

Abstract
The reeler mutation in mice produces an especially well characterized disorder, with systematically abnormal migration of cerebral cortical neurons. The reeler gene encodes a large protein, termed Reelin, that in the cortex is synthesized and secreted exclusively in the Cajal-Retzius neurons of the cortical marginal zone (D’Arcangelo et al., 1995). In reeler mutant mice, loss of Reelin protein is associated with a systematic loss of the normal, “inside-out” sequence of neurogenesis in the cortex: neurons are formed in the normal sequence but become localized in the cortex in a somewhat inverted, although relatively disorganized “outside-in” pattern. Here we show that the scrambler mutant mouse exhibits a loss of lamination in the cortex and hippocampus that is indistinguishable from that seen in the reeler mouse. We use BrdU birthdating studies to show that scrambler cortex shows a somewhat inverted “outside-in” sequence of birthdates for cortical neurons that is similar to that previously described in reeler cortex. Finally, we perform staining with the CR-50 monoclonal antibody (Ogawa et al., 1995), which recognizes the Reelin protein (D’Arcangelo et al., 1997). We show that Reelin immunoreactivity is present in the scrambler cortex in a normal pattern, suggesting that Reelin is synthesized and released normally. Our data suggest that scrambler is a mutation in the same gene pathway as the reeler gene (Relnrl) and is most likely downstream of Relnrl.
Keywords: cortex, neuronal migration, genetics, reeler, migration, scrambler, development, mdab1, Reelin
The reeler mouse is one of the most thoroughly characterized mouse mutants and has produced a fascinating paradigm for understanding neuronal migration in the cerebral cortex. First described more than 45 years ago (Falconer, 1951), the mutant mice have an ataxic, “reeling” gait, abnormalities in cerebellar development, and a malformation of the cortex (Falconer, 1951;Hamburgh, 1960, 1963). In reeler mice, cortical neurons have a normal sequence of neurogenesis but are abnormal in their subsequent migration and lamination. Instead of the normal “inside-out” sequence of lamination, cortical neurons in reeler assume a much less precise and roughly inverted “outside-in” sequence (Fig.1).
Fig. 1.
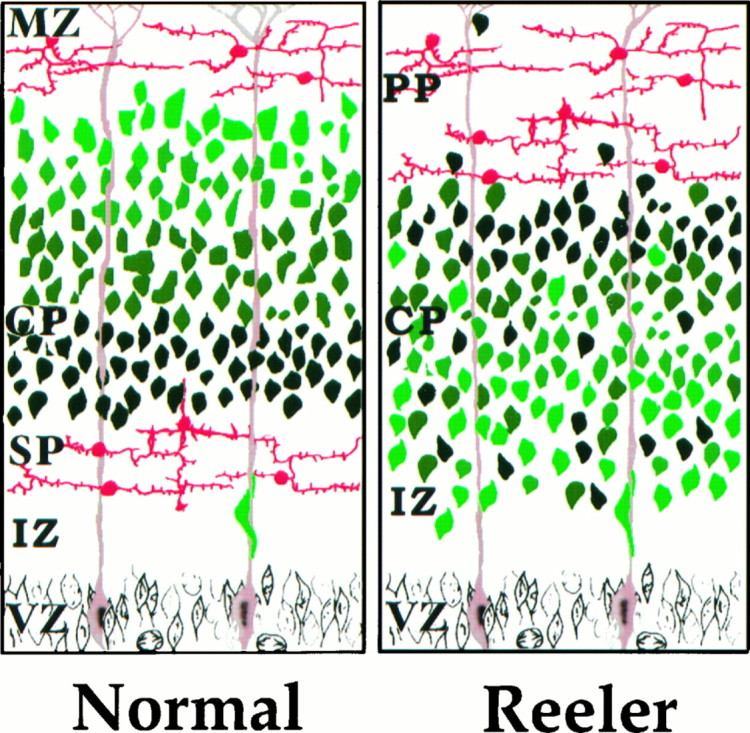
Summary of defects in reeler neocortical development. This schematic diagram of neocortical development illustrates the major defects in the reeler mouse cortex. First, the correspondence of age and position of neurons is roughly reversed in reeler neocortex relative to normal. In the normal cortex, the oldest cortical plate (CP) neurons (dark green) lie deeper in the cortex than cortical plate neurons formed later in development (light green). In the reeler cortex, older CP neurons (dark green) tend to settle in the more superficial aspect of the cortical plate zone than younger CP neurons (light green). Second, the layering of neurons by age is not as well preserved in reeler mice as in normal. Third, the preplate (PP) is not split into subplate (SP) and marginal zone (MZ) in reeler neocortex. Whereas the preplate layer of neurons (red) is split into two layers by migrating CP neurons in the normal cortex, in the reeler cortex these neurons remain as a single layer in the superficial aspect of the brain. The intermediate zone (IZ) and ventricular zone (VZ) appear normal in reeler mice.
Cortical development in the reeler mouse illustrates the fact that the cortex contains two cell populations with distinct developmental histories. The oldest cerebral cortical neurons form a precocious structure termed the primordial plexiform layer (Marin-Padilla, 1978) or preplate. Normally, later-born neurons divide this preplate into an outer marginal zone and an inner layer, termed the subplate (Allendoerfer and Shatz, 1994). In reeler mice, the cortical plate does not seem to divide the preplate normally (Fig. 1), based on analysis of axonal patterns (Caviness and Korde, 1981), histological observations (Caviness, 1977; Goffinet, 1979), immunohistochemical staining patterns (Ogawa et al., 1995), and cell birthdating (Caviness, 1982;Sheppard and Pearlman, 1997).
The recent cloning of the reeler gene (Relnrl, formerly rl) has been a most significant step in clarifying the mechanism of reeler gene function, although the precise role of the reeler gene remains elusive (D’Arcangelo et al., 1995; Hirotsune et al., 1995). The reeler gene encodes a 10,383 bp open reading frame that predicts a secreted protein, termed Reelin. In the cortex, the mRNA is limited to the Cajal-Retzius neurons that occupy the superficial layer of the preplate. Although Reelin is synthesized by the Cajal-Retzius neurons, the cells on which the secreted signal acts are unknown. Reelin may act directly on radial glia, cortical plate neurons, subplate neurons, or some combination. In reeler mice, radial glia show functional and morphological abnormalities (Pinto-Lord et al., 1982; Hunter-Schaedle, 1997), and reeler astrocytes show decreased ability to transform into radial glia in response to secreted factors (Hunter and Hatten, 1994,1995; Hunter-Schaedle, 1997). The nature of Reelin signaling would be greatly clarified by the identification of other mutations affecting cortical neuronal migration and lamination.
