

Abstract
Neuronal death after experimental traumatic brain injury (TBI) has features of both apoptosis and necrosis. Neurons in the peritrauma cortex, hippocampus, and dentate gyrus are particularly vulnerable. The apoptosis-suppressor gene bcl-2 is induced in brain after ischemia and epilepsy-induced injury and may serve to regulate neuronal death. We studied expression of bcl-2 mRNA and protein after experimental TBI in rats. To determine whether bcl-2 protein expression occurred in cells with evidence of apoptosis, triple-labeling studies were performed using (1) antibody against bcl-2, (2) bis-benzimide dye to examine gross nuclear morphology, and (3) terminal deoxynucleotidyl transferase-mediated biotin-dUTP nick-end labeling (TUNEL) to assess for DNA fragmentation. At 6 and 24 hr,bcl-2 mRNA was induced in ipsilateral peritrauma cortex, hippocampus, and dentate gyrus. By 72 hr the increase inbcl-2 mRNA was detected only in cortex. bcl-2 protein was induced at 8, 24, 72, and 168 hr in ipsilateral cortex and hippocampus. Cells expressing bcl-2 protein included neurons in the peritrauma cortex, hippocampus, hilus, and dentate gyrus. The gross nuclear morphology of neurons expressing bcl-2 appeared normal. Furthermore, biochemical evidence of DNA fragmentation, in a pattern characteristic of either apoptosis or necrosis, was seldom seen in neurons expressing bcl-2 protein (bcl-2 colocalized with TUNEL in 0–2% of TUNEL-positive cells observed). These data suggest that bcl-2 may play an important role in the regulation of neuronal death after TBI, and they support a role for bcl-2 as an inducible neuroprotective gene.
Keywords: apoptosis, bcl-2, hippocampus, neuron, rat, traumatic brain injury
Apoptosis, a form of programmed cell death, plays an important role in embryogenesis and in the normal development and maintenance of many adult tissues (Saunders, 1966; Kerr et al., 1972). Apoptosis is tightly regulated by the expression or activation of several genes and proteins (Stellar, 1995). The proto-oncogene bcl-2 is one such gene that was first cloned in a b-cell lymphoma line (Vaux et al., 1988). bcl-2 is a member of a family of related genes encoding proteins that either promote (e.g., bax, bcl-xS) or suppress (e.g., bcl-2, bcl-xL) programmed cell death (Hockenbery, 1995). bcl-2 is expressed in cells that survive fetal development (LeBrun et al., 1993; Novack and Korsmeyer, 1994), and it inhibits programmed cell death in manyin vitro systems (Vaux et al., 1988; Hockenbery et al., 1990; Sentman et al., 1991). In the nervous system, bcl-2protects against various stimuli that induce apoptotic neuronal death (Garcia et al., 1992; Allsopp et al., 1993; Batistatou et al., 1993;Kane et al., 1993; Zhong et al., 1993).
Both apoptotic and necrotic neuronal death occur after experimental traumatic brain injury (TBI). Rink et al. (1995) demonstrated that selectively vulnerable regions with neurons that undergo apoptotic death after fluid-percussion injury include cortex, CA3 hippocampus, and dentate gyrus. We have shown a similar pattern of DNA fragmentation and delayed neuronal death using a model of controlled cortical impact (CCI) with a secondary hypoxemic insult (Clark et al., 1997). Although the genetic regulation of delayed neuronal death after TBI remains undefined, bcl-2 and related proteins are expressed in models of ischemia and epilepsy in surviving neurons (Shimazaki et al., 1994;Chen et al., 1995; Krajewski et al., 1995; Graham et al., 1996; Chen et al., 1997). Because neuronal death after TBI may share similar, as well as distinct, mechanisms with ischemia and epilepsy-induced injury, we hypothesized that bcl-2 would be expressed after TBI. In this study, we report expression of bcl-2 mRNA and protein in neurons after TBI, using a model that simulates the clinical condition of TBI, secondary insult, and resuscitation (Clark et al., 1997). To determine whether bcl-2 expression is associated with suppression of neuronal death after TBI, we used multicolor immunocytochemical methods to examine whether neurons expressing bcl-2 protein had gross morphological or biochemical evidence of apoptosis.
MATERIALS AND METHODS
Model of TBI. All studies were approved by the University of Pittsburgh Animal Care and Use Committee. Virus-free, adult male Sprague Dawley rats (n = 65) had free access to food and water before and after surgery. Anesthesia was induced with 4% isoflurane (Anaquest, Memphis, TN) in O2. The trachea was intubated with a 14 gauge angiocatheter, and the lungs were mechanically ventilated with 2.0% isoflurane/66% N2O/balance O2. A femoral arterial catheter was inserted for continuous monitoring of blood pressure, arterial blood sampling, and administration of pancuronium bromide (0.1 mg · kg−1 · hr−1) (Elkins-Sinn, Cherry Hill, NJ). A rectal probe was inserted to monitor core temperature.
TBI was performed using the CCI model (Dixon et al., 1991; Kochanek et al., 1995). To simulate the clinical setting of TBI, secondary insult, and resuscitation, we used the following previously described protocol (Clark et al., 1997). Briefly, a craniotomy was made over the left parietal cortex. A temperature probe (Physiotemp, Clifton, NJ) was inserted through a burr hole into the left parietal cortex to monitor brain temperature. Rats were then warmed to a brain temperature of 37 ± 0.5°C and allowed to equilibrate under anesthesia (1.1% isoflurane/66% N2O/balance O2) for 30 min. After removal of the bone flap, injury was produced using the CCI device (Dixon et al., 1991) with minor modification (Kochanek et al., 1995). For all studies, depth of penetration of 2.5 mm, a velocity of 4.0 ± 0.2 m/sec, and a duration of deformation of 50 msec were used. To produce moderate hypoxemia, air and oxygen were blended to achieve an FiO2 of 11% (1.1% isoflurane/74% N2O/19% air/6% O2) 1 min after CCI. This produces a PaO2 in the rats of 44 ± 1 mm Hg and a 40% reduction in mean arterial blood pressure by 30 min (Clark et al., 1997). Arterial blood gas tensions were also obtained at 10 and 25 min after trauma. The bone flap was replaced and sealed with dental cement (Koldmount, Vernon Benshoff Co., Albany, NY), and the scalp incision was closed. Hypoxemia was maintained for a total of 30 min. Anesthesia was discontinued, and rats were resuscitated with 100% oxygen until they awoke. Rats were then extubated and placed in 100% oxygen for an additional 30 min before being returned to their cages.
Study protocol. To study the expression of bcl-2mRNA, naive rats and rats at 6, 24, and 72 hr after TBI were killed, and brain tissue was analyzed using in situ hybridization (n = 4 per group). Northern blot analysis was performed to confirm the specificity of the oligonucleotide probe used forin situ hybridization. To study the expression of bcl-2 protein, naive rats and rats at 8, 24, 72, and 168 hr after TBI were killed, and brain tissue was analyzed using Western blot analysis (n = 3–5 per group) and immunocytochemistry (n = 2–4 per group). For Western blot analysis, controls also included rats made hypoxemic but not traumatized. For immunocytochemistry, controls also included sham-operated (24 hr) rats. To determine whether bcl-2 protein expression occurred in cells with gross evidence of apoptosis, triple-labeling studies were performed using (1) antibody against bcl-2, (2) bis-benzimide dye (similar to Hoechst 33258 dye) to examine nuclear morphology, and (3) terminal deoxynucleotidyl transferase-mediated biotin-dUTP nick-end labeling (TUNEL) to assess for DNA fragmentation. To examine the time course of DNA fragmentation, TUNEL alone was performed on separate brain sections from naive rats and in rats at 6, 24, and 72 hr after TBI (n = 4 per group).
