

Abstract
The effect of partial hippocampal kindling, a model of temporal lobe seizure, on monosynaptic inhibition mediated by GABA was studied. Kindled rats were given 15 nonconvulsive hippocampal afterdischarges, and control rats were given low frequency or no stimulations. At 1–2 d after kindling, paired-pulse depression (PPD) of the IPSCs recorded in CA1 neurons in vitro was significantly smaller in kindled as compared with control rats. The difference in PPD persisted for at least 21 d after kindling. The decrease in PPD of the IPSCs after partial hippocampal kindling was likely caused by a reduced GABA autoinhibition after downregulation of presynaptic GABABreceptors. The GABAB antagonist CGP35348 (1 mm) suppressed PPD of the IPSCs more strongly in control than in kindled rats. Direct activation of the presynaptic GABAB receptors by baclofen suppressed the monosynaptic IPSCs significantly more in control than in kindled rats. The decay rate of a single-pulse IPSC was faster in kindled than in control rats on day 1 or day 21 after partial kindling. The difference in IPSC decay between kindled and control rats was found with or without a GABAB receptor antagonist. The low efficacy of the presynaptic GABAB receptors in kindled rats may provide compensatory stabilization of the postsynaptic membrane against further seizures or plasticity.
Keywords: inhibitory postsynaptic current, presynaptic inhibition, GABAB receptors, paired-pulse depression, kindling, seizures, hippocampus
The modulation of inhibition mediated by GABA is important for normal brain functions (Krnjević, 1991). A local and transient loss of inhibition may mediate physiological plasticity and memory storage (Davies et al., 1991; Mott and Lewis, 1991; Wilson and McNaughton, 1993; Buzsaki et al., 1994). GABAergic interneurons are subjected to various local and extrinsic control (Freund and Buzsaki, 1996). In addition, GABA release is controlled by autoinhibition via presynaptic GABABreceptors (Deisz and Prince, 1989; Thompson and Gahwiler, 1989; Davies et al., 1990, 1991; Nathan and Lambert, 1991; Silvilotti and Nistri, 1991; Bowery, 1993; Lambert and Wilson, 1994; Olpe et al., 1994; Pitler and Alger, 1994; Misgeld et al., 1995; Kaupmann et al., 1997). However, it is not known whether the presynaptic GABAB receptors are themselves regulated by neuronal activity.
GABA neurotransmission has long been associated with seizure activity. GABAA antagonists induce seizures (Gale, 1992;Schwartzkroin, 1993), and drugs that enhance GABA neurotransmission are effective anticonvulsants (Macdonald and McLean, 1986). Acute seizures suppress GABA function (Stelzer et al., 1987; Kapur et al., 1989). However, a decrease of GABA function in chronic epilepsy is still controversial (Babb et al., 1989; Engel, 1995).
In the kindling model of epilepsy, repeated stimulations are delivered to the brain that progressively evoke longer afterdischarges (ADs) and more severe convulsions (Goddard et al., 1969; McNamara et al., 1993). Full kindling refers to stimulations that evoked generalized tonic–clonic convulsions, whereas partial kindling evoked no convulsions. Partial hippocampal kindling is a good model of human temporal lobe seizures, which may not result in convulsions. Furthermore, the AD evoked by partial hippocampal kindling was mainly restricted to structures closely connected to the hippocampus (Leung et al., 1997), and no cell loss was detected (Cavazos et al., 1994). After hippocampal or amygdala kindling, paired-pulse inhibition of population spikes was increased in the dentate gyrus (Tuff et al., 1983; Oliver and Miller, 1985a; Zhao and Leung, 1992) but decreased in CA1 (Kamphuis et al., 1988; Zhao and Leung, 1991). The different paired-pulse response in the dentate gyrus and CA1 after kindling is consistent with the change in GABAA receptor binding (Titulaer et al., 1994, 1995), but not with the change in GABA immunoreactivity (Kamphuis et al., 1989) or GABA release (Kamphuis et al., 1991). In addition, EPSPs showed an increase in paired-pulse facilitation after kindling (Zhao and Leung, 1991, 1993).
The purpose of this study was to characterize the paired-pulse monosynaptic inhibitory response. At the time we began our study, the effect of kindling on monosynaptic inhibition and GABA autoinhibition was not known. Recently, Buhl et al. (1996) found a decrease in paired-pulse depression (PPD) of the IPSCs in dentate gyrus granule cells after full kindling of the perforant path. This study documents a similar finding in CA1 neurons after partial hippocampal kindling. In addition, we show that the kindling-induced decrease in PPD of the IPSCs was specifically mediated by a downregulation of presynaptic GABAB autoreceptors and lasted 3 weeks after partial kindling.
A brief summary of the work has been published previously (Wu and Leung, 1996; Leung et al., 1997).
MATERIALS AND METHODS
The kindling procedures and the in vitro slice preparation were reported previously (Leung et al., 1994). Briefly, repeated electrical stimulations were delivered through chronic, indwelling electrodes implanted in stratum radiatum of hippocampal CA1 of male Long–Evans rats. Experimental rats were given the kindling stimulus (1 sec, 100 Hz, pulses of 0.1 msec). About half of the control rats were given low-frequency stimulations (LFSs), each consisting of 100 pulses at 0.167 Hz, and the others were implanted with electrodes but not given any stimulations. There was no significant difference between the responses of the two types of control rats, and their data were combined into a single control group. ADs/LFSs were given hourly, 5 per day, for a total of 15 ADs/LFSs in 3 d.
On day 1–2 (day 1 group) or day 21–23 (day 21 group) after the last kindling or control treatment, the rat was anesthetized with halothane and decapitated. Most kindled and control rats were paired, and the experiments were done on consecutive days, with the experimenter unaware of the stimulation history of each rat. The hippocampus on the stimulated side was dissected out and 400-μm-thick transverse slices were obtained from the midseptotemporal level (Leung et al., 1994). The slices were placed in an interface chamber and perfused with artificial cerebrospinal fluid at 32 ± 1°C of the following composition (in mm): NaCl 124, KCl 5, NaH2PO4.H2O 1.25, MgSO4.7 H2O 2, CaCl2.6 H2O 2, NaHCO3 26, and glucose 10. A concentric bipolar stimulating electrode was placed in stratum radiatum in CA1 (on the CA3 side) ∼0.3 mm from the recording site. Sharp micropipettes (3m potassium acetate; impedance 60–100 MΩ) were used to impale CA1 neurons in the pyramidal cell layer. QX-314 (167 mm) was added to the potassium acetate in the micropipette to block fast Na+ channels (Connors and Prince, 1982; Leung and Yim, 1991) and GABAB responses (Nathan et al., 1990; Andrade, 1991; Otis and Mody, 1992). To isolate the IPSC without excitation, an NMDA antagonistd-2-amino-5-phosphonopentanoic acid (d-AP5; 20 μm) and a non-NMDA antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 20 μm) were added to the perfusate. Single-electrode voltage clamp was made by an Axoclamp 2A amplifier at 4.5–5.0 kHz switching frequency, and the voltage before the sample-and-hold device was monitored to ensure that the transients had decayed before being sampled. Because inhibitory synapses were predominantly located near the cell body, lack of space clamp (Rall and Segev, 1985) was not a significant problem. Paired-pulse stimulations were given at 0.12 Hz, 2 or 4 × threshold intensity (0.2 msec duration), and an interpulse interval (IPI) of 50–500 msec, with the cell clamped at a potential of −40 to −120 mV. The threshold intensity was determined as the lowest intensity that evoked a detectable field response in each slice before the perfusion of glutamate antagonists, and typically only one (occasionally two) neuron was recorded per slice. The potentials were amplified, digitized at 10 KHz, averaged (n = 4), and stored on line by a custom program. In some experiments, GABAB receptor antagonist CGP35348 (1 mm) or GABAB receptor agonist baclofen (1–10 μm) was added to the medium containing CNQX and d-AP5.
In voltage clamp, PPD was evaluated by the ratio of the peak of the IPSC evoked by the second pulse divided by that evoked by the first pulse. All IPSC measures were evaluated at −50 mV holding potential if not otherwise specified. In current clamp, PPD was evaluated by the ratio of the peak IPSP evoked by the second pulse to that evoked by the first pulse. The reversal potential of the IPSC was estimated by a linear interpolation of the two IPSCs (recorded every 10 mV of the holding potential) yielding positive and negative responses, respectively. The latter reversal potential was within a few millivolts of the estimate given by linear regression analysis of the IPSC versus the holding potential (range −50 to −100 mV).
