiration time was set at 0.1 sec, the respiratory rate at 120/min, and the inspiratory flow at ∼250 ml/min. The oxygen concentration in the mixture was adjusted to obtain an arterial PO2of 150–170 mmHg. End-tidal CO2 was monitored continuously using a CO2 analyzer (Capstar-100, CWI Inc.). The sample flow rate of the CO2 analyzer was set at 10 cc/min. Throughout the experiment, end-tidal CO2 was maintained at 2.6–2.7%, which corresponds to a PCO2 of 33–35 mmHg, as determined in preliminary studies in which end-tidal CO2 was compared with arterial PCO2measured by a blood gas analyzer.
MCA occlusion. For monitoring of the changes in CBF produced by MCA occlusion, two laser–Doppler flow probes (Vasamedic, St. Paul, MN) were inserted through burr holes placed in the center (3.5 mm lateral to the midline and 1 mm caudal to bregma) and the periphery (1.5 mm lateral to the midline and 1.7 mm rostral to lambda) of the ischemic territory (Chan et al., 1993). The location of the probe was selected in preliminary experiments to correspond to the region of brain that is spared from infarction in the iNOS knockouts. After placement of the probes, the MCA was occluded, and CBF was monitored for 90 min. CBF data are expressed as percentage of the preocclusion value.
Cerebrovascular reactivity to hypercapnia. Techniques for testing cerebral vascular reactivity to hypercapnia in mice were similar to those described previously in rats (Iadecola, 1992; Iadecola and Zhang, 1996). Mice were anesthetized and instrumented as described above. CBF was monitored continuously in the frontoparietal cortex with a laser–Doppler probe. After stabilization of arterial pressure and blood gases, CO2 was introduced into the circuit of the ventilator, and the increase in CBF produced by hypercapnia was monitored. CO2 administration was discontinued after the CBF increase reached a plateau (usually 2–3 min).
Data analysis
Data in text and figures are expressed as mean ± SE. Multiple comparisons were statistically evaluated by the ANOVA and Tukey’s test. Two-group comparisons were analyzed by the two-tailedt test for independent samples. Neurological scores were evaluated by nonparametric statistical procedures. Two group comparisons were analyzed by the Mann–Whitney U analysis (Huang et al., 1994; Yang et al., 1994), and multiple contrasts were analyzed by the Kruskal–Wallis ANOVA followed by the Tukey–Kramer test (Systat, Evanston, IL). For all procedures, probability values of <0.05 were considered statistically significant.
RESULTS
iNOS expression in wild-type and null mice
In wild-type mice, occlusion of the MCA induced expression of iNOS message (n = 4/time point) and enzymatic activity (n = 4/time point) (Fig.1). mRNA expression began between 24 and 48 hr, peaked at 96 hr, and subsided 7 d after MCA occlusion (Fig.1A). iNOS immunoreactivity was observed in mono- and polynuclear inflammatory cells invading the infarct border (Fig.2B). In contrast, MCA occlusion did not induce expression of message or protein in iNOS knockouts (Fig. 2A,B).
Fig. 1.
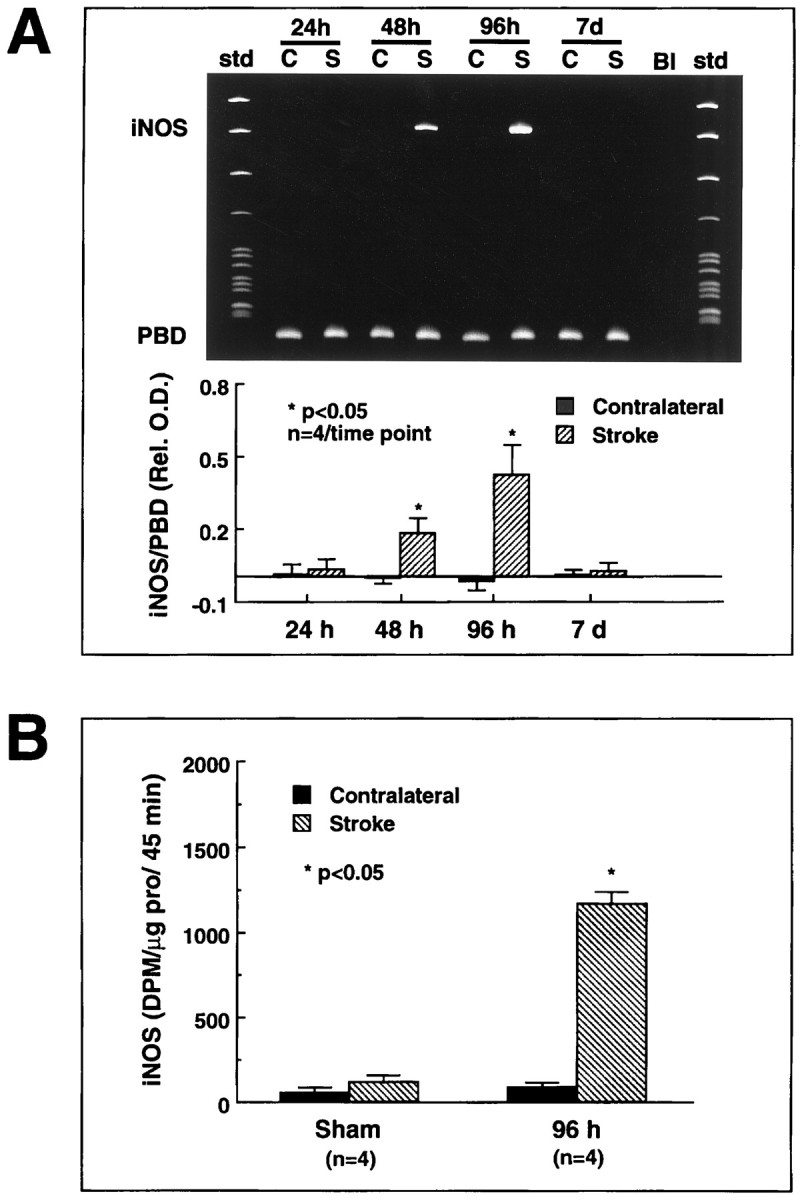
Expression of iNOS message and enzymatic activity after cerebral ischemia in B6 mice. A, Expression of iNOS message after focal cerebral ischemia. iNOS mRNA was determined by RT-PCR. A strong iNOS signal is observed at 48 and 96 hr after ischemia (top panel). The bottom panelshows the time course of the ratio of the optical density of the iNOS band by the density of the band corresponding to porphobilinogen deaminase (PBD), a housekeeping gene used for normalization. Significant elevation is observed at 48 and 96 hr after ischemia (p < 0.05; ANOVA and Tukey’s test). No expression is observed at 24 hr or 7 d (p > 0.05). B, iNOS enzymatic activity after cerebral ischemia. iNOS activity was measured using the citrulline conversion assay of Bredt and Snyder modified for detection of calcium-independent arginine-to-citrulline conversion, i.e., iNOS activity (Ross and Iadecola, 1996). Virtually no iNOS activity was observed in sham-operated mice. Substantial iNOS activity developed in the ischemic region 96 hr after MCA occlusion. The 96 hr time point was chosen because at this time iNOS message is maximal. These findings suggest that cerebral ischemia leads to expression of iNOS message and enzymatic activity in the injured brain.