In situ hybridization. Rats were anesthetized as described above and decapitated. Brains were quickly removed, rapidly frozen in pentane at −20°C, and stored at −80°C. Sections (20 μm) were cut on a cryostat, mounted on Probe-On slides (Fisher, Pittsburgh, PA), and processed for in situ hybridization as described previously (Graham et al., 1996). Briefly, a 40-mer antisense oligodeoxynucleotide probe complementary to bcl-2 was constructed. The sequence used was 5′-CACTGAATGCTCTCCGGTACCGCAGTTCAAACTCATCGCC-3′. A search of the GenBank database revealed no significant homology with other genes. In addition, we have previously confirmed the specificity of this probe for bcl-2 using Northern blot analysis (Graham et al., 1996) and the sense control (Chen et al., 1996, 1997). The oligodeoxynucleotide probes were labeled with α-[35S]dATP using terminal deoxynucleotidyltransferase (TdT) (Life Technologies, Gaithersburg, MD) and hybridized (1× 107 cpm/ml) at 42°C for 18 hr with the slides. After hybridization, the sections were rinsed in 1 × saline-sodium citrate (SSC) (150 mm NaCl, 15 mm sodium citrate, pH 7.4) at 55°C for 60 min with several changes of 1× SSC, rinsed again at room temperature for 60 min, dehydrated, and exposed to Kodak SB-5 film (Eastman Kodak, Rochester, NY) for 3 weeks. Slides from control and traumatized brain were hybridized together and developed on the same film. Relative changes in mRNA expression were semiquantified in hippocampal CA1 and CA3 regions, dentate gyrus, and ipsilateral cortex by measuring the ratio of the optical density in each region in rats after TBI compared with naive controls using an image analysis system (MCID, St. Catherines, Ontario, Canada). Cellular localization was evaluated by coating slides with Kodak NTB-2 emulsion. The sections were exposed at 4°C for 5 weeks, developed in D-19, and counterstained with cresyl violet.
Northern blot analysis. The specificity of the oligodeoxynucleotide probe was examined by Northern blot analysis. Naive rats and rats at 2, 6, and 24 hr after TBI (n = 2/group) were anesthetized as described above and then perfused transcardially with ice-cold saline. Brains were removed, and cortical tissue including and adjacent to the contusion was isolated. Total RNA was extracted from each sample using an RNA isolation kit (Promega, Madison, WI). Twenty micrograms of RNA/lane were electrophoresed through a 1% agarose-2.2 m formaldehyde gel and transferred to nylon membranes (Hybond-N, Amersham, Arlington Heights, IL). The same oligodeoxynucleotide probe used for in situhybridization was 3′ end-labeled with 40 μCi of α-[32P]dATP (specific activity > 106 Ci/mol) using TdT. Unincorporated nucleotides were separated by P-60-Sepharose columns. The labeled probe was denatured by heat and hybridized to the membranes at 42°C overnight. The washing procedure was performed under high stringency conditions in 2× SSC/0.1% SDS (three times for 45 min each) at 52°C. The membranes were then exposed to Kodak X-OMAT AR film (Eastman Kodak) using intensifier screens at −80°C for 24 hr. To control for variation in the amount of RNA in individual samples, thebcl-2 probe was stripped off the membranes in a solution containing 0.1× SSC/0.5% SDS at 100°C for 15 min; the membranes were then rehybridized with an oligonucleotide probe corresponding to 18S RNA.
Western blot analysis. Rats were anesthetized as described above and then perfused transcardially with ice-cold saline. Brains were removed and dissected to remove the ipsilateral hippocampus, cortex including and adjacent to the contusion, and cortex distal to the contusion. Each sample was homogenized in lysis buffer containing 0.1 m NaCl, 0.01 m Tris, and 0.1 mmEDTA, and the protease inhibitors chymostatin 2 μg/ml, leupeptin 2 μg/ml, pepstatin 2 μg/ml, and phenylmethylsulfonylfluoride 100 μg/ml. Lysates were centrifuged at 14,000 rpm for 30 min at 4°C and boiled in loading buffer for 3 min. Fifty microgram protein samples were loaded on a 12% SDS-polyacrylamide gel, separated electrophoretically, and transferred to a Hybond-N membrane (Amersham) overnight. The transferred membrane was incubated in the primary antibody against bcl-2 (Dako, Carpinteria, CA) at a 1:200 dilution at room temperature for 1 hr. After it was washed three times in PBS containing 0.1% Tween 20, the secondary antibody was applied at a 1:5000 dilution for 1 hr. The blot was washed in PBS containing 0.1% Tween 20 three times over 25 min and then incubated in commercial enhanced chemiluminescence reagents (Western Exposure Chemiluminescence Detection System, Clontech Laboratories, Palo Alto, CA) and exposed to Fuji RX film (Fuji, Tokyo, Japan). Autoradiogram signals were quantified by a gel densitometric scanning program (MCID). Densitometric values for bcl-2 were normalized to background values obtained on the same lane.
Immunocytochemistry and TUNEL. Rats were anesthetized as described above and then perfused transcardially with 200 ml heparinized saline (8 U/ml) followed by 500 ml 2% paraformaldehyde. Brains were removed and further immersion-fixed in 2% paraformaldehyde for 30 min, rinsed three times in PBS, and then placed in 30% sucrose at 4°C before they were snap-frozen in 2-methylbutane in liquid nitrogen. Brains were cut into 5 μm sections using a cryostat, mounted, and kept frozen until use. After removal from the cryostat chamber, slides were washed three times with PBS and then washed three times in PBS containing 0.5% bovine serum albumin and 0.15% glycine (buffer A). Nonspecific activity was blocked with 5% normal goat serum in buffer A. Brain sections were then incubated at room temperature for 1 hr in a 1:100 dilution of mouse monoclonal antibody against human bcl-2 (Dako). Sections were washed three times in buffer A and then incubated for 1 hr in a 1:3000 dilution of goat anti-mouse Cy3.18 immunoconjugate (Jackson Immunochemicals, West Grove, PA). Sections were then washed six times (5 min/wash) in buffer A, mounted in gelvatol, and coverslipped for light microscopy. A Nikon FXA light microscope equipped for epifluorescent illumination and differential interference contrast was used for observation. Fluorescent images were collected using an integrating three-chip Sony color video camera (700 × 600 pixels) equipped with a color frame grabber board. All images were collected while integrating at four frames per second.
For triple-label experiments, TUNEL was performed on additional sections using a modification of the technique reported by Gavrieli et al. (1992). Sections were washed twice with PBS, incubated in cold methanol for 30 min, washed twice with PBS, and then incubated at 37°C for 1 hr in buffer containing TdT and fluorescein-conjugated 12-dUTP (Boeringer Mannheim, Indianapolis, IN). Sections were then processed for bcl-2 immunocytochemistry as described above. To assess nuclear morphology, bis-benzimide (Sigma, St. Louis, MO; similar to Hoechst 33258) diluted in sterile water was applied to some sections for 30 sec before coverslipping. Sections were examined by fluorescence microscopy, and images were collected as described above using excitation/emission wavelengths of 550/565 (red), 494/520 (green), and 346/460 (blue) λ for bcl-2, TUNEL, and bis-benzimide, respectively. In sections from each specimen, the primary antibody was omitted to assess for nonspecific binding of the secondary antibody.
The bcl-2 antibody used for Western analysis and immunocytochemistry was generated against a bcl-2 peptide sequence of human origin (amino acids 41–54). Because we used this antibody to identify bcl-2 in rat tissue, we performed additional control experiments. We obtained a synthetic peptide that corresponds to amino acids 41–53 of the rat bcl-2 protein (Biosynthesis, Lewisville, TX). This sequence (RAAPTPGIFSFQP) has 77% homology to the corresponding human sequence, differing at positions 41, 45, and 51. The peptide (1, 5, and 10 μg) was loaded on a 16.5% Tris-tricine gel, and Western analysis was performed using the bcl-2 antibody as described above. Preabsorption studies were also performed by adding 10 μg of the synthetic peptide to the primary antibody solution before immunocytochemistry. Immunoreactivity in brain sections 24 hr after TBI with and without preabsorption of the primary antibody was compared.