Statistical differences between measures in different groups were tested by a nonparametric Wilcoxon test, using either cells or rats as the basic unit. For “per cell” statistics, all of the cells studied were included; however, to minimize bias from a single rat, no more than four cells were selected from the same rat. For the “per rat” statistics, the average measure for all of the cells recorded for one rat (n = 1–6 cells) was used, and the sample number was the number of rats. In experiments in which repeated time measures were made, e.g., after drug perfusion, a repeated-measure ANOVA was applied, followed by post hoc t tests.
The half-peak durations of the IPSC1 [t0.5(1)] and IPSC2 [t0.5(2)] was estimated as the time interval from the onset of an IPSC (∼2.4 msec after a stimulus) to the time the IPSC decayed to half the peak value. The falling phase of the IPSC was fitted by a single exponential IPSC(t) =Io e−t/τ, with initial amplitude Io and time constant τ. The time constants of the first- and second-pulse-evoked IPSC were named τ1 and τ2 respectively; the values reported here were derived from regression yielding R2 > 0.85. In most neurons, the single exponential decay was able to fit the IPSC well for a duration of 80 msec after the IPSC peak, and reducing the fitted duration did not substantially change τ. In some neurons (∼15%), the τ value was estimated using a IPSC duration of 50–80 msec, if the shorter duration improved the fit (as estimated byR2).
A nonparametric technique was also used to compare IPSC durations between ensemble of neurons recorded in different conditions. The mean IPSC of each recorded neuron was normalized by the first IPSC peak, and then the normalized IPSCs from different neurons were averaged at fixed times (0.1 msec bins for 250 msec duration). The ensemble SEM at each bin was also calculated. Because the IPSCs from different neurons were triggered at a fixed time by a digital pulse generator (Master 8, A.M.P.I., Jerusalem), their stimulation pulses were precisely lined up in time. When the ensemble averages of two conditions were compared, the lack of overlap between the SEMs of the two ensembles (compare Fig.2) suggests a statistical difference between the ensembles, which was more precisely evaluated by a repeated-measure ANOVA followed by apost hoc t test, using samples at every 5–10 msec intervals.
Fig. 2.

Ensemble averages of IPSCs from neurons recorded in CNQX and d-AP5 medium. For each pair of traces, thetop trace is the ensemble mean + 1 SEM and thebottom trace is the ensemble mean − 1 SEM. The kindled group is blue, and the control group isred. Day 1 (A) and day 21 (B) averages of kindled and control groups, without GABAB antagonist CGP35348. C, Day 1 groups recorded with CGP35348. All IPSCs were recorded using 2 × threshold stimulus intensity, 100 msec interpulse interval, and −50 mV holding potential. For each neuron, the recorded trace was normalized by the first-pulse IPSC peak, and then all neurons in the same group were combined in the ensemble average (see Materials and Methods). Number of neurons in kindled (K) and control (C) groups:A, C = 14, K = 16; B, C = 11, K = 18; C, C = 17, K = 17. Note faster decay of the first-pulse IPSC in the kindled as compared with control group under all conditions.
RESULTS
Kindling and CA1 cell characteristics
The duration of the hippocampal AD, recorded in the homotopic CA1 contralateral to the stimulated CA1, increased with the number of ADs delivered, similar to previous results (Leung et al., 1994, their Fig. 1). In the group of nine kindled rats used for day 1 recordings (data in Table 1), the first AD measured 31.5 ± 4.2 sec (mean ± SEM) and the fifteenth (last) was 93.7 ± 17.9 sec. The increase in AD duration from the first to the last AD was statistically significant. Similar results were obtained in other kindled rats. No behavioral convulsion was observed during the ADs.
Table 1.
Measures of monosynaptic IPSCs on day 1 after partial kindling or control procedures
| 2 × threshold | 4 × threshold | |||
|---|---|---|---|---|
| Kindled (21) | Control (16) | Kindled (11) | Control (13) | |
| Peak IPSC1 (nA) | 0.8 ± 0.07 | 0.98 ± 0.18 | 0.94 ± 0.13 | 1.08 ± 0.21 |
| Peak IPSC2 (nA) | 0.68 ± 0.07 | 0.68 ± 0.16 | 0.81 ± 0.12 | 0.75 ± 0.18 |
| Paired-pulse depression | 0.85 ± 0.031-165 | 0.63 ± 0.04 | 0.85 ± 0.031-160 | 0.65 ± 0.05 |
| Half-duration IPSC1 | 46.1 ± 1.71-160 | 57.7 ± 3.2 | 31.8 ± 2.11-160 | 45.5 ± 3.2 |
| t0.5(1) (msec) | ||||
| Half-duration IPSC2 | 42.6 ± 1.6 | 42.3 ± 2.0 | 27.7 ± 1.7 | 30.4 ± 2.2 |
| t0.5(2) (msec) | ||||
| Time constant IPSC1 | 35.2 ± 3.3* | 49.9 ± 5.4 (15) | ||
| τ1 (msec) | ||||
| Time constant IPSC2 | 39.9 ± 5.3 | 44.8 ± 5.9 (15) | ||
| τ2 (msec) | ||||
IPSCs were recorded at −50 mV holding potential, after paired-pulse stimulation (100 msec interpulse interval) of stratum radiatum in a medium with 20 μm CNQX and 20 μmd-AP5, at either 2 × or 4 × threshold stimulus intensity. Paired-pulse depression was estimated by the ratio (peak IPSC2/peak IPSC1).
F1-165: p < 0.001;
F1-160: p < 0.01;
p < 0.05 kindled different from control group for the same stimulus intensity.
The resting membrane potential from cells recorded at the CA1 cell layer was −58.3 ± 0.7 mV (mean ± SEM;n = 138); resting membrane potential was not different between kindled and control groups. The input resistance was 61.5 ± 5.5 MΩ (n = 22). With QX-314 in the recording micropipette, slow, presumed Ca2+ spikes of >10 msec duration could be evoked if a neuron was depolarized to approximately −45 mV (Leung and Yim, 1991).
PPD of the IPSCs 1 d after partial hippocampal kindling
Monosynaptic IPSCs/IPSPs were obtained by direct stimulation of the stratum radiatum in the presence of CNQX and d-AP5 (see Materials and Methods). The recorded IPSC was mediated by GABAA receptors, because it was abolished by GABAA antagonist bicuculline (not shown). No postsynaptic GABAB current was detected, and the amplitude of the single-pulse IPSCs was not decreased by GABAB antagonist CGP35348 (Fig. 1A). In neurons from control rats recorded after paired-pulse stimulation, the amplitude and duration of the IPSC after the second pulse (IPSC2) was smaller than the respective measure of the first-pulse IPSC (IPSC1) (Fig. 1A), as reported previously (McCarren and Alger, 1985; Davies et al., 1990; Lambert and Wilson, 1994; Olpe et al., 1994). In control neurons, the PPD ratio (peak IPSC2/peak IPSC1) was 0.63 ± 0.04 (n = 16) at an IPI of 100 msec (holding potential of −50 mV and using 2 × threshold intensity stimuli, i.e., 50 ± 1 μA, 0.2 msec duration pulses). In neurons from kindled rats, IPSC2 was strikingly similar in size and duration to IPSC1 (Fig. 1A). PPD was significantly smaller (p < 0.01) in kindled than in control rats at an IPI of 50, 100, and 150 msec but not 500 msec (Fig.1B; Table 1). There was no difference in the stimulus intensities used to evoke the IPSCs in the kindled and control groups. The latency to reach the IPSC1 or IPSC2 peak did not differ between control and kindled rats (12 ± 1 msec). Also, the peak amplitudes of the monosynaptic IPSCs after single-pulse radiatum stimulation were not significantly different (p > 0.3; Wilcoxon) between the two groups (Table 1).
Fig. 1.