A recently discovered mouse mutation, scrambler (scm), produces a phenotype similar to the reeler phenotype (Sweet et al., 1996). Homozygous scm/scm mice are behaviorally similar to reeler homozygotes, showing a reeling, ataxic gait that is evident after 2 weeks of age. However, the scrambler gene was mapped to chromosome 4, whereas Relnrl maps to chromosome 5 (Sweet et al., 1996). The cerebral cortex ofscm homozygotes resembles that ofRelnrlhomozygotes in its relative disorganization. The goal of this work was to analyze neurogenesis of the scm cortex and compare it with that ofRelnrl. Furthermore, we used cell type-specific markers to determine whetherRelnrl expression was affected inscm/scm mice.
MATERIALS AND METHODS
Mice and breeding. The scrambler mice were maintained in a virus-free mouse colony using standard procedures and treated according to protocols reviewed and approved by the institutional review boards of Harvard Medical School and Beth Israel Hospital. The scrambler mutation arose at The Jackson Laboratory in the inbred strain DC/Le, which carries the semidominant mutation dancer (Dc). The scrambler mutation was separated from dancer by outcrossing a male (+/Dc, +/scm) to a C3HeB/FeJ female, intercrossing the normal F1 progeny, and recovering the scrambler phenotype in the F2 progeny. The present colony was derived by continued inbreeding from the single outcross to the C3HeB/FeJ inbred strain (Sweet et al., 1996). We have also found that the mutation is penetrant on a mixed B6/C3H background. Heterozygous animals (+/scm) are phenotypically normal. Homozygousscm/scm females breed reasonably well, although it can take weeks for impregnation and the offspring often require care by foster mothers. Homozygous pups must be left with the mother until at least 6–8 weeks of age to survive and breed. Homozygous males do not breed under normal circumstances. Timed pregnancies were prepared by mating homozygous (scm/scm) dams to heterozygous (+/scm) males, or by breeding heterozygotes of both sexes. The day of discovery of the vaginal plug was counted as embryonic day (E) 0.
BrdU birthdating. The dams were injected intraperitoneally with ∼1.5 mg of bromodeoxyuridine (BrdU) (Sigma, St. Louis, MO) diluted in 7 mm NaOH (to 15 mg/ml) on E10, E11, E12, E14, E15, E16, E17, and E18. Pups were terminally anesthetized several weeks after birth and perfused with ethanol (95%) and acetic acid (3:1) or 4% paraformaldehyde. Brains were removed and post-fixed overnight with the same fixative. Brains were then dehydrated through a graded ethanol series and cleared in xylenes before they were embedded in paraffin. Homozygous and heterozygous hemispheres were generally bisected along the midline and combined in a single paraffin block for parasagittal sections; 13 μm sagittal serial sections were cut on a microtome and a 1/5 series of sections was mounted on gelatin-coated slides.
Sections were deparaffinized in xylenes and then hydrated through a graded ethanol series before being prepared for BrdU immunocytochemistry following standard procedures (Takahashi et al., 1992). Slides were washed with PBS, pH 7.4, several times and denatured with 2N HCl (20 min at 37°C). After a quick rinse with 0.1m PBS, pH 8.4, sections were incubated overnight in mouse anti-BrdU antibody (1:100; Becton Dickinson, San Jose, CA) and blocking solution (PBS, pH 7.4, 3% normal horse serum, 0.02% sodium azide, 0.3% Triton X-100) at 4°C. Sections were washed in PBS and further processed with the Vector ABC Labeling Kit Elite (Vector Labs, Burlingame, CA). The labeling was demonstrated with 0.05% diaminobenzidine and 0.01% H2O2. Some sections were counterstained with cresyl violet.
Sections were viewed under a Zeiss microscope using bright-field optics. Labeled cells were identified by the presence of uniform brown nuclear staining. Camera lucida drawings were made of an area of the parietal cortex dorsal to the hippocampus using a camera lucida device with a 10× objective. The same approximate region was drawn for every tenth section in the series (or, if the region was grossly damaged during processing, the closest available section in the series). Special care was taken to match, as closely as possible, mediolateral regions between +/scm and scm/scm littermates. This was facilitated by the fact that matching hemispheres for littermates were embedded in the same paraffin block and therefore were processed side by side. This process was performed for onescm/scm brain and one normal littermate’s brain from all litters labeled with BrdU at E12, E15, E16, and E18 (see Table 1). Twoscm/scm mice from an additional litter were also labeled with BrdU at E12 (without matching control littermates) and processed in similar manner. With the exception of brains labeled with BrdU at E18, from which only two sets of plots were obtained, camera lucida drawings were made from 4–16 sets of sections, totaling 134 camera lucida drawings.
Table 1.
BrdU injections
| Age | Animals analyzed (litters) | |
|---|---|---|
| +/scm or +/+ | scm/scm | |
| E10 | 8 (3) | 8 (2) |
| E11 | 6 (2) | 6 (2) |
| E12 | 2 (2) | 4 (3) |
| E14 | 4 (1) | 3 (1) |
| E15 | 2 (2) | 2 (2) |
| E16 | 6 (3) | 4 (3) |
| E17 | 0 (0) | 3 (3) |
| E18 | 3 (3) | 3 (3) |
| P2 | 0 (0) | 2 (2) |
The table lists the number of normal and mutant mice analyzed as adults for each given injection time point. The number of independent litters from which animals were derived is indicated in parentheses.
Summary patterns of labeled neurons were plotted on vertical lines representing the thickness of the cortex (white matter to pia) following the method of Rakic (1973). Briefly, a series of vertical lines were drawn on the camera lucida plots, spaced closely enough to reduce the difference in cortical thickness between adjacent lines. Ticks were drawn on each vertical line to mark the vertical location of nearby BrdU-labeled neurons. All vertical lines for a particular camera lucida drawing were scaled to the same size and combined into a single line. This process was performed for one set of paired normal and mutant sections from each litter labeled with BrdU at E12, E15, and E16 (see Table 1). The same approximate mediolateral region was examined in this way in all brains. A good match in mediolateral region between normal/mutant littermates was facilitated by the fact that both hemispheres in the set were embedded in the same paraffin block and therefore processed side by side.