Fig. 2.
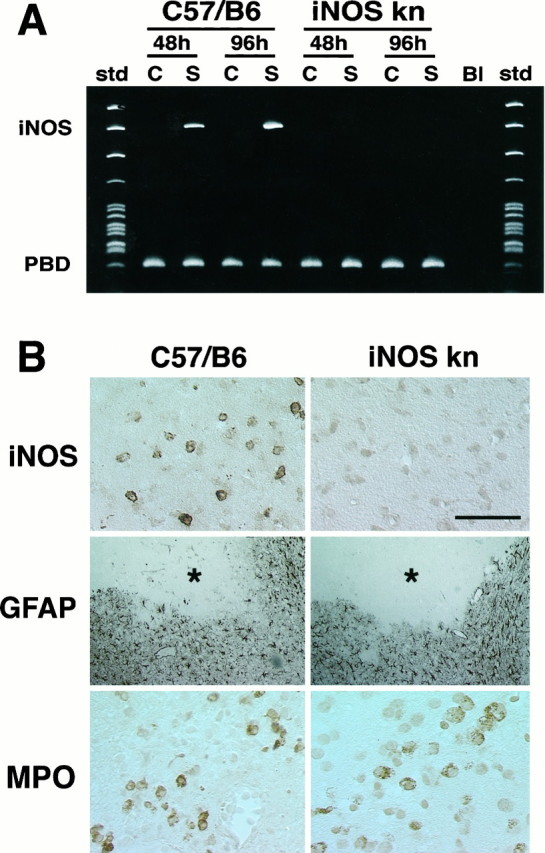
A, iNOS message detected by RT-PCR in wild-type mice (C57/B6) and iNOS knockouts at 48 and 96 hr after ischemia. No iNOS signal is detected in the ischemic brain at either time points. B, Immunoreactivity foriNOS, glial fibrillary acidic protein (GFAP), or myeloperoxidase (MPO) in iNOS null mice and controls (C57/B6) 96 hr after MCA occlusion. Cerebral ischemia is followed by expression of iNOS immunoreactivity in C57/B6 mice but not in iNOS knockouts (top panels). Immunoreactivity for the astrocytic marker GFAP (middle panels) is comparable between iNOS null mice and wild-type controls. The asterisk indicates the infarcted area. GFAP expression occurs at the border of the lesion. To detect infiltrating inflammatory cells in the post-ischemic brain, immunocytochemistry for MPO, an enzyme expressed in polymorphonuclear cells infiltrating the ischemic brain, was performed. Infiltrating myeloperoxidase-positive cells are observed both in iNOS null mice and in controls (bottom panels). Scale bar (shown intop right panel in B): topand bottom panels, 50 μm; middle panel, 250 μm.
To determine whether other inflammation-related genes were upregulated in the iNOS null mice, the mRNA expression of cyclooxygenase-2 (COX-2) was studied by RT-PCR. COX-2 is a prostaglandin-synthesizing enzyme that is induced in inflammatory states, including post-ischemic inflammation (Nogawa et al., 1997). Forty-eight hours after MCA occlusion, COX-2 mRNA was upregulated equally in iNOS null mice (0.25 ± 0.05 relative OD; n = 4) and in wild-type controls (0.23 ± 0.06; n = 4; p> 0.05; t test). The infiltration of neutrophils, as revealed by myeloperoxidase immunocytochemistry, and the expression of the astroglial marker GFAP were comparable in iNOS null mice and wild-type controls (Fig. 2B). These data indicate that iNOS expression does not occur in the post-ischemic brain of iNOS null mice, whereas COX-2 expression and the cellular reaction to cerebral ischemia are not affected.
Infarct volume in wild-type and iNOS null mice
We then studied the outcome of cerebral ischemia in iNOS null mice. The MCA was occluded, and the volume of the infarct was measured 96 hr later. Infarct volume was 28% smaller in iNOS knockouts than in wild-type controls (C57/B6 strain) (Figs.3, 4). The genetic background of iNOS knockouts is mixed, including both the C57/B6 and SV129 strains (MacMicking et al., 1995). To examine the possibility that the reduction in ischemic damage was related to a reduced susceptibility to ischemic injury of the SV129 strain, we compared stroke size in iNOS null mice and SV129 mice. Cortical infarct volumes (corrected for swelling) in iNOS null mice (18 ± 1 mm3; n = 6) were also smaller than those of SV129 mice (23 ± 0.6 mm3;n = 6; p < 0.05).