Kindled seizures reduced PPD of GABAAmediated IPSCs recorded at a holding potential of −50 mV.A, Examples of paired-pulse IPSCs in CA1 neurons from control and kindled rat, evoked by stratum radiatum stimuli of 2 × threshold intensity in the presence of CNQX and d-AP5, before and after perfusion with a CGP35348 (1 mm) medium. Peaks of IPSC1 and IPSC2 indicated by peak1 andpeak2, respectively. Resting membrane potential of control and “kindled” neuron was −59 and −60 mV, respectively.Filled circles indicate shock artifacts.B, Plot of the ratio of the peak IPSC evoked by the second pulse (peak IPSC2) to that evoked by the first pulse (peak IPSC1), in the absence of CGP35348, as a function of interpulse interval. Error bars are one SEM. Control group (C) = 8 neurons and kindled group (K) = 10 neurons, except for 500 msec interpulse interval data, where C = 6 and K = 4 neurons. ** (p < 0.01) indicates significant difference between control and kindled groups of neurons as assessed by the nonparametric Wilcoxon test.
If the stimulus intensity was doubled from 2 to 4 × threshold, IPSCs increased slightly, but the PPD ratio was not significantly changed (Table 1). The difference between the kindled and control groups was also significant (p < 0.01; Wilcoxon) at 4 × threshold intensity (Table 1).
The time course of the IPSCs appeared to be different in kindled as compared with control rats (Fig. 1A). The duration of the IPSC1 [t0.5(1)] was significantly longer in control than in kindled rats (Table 1). Time constant τ1 was estimated to be ∼50 msec in the control group, a value similar to that estimated by Roepstorff and Lambert (1994) or the slower time constant of Pearce (1993), and significantly larger (p < 0.05) than that in the kindled group (Table 1). The ensemble averages (normalized by the peak IPSC1 of each neuron) show that the average IPSC1 in the kindled group decayed much more rapidly than that of the control group (Fig.2A). The ensemble averages of IPSC1 were significantly different from each other at 30–100 msec after the pulse (p < 0.05;post hoc t test after significant group, time, and group × time effects with repeated-measure ANOVA). Other than the difference near the peak of the IPSC2 (10–30 msec after the second pulse), the late time course of the IPSC2 was not different between the ensemble averages of the kindled and control groups (p > 0.05; post hoc t test) (Fig. 2A).
The differences between kindled and control groups were robust and were shown using either per cell or per rat statistics (see Materials and Methods). Per rat statistics also revealed significant differences between kindled (n = 9) and control rats (n = 9) in the PPD, t0.5(1), and τ1 measures.
Current-clamp recordings on day 1 after kindling–control procedures
Some neurons were also recorded in a discontinuous current-clamp mode, and the results were congruent with the voltage clamp data. Radiatum stimulation at a prepulse membrane potential of −50 mV (maintained by current injection) yielded a ratio of IPSP peaks that was significantly larger (p < 0.01) in the kindled than in the control group. Similar results were found at 2 and 4 × threshold stimulus intensity (Table2).
Table 2.
Ratio of the IPSP peaks evoked by paired pulses at 100 msec interpulse interval, recorded in discontinuous current clamp mode at −50 mV membrane potential on day 1 or 21 after kindling/control procedures. Stimulation was at either 2 × or 4 × threshold stimulus intensity
| 2 × threshold | 4 × threshold | |||
|---|---|---|---|---|
| Kindled | Control | Kindled | Control | |
| Day 1 | 0.84 ± 0.04 (16)* | 0.67 ± 0.05 (16) | 0.82 ± 0.03 (11)* | 0.61 ± 0.05 (15) |
| Day 21 | 0.70 ± 0.05 (13) | 0.53 ± 0.11 (5) | ||
p < 0.01, difference between kindled and control groups.
IPSCs/IPSPs on day 21 after kindling
At 21 d after kindling, neurons from kindled rats also showed significantly smaller PPD than did neurons from control rats (p < 0.01; Fig. 2B). At 100 msec IPI, the PPD ratio was 0.77 ± 0.05 (n = 18) in neurons of kindled rats as compared with 0.55 ± 0.06 (n = 11) in neurons of control rats (Table3). Interestingly, IPSC1 amplitudes were similar among the day 21 neurons, and mean IPSC2 amplitude was larger in the kindled than the control group (Table 3). However, at 500 msec IPI, no significant difference in PPD was found between kindled and control groups (Table 3).
Table 3.
Measures of IPSCs recorded on day 21 after kindling/control procedures at −50 mV holding potential, in a medium with 20 μm CNQX and 20 μmd-AP5
| 100 msec IPI | 500 msec IPI | |||
|---|---|---|---|---|
| Kindled (18) | Control (11) | Kindled (14) | Control (6) | |
| IPSC1 peak (nA) | 0.72 ± 0.07 | 0.69 ± 0.07 | 0.65 ± 0.08 | 0.72 ± 0.10 |
| IPSC2 peak (nA) | 0.51 ± 0.043-150 | 0.37 ± 0.05 | 0.57 ± 0.06 | 0.60 ± 0.09 |
| Paired-pulse depression | 0.77 ± 0.053-160 | 0.55 ± 0.06 | 0.90 ± 0.02 | 0.85 ± 0.05 |
| Half-duration IPSC1 | 43.8 ± 3.33-160 | 62.0 ± 4.8 | 44.1 ± 3.2 | 66.2 ± 7.6 |
| t0.5(1) (msec) | ||||
| Half-duration IPSC2 | 32.9 ± 1.3 | 36.3 ± 1.1 | 37.8 ± 2.2 | 50.9 ± 3.1 |
| t0.5(2) (msec) | ||||
| Time constant IPSC1 | 38.1 ± 4.63-160 | 64.0 ± 7.2 | ||
| τ1 (msec) | ||||
| Time constant IPSC2 | 37.2 ± 4.23-150 | 60.2 ± 7.5 | ||
| τ2 (msec) | ||||
Stimulation was at 2 × threshold stimulus intensity and either 100 or 500 msec interpulse interval (IPI).
F3-160: p < 0.01;
F3-150: p < 0.05, kindled different from control groups for the same IPI.
The difference in IPSC decay between kindled and control rats also persisted 21 d after partial hippocampal kindling (Fig.2B). The measures t0.5(1), τ1, and τ2 were significantly smaller in the kindled than the control group (Table 3). Ensemble averages of the IPSCs (Fig.2B) revealed that the decay time course of IPSC1 or IPSC2 was faster in the kindled than the control group at >20 msec latency (p < 0.01; post hoc ttest after repeated-measure ANOVA, which gave significant group, time, and group × time effects).
When statistics per rat were performed, the day 21 kindled rats (n = 6) also showed significantly smallert0.5(1), τ1 (p < 0.05; Wilcoxon) than a pooled control group consisting of five day 21 control rats and nine day 1 controls. The control rats were pooled because there was no difference in any measure between the day 21 and day 1 control rats. PPD was also significantly different (p < 0.05) in day 21 kindled rats than in the pooled control rats.
IPSC reversal potentials and holding potentials
The reversal potentials of IPSC1 (RP1) were not significantly different between cells from day 1 kindled (n = 8 cells) and control groups (n = 13 cells) or between day 21 kindled (n = 15) and control groups (n = 12). Similarly, the reversal potential of IPSC2 (RP2) was not different between kindled and control groups, on either day 1 or day 21. However, when RP2 was compared with RP1 in the same neuron (Fig. 3), RP2 was more positive than RP1 by 4.3 ± 1.1 mV (n = 13) in control day 1 neurons and by 2.2 ± 0.6 mV in kindled day 1 neurons (n = 8). In the day 1 groups, RP1 and RP2 were 82.3 ± 7.1 mV and 78.3 ± 6.8 mV (n = 13), respectively, in the control group, as compared with 84.0 ± 3.3 mV and 82.9 ± 3.3 mV (n = 8) in the kindled group. The slight but significant (paired Wilcoxon; p< 0.01) depolarizing shift of RP2 with respect to RP1 was the main reason that PPD appeared to be weaker or absent at holding potentials of −80 to −110 mV (Fig. 3A). When the chord conductance of IPSC1 (GI1) and IPSC2 (GI2) was estimated at each holding potential [by peak IPSC/(holding minus reversal potential)], the paired conductance ratio (GI2/GI1) was similar across holding potentials for a particular group of neurons. The conductance ratio (GI2/GI1) was significantly different (p < 0.05) between day 1 kindled and control neurons at all holding potentials used (−50 to −120 mV). At −50 mV holding potential, the conductance ratio was ∼15% higher than peak IPSC ratio, but the difference between day 1 kindled and control groups was similarly robust (p < 0.001) using either ratio of conductances or ratio of peak currents.