To examine the time course of DNA fragmentation, additional TUNEL was performed on separate brain sections using a diaminobenzidine colorometric method for light-microscopic analysis as described previously (Clark et al., 1997). Briefly, frozen 20 μm coronal sections through the anterior hippocampus were cut using a cryostat and mounted on glass slides. Sections were post-fixed in 10% neutral buffered formalin for 10 min at room temperature followed by ethanol/acetic acid (2:1) for 5 min at −20°C. Sections were then incubated in 3% Triton X-100 (Sigma) at room temperature for 1 hr and then placed in 3% H2O2 and 30% methanol in PBS for 20 min. Sections were then incubated in 300 U/ml TdT and 20 nmol/ml biotin-16-dUTP (Boeringer Mannheim) in 1 ml 1× TdT buffer at 37°C for 90 min, washed with PBS three times, incubated in avidin–biotin complex (ABC standard kit, Vector Labs, Burlingame, CA), and TUNEL-visualized with diaminobenzidine (DAB substrate kit, Vector Labs). The number of TUNEL-positive cells/400× field were counted in the dentate gyrus, the CA1 and CA3 regions of the hippocampus, and the peritrauma cortex (four representative 400× fields were counted in cortex) for each section. After quantification of TUNEL-positive cells, coverslips were removed, and sections were counterstained lightly with cresyl violet and recoverslipped.
Statistical analysis. All data are presented as mean ± SEM. Comparisons of bcl-2 mRNA expression, bcl-2 protein expression, and TUNEL-positive cells at different times after TBI versus controls were made using Kruskal–Wallis and Dunn’s test (or Dunnett’s test if all groups were of equal size). A pvalue < 0.05 was considered significant.
RESULTS
bcl-2 mRNA Expression After TBI
Figure 1 shows representative autoradiographs after bcl-2 in situ hybridization.bcl-2 mRNA expression was increased after TBI compared with naive control brains. The increase in bcl-2 mRNA was detected primarily in the ipsilateral hemisphere. At 6 hr there were modest increases in relative bcl-2 mRNA levels in ipsilateral cortex (1.3-fold), dentate gyrus (twofold), and the CA3 hippocampal region (1.4-fold) compared with naive controls (Fig.2). At 24 hr, there were modest increases in relative bcl-2 mRNA levels in ipsilateral cortex (1.5-fold), dentate gyrus (1.4-fold), and the CA1 (1.5-fold) and CA3 (1.threefold) hippocampal regions compared with naive controls (Fig.2). By 72 hr the increase in bcl-2 mRNA was seen in the ipsilateral cortex (1.5-fold) but not the ipsilateral hippocampal regions, compared with naive controls. A statistical difference in relative bcl-2 mRNA levels was detected in the CA1 hippocampal region at 24 hr and in the dentate gyrus (p < 0.05); however, the time when the difference occurred in the dentate gyrus could not be determined by multiple comparisons. bcl-2 mRNA was detected primarily in cellular layers of the dentate gyrus, hilus, CA1 and CA3 hippocampal regions, and cells in cortex (Fig. 3). Many of the cells in which bcl-2 mRNA was detected seemed to be neurons. At 24 and 72 hr, neuronal loss was seen in the CA3 hippocampal region, cortex, and dentate gyrus. A relative increase inbcl-2 mRNA was also detected in the hippocampal white matter and the leptomeninges between the hippocampus and thalamus.
Fig. 1.
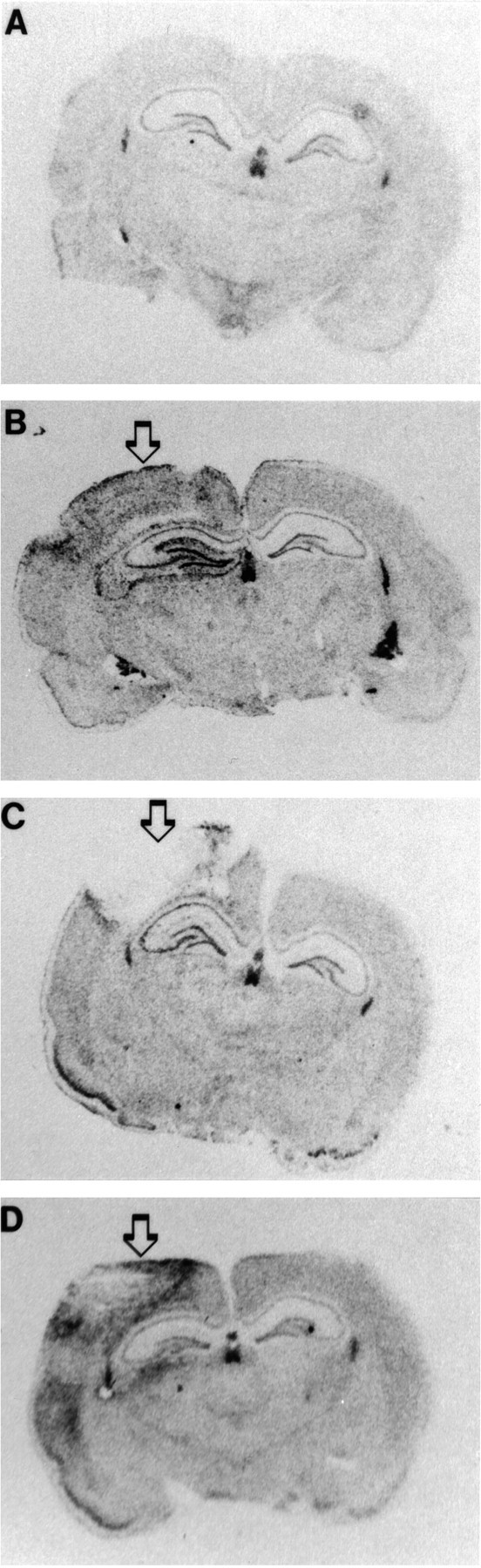
bcl-2 in situ hybridization. Autoradiograms through the level of anterior hippocampus in a naive rat and rats at 6, 24, and 72 hr after TBI (arrows mark the site of impact). bcl-2 mRNA is increased in the peritrauma cortex and ipsilateral hippocampus after TBI versus naive control.
Fig. 2.

Relative changes in bcl-2mRNA in dentate gyrus, CA3 hippocampus, CA1 hippocampus, and cortex in rats at 6, 24, and 72 hr after TBI versus naive controls (n = 4/group). Data are presented as mean ± SEM and represent fold changes in sections from rats after TBI versus naive controls in the ipsilateral (closed circles) and contralateral (open circles) hemispheres. There was a significant difference in bcl-2 mRNA levels in CA1 hippocampus at 24 hr versus control (p < 0.05; Kruskal–Wallis and Dunn’s test). There was also a difference inbcl-2 mRNA levels in the dentate gyrus (p < 0.05; Kruskal–Wallis test), but the time when this difference occurred could not be determined by multiple comparisons.
Fig. 3.

bcl-2 in situ hybridization dipped-emulsion photomicrographs of the ipsilateral dentate–hilar area from a rat 6 hr after TBI (B, D) and a naive control rat (A, C). An increase in signal intensity is seen over cellular layers in these regions after TBI (arrows). Scale bar, 50 μm.
Figure 4 shows a representative Northern blot using the bcl-2 antisense oligonucleotide. A single band of the 2.7 kb size corresponding to the bcl-2transcript (Negrini et al., 1987) was detected in the ipsilateral cortex at 2, 6, and 24 after TBI.
Fig. 4.