Fig. 3.
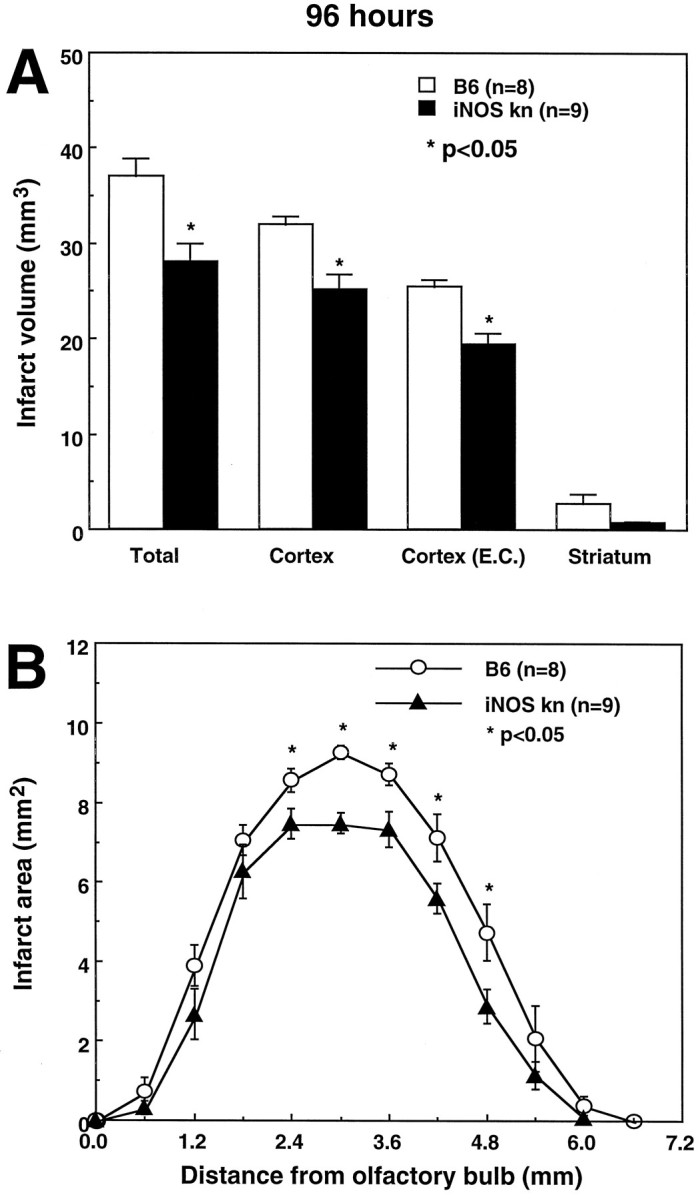
A, Infarct volume in wild-type controls (C57/B6) and iNOS knockouts killed 96 hr after occlusion of the MCA. Infarct size is smaller in iNOS knockouts (p < 0.01; t test). The difference persists after correction for ischemic swelling [Cortex (E.C.)]. B, Distribution of the infarcted areas at different rostrocaudal levels in wild-type controls (C57/B6) and iNOS knockouts. The area of infarction is smaller in the iNOS knockouts at all rostrocaudal levels. Values for iNOS null mice and controls were compared at each rostrocaudal level by the unpairedt test.
Fig. 4.

Representative brain sections illustrating the distribution of the ischemic damage in wild-type mice (C57/B6) and iNOS knockouts 96 hr after MCA occlusion. The area of infarction involves the cerebral cortex almost exclusively. Note that the infarct is smaller in the knockouts and that the area spared from infarction involves the peripheral regions of the infarct. This peripheral region represents the so-called ischemic penumbra.
The observation that iNOS expression occurs >24 hr after cerebral ischemia (Fig. 1) suggests that iNOS may play a role in the delayed evolution of the damage. To provide evidence in support of this hypothesis, infarct volume was determined in iNOS null mice killed 24 hr after MCA occlusion. At this time, iNOS is not yet expressed in the post-ischemic brain of wild-type controls (Fig. 1A). At 24 hr after MCA occlusion, stroke volume in iNOS null mice was not reduced compared with wild-type controls (Fig.5A). Therefore, the reduction in infarct size in iNOS knockouts is observed 96 hr but not 24 hr after MCA occlusion.
Fig. 5.