Fig. 3.

Average IPSC (n = 4 sweeps) versus voltage relation, in a medium with CNQX and d-AP5 but without CGP35348, at −50 to −110 mV holding potentials for a neuron from (A) day 1 control and (B) day 1 kindled rat. Outward rectification was seen in both neurons. A depolarizing shift of the reversal potential for IPSC2 as compared with IPSC1 is shown in A. At all holding potentials, PPD of the IPSCs was observed in Abut not in B.
Effect of GABAB antagonist on paired IPSCs
To investigate the role of presynaptic GABAB receptors in the PPD of IPSCs, GABAB receptor antagonist CGP35348 (1 mm) was added to the medium with CNQX andd-AP5. Recordings were made 1 d after kindling and control procedures. In a medium with CGP35348, the mean PPD at 100 msec IPI was slightly smaller in the kindled as compared with the control group (Fig. 2C; Table 4), but the difference between groups was not statistically significant (p = 0.09).
Table 4.
IPSC measures recorded in a CGP35348 (1 mm) medium with CNQX and d-AP5, 1 day after kindling/control procedures, using 100 msec interpulse interval and −50 mV holding potential, at 2 × or 4 × threshold stimulus intensity
| 2 × threshold | 4 × threshold | |||
|---|---|---|---|---|
| Kindled (17) | Control (17) | Kindled (16) | Control (15) | |
| Peak IPSC1 (nA) | 0.99 ± 0.09 | 0.92 ± 0.10 | 1.00 ± 0.07 | 0.89 ± 0.10 |
| Peak IPSC2 (nA) | 0.94 ± 0.08 | 0.76 ± 0.08 | 0.93 ± 0.07 | 0.75 ± 0.09 |
| Paired-pulse depression | 0.96 ± 0.02 | 0.89 ± 0.04 | 0.92 ± 0.03 | 0.86 ± 0.03 |
| Half-duration IPSC1 | 45.0 ± 3.34-150 | 58.7 ± 3.5 | 49.7 ± 3.4 | 56.2 ± 2.6 |
| t0.5(1) (msec) | ||||
| Half-duration IPSC2 | 49.1 ± 3.4 | 55.6 ± 1.5 | 51.7 ± 2.2 | 54.7 ± 2.0 |
| t0.5(2) (msec) | ||||
| Time constant IPSC1 | 30.0 ± 2.9 (16)4-160 | 53.1 ± 6.1 | ||
| τ1 (msec) | ||||
| Time constant IPSC2 | 33.1 ± 2.6 (16)4-150 | 61.9 ± 6.4 | ||
| τ2 (msec) | ||||
F4-160: p < 0.01;
F4-150: p < 0.05 kindled different from control group for the same stimulus intensity.
The faster IPSC decay in the kindled than in the control group of neurons persisted in a medium with CGP35348. In the latter medium,t0.5(1) and τ1 were significantly different between kindled and control groups (Table 4), and in addition, a significant difference was found for τ2 (Table 4). Significant differences (p ≤ 0.05; Wilcoxon) int0.5(1), τ1, and τ2 between kindled (n = 6 rats) and control (n = 8 rats) groups were also found if per rat statistics were used.
The ensemble averages (Fig. 2C) reveal the differences graphically. The IPSC1 ensemble averages of control and kindled groups were statistically different at 20–100 msec after the first pulse (p < 0.01; post hoc t test, after repeated-measure ANOVA revealed significant group, time, and group × time effects). In a medium with CGP35348, IPSC2 ensemble averages were also different between control and kindled groups at 20–140 msec after the second pulse (p < 0.01;post hoc t test after repeated-measure ANOVA yielding significant group, time, and group × time effects).
Single neurons were assessed before, during, and after washout of 1 mm CGP35348 (Fig. 1A). In the day 1 control group, CGP35348 decreased PPD and increased IPSC2 duration in all five neurons (Fig. 1A; p < 0.05; paired Wilcoxon). In the same group, CGP35348 had no consistent effect on IPSC amplitudes, but it increased τ1 ort0.5(1) in four of five neurons (Fig.1A; 0.05 < p < 0.1). In eight neurons from the day 1 kindled group, CGP35348 decreased PPD significantly (p < 0.05; Fig.1A), although the PPD decrease was smaller than that in the control group. This suggests that the GABABantagonist was still effective in blocking autoinhibition in the kindled group, but its effect was reduced as compared with the control group. CGP35348 increased t0.5(1) and τ1 significantly (p < 0.05) in the day 1 kindled group of neurons.
Effects of GABAB agonist baclofen on paired IPSCs
A decrease in GABA release and an increase in uptake may limit the activation of the presynaptic GABAB receptors (Isaacson et al., 1993; Roepstorff and Lambert, 1994). Thus, the GABABagonist baclofen (10 μm in the bath) was used to directly activate the presynaptic GABAB receptors (Davies et al., 1990; Thompson and Gahwiler, 1992; Misgeld et al., 1995). Baclofen reduced IPSC1 in neurons from both control and kindled rats, but the reduction was significantly stronger in control than in kindled rats (Fig. 4A). The effect of baclofen was detected at 5 min after perfusion and was near maximal by 10 min (Fig. 4B). At 15 min after baclofen, the peak IPSC1 of the control group was 23 ± 3% of the baseline (seven neurons), whereas that of the kindled group was 44 ± 5% (nine neurons) (Fig. 4B). The effect of baclofen was predominantly on presynaptic inhibition because QX-314 in the micropipette already blocked the postsynaptic GABABconductance (Nathan et al., 1990; Andrade, 1991).
Fig. 4.
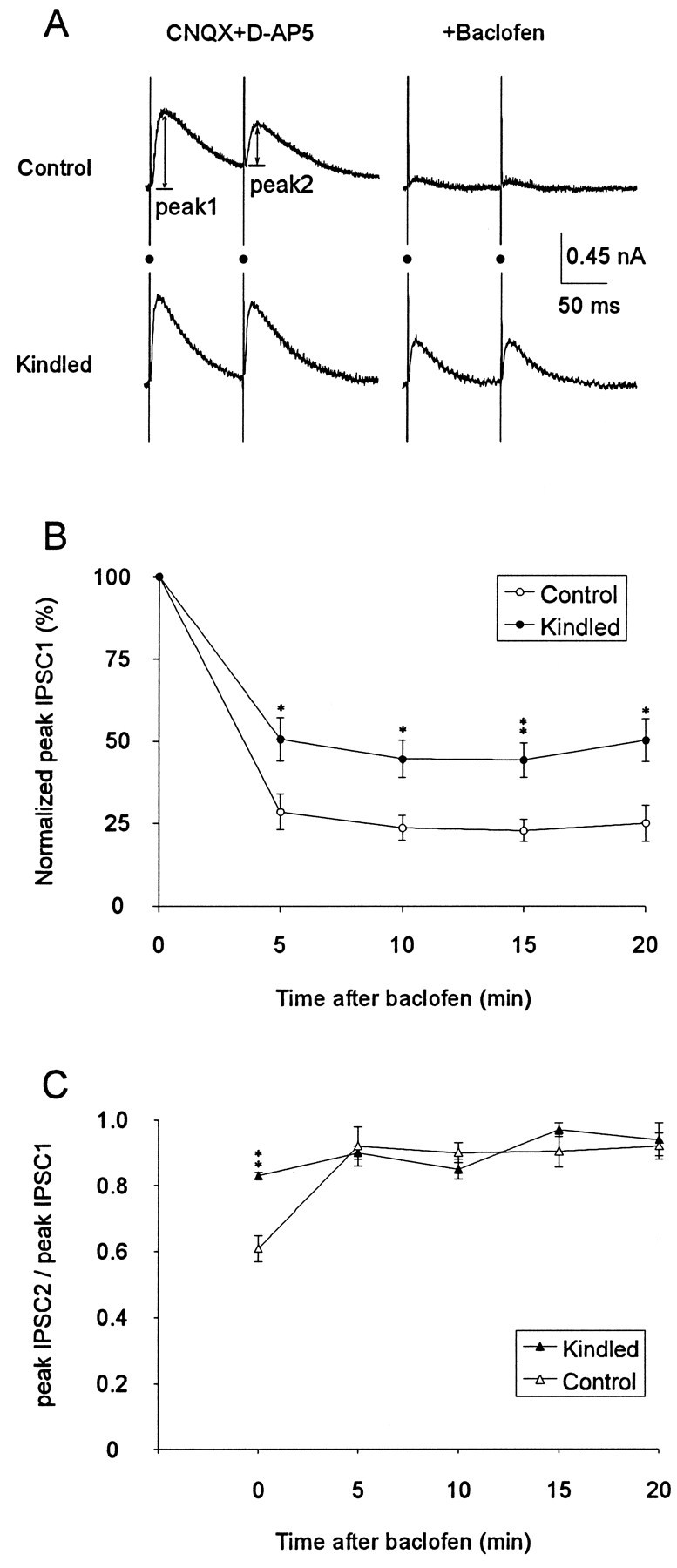
Partial hippocampal kindling reduced the effect of GABAB agonist baclofen in suppressing IPSCs.A, Examples of paired-pulse IPSCs in CA1 neurons from control and kindled rat, recorded at −50 mV holding potential, before and after the perfusion of 10 μm baclofen (added to the CNQX and d-AP5 medium). Resting membrane potential for control and “kindled” neuron was −60 and −63 mV, respectively.Filled circles indicate shock artifacts.B, Baclofen reduced the first IPSC in neurons from both kindled and control rats, but the effect was significantly larger in control as compared with kindled rats. C, The difference in the PPD (ratio of IPSC peaks) between kindled and control rats was abolished after 5 min of baclofen perfusion. * (p < 0.05), ** (p < 0.01) indicate significant difference between kindled and control groups (Wilcoxon) at a fixed time after baclofen.