Northern blot analysis of bcl-2mRNA and 18S rRNA in ipsilateral cortex from naive control brain and brains 2, 6, and 24 hr after TBI (representative samples fromn = 2/group).
bcl-2 protein expression after TBI
Western blot analysis showed that bcl-2 protein is increased in traumatized cortex and ipsilateral hippocampus after TBI compared with naive and hypoxemia-alone controls. Figure5 shows protein bands at the predicted 26 kDa size for the bcl-2 gene product. In traumatized cortex, bcl-2 protein was increased 22.6-fold at 8 hr (p< 0.05 vs control), 19.3-fold at 24 hr, 19.7-fold at 72 hr (p < 0.05 vs control), and 15-fold at 168 hr compared with naive controls. In ipsilateral hippocampus bcl-2 protein was increased 2.5-fold at 8 hr, 5.9-fold at 24 hr, 5.3-fold at 72 hr, and 2.4-fold at 168 hr compared with naive controls (p = 0.07). bcl-2 protein expression was not increased in distal cortex of rats after TBI compared with naive controls (data not shown). Hypoxemia alone did not induce bcl-2 protein expression in cortex or hippocampus compared with naive controls (Fig.5).
Fig. 5.

Western blot analysis of bcl-2 protein in (A) cortex and hippocampus from naive control brains and brains 8, 24, 72, and 168 hr after TBI. bcl-2 immunoreactivity is increased in both cortex and hippocampus after TBI at the predicted molecular size (26 kDa). B, Hypoxemia alone (FiO2= 0.11 × 30 min) did not increase bcl-2 immunoreactivity at 24 hr compared with naive rats in cortex or hippocampus. Data are presented as mean ± SEM and represent brain tissue samples fromn = 3–5 rats/group. There was a significant difference in bcl-2 protein levels in cortex at 8 and 72 hr (p < 0.05; Kruskal–Wallis and Dunn’s test). There was a trend toward an increase in bcl-2 protein levels in hippocampus (p = 0.07; Kruskal–Wallis test).
bcl-2 protein was detected in many cells in the peritrauma cortex, ipsilateral dentate gyrus, ipsilateral CA3 hippocampus, and hilus at all time points assessed after TBI (Fig.6). Many cells expressing bcl-2 had the morphological appearance of neurons. To determine the relative number of cells expressing bcl-2 within a region of interest, fluorescent images were overlayed on differential interference contrast images (Fig. 7). Within each region both immunopositive and immunonegative neurons were seen. Approximately half of the neurons within CA3 hippocampus, dentate gyrus, and hilus expressed bcl-2, with the amount of expression varying between rats. Maximal bcl-2 protein expression in hippocampus and dentate gyrus as detected by immunocytochemistry appeared to occur at 24 and 72 hr, consistent with results of Western blot analysis. bcl-2 protein was also detected in glia and blood vessels and occasionally in neurons in CA1 hippocampus (data not shown). Immunofluorescence for bcl-2 protein was minimally detected in the superficial cortex of sham-operated rats and the contralateral hemispheres of rats after TBI. bcl-2 labeling was not detected in sections incubated without the primary antibody (Fig.6E,F). Preabsorption with the corresponding rat peptide sequence reduced, but did not completely block, the amount of immunoreactivity seen in brain sections 24 hr after TBI compared with nonpreabsorbed sections (Fig. 6G,H). Western blot analysis of the peptide corresponding to amino acids 41–53 of the rat bcl-2 protein showed that the bcl-2 antibody immunoreacted with the 1.4 kDa synthesized rat peptide (data not shown).
Fig. 6.
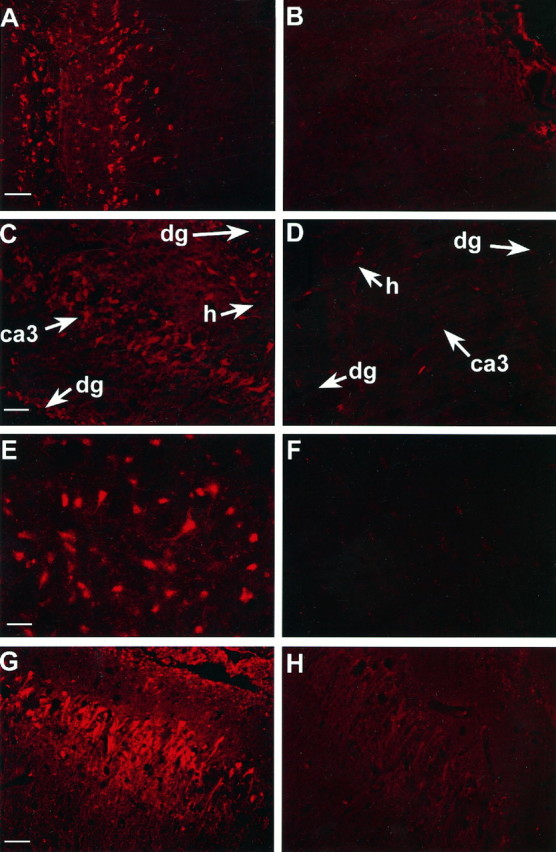
Immunofluorescent images of bcl-2 protein in brain sections 24 hr after TBI or sham-operation (bcl-2 labeling =red). bcl-2 immunoreactivity is increased in neurons in cortex, CA3 hippocampus, dentate gyrus, and hilus after TBI versus sham-operation. A, Ipsilateral cortex after TBI.B, Ipsilateral cortex after sham-operation.C, Ipsilateral hippocampus after TBI. D, Ipsilateral hippocampus after sham-operation. E,F, Consecutive brain sections incubated with (E) and without (F) primary antibody. G, H, Consecutive brain sections incubated with primary antibody (G) or primary antibody preabsorbed with rat bcl-2 peptide (H). ca3, CA3 hippocampus;dg, dentate gyrus; h, hilus. Scale bars:A–D, 50 μm; E, G, 25 μm; G, H, 100 μm.
Fig. 7.
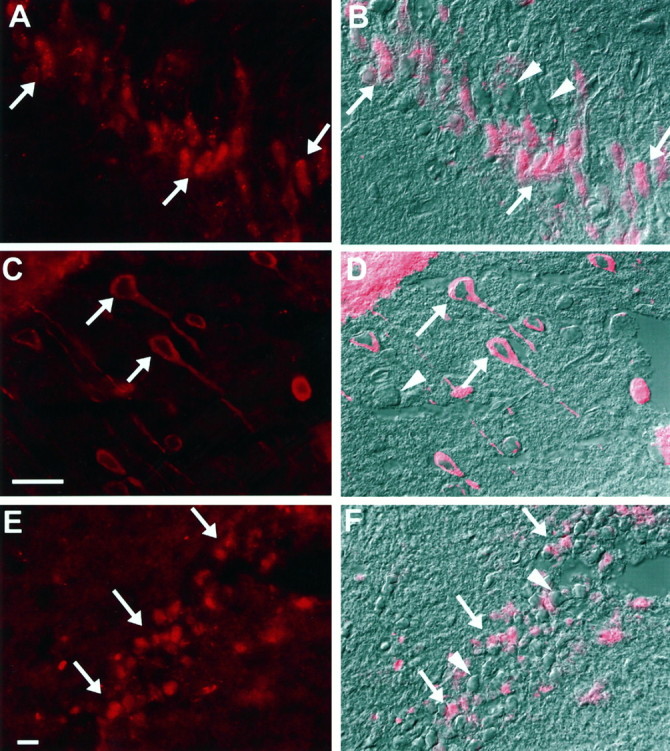
Immunofluorescent and differential interference contrast (DIC) images showing bcl-2 immunoreactivity in neurons after TBI in rats (bcl-2 labeling = red).A, B, CA3 hippocampus 24 hr after TBI (A, fluorescent image; B, DIC image with fluorescent overlay). C, D, Cortex 72 hr after TBI (C, fluorescent image; D, DIC image with fluorescent overlay). E, F, Dentate gyrus 72 hr after TBI (E, fluorescent image;F, DIC image with fluorescent overlay). Note both bcl-2 immunopositive (arrows) and immunonegative (arrowheads) neurons in each region. Scale bars:A, B, E, F, 25 μm; C,D, 25 μm.