CR-50 and α-calretinin immunocytochemistry. Litters from matings between heterozygous males and females were deeply anesthetized and perfused with 4% paraformaldehyde 0–3 d after birth. Brains were removed and post-fixed in the same fixative overnight. Brains were then processed for cryostat sectioning, and 10–20 μm sagittal sections were obtained. Hemispheres from normal (+/+ or +/scm) andscm/scm littermates were embedded in the same block so that sections from the same approximate mediolateral region could be processed side by side on the same microscope slide. CR-50 immunocytochemistry was performed after Ogawa et al. (1995). Sections were preincubated for 30 min with blocking solution (PBS, pH 7.4, 3% normal goat serum, 0.02% sodium azide, 0.3% Triton X-100). After sections were washed in PBS, they were incubated with CR-50 (1:100; a generous gift from Dr. Masaharu Ogawa) or α-calretinin (1:1000; Chemicon, Temecula, CA) overnight at 4°C. After PBS washes, sections were incubated with the secondary antibody. CR-50 binding was revealed with goat anti-mouse antibody conjugated with fluorescein isothiocyanate (FITC) (diluted 1:200; Jackson ImmunoResearch Labs, West Grove, PA) or with Cy3 (diluted 1:600; Jackson ImmunoResearch Labs). α-calretinin binding was revealed with goat anti-rabbit conjugated with FITC (diluted 1:200; Jackson ImmunoResearch Labs) or with Cy3 (diluted 1:800; Jackson ImmunoResearch Labs).
RESULTS
Phenotype of the mouse
The scm/scm mice were generally noticeably smaller than their +/+ or +/scm littermates, although the size difference was not clearly evident until after 2 weeks of age. They showed an ataxic, shambling gait that was first evident at about 2 weeks of age, often accompanied by trembling. This behavior was indistinguishable, after casual observation, from the behavior of reeler mice obtained from The Jackson Laboratory and maintained in the same colony. Viability was somewhat reduced, with about one fourth of the homozygous mutant animals dying spontaneously by adulthood. Survival was increased substantially by delaying weaning until 5–6 weeks of age and culling unaffected pups from the litters.
Histological observations
As noted previously by Sweet et al. (1996), cresyl violet staining of scm/scm brains demonstrated a virtual absence of detectable lamination in the cerebral cortex (Fig.2). Beneath the pial surface, the normal cortex shows a cell-sparse layer designated layer I. Inscm/scm homozygotes, there is an increased cell density in the region just beneath the pial surface, with no clear definition of layer I. In the normal mouse, the remaining cortical layers can be defined on the basis of the differing amounts of large pyramidal neurons, small pyramidal neurons, and nonpyramidal neurons. In the scm/scm and rl/rl cortex, the large pyramidal neurons are scattered throughout the width of the cortex (Fig. 2D,F,arrows). The abnormalities of cortical lamination present in the scm/scm mouse bear a very strong similarity to those displayed by theRelnrl/Relnrlbrain (Fig. 2).
Fig. 2.
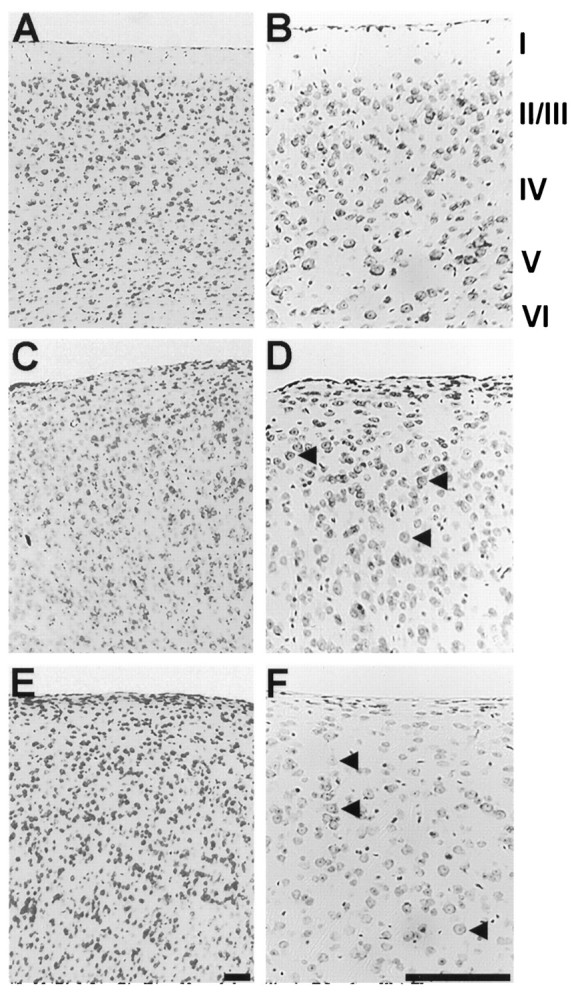
Histological appearance of the normal, reeler, and scrambler cortex. In A and B, photomicrographs of cresyl violet-stained, sagittal sections illustrate the typical six-layered structure of the normal cortex (although layersII/III are not sharply distinct). The sagittal sections shown in C and D demonstrate the characteristic lamination abnormality in the reeler mouse cortex.E and F show sagittal sections from the cortex of the scrambler mouse. As in reeler, the layers are not clearly distinct, and the cortex exhibits a lower overall level of cytoarchitectonic organization. The marginal zone (layer I), as defined by a low abundance of cell nuclei, is not well formed. Large pyramidal neurons can be seen through all layers of the cortex (arrowheads in D, F) and are somewhat more common at more superficial locations, as in reeler. Scale bars, 400 μm.
The hippocampus of scm/scm is highly disorganized compared with the normal hippocampus (Fig.3). In the normal hippocampus, the CA fields show a sharply laminated pyramidal layer. In contrast, both scrambler and reeler mice show a poor segregation of cell-rich and cell-poor laminae, with a semblance of two rows of pyramidal cells (Fig. 3B,C) (Stanfield and Cowan, 1979a,b). At some levels the dentate gyrus is not clearly evident (Fig.3C), although at other levels of the hippocampus a well formed dentate gyrus can be seen (data not shown). Thescm/scm cerebellum also displays abnormalities similar toRelnrl/Relnrl(Sweet et al., 1996). The cerebellar phenotype is described elsewhere (Goldowitz et al., 1997).