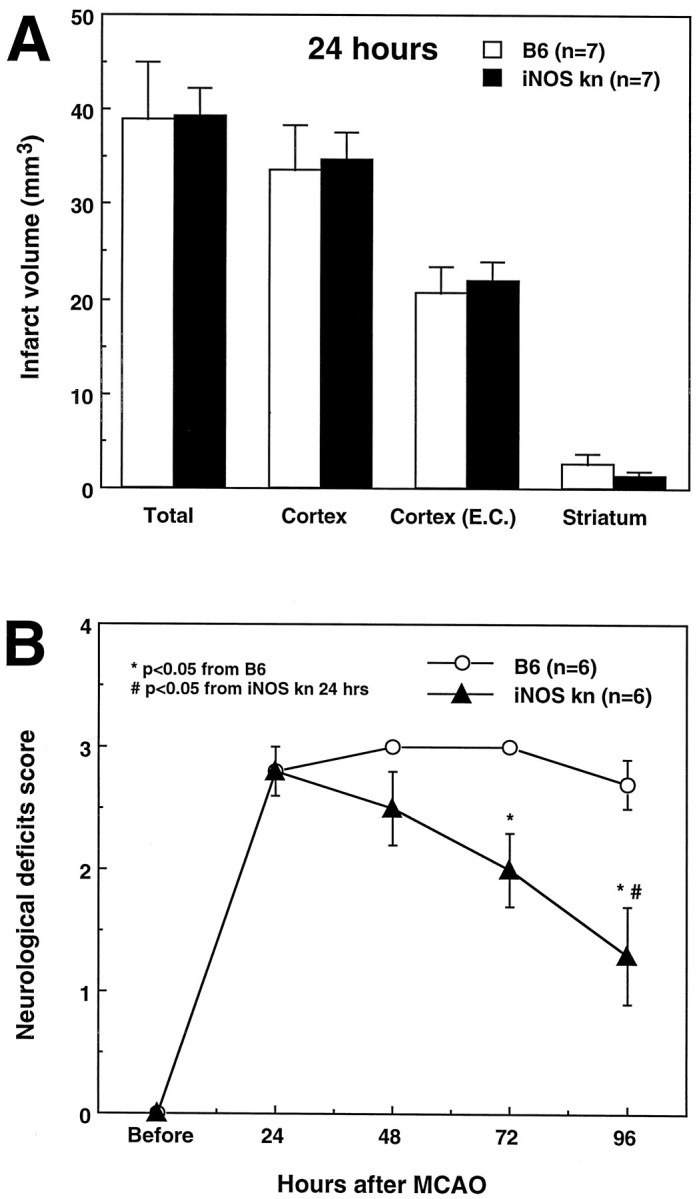
A, Infarct volume in wild-type controls (C57/B6) and iNOS knockouts killed 24 hr after occlusion of the MCA. Data were corrected for ischemic swelling [Cortex (E.C.)]. Infarct volume is not different between wild-type mice and iNOS knockouts (p > 0.05;t test). B, Neurological deficits in wild-type mice (C57/B6) and iNOS knockouts. Mice were examined before induction of ischemia and at 24 hr intervals until the time they were killed 96 hr after MCA occlusion. Higher neurological scores indicate more severe deficits. Neurological scores were not different 24 hr after MCA occlusion (p > 0.05; Mann–Whitney U test). However, although the scores remained stable in wild-type mice during the following 96 hr (p > 0.05; Kruskal–Wallis ANOVA), they improved markedly in the iNOS knockouts (p< 0.05 from wild-type mice at 96 hr and p < 0.05 from 24 hr).
Motor deficits in wild-type and iNOS null mice
To determine whether the reduction in infarct size corresponds to a functional improvement, mice were evaluated using a neurological scale for rodents (Huang et al., 1994; Yang et al., 1994). Twenty-four hours after ischemia, neurological scores did not differ between wild-type controls (C57/B6) and knockouts (Fig. 5B) (p > 0.05). Although the deficits remained stable between 24 and 96 hr in controls (p > 0.05; Kruskal–Wallis ANOVA), in iNOS knockouts the deficits improved steadily. At 96 hr after MCA occlusion, the deficits were significantly smaller than those of wild-type mice (p < 0.05; Mann–Whitney U test). Therefore, the iNOS knockouts have a better functional outcome after MCA occlusion.
CBF in wild-type and iNOS null mice
In these experiments, the effect of MCA occlusion on CBF and the CBF reactivity to hypercapnia were studied in wild-type and iNOS null mice. MCA occlusion resulted in a comparable flow reduction in wild-type mice (C57/B6) and iNOS knockouts both in the center and at the periphery of the ischemic territory (Fig.6). Similarly, in intact mice, the increase in CBF produced by hypercapnia (PCO2 = 50–60 mmHg) did not differ between wild-type controls and iNOS null mice (wild type, +74 ± 3%; null mice, +71 ± 3% of baseline; p > 0.05; n = 6/group). These data indicate that the degree of ischemia induced by MCA occlusion and the vascular reactivity to hypercapnia are comparable between wild-type mice and iNOS knockouts.
Fig. 6.
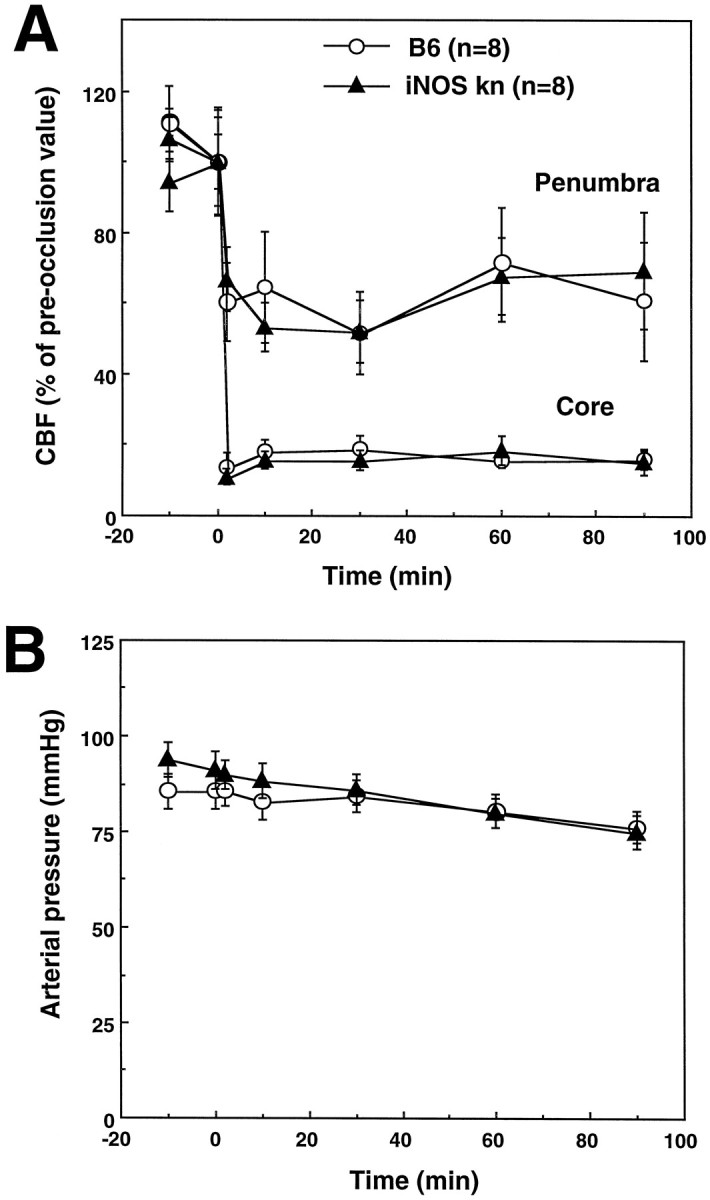
A, Effect of MCA occlusion on CBF in iNOS knockouts and wild-type controls (C57/B6). The reduction in CBF produced by MCA occlusion is similar in the two groups both in the center of the ischemic territory (Core) and at the periphery (Penumbra). B, Arterial pressure of the mice before and after MCA occlusion. Arterial pressure did not differ between controls and iNOS knockouts (p > 0.05).