Time course and gross nuclear morphology of cells with evidence of DNA fragmentation after TBI
The time course of TUNEL-positive cells in the ipsilateral peritrauma cortex, dentate gyrus, and CA1 and CA3 regions of the hippocampus is shown in Figure 8. The number of TUNEL-positive cells was increased and was maximal at 24 hr in each region assessed (p < 0.05 vs control). In the CA1 and CA3 regions of the hippocampus, the number of surviving neurons without evidence of DNA fragmentation was quantified. In naive rats there were 91.7 ± 3.6 and 63.7 ± 2.2 neurons/400× field in CA1 and CA3, respectively. By 72 hr, there were 71.9 ± 9.6 and 35.9 ± 4.0 neurons/400× field in CA1 (p = 0.12 vs control) and CA3 (p < 0.05 vs control), respectively. Many TUNEL-positive cells were also seen in the ipsilateral hilus (at 6, 24, and 72 hr) and thalamus (at 24 and 72 hr). TUNEL-positive cells were not observed in the contralateral hemispheres at any time point.
Fig. 8.

Time course of TUNEL-positive cells after TBI. TUNEL-positive cells/400× field were counted in the peritrauma cortex, dentate gyrus, and CA1 and CA3 regions of the hippocampus (in the cortex, 4 representative 400× fields were counted). Mean ± SEM;n = 4/group; *p < 0.05 versus control; Kruskal–Wallis and Dunnett’s test. DG, Dentate gyrus; CA3, CA3 hippocampus; CA1, CA1 hippocampus.
TUNEL-positive cells displayed morphophological characteristics suggestive of either apoptosis or necrosis (Fig.9) or both. In some TUNEL-positive cells a condensed, clumped, and pyknotic nuclear pattern was seen (suggestive of apoptosis), whereas in others a diffuse pattern of TUNEL staining without shrinkage of the nuclei and with occasional extravasation of TUNEL labeling into the cytoplasm (suggestive of necrosis) was observed. In the dentate gyrus the pattern suggestive of apoptotic death was most commonly observed. In the CA3 and CA1 regions of the hippocampus and hilus, the pattern suggestive of necrotic death was seen most often. In the cortex and thalamus, both patterns were consistently observed. In many TUNEL-positive cells, a characteristic pattern of apoptosis versus necrosis could not be discriminated.
Fig. 9.
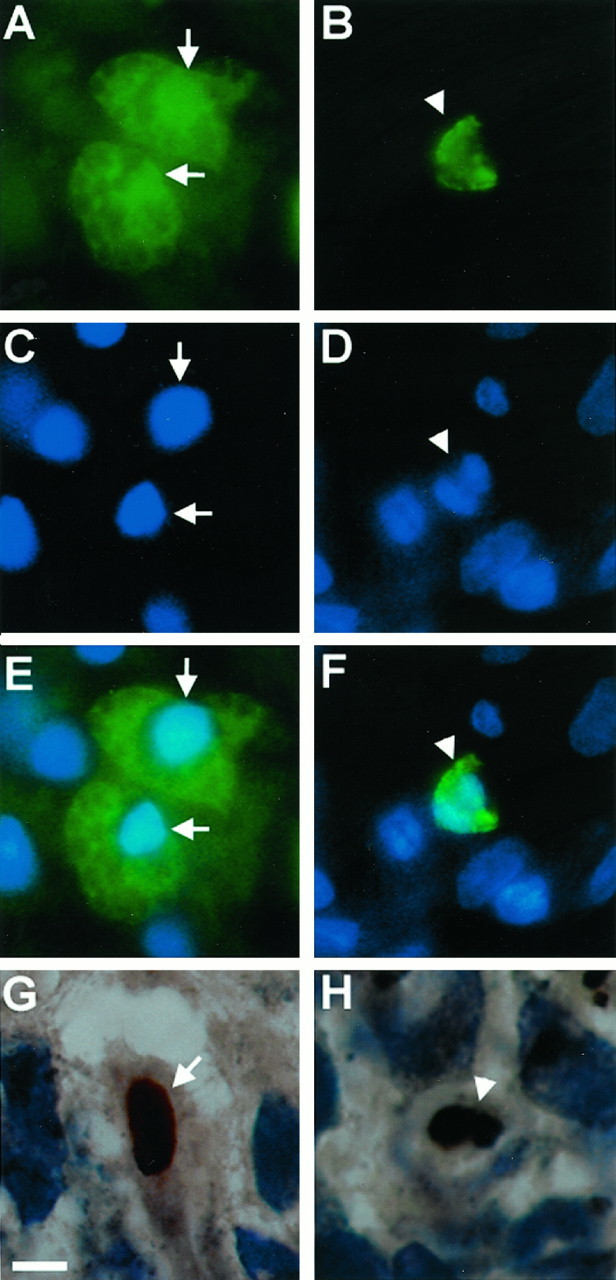
Immunofluorescent images and photomicrographs showing nuclear morphologies characteristic of both necrosis (A, C, E, G) and apoptosis (B, D, F, H) after TBI (TUNEL-FITC = green; TUNEL-diaminobenzidine = brown; bis-benzimide = blue). Note TUNEL-positive cells demonstrating a condensed, pyknotic, and clumped nuclear pattern suggestive of apoptosis (arrowheads) and TUNEL-positive cells with a diffuse pattern of TUNEL staining without shrinkage of the nuclei and with occasional extravasation of TUNEL labeling into the cytoplasm, suggestive of necrosis (arrows). A, C, E, G, Ipsilateral CA3 hippocampus at 24 hr. B, D, F, Ipsilateral cortex at 24 hr. H, Ipsilateral dentate gyrus at 24 hr. Scale bar, 10 μm.
bcl-2 protein expression in neurons without evidence of DNA fragmentation and gross nuclear damage after TBI
Neurons expressing bcl-2 protein rarely demonstrated gross nuclear or biochemical evidence of apoptosis after TBI. Bis-benzimide (similar to Hoechst) staining of nuclear material demonstrated both a normal gross nuclear morphology and distribution of nuclear material in all bcl-2-positive neurons observed (Fig.10). In contrast, there were many bcl-2-negative cells in selectively vulnerable regions (CA3 hippocampus, dentate gyrus, and cortex) with condensed nuclear material and shrunken nuclei. With rare exception, neurons expressing bcl-2 protein were not concurrently TUNEL-positive. Less than 2% of all TUNEL-positive cells observed, displaying characteristics of either apoptosis or necrosis, also expressed bcl-2 (Fig. 10).
Fig. 10.

Immunofluorescent images of bcl-2 protein, TUNEL, and bis-benzimide dye in brain sections after TBI (bcl-2 labeling = red; TUNEL = green; bis-benzimide = blue). Note many TUNEL-positive neurons (arrowheads) in these selectively vulnerable regions after TBI; bcl-2 immunoreactivity persists in some cells (arrows) but is not seen in cells with detectable DNA fragmentation (TUNEL). Bis-benzimide staining demonstrates normal nuclear morphology in bcl-2-labeled neurons (arrows) in contrast to TUNEL-positive neurons, which show a condensed and shrunken nucleus (arrowheads). A, Ipsilateral CA3 hippocampus at 7 d (inset shows higher magnification). B–D, Ipsilateral cortex at 24 hr.E, Ipsilateral CA3 hippocampal neurons at 24 hr.F, Ipsilateral hilar neurons at 24 hr. Scale bars:A, 50 μm; C–F, 20 μm.