Before the perfusion of baclofen, PPD was 0.61 ± 0.04 (n = 7) in control neurons and 0.83 ± 0.01 (n = 9) in “kindled” neurons. The difference in PPD between kindled and control neurons was significant (p < 0.01), confirming the result of the earlier experiment (Table 1). Baclofen decreased PPD in control neurons significantly to 0.92 ± 0.06 (n = 7; repeated-measure ANOVA; p < 0.01), consistent with the previous literature (Davies et al., 1990; Mott and Lewis, 1991). In contrast, baclofen did not significantly change PPD in the kindled rats (Fig. 4C).
DISCUSSION
PPD of IPSCs in CA1
The data on PPD in CA1 neurons in the control rats were in accordance with those reported previously (Davies et al., 1990; Davies and Collingridge, 1993). A notable exception is the finding that the reversal potential of IPSC2 (RP2) was significantly more depolarized than that of IPSC1 (RP1) by ∼4 mV, similar to the results of McCarren and Alger (1985). Davies et al. (1990) stated that PPD “was not associated with any change in reversal potential of the IPSC,” but the resolution of their measures was unclear (see their Fig. 4). At −50 mV holding potential, the reversal potential shift contributed ∼15% to the PPD, and this relatively small contribution did not change the main results of this study or those of the previous studies (Davies et al., 1990; Davies and Collingridge, 1993). The shift in reversal potential may indicate a change in intracellular Cl− concentration (McCarren and Alger, 1985;Thompson and Gahwiler, 1989).
Downregulation of presynaptic GABAB autoreceptors
As shown previously by Davies et al. (1990), PPD of the IPSC peak amplitudes is mainly a measure of presynaptic inhibition of GABA release. The larger ratio of paired-pulse IPSC amplitudes (and durations) in kindled as compared with control rats, at day 1 or day 21 after partial hippocampal kindling, suggests that GABA autoinhibition was weaker in kindled than in control rats. We infer that the weaker autoinhibition was mediated by a decreased efficacy or downregulation of the presynaptic GABAB receptors on GABA terminals. The PPD of IPSCs in control rats was strongly suppressed by GABAB antagonist CGP35348 or GABAB agonist baclofen (by occlusion), but the PPD of IPSCs in kindled rats was relatively insensitive to GABAB agonist or antagonist. Exogenously applied GABAB agonist baclofen suppressed the first-pulse IPSC in control rats much more than that in kindled rat, confirming that presynaptic GABAB receptor efficacy was reduced in kindled as compared with control rats.
The decrease in presynaptic GABAB receptor efficacy may be caused by a downregulation of the number of GABAB receptors or a decrease in the coupling of GABAB receptors to guanine-nucleotide-binding proteins (Pitler and Alger, 1994; Misgeld et al., 1995; Kaupmann et al., 1997) or to presynaptic Ca2+ currents (Pfrieger et al., 1994; Wu and Saggau, 1997). In the presence of a GABAB antagonist, no significant difference in PPD was detected between kindled and control groups, which does not suggest a role for non-GABABmediated presynaptic inhibition (Lambert and Wilson, 1993; Wilcox and Dichter, 1994).
Effects of GABAB antagonist and kindling on the IPSC time course
The decay rate of IPSC1 was slowed by GABAB antagonist CGP35348, whereas the peak amplitude was not consistently changed. CGP35348 has little affinity for the GABAA receptor (Olpe et al., 1990), and suppression of residual postsynaptic GABAB currents does not account for the CGP35348 effect, because it should decrease rather than increase the late time course of IPSC1. The mechanism by which CGP35348 slowed the IPSCs is not known. It is possible that presynaptic GABAB inhibition after a single pulse may contribute to a shutdown of further GABA release (20 msec after the afferent stimulus). This is apparently the first time that a GABAB antagonist has been shown to affect the kinetics of a single evoked IPSC. It has not been reported before perhaps because of overlap with the postsynaptic GABABcurrents (Davies et al., 1990; Wan et al., 1996) or a lack of quantitative analysis of the IPSCs.
The decay rate of the IPSC was faster in kindled as compared with control rats, best characterized by smaller τ1 values in the kindled than in the control group, and this effect persisted at least 21 d after partial hippocampal kindling. The effect was found in the presence of GABAB antagonist CGP35348, suggesting that it was independent of GABAB receptors. The mechanism underlying the difference in IPSC decay is not known. The release of GABA may be less sustained in kindled as compared with control rats. GABA uptake may be increased in the kindled rat, and a change in GABA uptake would affect the decay more than the peak of the IPSC (Isaacson et al., 1993; Roepstorff and Lambert, 1994). Changes in GABAA receptor subunits or phosphorylation (Stelzer, 1992;Macdonald and Olsen, 1994) may affect the IPSC kinetics, and GABAA receptor subunit changes have been reported in CA1 after hippocampal kindling, although the CA1 changes were small and did not last more than a few days (Clark et al., 1994; Kokaia et al., 1994;Kamphuis et al., 1995). Otis et al. (1994) found no change in the time course of miniature IPSCs in dentate granule cells after full kindling, but the decay time constant of miniature IPSCs (Collingridge et al., 1984; Ropert et al., 1990; Otis and Mody, 1992) is considerably smaller than that of evoked IPSCs (Pearce, 1993; Roepstorff and Lambert, 1994; this study).
Effect of kindling on neuronal excitation and inhibition
The loss of presynaptic GABAB receptor efficacy on GABA terminals after hippocampal kindling may be ubiquitous and pervasive, as suggested by the decrease in PPD of the IPSCs in the dentate gyrus after convulsive kindled seizures (Buhl et al., 1996). Because kindling induces the same change in PPD of the IPSCs but different changes of paired-pulse spike response in CA1 and the dentate gyrus (see introductory remarks), PPD of IPSCs cannot be the crucial factor involved in the paired-pulse population spike response. In CA1 neurons in vitro, the magnitude of IPSC2 was not a significant factor in the generation of an early latency spike after the second pulse (Leung and Fu, 1994). However, a faster decay of IPSC1 may account partly for the increased paired-pulse facilitation of the population spike in CA1 neurons, which persisted for approximately 6 weeks after partial hippocampal kindling (Leung et al., 1994).
We found no significant reduction of the peak of the monosynaptic IPSC1 in CA1 neurons after partial kindling. Similarly, Oliver and Miller (1985b) found no change in CA1 neuronal excitability during recurrent inhibition after kindling. Titulaer et al. (1994, 1995) reported a decrease in postsynaptic GABAA receptor binding of 10–25% in CA1, which may not be manifested in the IPSCs. The complexity of the inhibitory circuitry and its modulation cannot be determined by the monosynaptic IPSCs alone. Seizures have been suggested to selectively shut down afferent pathways to the GABAergic interneuron (Sloviter, 1991; Bekenstein and Lothman, 1993; Mangan et al., 1995). Preliminary studies (L. S. Leung and D. Zhao, unpublished observations) revealed that partial hippocampal kindling induced a suppression of PPD of disynaptic inhibition but no apparent loss of single-pulse feedforward inhibition.