Fig. 3.
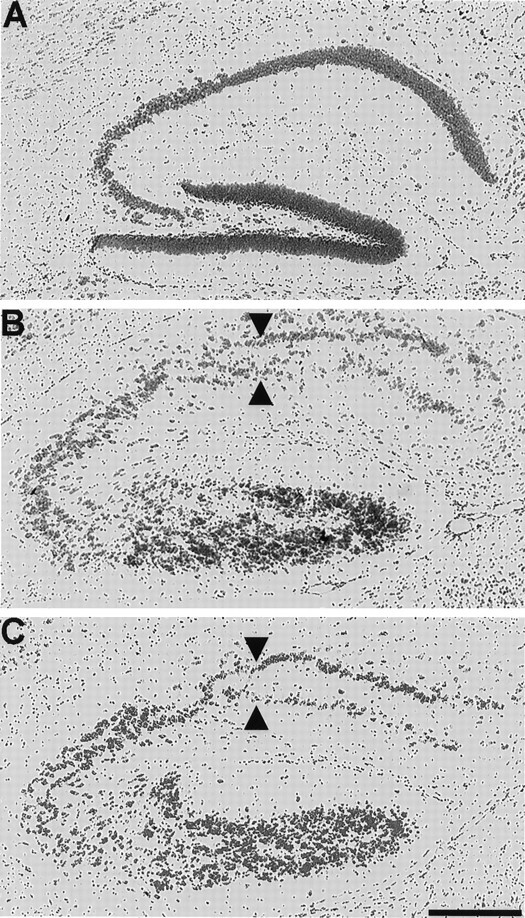
Histological appearance of the normal, reeler, and scrambler hippocampus. The sagittal section shown in Aexemplifies the lamination pattern observed in the normal mouse hippocampus. The sagittal section shown in B illustrates the abnormal lamination of the reeler hippocampus. Note the almost complete duplication of the pyramidal cell layer (arrowheads) and overall level of disorganization. (For a more detailed description of abnormalities in reeler hippocampus, seeStanfield and Cowan, 1979a,b.) In C, a sagittal section from the scrambler cortex shows a disruption and partial duplication of the pyramidal cell layer (arrowheads) quite similar to that observed in the hippocampus of the reeler mouse. Scale bar, 400 μm.
BrdU birthdating
One of the hallmarks of theRelnrl/Relnrlphenotype is the roughly inverted gradient of neuronal birth among the neurons that form the cortical plate (Caviness, 1982). To find out whether scm/scm mice would resembleRelnrl/Relnrlin this respect, we performed a series of birthdating experiments by injecting pregnant dams with BrdU at various ages during neurogenesis (Table1). Figure4 illustrates the results of typical experiments.
Fig. 4.

Patterns of BrdU labeling in normal and scrambler mouse cortex. A shows large neurons inscm/scm cortex labeled with BrdU at E12 (arrowheads), indicating that the largest cortical neurons (characteristic of layer V), although abnormally positioned, are still among the older neurons in the cortex. In Band C, labeling is illustrated in sagittal sections taken from animals that received BrdU at E16 and were analyzed as adults. B shows +/scm neocortex. Note the neat organization of the labeled neurons at the superficial aspect of the cortex in an area corresponding to layers II/III. Cshows scm/scm neocortex labeled with BrdU on E16. Note the much wider distribution of labeled neurons, with the majority located in the deeper half of the cortex. This observation closely matches results from similar studies inRelnrl/Relnrlcortex (Caviness, 1982). Scale bars, 100 μm.
Neurons in +/scm mice labeled at E12 with BrdU were generally confined to the inferior aspect of the cortex, roughly corresponding to layer VI (Fig. 5). In contrast, BrdU-labeled cells in a scm/scm littermate were spread widely throughout the thickness of the cortex (Fig. 5) and did not form a coherent layer. A majority of neurons labeled with BrdU occupied the superficial aspect of the scm/scm cortex, corresponding to the normal location of layers I–IV, with a smaller but still significant number of neurons in the region normally corresponding to layer VI. The same overall pattern of distribution was observed at several different rostrocaudal and mediolateral levels, with only minor differences, in all sections examined. Two brains obtained from a different litter showed the same qualitative results (Table 1).
Fig. 5.
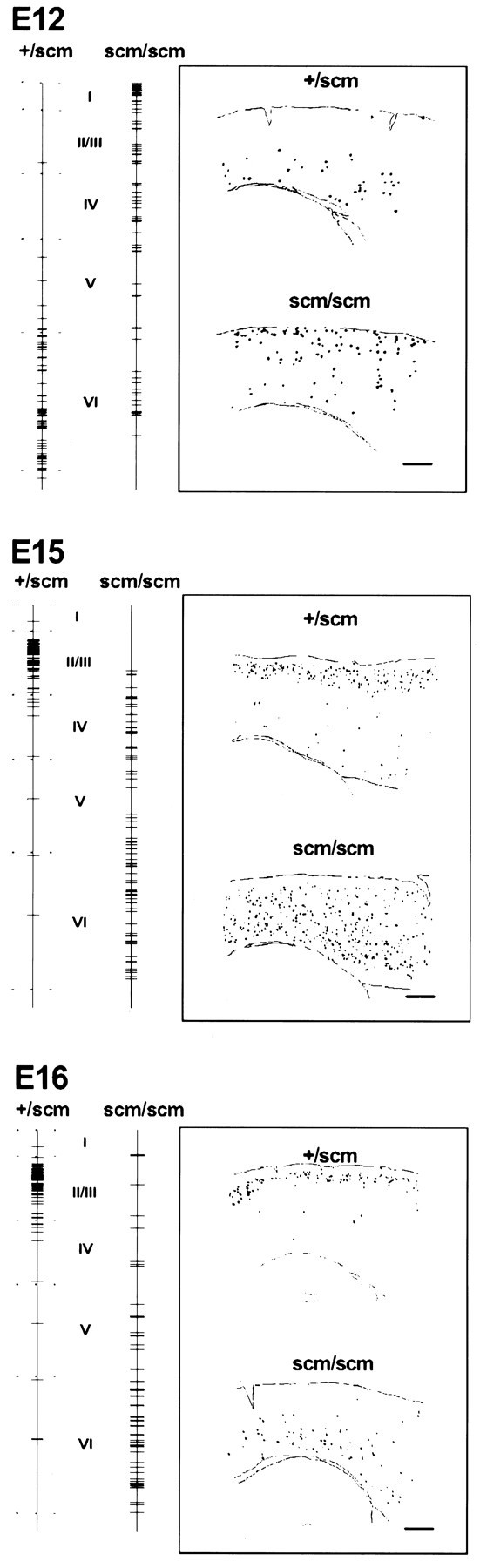
Summary patterns of BrdU labeling after injection at E12, E15, and E16. The positions of BrdU-labeled cells are represented by horizontal lines drawn on avertical line extending the thickness of the cortex from pia to white matter. Each tick mark on thevertical line represents a single BrdU-labeled neuron. Each line combines the vertical position of all BrdU-labeled neurons from the camera lucida drawing of a region of the parietal cortex, lying dorsal to the hippocampus, of a single sagittal section. (See Materials and Methods for details.) The approximate boundaries of each layer are indicated for normal cortex. For each labeling age, samples of camera lucida drawings from BrdU-labeled normal and scm/scm brain sections are displayed in the insets. Scale bars, 400 μm.