DISCUSSION
The major findings of this study are that (1) the apoptosis-suppressor gene bcl-2 and its translated protein are induced in surviving neurons in regions selectively vulnerable to apoptotic cell death after TBI, and (2) with rare exception, bcl-2 protein expression occurs in neurons without evidence of apoptosis (or overt necrosis), as determined by co-labeling with TUNEL and observation of gross nuclear morphology. Although a cause and effect relationship was not demonstrated, these data suggest that bcl-2may play an important role in inhibiting (or delaying) apoptotic cell death after TBI, and they support a role for bcl-2 as an inducible neuroprotective gene.
bcl-2 protein is induced at 8 hr and maintained up to 7 d in surviving neurons in peritrauma cortex, CA3 hippocampus, dentate gyrus, and hilus after TBI (Figs. 6, 7, 10). CA3 hippocampus, dentate gyrus, and hilus are some of the regions particularly vulnerable to neuronal death after TBI (Cortez et al., 1989; Lowenstein et al., 1992; Dietrich et al., 1994; Clark et al., 1997). In our model of TBI, roughly half of the neurons within these regions were bcl-2 immunoreactive. Importantly, this heterogeneous pattern and degree of bcl-2 expression mirrors the pattern of neuronal survival (∼50% of CA3 neurons by 1 week) seen in this model of TBI plus secondary insult (Clark et al., 1997), suggesting that bcl-2 expression may prevent neuronal death in individual cells within these regions. Evidence in support of this hypothesis includes studies showing (1) that overexpression of bcl-2 produced by viral transfection reduces neuropathology after focal ischemia (Linnik et al., 1995;Lawrence et al., 1996) and (2) that transgenic mice overexpressing bcl-2 have reduced infarction size after focal cerebral ischemia compared with wild-type mice (Martinou et al., 1994).
bcl-2 mRNA is induced within 6 hr after TBI in peritrauma cortex and hippocampus. Regionally, an increase in bcl-2 protein followed increases in bcl-2 mRNA expression in cortex, dentate gyrus, and CA3 hippocampus after TBI. In CA1 hippocampus,bcl-2 mRNA was also expressed but bcl-2 protein was not detected. After cerebral ischemia, bcl-2 mRNA is induced but not translated in CA1 neurons, which are selectively vulnerable and die (Krajewski et al., 1995; Chen et al., 1997). After kainic acid injury,bcl-2 mRNA is induced but not translated in CA3 neurons, which are selectively vulnerable and die (Graham et al., 1996). Thus, after ischemia and epilepsy a role for the translational block ofbcl-2 in selective vulnerability is supported. After TBI, the lack of bcl-2 protein translated in CA1 is consistent with the occurrence of a sublethal insult in this area, because neurons in CA1 are less vulnerable than neurons in CA3 in this model (Clark et al., 1997). Thus, translational block of bcl-2 in this model of TBI does not herald cell death.
With rare exception, bcl-2-expressing neurons did not show evidence of apoptosis in the two methods of detection that were used. TUNEL is a marker of DNA fragmentation and damage that supports but does not confirm apoptotic cell death (Gavrieli et al., 1992;Charriaut-Marlangue and Ben-Ari, 1995). The temporal pattern of TUNEL-positive cells in selectively vulnerable regions was similar to the temporal pattern of bcl-2 mRNA induction (Figs. 2, 8). The early time course of TUNEL-positive cells was also similar to the initial increase in bcl-2 protein levels in both cortex and hippocampus; however, the number of TUNEL-positive cells decreased at 72 hr, whereas relative bcl-2 protein levels were not reduced (Figs. 5,8). Importantly, relative bcl-2 protein levels in hippocampus did not decrease at 72 hr despite a reduction in CA1 and CA3 hippocampal cell counts (decreased by 21.5% and 43.6% vs control, respectively). We also examined the gross nuclear morphology of neurons expressing bcl-2 using bis-benzimide dye. Bis-benzimide stains nuclear material and has been used to identify cells undergoing apoptosis (Chrest et al., 1993). Because bcl-2 suppresses apoptotic neuronal death in many in vitro systems (Garcia et al., 1992; Allsopp et al., 1993;Batistatou et al., 1993; Kane et al., 1993; Zhong et al., 1993) and neurons expressing bcl-2 showed normal gross nuclear morphology and were not TUNEL-positive as found in this study, we speculate that bcl-2 protects neurons from endonuclease activation, DNA fragmentation, and nuclear damage. Alternative explanations include the following: (1) all viable neurons express bcl-2 in response to stress and bcl-2 is not directly protective, and (2) in dying neurons all protein synthesis is inhibited and/or proteins are denatured by proteases, making detection of bcl-2 or other proteins unlikely [although in a cardiac arrest model in rats, Krajewski et al. (1995) reported colocalization with TUNEL and Bax (a pro-apoptotic gene product) in dying neurons]. bcl-2 can also protect neurons from necrotic cell death (Kane et al., 1995). Consistent with this possibility, neurons that were TUNEL-positive with a predominantly necrotic morphology also did not express bcl-2 in our study (although we have not excluded the possibility that bcl-2 can be expressed in TUNEL-negative, necrotic neurons). Further study is needed to determine whether bcl-2 plays an active role in the prevention of apoptotic and/or necrotic neuronal death after TBI.
Cells with evidence of DNA fragmentation displayed morphophological characteristics suggestive of both apoptosis and necrosis after TBI (Fig. 9). In some cells a condensed, pyknotic nucleus with clumping of nuclear material was seen (suggestive of apoptosis), whereas in others a diffuse pattern of TUNEL staining without shrinkage of the nuclei and with occasional extravasation of TUNEL labeling into the cytoplasm (suggestive of necrosis) was observed. This is similar to our previous report using this model of TBI with secondary insult (Clark et al., 1997). Using a fluid–percussion injury model without a controlled secondary insult, Rink et al. (1995) were the first to report TUNEL positivity in CA3, dentate, and cortical neurons with characteristics of both necrotic (including cytoplasmic TUNEL staining) and apoptotic death after TBI. Because of differences in the methods of quantitation, it is difficult to compare the degree of apoptosis and necrosis in our model compared with the fluid–percussion model, although it is clear that both apoptosis and necrosis occur in selectively vulnerable neurons in both models of TBI. Neuronal death by both apoptosis and necrosis after brain injury in vivo has also been comprehensively described in models of excitotoxicity (Portera-Cailliau et al., 1997a,b) and ischemia (Deshpande et al., 1992; Li et al., 1995;Nitatori et al., 1995). The studies by Portera-Cailliau et al. (1997a,b) provide evidence for an “apoptosis–necrosis continuum” that depends on neuronal maturity and glutamate receptor subtype. Thus, it is possible that the regional differences in cellular morphologies seen after TBI, i.e., a predominant apoptotic pattern in dentate gyrus versus a predominant necrotic pattern in CA3 hippocampus, may reflect regional differences in non-NMDA and NMDA receptor subtype activation.
bcl-2 may suppress apoptosis via several mechanisms. Recent studies suggest that bcl-2 may prevent apoptosis by blocking cytochrome c release from mitochondria (Kluck et al., 1997; Yang et al., 1997). The localization of bcl-2 in the mitochondrial membrane (Hockenbery et al., 1990) and the ability of bcl-2 to prevent apoptotic cell death induced by oxidant stress in several in vitro systems (Hockenbery et al., 1993; Kane et al., 1993; Zhong et al., 1993;Lawrence et al., 1996) suggests that bcl-2 may exert its protective effects either by acting as an antioxidant or by reducing free radical production. A role for bcl-2 in the regulation of calcium homeostasis is also supported (Lam et al., 1994). In a model of excitoxic neuronal injury, transforming growth factor β overexpression induced bcl-2 expression and protected rat hippocampal neurons in culture (Prehn et al.,1994). Nitric oxide sustained bcl-2 levels in lymphocytes and prevented programmed cell death after treatment with cyclic GMP analogs (Genaro et al., 1995). bcl-2 may exert its effects by regulating other gene products that promote apoptosis, such as forming heterodimers with the pro-apoptotic protein bax (Oltvai et al., 1993; Yin et al., 1994) or by inhibiting effects of cysteine protease activation (Miura et al., 1993; Kumar et al., 1994; Srinivasan et al., 1996). Recent studies show that bcl-2 family genes interact with CED-4, aCaenorhabditis elegans death-promoting gene, and alter its cellular distribution (Wu et al., 1997). Finally, bcl-2 may play a role in regulating other important mechanisms of cell function (Fernandez-Sarabia and Bischoff, 1993; Marvel et al., 1994; Ryan et al., 1994). Free radical damage (Kontos and Wei, 1986), loss of cellular calcium homeostasis (Young, 1992), excitotoxicity (McIntosh, 1993), and induction of genes encoding cysteine proteases (Yakolev et al., 1996) are all implicated in the evolution of neuronal damage after TBI. Finally, other bcl-2 gene family members, includingbcl-xL,bcl-xS, andbcl-x-β, likely play a role in facilitating neuronal survival after brain injury (Boise et al., 1993; Gottschalk et al., 1994; Gonzalez-Garcia et al., 1995; Krajewski et al., 1995; Chen et al., 1997).