Significance of GABAB autoreceptor regulation
This is the first time that neural activity, in this case electrographic seizure, has been shown to regulate autoinhibition specifically through the presynaptic GABAB receptors. Buhl et al. (1996) showed a suppression of PPD of the IPSCs in dentate granule cells 1–2 d after full kindling (15 stage 5 seizures) of the perforant path but did not show the involvement of GABABreceptors. Downregulation of presynaptic GABAB receptors was proposed after kainic acid seizures, but not for the autoreceptors on GABAergic terminals (Haas et al., 1996). A decrease in efficacy of presynaptic GABAB receptors on glutamatergic terminals was shown in the basolateral amygdala after amygdala kindling (Asprodini et al., 1992) and in CA1 after partial hippocampal kindling (Wu and Leung, 1997).
Presynaptic GABAB receptors and autoinhibition may be of great but as yet unproven significance (Bowery, 1993; Misgeld et al., 1995). Release studies (Waldmeier et al., 1993) indicate that “basal release alone already substantially activated the autoreceptor,” whereas no clear presynaptic GABAB effects have been demonstrated in vivo (Olpe et al., 1993; Misgeld et al., 1995). An important function of presynaptic GABAB receptors on GABA terminals may be to regulate the amount of postsynaptic inhibition and thus control the postsynaptic excitability that may lead to paroxysmal activity (Ben-Ari et al., 1979). Microinfusion of baclofen in the hippocampus of behaving rats induced seizure activity (Vaurio et al., 1996), which may be mediated by disinhibition through presynaptic GABAB receptors or postsynaptic GABAB receptors on inhibitory interneurons (Mott et al., 1989). The downregulation of GABAB receptors after seizures (Haas et al., 1996; this study) may thus serve to prevent more seizures and contribute to the refractory period after seizures (Engel, 1995). A possible trade-off is that normal physiological plasticity such as long-term potentiation may also be blocked (Hesse and Teyler, 1976; Wu and Leung, 1997).
Footnotes
This work was supported by National Institutes of Health Grant NS-25383, a grant from Natural Sciences and Engineering Research Council, and an internal grant from the London Health Sciences Centre. We thank B. Shen, K. Canning, and K. Wu for technical assistance and Drs. Peter Cain, Peter Carlen, Karen Gale, Rick McLachlan, Steve Sims, and Kevin Canning for comments and discussions. We thank Novartis, Basel, Switzerland, for the gift of CGP35348 and Astra (Westborough) for QX-314.
Correspondence should be addressed to Dr. L. Stan Leung, Department of Clinical Neurological Sciences, University Campus, London Health Sciences Centre, The University of Western Ontario, London, Ontario, Canada N6A 5A5.
REFERENCES
- 1.Andrade RA. Blockade of neurotransmitter-activated K+ conductance by QX-314 in the rat hippocampus. Eur J Pharmacol. 1991;199:259–262. doi: 10.1016/0014-2999(91)90467-5. [DOI] [PubMed] [Google Scholar]
- 2.Asprodini EK, Rainnie DG, Shinnick-Gallagher P. Epileptogenesis reduces the sensitivity of presynaptic gamma-aminobutyric acid B receptors on glutamatergic afferents in the amygdala. J Pharmacol Exp Ther. 1992;262:1011–1021. [PubMed] [Google Scholar]
- 3.Babb TL, Pretorius JK, Kupfer WR, Crandall PH. Glutamate decarboxylase-immunoreactive neurons are preserved in human epileptic hippocampus. J Neurosci. 1989;9:2562–2574. doi: 10.1523/JNEUROSCI.09-07-02562.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bekenstein JW, Lothman EW. Dormancy of inhibitory interneurons in a model of temporal lobe epilepsy. Science. 1993;259:97–100. doi: 10.1126/science.8093417. [DOI] [PubMed] [Google Scholar]
- 5.Ben-Ari Y, Krnjevic K, Reinhardt W. Hippocampal seizures and failure of inhibition. Can J Physiol Pharmacol. 1979;57:1462–1466. [Google Scholar]
- 6.Bowery NG. GABAB receptor pharmacology. Annu Rev Pharmacol Toxicol. 1993;33:109–147. doi: 10.1146/annurev.pa.33.040193.000545. [DOI] [PubMed] [Google Scholar]
- 7.Buhl EH, Otis TS, Mody I. Zinc-induced collapse of augmented inhibition by GABA in a temporal lobe epilepsy model. Science. 1996;271:369–373. doi: 10.1126/science.271.5247.369. [DOI] [PubMed] [Google Scholar]
- 8.Buzsaki G, Bragin A, Chrobak JJ, Nadasdy Z, Sik A, Hsu M, Ylinen A. Oscillatory and intermittent synchrony in the hippocampus: relevance to memory trace formation. In: Buzsaki G, Llinas R, Singer W, Bethoz A, Christian T, editors. Temporal coding in the brain. Springer; New York: 1994. pp. 145–172. [Google Scholar]
- 9.Cavazos JE, Das I, Sutula TP. Neuronal loss induced in limbic pathways by kindling: evidence for induction of hippocampal sclerosis by repeated brief seizures. J Neurosci. 1994;14:3106–3121. doi: 10.1523/JNEUROSCI.14-05-03106.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark M, Massenburg GS, Weiss SR, Post RM. Analysis of the hippocampal GABA-A receptor system in kindled rats by autoradiographic and in situ hybridization techniques: contingent tolerance to carbamazepine. Brain Res Mol Brain Res. 1994;26:309–319. doi: 10.1016/0169-328x(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 11.Collingridge GL, Gage PW, Robertson B. Inhibitory post-synaptic currents in rat hippocampal CA1 neurons. J Physiol (Lond) 1984;356:551–564. doi: 10.1113/jphysiol.1984.sp015482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Connors BW, Prince DA. Effects of local anesthetic QX-314 on the membrane properties of hippocampal pyramidal neurons. J Pharmacol Exp Ther. 1982;220:476–481. [PubMed] [Google Scholar]
- 13.Davies CH, Collingridge GL. The physiological regulation of synaptic inhibition by GABA(B) autoreceptors in rat hippocampus. J Physiol (Lond) 1993;472:245–265. doi: 10.1113/jphysiol.1993.sp019945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davies CH, Davies SN, Collingridge GL. Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. J Physiol (Lond) 1990;424:513–531. doi: 10.1113/jphysiol.1990.sp018080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies CH, Starkey SJ, Pozza MF, Collingridge GL. GABAB autoreceptors regulate the induction of LTP. Nature. 1991;349:609–611. doi: 10.1038/349609a0. [DOI] [PubMed] [Google Scholar]
- 16.Deisz RA, Prince DA. Frequency-dependent depression of inhibition in guinea-pig neocortex in vitro by GABAB receptor feed-back on GABA release. J Physiol (Lond) 1989;412:513–541. doi: 10.1113/jphysiol.1989.sp017629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Engel J., Jr Inhibitory mechanisms of epileptic seizure generation. Adv Neurol. 1995;67:157–171. [PubMed] [Google Scholar]
- 18.Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6:345–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 19.Gale K. Role of GABA in the genesis of chemoconvulsant seizures. Toxicol Lett. 1992;64/65:417–428. doi: 10.1016/0378-4274(92)90215-6. [DOI] [PubMed] [Google Scholar]
- 20.Goddard GV, McIntyre DC, Leech CK. A permanent change in brain function resulting from daily electrical stimulation. Exp Neurol. 1969;25:295–330. doi: 10.1016/0014-4886(69)90128-9. [DOI] [PubMed] [Google Scholar]