Large, BrdU-labeled neurons were found throughout the thickness of thescm/scm cortex after labeling at E12 (Fig.4A). Because the large pyramidal neurons of layer V normally undergo their last mitosis at around E12–E14, the large BrdU-labeled neurons observed at E12 probably correspond to neurons normally destined for layer V. The larger neurons appear to be among the older cortical plate neurons in both normal and scm/scmmice, suggesting that the scrambler mutation affects neuronal migration without drastically altering the sequence of cortical neurogenesis.
Mice injected on E14 contained BrdU-labeled neurons throughout the width of the cortex. No noticeable differences were observed betweenscm/scm and normal littermates (data not shown). The widespread localization of cells labeled with BrdU at E14 was probably caused by the fact that neurogenesis peaks around this time, and the widespread distribution of BrdU-labeled cells accounts for the lack of clear differences between mutant and normal littermates.
Neurons labeled with BrdU at E15 occupied an area corresponding to layers II/III in +/scm cortex (Fig. 5). Inscm/scm, cells labeled with BrdU were scattered throughout most of the cortex, but tended to be more common in the deeper half of the cortex. A similar pattern was observed at several different rostrocaudal and mediolateral levels. In a +/scm cortex from a second litter, the neurons labeled with BrdU were found mostly in layers V and VI, whereas scm/scm cortex contained BrdU-labeled neurons scattered throughout all layers, except layer I, with no clear peak of distribution (data not shown). The pattern of BrdU-labeled neurons of both normal and scm/scm animals from this second litter was more typical of labeling at E14, suggesting that the second litter was somewhat younger than E15 at the time of the BrdU injection.
In +/scm mice, most cells labeled with BrdU at E16 were confined to layers II/III (Figs. 4B, 5). In sharp contrast, scm/scm littermates had BrdU-labeled neurons in the deeper aspect of the cortex, with the majority in an area corresponding to the normal location of layer VI (Figs.4C, 5). Again, similar patterns were seen at several different rostrocaudal and mediolateral levels. Furthermore, the results were reproduced in brains obtained from two additional litters, with only minor differences (data not shown).
By E17–18, neurogenesis had clearly tapered off; very few labeled neurons could be observed in either scm/scm or normal mice when these animals were analyzed as adults (data not shown). This is consistent with previous studies of neurogenesis that indicate that most neurons are born on or before E16 and that very little neurogenesis occurs after E16. Neurons labeled after E16, in addition to being few in number, were also scattered throughout the thickness of the cortex, and no clear differences were observed betweenscm/scm and normal littermates.
It is important to note that the period of neurogenesis appears the same in normal and scrambler mice. A few neurons were labeled with BrdU as early as E10 in both scm/scm and normal cortex (data not shown), indicating the onset of neurogenesis. At around E12, neurogenesis initiates in the cortical plate, peaks around E14, and rapidly tapers off after E16, with little or no neurogenesis occurring after E18. No labeled cortical neurons could be observed in mice (mutant or normal) labeled on or after postnatal day (P) 0 (data not shown). Moreover, early BrdU injections (E12) labeled large cortical neurons in normal and scm/scm cortex, whereas later injections (E15, E16) labeled small neurons in both normal andscm/scm mice. Therefore, the sequence of neurogenesis appears normal in scm/scm mice, with the fundamental problem being the subsequent pattern of neuronal migration.
CR-50 immunocytochemistry
The Relnrlgene encodes the Reelin protein, which is expressed by Cajal-Retzius neurons in the marginal zone of the cerebral cortex (D’Arcangelo et al., 1995; Hirotsune et al., 1995). Relnrl/Relnrlmice lack a functional Reelin protein because of either a large-scale deletion of the gene or a point mutation that prevents Reelin secretion (D’Arcangelo et al., 1997). One possible explanation for the strong resemblance betweenRelnrl/Relnrlandscm/scm mice is that the normal scm gene is essential for normal Reelin expression, or the location or survival of Cajal-Retzius neurons themselves. To test this hypothesis, we used CR-50, a monoclonal antibody that recognizes an epitope present in Reelin (D’Arcangelo et al., 1997). We found that at P0–P1, when the scrambler phenotype is clearly discernible by lack of cerebellar foliation, CR-50 expression appears normal in scm/scmneocortex (Fig. 6). In bothscm/scm and normal littermates, the staining appeared to be cytoplasmic within the Cajal-Retzius cells, with an area corresponding to the nucleus showing faint staining. In addition, punctate, apparently extracellular staining could be observed on processes in the marginal zone of normal brains and the corresponding area of scm/scm mice. This type of staining is similar to previous descriptions of CR-50 staining (Ogawa et al., 1995). At later stages (P7 and P14), the marginal zone CR-50 immunoreactivity was much reduced and a few faintly stained cells were observed throughout other layers of the cortex in bothscm/scm pups and normal littermates (data not shown).
Fig. 6.