In conclusion, our results support a role for bcl-2 in the regulation of apoptotic neuronal death after TBI. Further investigation is warranted to determine whether augmenting bcl-2expression in individual neurons affects the degree of apoptotic and necrotic death and subsequent neuron survival after TBI.
Footnotes
This work was supported by Public Health Service Grants 1KO8 NS01946, 2P50 NS30318-04A1, and 5P30 HD28836, and by a grant from the Children’s Hospital of Pittsburgh. S.H.G was also supported in part by the Department of Veterans Medical Affairs Merit Review Program. P.M.K. was also supported in part by an Established Investigator Grant from the Society of Critical Care Medicine. We thank Dr. Roger P. Simon for helpful comments and critical review of this manuscript, Joanne Schiding and Scott Heineman for expert technical assistance, and Francie Seigfried for editorial assistance.
Correspondence should be addressed to Dr. Steven H. Graham, Department of Neurology, S-526 Biomedical Science Tower, Pittsburgh, PA 15261.
REFERENCES
- 1.Allsopp TE, Wyatt S, Paterson HF, Davies AM. The proto-oncogene bcl-2 can selectively rescue neurotrophic factor-dependent neurons from apoptosis. Cell. 1993;73:295–307. doi: 10.1016/0092-8674(93)90230-n. [DOI] [PubMed] [Google Scholar]
- 2.Batistatou A, Merry DE, Korsmeyer SJ, Greene LA. bcl-2 affects survival but not neuronal differentiation of PC12 cells. J Neurosci. 1993;13:4422–4428. doi: 10.1523/JNEUROSCI.13-10-04422.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Munez G, Thompson CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 4.Charriaut-Marlangue C, Ben-Ari Y. A cautionary note on the use of TUNEL stain to determine apoptosis. NeuroReport. 1995;7:61–64. [PubMed] [Google Scholar]
- 5.Chen J, Graham SH, Chan PH, Lan J, Zhou RL, Simon RP. bcl-2 is expressed in neurons that survive focal ischemia in the rat. NeuroReport. 1995;6:394–398. doi: 10.1097/00001756-199501000-00040. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Zhu RL, Nakayama M, Kawaguchi K, Jin K, Stetler RA, Simon RP, Graham SH. Expression of the apoptosis-effector gene, bax, is up-regulated in vulnerable hippocampal CA1 neurons following global ischemia. J Neurochem. 1996;67:64–71. doi: 10.1046/j.1471-4159.1996.67010064.x. [DOI] [PubMed] [Google Scholar]
- 7.Chen J, Graham SH, Nakayama M, Zhu RL, Jin K, Stetler RA, Simon RP. Apoptosis repressor genes bcl-2 and bcl-x-long are expressed in the rat brain following global ischemia. J Cereb Blood Flow Metab. 1997;17:2–10. doi: 10.1097/00004647-199701000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Chrest FJ, Buchholz MA, Kim YH, Kwon T-K, Nordin AA. Identification and quantitation of apoptotic cells following anti-CD3 activation of murine G0 T-cells. Cytometry. 1993;14:883–890. doi: 10.1002/cyto.990140806. [DOI] [PubMed] [Google Scholar]
- 9.Clark RSB, Kochanek PM, Dixon CE, Chen M, Heineman S, Marion DW, DeKosky ST, Graham SH. Early neuropathologic effects of mild or moderate hypoxemia after controlled cortical impact injury in rats. J Neurotrauma. 1997;14:179–189. doi: 10.1089/neu.1997.14.179. [DOI] [PubMed] [Google Scholar]
- 10.Cortez SC, Mcintosh TK, Noble LJ. Experimental fluid percussion brain injury: vascular disruption and neuronal and glial alterations. Brain Res. 1989;482:271–282. doi: 10.1016/0006-8993(89)91190-6. [DOI] [PubMed] [Google Scholar]
- 11.Deitrich WD, Alonso O, Busto R, Globus MYT, Ginsberg MD. Post-traumatic brain hypothermia reduces histopathological damage following concussive brain injury in the rat. Acta Neuropathol. 1994;87:250–258. doi: 10.1007/BF00296740. [DOI] [PubMed] [Google Scholar]
- 12.Deshpande J, Bergstedt K, Linden T, Kalimo H, Wieloch T. Ultrastructural changes in the hippocampal CA1 region following transient cerebral ischemia: evidence against programmed cell death. Exp Brain Res. 1992;88:91–105. doi: 10.1007/BF02259131. [DOI] [PubMed] [Google Scholar]
- 13.Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- 14.Fernandez-Sarabia MJ, Bischoff JR. bcl-2 associates with the ras-related protein R-ras p23. Nature. 1993;366:274–275. doi: 10.1038/366274a0. [DOI] [PubMed] [Google Scholar]
- 15.Garcia I, Martinou I, Tsujimoto Y, Martinou JC. Prevention of programmed cell death of sympathetic neurons by the bcl-2 proto-oncogene. Science. 1992;258:302–304. doi: 10.1126/science.1411528. [DOI] [PubMed] [Google Scholar]
- 16.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Genaro AM, Hortelano S, Alvarez A, Martinez AC, Bosca L. Splenic B lymphocyte programmed cell death is prevented by nitric oxide release through mechanisms involving sustained bcl-2 levels. J Clin Invest. 1995;95:1884–1890. doi: 10.1172/JCI117869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalez-Garcia M, Garcia I, Ding L, O’Shea S, Boise LH, Thompson CB, Munez G. bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc Natl Acad Sci USA. 1995;92:4304–4308. doi: 10.1073/pnas.92.10.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gottschalk AR, Boise LH, Thompson CB, Quintans J. Identification of immunosuppressant-induced apoptosis in a murine B-cell line and its prevention by bcl-x but not bcl-2. Proc Natl Acad Sci USA. 1994;91:7350–7354. doi: 10.1073/pnas.91.15.7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graham SH, Chen J, Stetler RA, Zhu RL, Jin KL, Simon RP. Expression of proto-oncogene bcl-2 is increased in the rat brain following kainate-induced seizures. Restorative Neurol Neurosci. 1996;9:243–250. doi: 10.3233/RNN-1996-9407. [DOI] [PubMed] [Google Scholar]
- 21.Hockenbery DM. bcl-2, a novel regulator of cell death. BioEssays. 1995;17:631–638. doi: 10.1002/bies.950170709. [DOI] [PubMed] [Google Scholar]
- 22.Hockenbery DM, Nunez G, Milliman CL, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 23.Hockenbery DM, Oltvai ZN, Yin X-M, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 24.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 25.Kane DJ, Ord T, Anton R, Bredesen DE. Expression of bcl-2 inhibits necrotic neural cell death. J Neurosci Res. 1995;40:269–275. doi: 10.1002/jnr.490400216. [DOI] [PubMed] [Google Scholar]
- 26.Kerr JFR, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 28.Kochanek PM, Marion DW, Zhang W, Schiding JK, White M, Palmer AM, Clark RS, O’Malley ME, Styren SD, Ho C. Severe controlled cortical impact in rats: assessment of cerebral edema, blood flow, and contusion volume. J Neurotrauma. 1995;12:1015–1025. doi: 10.1089/neu.1995.12.1015. [DOI] [PubMed] [Google Scholar]
- 29.Kontos HA, Wei EP. Superoxide production in experimental brain injury. J Neurosurg. 1986;64:803–807. doi: 10.3171/jns.1986.64.5.0803. [DOI] [PubMed] [Google Scholar]