- 21.Haas KZ, Stanton EF, Moshe SL, Stanton PK. Kainic acid-induced seizures enhance dentate gyrus inhibition by downregulation of GABAB receptors. J Neurosci. 1996;16:4250–4260. doi: 10.1523/JNEUROSCI.16-13-04250.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hesse GW, Teyler TJ. Reversible loss of hippocampal long term potentiation following electroconvulsive seizures. Nature. 1976;264:562–564. doi: 10.1038/264562a0. [DOI] [PubMed] [Google Scholar]
- 23.Isaacson JS, Solis JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- 24.Kamphuis W, Lopes da Silva FH, Wadman WJ. Changes in local evoked potentials in the rat hippocampus (CA1) during kindling epileptogenesis. Brain Res. 1988;440:205–215. doi: 10.1016/0006-8993(88)90988-2. [DOI] [PubMed] [Google Scholar]
- 25.Kamphuis W, Huisman E, Wadman WJ, Lopes da Silva FH. Decrease in GABA immunoreactivity and alteration of GABA metabolism after kindling in the rat hippocampus. Exp Brain Res. 1989;74:375–386. doi: 10.1007/BF00248871. [DOI] [PubMed] [Google Scholar]
- 26.Kamphuis W, Huisman E, Veerman MJ, Lopes da Silva FH. Development of changes in endogenous GABA release during kindling epileptogenesis in rat hippocampus. Brain Res. 1991;545:33–40. doi: 10.1016/0006-8993(91)91266-4. [DOI] [PubMed] [Google Scholar]
- 27.Kamphuis W, De Rijk TC, Lopes da Silva FH. Expression of GABAA receptor subunit MRNAs in hippocampal pyramidal and granular neurons in the kindling model of epileptogenesis: an in situ hybridization study. Brain Res Mol Brain Res. 1995;31:33–47. doi: 10.1016/0169-328x(95)00022-k. [DOI] [PubMed] [Google Scholar]
- 28.Kapur J, Stringer JL, Lothman EW. Evidence that repetitive seizures in the hippocampus cause a lasting reduction of GABAergic inhibition. J Neurophysiol. 1989;61:417–426. doi: 10.1152/jn.1989.61.2.417. [DOI] [PubMed] [Google Scholar]
- 29.Kaupmann K, Huggel K, Held J, Flor PJ, Bischoff S, Mickel SJ, McMaster G, Angst C, Bittiger H, Froestl W, Bettler B. Expression cloning of GABA-B receptors uncovers similarity to metabotropic glutamate receptors. Nature. 1997;386:239–246. doi: 10.1038/386239a0. [DOI] [PubMed] [Google Scholar]
- 30.Kokaia M, Pratt GD, Elmer E, Bengzon J, Fritschy JM, Kokaia Z, Lindvall O, Mohler H. Biphasic differential changes of GABAA receptor subunit mRNA levels in dentate gyrus granule cells following recurrent kindling-induced seizures. Brain Res Mol Brain Res. 1994;23:323–332. doi: 10.1016/0169-328x(94)90242-9. [DOI] [PubMed] [Google Scholar]
- 31.Krnjević K. Significance of GABA in brain function. In: Tunnicliff G, Raess BU, editors. GABA mechanisms in epilepsy. Wiley; New York: 1991. pp. 47–87. [Google Scholar]
- 32.Lambert NA, Wilson WA. Heterogeneity in presynaptic regulation of GABA release from hippocampal inhibitory neurons. Neuron. 1993;11:1057–1067. doi: 10.1016/0896-6273(93)90219-h. [DOI] [PubMed] [Google Scholar]
- 33.Lambert NA, Wilson WA. Temporally distinct mechanisms of use-dependent depression at inhibitory synapses in rat hippocampus in vitro. J Neurophysiol. 1994;72:121–130. doi: 10.1152/jn.1994.72.1.121. [DOI] [PubMed] [Google Scholar]
- 34.Leung LS, Fu X-W. Factors affecting paired-pulse facilitation in CA1 neurons in vitro. Brain Res. 1994;650:75–84. doi: 10.1016/0006-8993(94)90209-7. [DOI] [PubMed] [Google Scholar]
- 35.Leung LS, Yim CY. Intrinsic membrane potential oscillations in hippocampal neurons in vitro. Brain Res. 1991;553:261–274. doi: 10.1016/0006-8993(91)90834-i. [DOI] [PubMed] [Google Scholar]
- 36.Leung LS, Zhao D, Shen B. Long-lasting effects of partial hippocampal kindling on hippocampal physiology and function. Hippocampus. 1994;4:696–704. doi: 10.1002/hipo.450040607. [DOI] [PubMed] [Google Scholar]
- 37. Leung LS, Wu C, Wu K, Shen B, Sutherland R, Zhao D. Long-lasting behavioral and electrophysiological effects induced by partial hippocampal kindling. Kindling 5 Corcoran ME, Moshe S. 1997. Plenum; New York, in press. [Google Scholar]
- 38.Macdonald RL, McLean MJ. Anticonvulsant drugs: mechanisms of action. Adv Neurol. 1986;44:713–736. [PubMed] [Google Scholar]
- 39.Macdonald RL, Olsen RW. GABAA receptor channels. Annu Rev Neurosci. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- 40.Mangan PS, Rempe DA, Lothman EW. Changes in inhibitory neurotransmission in the CA1 region and dentate gyrus in a chronic model of temporal lobe epilepsy. J Neurophysiol. 1995;74:829–840. doi: 10.1152/jn.1995.74.2.829. [DOI] [PubMed] [Google Scholar]
- 41.McCarren M, Alger BE. Use-dependent depression of IPSPs in rat hippocampal pyramidal cells in vitro. J Neurophysiol. 1985;53:557–571. doi: 10.1152/jn.1985.53.2.557. [DOI] [PubMed] [Google Scholar]
- 42.McNamara JO, Bonhaus DW, Shin C. The kindling model of epilepsy. In: Schwartzkroin PA, editor. Epilepsy: models, mechanisms, and concepts. Cambridge UP; Cambridge, UK: 1993. pp. 27–47. [Google Scholar]
- 43.Misgeld U, Bijak M, Jarolimek W. A physiological role for GABAB receptors and the effects of baclofen in the mammalian central nervous system. Prog Neurobiol. 1995;46:423–462. doi: 10.1016/0301-0082(95)00012-k. [DOI] [PubMed] [Google Scholar]
- 44.Mott DD, Lewis DV. Facilitation of the induction of long-term potentiation by GABAB receptors. Science. 1991;252:1718–1720. doi: 10.1126/science.1675489. [DOI] [PubMed] [Google Scholar]
- 45.Mott DD, Bragdon AC, Lewis DV, Wilson WA. Baclofen has a proepileptic effect in the rat dentate gyrus. J Pharmacol Exp Ther. 1989;249:721–725. [PubMed] [Google Scholar]
- 46.Nathan T, Jensen MS, Lambert JD. The slow inhibitory postsynaptic potential in rat hippocampal CA1 neurones is blocked by intracellular QX-314. Neurosci Lett. 1990;110:309–313. doi: 10.1016/0304-3940(90)90865-7. [DOI] [PubMed] [Google Scholar]
- 47.Nathan T, Lambert JD. Depression of the fast IPSP underlies paired-pulse facilitation in area CA1 of the rat hippocampus. J Neurophysiol. 1991;66:1704–1715. doi: 10.1152/jn.1991.66.5.1704. [DOI] [PubMed] [Google Scholar]
- 48.Oliver MW, Miller JJ. Analysis of inhibitory processes in the dentate gyrus following kindling-induced epilepsy. Exp Brain Res. 1985a;57:443–447. doi: 10.1007/BF00237830. [DOI] [PubMed] [Google Scholar]
- 49.Oliver MW, Miller JJ. Inhibitory processes of hippocampal CA1 pyramidal neurons following kindling-induced epilepsy in the rat. Can J Physiol Pharmacol. 1985b;63:872–878. doi: 10.1139/y85-143. [DOI] [PubMed] [Google Scholar]
- 50.Olpe HR, Karlsson G, Pozza MF, Greiner K, Brugger F, Steinmann MW, van Riezen H, Fagg G, Hall RG, Froestl W, Bittiger H. CGP35348: a centrally active blocker of GABAB receptors. Eur J Pharmacol. 1990;187:27–38. doi: 10.1016/0014-2999(90)90337-6. [DOI] [PubMed] [Google Scholar]
- 51.Olpe HR, Steinmann MW, Ferrat T, Pozza MF, Greiner K, Brugger F, Froestl W, Mickel SJ, Bittiger H. The actions of orally active GABAB receptor antagonists on GABAergic transmission in vivo and in vitro. Eur J Pharmacol. 1993;233:179–186. doi: 10.1016/0014-2999(93)90048-m. [DOI] [PubMed] [Google Scholar]