CR-50 staining in the scrambler cortex.A and B show CR-50 immunocytochemistry on a sagittal section of normal P0 cortex at low and high magnifications, respectively. C and D show CR-50 immunocytochemistry on a sagittal section of P0 cortex from ascm/scm littermate at similar magnifications. Scale bars, 100 μm.
Calretinin, a calcium binding protein, also serves as a marker for Cajal-Retzius cells in the immature cortex of the mouse (Soriano et al., 1994; Ogawa et al., 1995). In both scm/scmand normal littermates, calretinin-like immunoreactivity is confined to a layer immediately below the pial surface (data not shown). Thus, like CR-50, calretinin immunoreactivity suggests the presence of normal Cajal-Retzius cells in the scrambler mouse cortex.
DISCUSSION
Scrambler copies the cerebral cortical phenotype of the reeler mouse
Our data show that the cerebral cortical phenotype and pattern of neuronal cytogenesis of the scrambler mouse are remarkably similar to that of the reeler mouse, despite the fact that the two genes are located on different chromosomes (Sweet et al., 1996; Ware et al., 1997). Both mutants show severe disruption of cerebral cortical as well as hippocampal lamination. Both mutants show cerebellar hypoplasia as well (Sweet et al., 1996; Goldowitz et al., 1997). In both mutants, the sequence of cortical neurogenesis appears normal, extending from E10 to E18, and with larger neurons that normally populate deep cortical layers being born early. However, both mutants show abnormal subsequent migration and lamination of postmitotic neurons, with the neat layering of cortical neurons born at similar developmental times being absent in both mutants. Moreover, in both reeler and scrambler, there is a tendency for the vertical distribution of neurons born at a given age to be inverted relative to normal.
In reeler mice, the first observable abnormality is a failure of the preplate to be split into two layers by migrating cortical plate neurons (Ogawa et al., 1995; for review, see Caviness et al., 1988;Goffinet, 1992) (Fig. 1). The abnormal, and partially inverted, lamination of cortical plate neurons presumably represents a secondary effect of the inability of newer neurons to migrate past older, postmigratory neurons. In the present study, we have not shown directly that the preplate fails to split normally in scrambler mice, because we have not analyzed the short survival times that would be necessary to determine this. However, the very close similarity between reeler and scrambler birthdates strongly suggests the same fundamental pathogenesis.
Scrambler and reeler appear to differ in one key aspect, namely that scrambler mice show apparently normal expression of Reelin protein, based on CR-50 immunocytochemistry. In reeler mice, Reelin protein is either absent or abnormally localized (D’Arcangelo et al., 1997), whereas our data suggest that in scrambler mice Reelin protein is expressed normally. These data suggest that scm may be downstream of Reelin. Furthermore, although RC2—a monoclonal antibody that recognizes an epitope present in radial glia fibers—reveals a morphological abnormality in radial glia fibers in the reeler mouse (Hunter-Schaedle, 1997), preliminary observations of RC2 staining on scrambler cortex revealed no abnormalities in radial glia fibers (K. Hunter-Schaedle, J. L. Gonzalez, C. J. Russo, and C. A. Walsh, unpublished observations).
Evidence supporting the downstream action of the scm gene product, relative to Reelin, comes from the recent identification of the scrambler gene. The scrambler mutation disrupts the mouse disabled 1 (mdab1) gene by causing aberrant splicing ofmdab1 to an intracisternal A-particle retrotransposon element (Sheldon et al., 1997; Ware et al., 1997). Furthermore, engineered mutations in mdab1 appear to cause a phenotype indistinguishable from scrambler (Howell et al., 1997b). Originally identified by virtue of its physical binding to Src, the mDab1 protein also binds other nonreceptor tyrosine kinases such as Fyn and Abl (Howell et al., 1997a). mDab1 is a phosphoprotein with a phosphotyrosine binding domain that is intensely expressed in developing CNS neurons (Howell et al., 1997a). The Drosophila disabled gene, of which mdab1 is a vertebrate homolog, was identified by virtue of its genetic interaction with the Drosophila abl gene (Dabl). Therefore, interactions between mDab1 and Abl, or related proteins, are likely to be involved in the migration of cortical plate neurons.
Other genes regulating neuronal migration
The putative relationship between mDab1 and Abl suggests a possible link between scrambler and human mutations affecting neuronal migration in the cortex. Positional cloning of the double cortex/X-linked lissencephaly gene (DC/XLIS) has recently shown that it encodes a novel protein, termed Doublin, that represents a potential substrate for phosphorylation by the Abl protein (J. G. Gleeson, K. M. Allen, J. W. Fox, E. D. Lamperti, S. Berkovic, I. Scheffer, W. B. Dobyns, S. Minnerath, M. E. Ross, and C. A. Walsh, unpublished observations). Lissencephaly is a severe migrational disturbance in which the majority of cortical neurons migrate successfully out of the ventricular zone, but then arrest along the migratory route to the cortex (Dobyns and Truwit, 1995). Complete absence of cortical gyri (giving rise to the smooth-surfaced brain described by the term lissencephaly) is thought to be a secondary result. Similarities between lissencephaly and the reeler and scrambler phenotype include the fact that neurons in the lissencephalic cortex do migrate, because they are not arrested in the germinal zone as is seen in other migrational disturbances (e.g., Eksioglu et al., 1996). As in reeler and scrambler mutant mice, neurons migrate most of the way to the cortex, but arrest short of the appropriate location.
Another gene causing human lissencephaly presents a histological phenotype indistinguishable from DC/XLIS (Ross et al., 1997). This gene, called LIS1, is located on chromosome 17, and has been identified as encoding a regulatory subunit of platelet-activating factor acetylhydrolase containing eight WD40 repeats with homology to β-subunits of heterotrimeric G-proteins (Reiner et al., 1993; Hattori et al., 1994). Nonreceptor tyrosine kinases such as Abl have been shown to phosphorylate the α-subunit of several different heterotrimeric G-proteins (Neer, 1995). Although the relationship between LIS1, Doublin, and mDab1 is completely unknown, an interaction between LIS1 and Abl via a Gα subunit is an intriguing possibility.