- 30.Krajewski S, Mai JK, Krajewska M, Sikorska M, Mossakowski MJ, Reed JC. Upregulation of Bax protein levels in neurons following cerebral ischemia. J Neurosci. 1995;15:6364–6376. doi: 10.1523/JNEUROSCI.15-10-06364.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar S, Kinoshita M, Noda M, Copeland NG, Jenkins NA. Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1β converting enzyme. Genes Dev. 1994;8:1613–1626. doi: 10.1101/gad.8.14.1613. [DOI] [PubMed] [Google Scholar]
- 32.Lam M, Dubyak G, Chen L, Nunez G, Miesfeld RL, Distelhorst CW. Evidence that bcl-2 represses apoptosis by regulating endoplasmic reticulum-associated calcium fluxes. Proc Natl Acad Sci USA. 1994;91:6569–6573. doi: 10.1073/pnas.91.14.6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawrence MS, Ho DY, Sun GH, Steinberg GK, Sapolsky RM. Overexpression of bcl-2 with herpes simplex virus vectors protects CNS neurons against neurological insults in vitro and in vivo. J Neurosci. 1996;16:486–496. doi: 10.1523/JNEUROSCI.16-02-00486.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.LeBrun DP, Warnke RA, Cleary ML. Expression of bcl-2 in fetal tissues suggests a role in morphogenesis. Am J Pathol. 1993;142:743–753. [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Chopp M, Jiang N, Yao F, Zaloga C. Temporal profile of in situ DNA fragmentation after transient middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1995;15:389–397. doi: 10.1038/jcbfm.1995.49. [DOI] [PubMed] [Google Scholar]
- 36.Linnik MD, Zahos R, Geschwind MD, Federoff HJ. Expression of bcl-2 from a defective herpes simplex virus-1 vector limits neuronal death in focal cerebral ischemia. Stroke. 1995;26:1670–1675. doi: 10.1161/01.str.26.9.1670. [DOI] [PubMed] [Google Scholar]
- 37.Lowenstein DH, Thomas MJ, Smith DH, Mcintosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12:4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C, Huarte J. Overexpression of BLC-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 39.Marvel J, Perkins GR, Lopez Rivas A, Collins MKL. Growth factor starvation of bcl-2 overexpressing murine bone marrow cells induced refractoriness to IL-3 stimulation of proliferation. Oncogene. 1994;9:1117–1122. [PubMed] [Google Scholar]
- 40.McIntosh TK. Novel pharmacologic therapies in the treatment of experimental traumatic brain injury: a review. J Neurotrauma. 1993;10:215–261. doi: 10.1089/neu.1993.10.215. [DOI] [PubMed] [Google Scholar]
- 41.Miura M, Zhu H, Rotello R, Hartwieg EA, Yaun J. Induction of apoptosis in fibroblasts by IL-1β converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- 42.Negrini M, Silini E, Kozak C, Tsujimoto Y, Croce CM. Molecular analysis of mbcl-2: structure and expression of the murine gene homologous to the human gene involved in follicular lymphoma. Cell. 1987;49:455–463. doi: 10.1016/0092-8674(87)90448-x. [DOI] [PubMed] [Google Scholar]
- 43.Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Novack DV, Korsmeyer SJ. Bcl-2 protein expression during murine development. Am J Pathol. 1994;145:61–73. [PMC free article] [PubMed] [Google Scholar]
- 45.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 46.Portera-Cailliau C, Price DL, Martin LJ. Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphologic continuum. J Comp Neurol. 1997a;378:70–87. [PubMed] [Google Scholar]
- 47.Portera-Cailliau C, Price DL, Martin LJ. Non-NMDA and NMDA receptor-mediated excitotoxic neuronal deaths in adult brain are morphologically distinct: further evidence for an apoptosis-necrosis continuum. J Comp Neurol. 1997b;378:88–104. [PubMed] [Google Scholar]
- 48.Prehn JHM, Bindokas VP, Marcuccilli CJ, Krajewski S, Reed JC, Miller RJ. Regulation of neuronal bcl-2 protein expression and calcium homeostasis by transforming growth factor type β confers wide-ranging protection on rat hippocampal neurons. Proc Natl Acad Sci USA. 1994;91:12599–12603. doi: 10.1073/pnas.91.26.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rink A, Fung K-M, Trojanowksi JQ, Lee VMY, Neugebauer E, McIntosh TK. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- 50.Ryan JJ, Prochownik E, Gottlieb CA, Apel IJ, Merino R, Munez G, Clarke MF. c-myc and bcl-2 modulate p53 function by altering p53 subcellular trafficking during the cell cycle. Proc Natl Acad Sci USA. 1994;91:5878–5882. doi: 10.1073/pnas.91.13.5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sato T, Irie S, Krajewski S, Reed JC. Cloning and sequencing of a cDNA encoding the rat bcl-2 protein. Gene. 1994;140:291–292. doi: 10.1016/0378-1119(94)90561-4. [DOI] [PubMed] [Google Scholar]
- 52.Saunders JW. Death in embryonic systems. Science. 1966;154:604–612. doi: 10.1126/science.154.3749.604. [DOI] [PubMed] [Google Scholar]
- 53.Sentman CL, Shutter RH, Hockenbery D, Kanagawa O, Korsmeyer SJ. bcl-2 inhibits multiple forms of apoptosis but not negative selection in thymocytes. Cell. 1991;67:879–888. doi: 10.1016/0092-8674(91)90361-2. [DOI] [PubMed] [Google Scholar]
- 54.Shimazaki K, Ishida A, Kawai N. Increase in bcl-2 oncoprotein and the tolerance to ischemia-induced neuronal death in the gerbil hippocampus. Neurosci Res. 1994;20:95–99. doi: 10.1016/0168-0102(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 55.Srinivasan A, Foster LM, Testa MP, Ord T, Keane RW, Bredesen DE, Kayalar C. Bcl-2 expression in neural cells blocks activation of ICE/CED-3 family proteases during apoptosis. J Neurosci. 1996;16:5654–5660. doi: 10.1523/JNEUROSCI.16-18-05654.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 57.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 58.Wu D, Wallen HD, Nunez G. Interaction and regulation of subcellular localization of CED-4 by CED-9. Science. 1997;275:1126–1129. doi: 10.1126/science.275.5303.1126. [DOI] [PubMed] [Google Scholar]
- 59.Yakovlev AG, Knoblach SM, Rosenthal D, Smulson M, Faden AI. Upregulation of CPP32 cysteine protease associated with apoptosis in rat cortex following traumatic brain injury. Soc Neurosci Abstr. 1996;22:20. [Google Scholar]
- 60.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng T-I, Jones DP, Wang X. Prevention of apoptosis by bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 61.Yin X-M, Oltvai ZN, Korsmeyer SJ. BH1 and BH2 domains of bcl-2 are required for inhibition of apoptosis and heterodimerization with bax. Nature. 1994;369:321–323. doi: 10.1038/369321a0. [DOI] [PubMed] [Google Scholar]
- 62.Young W. Role of calcium in central nervous system injuries. J Neurotrauma. 1992;9:S9–S25. [PubMed] [Google Scholar]
- 63.Zhong L-T, Sarafian T, Kane DJ, Charles AC, Mah SP, Edwards RH, Bredesen DE. bcl-2 inhibits death of central neural cells induced by multiple agents. Proc Natl Acad Sci USA. 1993;90:4533–4537. doi: 10.1073/pnas.90.10.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]