DISCUSSION
We have demonstrated that knockout mice lacking the iNOS gene do not express iNOS message or protein after MCA occlusion and have smaller infarcts and reduced motor deficits in comparison to wild-type controls. The effect is not observed 24 hr after ischemia but becomes apparent at 96 hr. These data provide nonpharmacological evidence that iNOS expression contributes to the development of the tissue damage and neurological deficits resulting from cerebral ischemia.
The reduction in infarct size cannot be attributed to hemodynamic–vascular factors, because the CBF decrease produced by MCA occlusion and the reactivity of the cerebral circulation to hypercapnia did not differ between wild-type and knockouts. Similarly, the protection in the knockouts is not caused by a reduced inflammatory reaction, because the expression of another inflammation-related gene, COX-2, was not reduced in the knockouts. In addition, the degree of cellular infiltration and glial activation was comparable in wild-type and knockout mice. Strain differences in susceptibility to ischemic injury are also unlikely to play a role, because the infarct in the knockouts was smaller than that of both C57/B6 and SV129 mice. Therefore, the reduction in ischemic damage in the null mice is likely to result from the absence of iNOS expression and not from other factors. It must be pointed out, however, that comparisons with both parent strains individually do not completely rule out factors related to the mixed genetic background of the null mice (Gerlai, 1996). In addition, compensatory changes in the null mice secondary to the lack of the iNOS gene cannot be excluded.
The expression of iNOS begins >24 hr after induction of ischemia and peaks at 96 hr. Furthermore, the reduction in infarct size and the improvement in neurological deficits is observed at 96 hr but not at 24 hr after MCA occlusion. These data indicate that iNOS does not participate in the initiation of ischemic brain damage. Rather, the evidence suggests that iNOS is involved in the mechanisms of the delayed evolution of the damage that occurs after focal cerebral ischemia. Most studies to date have focused on pathogenic factors acting in the early stages of ischemic brain damage. Thus, glutamate excitotoxicity, calcium overload, and free radical damage have been extensively investigated as factors initiating ischemic brain death (Choi, 1990; Siesjo, 1994; Chan, 1996). Recent evidence, however, indicates that the ischemic damage continues to progress well after the onset of ischemia. For example, magnetic resonance imaging (MRI) and positron emission tomography (PET) studies have demonstrated that the area of irretrievable damage increases in size for days after the onset of ischemia (Heiss et al., 1992; Furlan et al., 1996; Marchal et al., 1996; Baird et al., 1997). This evidence has challenged the concept of a rigid therapeutic window of 4–6 hr and has raised the possibility that treatments can be instituted many hours after the onset of ischemia (Baron et al., 1995). The findings of the present study identify iNOS expression as one of the factors contributing to the delayed evolution of ischemic brain damage. In particular, they suggest that large amounts of NO produced by iNOS in inflammatory cells infiltrating ischemic tissue contribute to the expansion of the infarct that occurs in the post-ischemic period.
In apparent contrast with the hypothesis that ischemic brain damage evolves during the post-ischemic period is the observation that the infarct volume of wild-type mice, as estimated by the area of pallor in thionin-stained sections, does not increase between 24 and 96 hr after MCA occlusion (compare Figs. 3 and 5). Similarly, the neurological deficits do not worsen during this period. These findings, however, are not surprising, because the pale area at 24 hr represents brain tissue that is functionally compromised but not entirely necrotic (Garcia et al., 1995). Although necrosis develops rapidly in the center of the ischemic territory, it progresses at a much slower pace at the periphery of the infarct (Dereski et al., 1993; Garcia et al., 1993). MRI and PET studies have demonstrated that the volume of permanently damaged brain grows over several days after ischemia (Heiss et al., 1992; Baird et al., 1997). These imaging data, therefore, support the histological evidence of a slow progression of irreversible tissue damage at the periphery of the ischemic territory. The lack of progression of the neurological deficits between 24 and 96 hr can be attributed to the fact that the deficits are already maximal at 24 after MCA occlusion because of ischemia-induced brain dysfunction (Astrup et al., 1977). Although in wild-type mice the volume of pallor remains stable between 24 and 96 hr, in the knockouts, the volume at 96 hr is smaller than at 24 hr. The reduction in lesion size in the post-ischemic period indicates that the growth of the infarct is less pronounced in the knockouts. Importantly, the reduction in infarct volume is accompanied by an improvement in neurological deficits. The correspondence between reduction in infarct volume and neurological deficits in the knockouts indicates that the brain tissue spared from infarction is indeed functional.