- 52.Olpe HR, Steinmann MW, Greiner K, Pozza MF. Contribution of presynaptic GABA-B receptors to paired-pulse depression of GABA-responses in the hippocampus. Naunyn Schmiedebergs Arch Pharmacol. 1994;349:473–477. doi: 10.1007/BF00169135. [DOI] [PubMed] [Google Scholar]
- 53.Otis TS, Mody I. Modulation of decay kinetics and frequency of GABAA receptor-mediated spontaneous inhibitory postsynaptic currents in hippocampal neurons. Neuroscience. 1992;49:13–32. doi: 10.1016/0306-4522(92)90073-b. [DOI] [PubMed] [Google Scholar]
- 54.Otis TS, de Koninck Y, Mody I. Lasting potentiation of inhibition is associated with an increased number of gamma-aminobutyric acid type A receptors activated during miniature inhibitory postsynaptic currents. Proc Natl Acad Sci USA. 1994;91:7698–7702. doi: 10.1073/pnas.91.16.7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pearce RA. Physiological evidence for two distinct GABAA responses in rat hippocampus. Neuron. 1993;10:189–200. doi: 10.1016/0896-6273(93)90310-n. [DOI] [PubMed] [Google Scholar]
- 56.Pfrieger FW, Gottmann K, Lux HD. Kinetics of GABAB receptor mediated inhibition of calcium currents and excitatory synaptic transmission in hippocampal neurons in vitro. Neuron. 1994;12:97–107. doi: 10.1016/0896-6273(94)90155-4. [DOI] [PubMed] [Google Scholar]
- 57.Pitler TA, Alger BE. Differences between presynaptic and postsynaptic GABAB mechanisms in rat hippocampal pyramidal cells. J Neurophysiol. 1994;72:2317–2327. doi: 10.1152/jn.1994.72.5.2317. [DOI] [PubMed] [Google Scholar]
- 58.Rall W, Segev I. Space-clamp problems when voltage clamping branched neurons with intracellular microelectrodes. In: Smith TG Jr, editor. Voltage and patch clamping with microelectrodes. American Physiology Society; Bethesda, MD: 1985. pp. 191–215. [Google Scholar]
- 59.Roepstorff A, Lambert JDC. Factors contributing to the decay of the stimulus-evoked IPSC in rat hippocampal CA1 neurons. J Neurophysiol. 1994;72:2911–2926. doi: 10.1152/jn.1994.72.6.2911. [DOI] [PubMed] [Google Scholar]
- 60.Ropert N, Miles R, Korn H. Characteristics of miniature inhibitory postsynaptic currents in CA1 pyramidal neurones of rat hippocampus. J Physiol (Lond) 1990;428:707–722. doi: 10.1113/jphysiol.1990.sp018236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwartzkroin PA. Epilepsy: models, mechanisms, and concepts. Cambridge UP; Cambridge, UK: 1993. [Google Scholar]
- 62.Silvilotti L, Nistri A. GABA receptor mechanisms in the central nervous system. Prog Neurobiol. 1991;36:35–92. doi: 10.1016/0301-0082(91)90036-z. [DOI] [PubMed] [Google Scholar]
- 63.Sloviter RS. Permanently altered hippocampal structure, excitability, and inhibition after experimental status epilepticus in the rat: the “dormant basket cell” hypothesis and its possible relevance to temporal lobe epilepsy. Hippocampus. 1991;1:41–66. doi: 10.1002/hipo.450010106. [DOI] [PubMed] [Google Scholar]
- 64.Stelzer A. Intracellular regulation of GABAA receptor function. Ion Channels. 1992;3:83–136. doi: 10.1007/978-1-4615-3328-3_4. [DOI] [PubMed] [Google Scholar]
- 65.Stelzer A, Slater NT, ten Bruggencate G. Activation of NMDA receptors blocks GABAergic inhibition in an in vitro model of epilepsy. Nature. 1987;326:698–701. doi: 10.1038/326698a0. [DOI] [PubMed] [Google Scholar]
- 66.Thompson SM, Gahwiler BH. Activity-dependent disinhibition I. Repetitive stimulation reduces IPSP driving force and conductance in the hippocampus in vitro. J. Neurophysiol. 1989;61:501–511. doi: 10.1152/jn.1989.61.3.501. [DOI] [PubMed] [Google Scholar]
- 67.Thompson SM, Gahwiler BH. Comparison of the actions of baclofen at pre- and postsynaptic receptors in the rat hippocampus in vitro. J Physiol (Lond) 1992;451:329–345. doi: 10.1113/jphysiol.1992.sp019167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Titulaer M, Kamphuis W, Pool CW, van Heerikhuize JJ, Lopes da Silva FH. Kindling induces time-dependent and regional specific changes in the [3H] muscimol binding in the rat hippocampus: a semi-quantitative autoradiographic study. Neuroscience. 1994;59:817–826. doi: 10.1016/0306-4522(94)90286-0. [DOI] [PubMed] [Google Scholar]
- 69.Titulaer MN, Ghijsen WE, Kamphuis W, De Rijk TC, Lopes da Silva FH. Opposite changes in GABAA receptor function in the Ca1–3 area and fascia dentata of kindled rat hippocampus. J Neurochem. 1995;64:2615–2621. doi: 10.1046/j.1471-4159.1995.64062615.x. [DOI] [PubMed] [Google Scholar]
- 70.Tuff LP, Racine RJ, Adamec R. The effects of kindling on GABA-mediated inhibition in the dentate gyrus of the rat. I. Paired-pulse depression. Brain Res. 1983;277:79–90. doi: 10.1016/0006-8993(83)90909-5. [DOI] [PubMed] [Google Scholar]
- 71.Vaurio RG, Proctor MR, Gale K. GABAB activity within the hippocampus attenuates the propagation of limbic seizures. Soc Neurosci Abstr. 1996;22:2103. [Google Scholar]
- 72.Wilcox KS, Dichter MA. Paired pulse depression in cultured hippocampal neurons is due to a presynaptic mechanism independent of GABAB autoreceptor activation. J Neurosci. 1994;14:1775–1788. doi: 10.1523/JNEUROSCI.14-03-01775.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wilson MA, McNaughton BL. Dynamics of the hippocampal ensemble code for space. Science. 1993;261:1055–1057. doi: 10.1126/science.8351520. [DOI] [PubMed] [Google Scholar]
- 74.Waldmeier PC, Hertz C, Grunenwald C, Baumann PA. Autoreceptor-mediated regulation of GABA release: role of uptake inhibition and effects of novel GABABB antagonists. Naunyn Schmiedebergs Arch Pharmacol. 1993;347:514–520. doi: 10.1007/BF00166744. [DOI] [PubMed] [Google Scholar]
- 75.Wan FJ, Berton F, Madamba SG, Francesconi W, Siggins GR. Low ethanol concentrations enhance GABAergic inhibitory postsynaptic potentials in hippocampal pyramidal neurons only after block of GABAB receptors. Proc Natl Acad Sci USA. 1996;93:5049–5054. doi: 10.1073/pnas.93.10.5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu C, Leung LS. Suppression of paired-pulse depression of monosynaptic inhibitory postsynaptic currents (IPSCs) in CA1 neurons by partial hippocampal kindling. Soc Neurosci Abstr. 1996;22:2103. [Google Scholar]
- 77.Wu C, Leung LS. Presynaptic GABAB inhibition of monosynaptic IPSC and field EPSP in CA1 was decreased after partial hippocampal kindling. Soc Neurosci Abstr. 1997;23:2159. doi: 10.1523/JNEUROSCI.17-23-09261.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu L, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- 79.Zhao D, Leung LS. Effects of hippocampal kindling on paired-pulse response in CA1 in vitro. Brain Res. 1991;564:220–229. doi: 10.1016/0006-8993(91)91457-c. [DOI] [PubMed] [Google Scholar]
- 80.Zhao D, Leung LS. Hippocampal kindling induced paired-pulse depression in the dentate gyrus and paired-pulse facilitation in CA3. Brain Res. 1992;582:163–167. doi: 10.1016/0006-8993(92)90333-5. [DOI] [PubMed] [Google Scholar]
- 81.Zhao D, Leung LS. Partial hippocampal kindling increases paired-pulse facilitation and burst frequency in hippocampal CA1 neurons. Neurosci Lett. 1993;154:191–194. doi: 10.1016/0304-3940(93)90204-x. [DOI] [PubMed] [Google Scholar]