Recently, two engineered mutations in mice have generated phenotypes that resemble reeler and scrambler to a certain degree. Cdk5 expression in the nervous system is limited to postmitotic neurons of the PNS and CNS (Nikolic et al., 1996). Mice homozygous for mutations in thecdk5 gene die on the day of birth and show many neurological abnormalities (Ohshima et al., 1996). The cerebral cortex of thecdk5 −/− mutants shows a lack of obvious lamination and an excess of neurons in the marginal zone, highly reminiscent of the reeler/scrambler phenotype. It should be noted, however, that birthdating studies have not been performed to confirm the similarity. Engineered mutations in p35, which was identified on the basis of the binding of p35 to Cdk5 (Tsai et al., 1994), also result in brain abnormalities reminiscent of reeler and scrambler (Chae et al., 1997). As in reeler and scrambler, cortical plate neurons appear to retain their normal birthdate patterns but fail to migrate normally, resulting in a somewhat inverted and disorganized cortical layering. In contrast to the reeler and scrambler phenotype, the marginal zone in the p35 mutants is strikingly well formed. The exact relationship of Cdk5 and p35 to other genes involved in neuronal migration is not clear.
Process outgrowth and neuronal migration
Cortical plate neurons migrate to the cortex by extension of a leading, neurite-like process (Rakic, 1972). In light of this, it is interesting to note that many of the genes discussed here are implicated in both neuronal migration and neurite outgrowth (Table2). p35 and Cdk5 have been implicated in neuronal process outgrowth (Nikolic et al., 1996). TheDrosophila disabled gene (dab), as well as otherDrosophila genes that interact with Dabl, such asfax and prospero, have been implicated in genetic interactions necessary for normal axon outgrowth (Gertler et al., 1993). Furthermore, kinases such as Src have been implicated in axon outgrowth as well (Cox and Maness, 1992), which is interesting in light of the fact that mDab1 interacts physically with Src. Finally, Reelin has been shown to be important for early branching and collateral extension of entorhinohippocampal axons (Del Río et al., 1997). The potential roles for these genes in analogous or identical mechanisms for neuronal migration and neurite outgrowth suggest an emerging framework useful for the future study of cortical development.
Table 2.
Relative effect of gene mutations
| Species | Mutation | Layer I | Hippocampus | Cerebellum |
|---|---|---|---|---|
| Mouse | Reeler (Reelin) | +++ | +++ | +++ |
| Scrambler (mDab1) | +++ | +++ | ++ | |
| Cdk5 mutation | +++ | +++ | +? | |
| P35 mutation | − | ± | ± | |
| Human | LIS1 mutation | − | ++ | ± |
| DC/XLIS (Doublin) | − | ++ | ± |
Different mutations are associated with varying degrees of abnormality in the cortex, cerebellum, and hippocampus. See further details in Discussion.
Footnotes
J.L.G. was supported by a Ford Foundation fellowship. C.A.W. was supported by grants from the Klingenstein Foundation Fund, National Institutes of Health (NIH) Grants KO8-NS01520 and RO1-NS 32457, and the Mental Retardation Research Center Grant (NIH-P30-HD18655). C.A.W. is a Scholar of the Rita Allen Foundation. D.G. was supported by NIH Grant NS23475 and the University of Tennessee. M.T.D. and H.O.S. were supported by National Science Foundation Grant DBI 9502221 and NIH-National Center for Research Resources Grant RR01183. We thank M. Ogawa for the gift of CR-50 antiserum, R. Bronson for advice, K. Hunter for communicating data before publication, K. Allen for help with the mouse colony, and L. Garcia for assistance with histology.
Correspondence should be addressed to Dr. C. A. Walsh, Division of Neurogenetics, Beth Israel Deaconess Medical Center/Harvard Medical School, Harvard Institutes of Medicine, 77 Avenue Louis Pasteur, Boston, MA 02115.
REFERENCES
- 1.Allendoerfer KL, Shatz CJ. The subplate, a transient neocortical structure: its role in the development of connections between thalamus and cortex. Annu Rev Neurosci. 1994;17:185–218. doi: 10.1146/annurev.ne.17.030194.001153. [DOI] [PubMed] [Google Scholar]
- 2.Caviness VS., Jr . Reeler mutant mouse: a genetic experiment in developing mammalian cortex. In: Cowan WM, Ferrendelli JA, editors. Approaches to the cell biology of neurons, Vol 2. Society for Neuroscience; Bethesda, MD: 1977. pp. 27–46. [Google Scholar]
- 3.Caviness VS., Jr Neocortical histogenesis in normal and reeler mice: a developmental study based upon 3H-thymidine autoradiography. Brain Res. 1982;256:293–302. doi: 10.1016/0165-3806(82)90141-9. [DOI] [PubMed] [Google Scholar]
- 4.Caviness VS, Jr, Korde MG. Monoaminergic afferents to the neocortex: a developmental histofluorescence study in normal and Reeler mouse embryos. Brain Res. 1981;209:1–9. doi: 10.1016/0006-8993(81)91167-7. [DOI] [PubMed] [Google Scholar]
- 5.Caviness VS, Jr, Crandall JE, Edwards MA. The reeler malformation: implications for neocortical histogenesis. In: Peters A, Jones EG, editors. Cerebral neocortex. Plenum; New York: 1988. pp. 59–89. [Google Scholar]
- 6.Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 1997;18:29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- 7.Cox ME, Maness PF. Protein tyrosine kinases in nervous system development. Semin Cell Biol. 1992;3:117–126. doi: 10.1016/s1043-4682(10)80021-2. [DOI] [PubMed] [Google Scholar]
- 8.D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 9.D’Arcangelo G, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Curran T. Reelin is a secreted glycoprotein recognized by the CR-50 monoclonal antibody. J Neurosci. 1997;17:23–41. doi: 10.1523/JNEUROSCI.17-01-00023.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Del Río JA, Heimrich B, Borrell V, Förster E, Drakew A, Alcántara S, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Derer P, Frotscher M, Soriano E. A role for Cajal-Retzius cells and reelin in the development of hippocampal connections. Nature. 1997;385:70–74. doi: 10.1038/385070a0. [DOI] [PubMed] [Google Scholar]