Previous studies have indicated that NO is also involved in the acute stages of ischemic brain damage (Dawson et al., 1991; Nowicki et al., 1991; for review, see Iadecola, 1997). Shortly after induction of ischemia, NO is thought to be produced in response to activation of glutamate receptors, which in turn leads to sustained activation of neuronal NOS (Malinski et al., 1993; Lin et al., 1996). Accordingly, early treatment with inhibitors of neuronal NOS reduces ischemic damage, whereas knockout mice lacking the neuronal NOS gene have smaller infarcts (Huang et al., 1994; Yoshida et al., 1994; Nagafuji et al., 1995; Zhang et al., 1996). The present study suggests that NO plays a role also in the late stages of cerebral ischemia. Therefore, NO contributes to both the initiation and the progression of ischemic injury.
The mechanisms by which NO produced by iNOS contributes to ischemic brain damage are unknown. However, it is well established that large amounts of NO are cytotoxic, an effect exerted through multiple mechanisms. These include peroxynitrite-mediated oxidative damage, DNA damage, and energy failure resulting from inhibition of critical energy-producing enzymes and from poly(ADP-ribose)synthase activation (Beckman et al., 1990; Nguyen et al., 1992; Zhang et al., 1994; Gross et al., 1996). It is therefore likely that NO produced by iNOS in the late post-ischemic period affects the survival of potentially viable neurons at the infarct border. This suggestion is strengthened by the observation that iNOS expression worsens the neurotoxicity produced by activation of glutamate receptors or oxygen–glucose deprivation in neuronal cultures (Dawson et al., 1994; Hewett et al., 1994, 1996).
The findings of the present study have important implications for the pathophysiology and treatment of ischemic stroke. First, they provide evidence that ischemic brain damage evolves over a period of days and that retrievable brain tissue is present >12 hr after the onset of ischemia. This conclusion is also supported by recent imaging studies in humans demonstrating that ∼30% of ischemic brain tissue remains viable for >12 hr after the onset of ischemia (Furlan et al., 1996;Marchal et al., 1996). Second, the study identifies iNOS as one of the factors contributing to the post-ischemic evolution of the stroke and provides the rationale for therapeutic interventions based on iNOS inhibition. Treatments based on iNOS inhibition could be implemented in the late stages of cerebral ischemia (12–24 hr after stroke), at a time when current therapeutic approaches are no longer efficacious (Koroshetz and Moskowitz, 1996). iNOS inhibitors could complement interventions, such as thrombolysis or glutamate receptor inhibition, directed at the early stages of cerebral ischemia. Targeting both the early and late pathogenic components of cerebral ischemia is likely to be a successful strategy for the treatment of ischemic stroke.
Footnotes
This work was supported by grants from the American Heart Association and National Institutes of Health Grants NS34179 and NS35806. C.I. is an Established Investigator of the American Heart Association. We thank Ms. Colleen Forster for the histological analysis and Ms. Karen MacEwan for editorial assistance.
Correspondence should be addressed to Dr. C. Iadecola, Department of Neurology, University of Minnesota, Box 295 UMHC, 420 Delaware Street SE, Minneapolis, MN 55455.
REFERENCES
- 1.Adamson DC, Wildemann B, Sasaki M, Glass JD, McArthur JC, Christov VI, Dawson TM, Dawson VL. Immunologic NO synthase: elevation in severe AIDS dementia and induction by HIV-1 gp41. Science. 1996;274:1917–1921. doi: 10.1126/science.274.5294.1917. [DOI] [PubMed] [Google Scholar]
- 2.Astrup J, Symon L, Branston NM, Lassen NA. Cortical evoked potential and extracellular K+ and H+ at critical levels of brain ischemia. Stroke. 1977;8:51–57. doi: 10.1161/01.str.8.1.51. [DOI] [PubMed] [Google Scholar]
- 3.Bagasra O, Michaels FH, Zheng Y-M, Bobroski LE, Spitsin SV, Fu F, Tawadros R, Koprowski H. Activation of the inducible form of nitric oxide synthase in the brains of patients with multiple sclerosis. Proc Natl Acad Sci USA. 1995;92:12041–12045. doi: 10.1073/pnas.92.26.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baird AE, Benfield A, Schlaug G, Siewert B, Lovblad KO, Edelman RR, Warach S. Enlargement of human cerebral ischemic lesion volumes measured by diffusion-weighted magnetic resonance imaging. Ann Neurol. 1997;41:581–589. doi: 10.1002/ana.410410506. [DOI] [PubMed] [Google Scholar]
- 5.Baron J-C, von Kummer R, del Zoppo GJ. Treatment of acute ischemic stroke. Challenging the concept of a rigid and universal time window. Stroke. 1995;26:2219–2221. doi: 10.1161/01.str.26.12.2219. [DOI] [PubMed] [Google Scholar]
- 6.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunton VG, Grant MH, Wallace HM. Spermine toxicity in BHK-21/C13 cells in the presence of bovine serum: effect of aminoguanidine. Toxicol In Vitro. 1994;8:337–341. doi: 10.1016/0887-2333(94)90154-6. [DOI] [PubMed] [Google Scholar]
- 8.Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- 9.Chan PH, Kamii H, Yang G, Gafni J, Epstein CJ, Carlson E, Reola L. Brain infarction is not reduced in SOD-1 transgenic mice after a permanent focal cerebral ischemia. NeuroReport. 1993;5:293–296. doi: 10.1097/00001756-199312000-00028. [DOI] [PubMed] [Google Scholar]
- 10.Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- 11.Choi DW. Cerebral hypoxia: some new approaches and unanswered questions. J Neurosci. 1990;10:2493–2501. doi: 10.1523/JNEUROSCI.10-08-02493.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark RSB, Kochanek PM, Schwartz MA, Schiding JK, Turner DS, Chen M, Carlos TM, Watkins SC. Inducible nitric oxide expression in cerebrovascular smooth muscles and neutrophils after traumatic brain injury. Pediatr Res. 1996;39:784–790. doi: 10.1203/00006450-199605000-00007. [DOI] [PubMed] [Google Scholar]
- 13.Dalkara T, Irikura K, Huang Z, Panahian N, Moskowitz MA. Cerebrovascular responses under controlled and monitored physiological conditions in the anesthetized mice. J Cereb Blood Flow Metab. 1995;15:631–638. doi: 10.1038/jcbfm.1995.78. [DOI] [PubMed] [Google Scholar]
- 14.Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dawson VL, Brahmbhatt, Mong JA, Dawson TM. Expression of inducible nitric oxide synthase causes delayed neurotoxicity in primary mixed neuronal-glial cortical cultures. Neuropharmacology. 1994;33:1425–1430. doi: 10.1016/0028-3908(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 16.Dereski MO, Chopp M, Knight RA, Rodolosi LC, Garcia JH. The heterogeneous temporal evolution of focal ischemic neuronal damage in the rat. Acta Neuropathol. 1993;85:327–333. doi: 10.1007/BF00227730. [DOI] [PubMed] [Google Scholar]
- 17.Endoh M, Maiese K, Wagner J. Expression of the inducible form of nitric oxide synthase by reactive astrocytes after transient global ischemia. Brain Res. 1994;651:92–100. doi: 10.1016/0006-8993(94)90683-1. [DOI] [PubMed] [Google Scholar]
- 18.Furlan M, Marchal G, Viader F, Derlon J-M, Baron J-C. Spontaneous neurological recovery after stroke and the fate of the ischemic penumbra. Ann Neurol. 1996;40:216–226. doi: 10.1002/ana.410400213. [DOI] [PubMed] [Google Scholar]
- 19.Garcia JH, Yoshida Y, Chen H, Li Y, Zhang ZG, Lian J, Chen S, Chopp M. Progression from ischemic injury to infarct following middle cerebral artery occlusion in the rat. Am J Pathol. 1993;142:623–635. [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia JH, Liu KF, Ho KL. Neuronal necrosis after middle cerebral artery occlusion in Wistar rats progresses at different time intervals in the caudoputamen and the cortex. Stroke. 1995;26:636–642. doi: 10.1161/01.str.26.4.636. [DOI] [PubMed] [Google Scholar]
- 21.Gerlai R. Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci. 1996;19:177–181. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- 22.Gross SS, Wolin MS. Nitric oxide: pathophysiological mechanisms. Annu Rev Physiol. 1995;57:737–769. doi: 10.1146/annurev.ph.57.030195.003513. [DOI] [PubMed] [Google Scholar]
- 23.Gross WL, Bak MI, Ingwall JS, Arstall MA, Smith TW, Balligand J-L, Kelly RA. Nitric oxide inhibits creatine kinase and regulates rat heart contractile reserves. Proc Natl Acad Sci USA. 1996;93:5604–5609. doi: 10.1073/pnas.93.11.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hara E, Takahashi K, Tominaga T, Kumabe T, Kayama T, Suzuki H, Fujita H, Yoshimoto T, Shirato K, Shibahara S. Expression of heme oxygenase and inducible nitric oxide synthase mRNA in human brain tumors. Biochem Biophys Res Commun. 1996;224:153–158. doi: 10.1006/bbrc.1996.0999. [DOI] [PubMed] [Google Scholar]
- 25.Heiss W-D, Huber M, Fink GR, Herloz K, Pietrzyk U, Wagner R, Weinhard K. Progressive derangement of periinfarct viable tissue in ischemic stroke. J Cereb Blood Flow Metab. 1992;12:193–203. doi: 10.1038/jcbfm.1992.29. [DOI] [PubMed] [Google Scholar]
- 26.Hewett SJ, Csernansky CA, Choi DW. Selective potentiation of NMDA-induced neuronal injury following induction of astrocytic iNOS. Neuron. 1994;13:487–494. doi: 10.1016/0896-6273(94)90362-x. [DOI] [PubMed] [Google Scholar]
- 27.Hewett SJ, Muir JK, Lobner D, Symons A, Choi DW. Potentiation of oxygen-glucose deprivation-induced neuronal death after induction of iNOS. Stroke. 1996;27:1586–1591. doi: 10.1161/01.str.27.9.1586. [DOI] [PubMed] [Google Scholar]
- 28.Huang A, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 29.Iadecola C. Does nitric oxide mediate the increases in cerebral blood flow elicited by hypercapnia? Proc Natl Acad Sci USA. 1992;89:3913–3916. doi: 10.1073/pnas.89.9.3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iadecola C. Bright and dark sides of nitric oxide in ischemic brain damage. Trends Neurosci. 1997;20:132–138. doi: 10.1016/s0166-2236(96)10074-6. [DOI] [PubMed] [Google Scholar]
- 31.Iadecola C, Zhang F. Permissive and obligatory roles of NO in cerebrovascular responses to hypercapnia and acetylcholine. Am J Physiol. 1996;271:R990–R1001. doi: 10.1152/ajpregu.1996.271.4.R990. [DOI] [PubMed] [Google Scholar]
- 32.Iadecola C, Zhang F, Xu X. Inhibition of inducible nitric oxide synthase ameliorates cerebral ischemic damage. Am J Physiol. 1995a;268:R286–R292. doi: 10.1152/ajpregu.1995.268.1.R286. [DOI] [PubMed] [Google Scholar]
- 33.Iadecola C, Zhang F, Xu X, Casey R, Ross ME. Inducible nitric oxide synthase gene expression in brain following focal cerebral ischemia. J Cereb Blood Flow Metab. 1995b;15:378–384. doi: 10.1038/jcbfm.1995.47. [DOI] [PubMed] [Google Scholar]
- 34.Iadecola C, Zhang F, Casey R, Clark HB, Ross ME. Inducible nitric oxide synthase gene expression in vascular cells following transient focal cerebral ischemia. Stroke. 1996;27:1373–1380. doi: 10.1161/01.str.27.8.1373. [DOI] [PubMed] [Google Scholar]
- 35.Koroshetz WJ, Moskowitz MA. Emerging treatments for stroke in humans. Trends Pharmacol Sci. 1996;17:227–233. doi: 10.1016/0165-6147(96)10020-1. [DOI] [PubMed] [Google Scholar]
- 36.Lin SZ, Chiou AL, Wang Y. Ketamine antagonizes nitric oxide release from cerebral cortex after middle cerebral artery ligation in rats. Stroke. 1996;27:747–752. doi: 10.1161/01.str.27.4.747. [DOI] [PubMed] [Google Scholar]
- 37.Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24:117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- 38.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N, Chen H, Mudgett JS. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 39.Malinski T, Bailey F, Zhang ZG, Chopp M. Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1993;13:355–358. doi: 10.1038/jcbfm.1993.48. [DOI] [PubMed] [Google Scholar]
- 40.Marchal G, Beaudouin V, Rioux P, de la Sayette V, Le Doze F, Viader F, Derlon J-M, Baron J-C. Prolonged persistence of substantial volumes of potentially viable brain tissue after stroke. Stroke. 1996;27:599–606. doi: 10.1161/01.str.27.4.599. [DOI] [PubMed] [Google Scholar]
- 41.Nagafuji T, Sugiyama M, Muto A, Makino T, Miyauchi T, Nabata H. The neuroprotective effect of a potent and selective inhibitor of type I NOS (L-MIN) in a rat model of focal cerebral ischaemia. NeuroReport. 1995;6:1541–1545. doi: 10.1097/00001756-199507310-00019. [DOI] [PubMed] [Google Scholar]
- 42.Nguyen T, Brunson D, Crespi CL, Penman BW, Wishnok JS, Tannenbaum SR. DNA damage and mutation in human cells exposed to nitric oxide in vitro. Proc Natl Acad Sci USA. 1992;89:3030–3034. doi: 10.1073/pnas.89.7.3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nogawa S, Zhang F, Ross E, Iadecola C. Cyclooxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci. 1997;17:2746–2755. doi: 10.1523/JNEUROSCI.17-08-02746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nowicki JP, Duval D, Poignet H, Scatton B. Nitric oxide mediates neuronal death after focal cerebral ischemia in the mouse. Eur J Pharmacol. 1991;204:339–340. doi: 10.1016/0014-2999(91)90862-k. [DOI] [PubMed] [Google Scholar]
- 45.Ross ME, Iadecola C. Nitric oxide synthase expression in cerebral ischemia: neurochemical, immunocytochemical and molecular approaches. In: Packer L, editor. Methods in enzymology. Academic; Orlando, FL: 1996. pp. 408–426. [DOI] [PubMed] [Google Scholar]
- 46.Sessa A, Perin A. Diamine oxidase in relation to diamine and polyamine metabolism. Agents Actions. 1994;43:69–77. doi: 10.1007/BF02005768. [DOI] [PubMed] [Google Scholar]
- 47.Siesjo BK. Calcium-mediated processes in neuronal degeneration. Ann NY Acad Sci. 1994;747:140–161. doi: 10.1111/j.1749-6632.1994.tb44406.x. [DOI] [PubMed] [Google Scholar]
- 48.Sobey CG, Faraci FM. Effects of a novel inhibitor of guanylyl cyclase on dilator responses of mouse cerebral arterioles. Stroke. 1997;28:837–843. doi: 10.1161/01.str.28.4.837. [DOI] [PubMed] [Google Scholar]
- 49.Vodovotz Y, Lucia MS, Flanders KC, Chesler L, Xie QW, Smith TW, Weidner J, Mumford R, Webber R, Nathan C, Roberts AB, Lippa CF, Sporn MB. Inducible nitric oxide synthase in tangle-bearing neurons of patients with Alzheimer’s disease. J Exp Med. 1996;184:1425–1433. doi: 10.1084/jem.184.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, Xu D, Muller W, Moncada S, Liew FY. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature. 1995;375:408–411. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]
- 51.Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:165–170. doi: 10.1161/01.str.25.1.165. [DOI] [PubMed] [Google Scholar]
- 52.Yoshida T, Limmroth V, Irikura K, Moskowitz MA. The NOS inhibitor, 7-nitroindazole, decreases focal infarct volume but not the response to topical acetylcholine in pial vessels. J Cereb Blood Flow Metab. 1994;14:924–929. doi: 10.1038/jcbfm.1994.123. [DOI] [PubMed] [Google Scholar]
- 53.Zhang F, Iadecola C. Stimulation of the fastigial nucleus enhances EEG recovery and reduces tissue damage after focal cerebral ischemia. J Cereb Blood Flow Metab. 1992;12:962–970. doi: 10.1038/jcbfm.1992.133. [DOI] [PubMed] [Google Scholar]
- 54.Zhang F, Iadecola C. Infarct measurement methodology. J Cereb Blood Flow Metab. 1994;14:697–698. doi: 10.1038/jcbfm.1994.88. [DOI] [PubMed] [Google Scholar]
- 55.Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- 56.Zhang ZG, Reif D, Macdonald J, Tang WX, Kamp D, Gentile R, Shakespeare W, Murray R, Chopp M. ARL 17477, a potent and selective neuronal NOS inhibitor decreases infarct volume after transient middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab. 1996;16:599–604. doi: 10.1097/00004647-199607000-00009. [DOI] [PubMed] [Google Scholar]
- 57.Zimmerman GA, Meistrell MI, Bloom O, Cockroft KM, Bianchi M, Risucci D, Broome J, Farmer P, Cerami A, Vlassara H, Tracey KJ. Neurotoxicity of advanced glycation endproducts (AGEs) during focal stroke, and neuroprotective effects of aminoguanidine. Proc Natl Acad Sci USA. 1995;92:3744–3748. doi: 10.1073/pnas.92.9.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]