- 11.Dobyns WB, Truwit CL. Lissencephaly and other malformations of cortical development: 1995 update. Neuropediatrics. 1995;26:132–147. doi: 10.1055/s-2007-979744. [DOI] [PubMed] [Google Scholar]
- 12.Eksioglu YZ, Scheffer IE, Cardenas P, Knoll J, DiMario F, Ramsby G, Berg M, Kamuro K, Berkovic G, Duyk GM, Parisi J, Huttenlocher PR, Walsh CA. Periventricular heterotopia: an X-linked dominant epilepsy locus causing aberrant cerebral cortical development. Neuron. 1996;16:77–87. doi: 10.1016/s0896-6273(00)80025-2. [DOI] [PubMed] [Google Scholar]
- 13.Falconer DS. Two new mutants, “trembler” and “reeler,” with neurological actions in the house mouse (Mus Musculus L.). J Genet. 1951;50:192–201. doi: 10.1007/BF02996215. [DOI] [PubMed] [Google Scholar]
- 14.Gertler FB, Hill KK, Clark MJ, Hoffmann FM. Dosage-sensitive modifiers of Drosophila abl tyrosine kinase function: prospero, a regulator of axonal outgrowth, and disabled, a novel tyrosine kinase substrate. Genes Dev. 1993;7:441–453. doi: 10.1101/gad.7.3.441. [DOI] [PubMed] [Google Scholar]
- 15.Goffinet AM. An early developmental defect in the cerebral cortex of the reeler mouse. A morphological study leading to a hypothesis concerning the action of the mutant gene. Anat Embryol (Berl) 1979;157:205–216. doi: 10.1007/BF00305160. [DOI] [PubMed] [Google Scholar]
- 16.Goffinet AM. The reeler gene: a clue to brain development and evolution. Int J Dev Biol. 1992;36:101–107. [PubMed] [Google Scholar]
- 17.Goldowitz D, Cushing RC, Laywell E, D’Arcangelo G, Sheldon M, Sweet H, Davisson M, Steindler D, Curran T. Cerebellar disorganization characteristic of reeler in scrambler mutant mice despite presence of Reelin. J Neurosci. 1997;17:8767–8777. doi: 10.1523/JNEUROSCI.17-22-08767.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamburgh M. Observations on the neuropathology of “reeler,” a neurological mutation in mice. Experientia. 1960;16:460–461. [Google Scholar]
- 19.Hamburgh M. Analysis of the postnatal developmental effects of “reeler,” a neurological mutation in mice. A study in developmental genetics. Dev Biol. 1963;8:165–185. doi: 10.1016/0012-1606(63)90040-x. [DOI] [PubMed] [Google Scholar]
- 20.Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K. Miller-Dieker lissencephaly gene encodes a subunit of brain platelet-activating factor acetylhydrolase. Nature. 1994;370:216–218. doi: 10.1038/370216a0. [DOI] [PubMed] [Google Scholar]
- 21.Hirotsune S, Takahara T, Sasaki N, Hirose K, Yoshiki A, Ohashi T, Kusakabe M, Murakami Y, Muramatsu M, Watanabe S, Nakao K, Katsuki M, Hayashizaki Y. The reeler gene encodes a protein with an EGF-like motif expressed by pioneer neurons. Nat Genet. 1995;10:77–83. doi: 10.1038/ng0595-77. [DOI] [PubMed] [Google Scholar]
- 22.Howell BW, Gertler FB, Cooper JA. Mouse disabled (mDab1): a Src binding protein implicated in neuronal development. EMBO J. 1997a;16:121–132. doi: 10.1093/emboj/16.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997b;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- 24.Hunter K, Hatten M. Regulation of the radial glia-astrocyte transformation pathway in the developing forebrain. Soc Neurosci Abstr. 1994;523.11:1276. [Google Scholar]
- 25.Hunter K, Hatten M. Radial glial cell transformation to astrocytes is bidirectional: regulation by a diffusible factor in embryonic forebrain. Proc Natl Acad Sci USA. 1995;92:2061–2065. doi: 10.1073/pnas.92.6.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hunter-Schaedle KE. Radial glial cell development and transformation are disturbed in reeler forebrain. J Neurobiol. 1997;33:459–472. doi: 10.1002/(sici)1097-4695(199710)33:4<459::aid-neu9>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 27.Marin-Padilla M. Dual origin of the mammalian neocortex and evolution of the cortical plate. Anat Embryol (Berl) 1978;152:109–126. doi: 10.1007/BF00315920. [DOI] [PubMed] [Google Scholar]
- 28.Neer E. Heterotrimeric G proteins: organizers of transmembrane signals. Cell. 1995;80:249–257. doi: 10.1016/0092-8674(95)90407-7. [DOI] [PubMed] [Google Scholar]
- 29.Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996;10:816–825. doi: 10.1101/gad.10.7.816. [DOI] [PubMed] [Google Scholar]
- 30.Ogawa M, Miyata T, Nakajima K, Yagyu K, Seike M, Ikenaka K, Yamamoto H, Mikoshiba K. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron. 1995;14:899–912. doi: 10.1016/0896-6273(95)90329-1. [DOI] [PubMed] [Google Scholar]
- 31.Ohshima T, Ward J, Huh C-G, Longenecher G, Veeranna, Pant H, Brady R, Martin L, Kulkarni A. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pinto-Lord MC, Evrard P, Caviness VS., Jr Obstructed neuronal migration along radial glial fibers in the neocortex of the reeler mouse: a Golgi-EM analysis. Brain Res. 1982;256:379–393. doi: 10.1016/0165-3806(82)90181-x. [DOI] [PubMed] [Google Scholar]
- 33.Rakic P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol. 1972;145:61–84. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- 34.Rakic P. Kinetics of proliferation and latency between final cell division and onset of differentiation of cerebellar stellate and basket neurons. J Comp Neurol. 1973;147:523–546. doi: 10.1002/cne.901470407. [DOI] [PubMed] [Google Scholar]
- 35.Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH. Isolation of a Miller-Dieker lissencephaly gene containing G-protein beta-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- 36.Ross ME, Allen KM, Srivastava AK, Featherstone T, Ross ME, Allen KM, Srivastava AK, Featherstone T, Gleeson JG, Hirsch B, Harding BN, Andermann E, Abdullah R, Berg M, Czapansky-Bielman D, Flanders DJ, Guerrini R, Motte J, Mira AP, Scheffer I, Berkovic S, Scaravilli F, King RA, Ledbetter DH, Schlessinger D, Dobyns WB, Walsh CA. Linkage and physical mapping of X-linked lissencephaly/SBH (XLIS): a gene causing neuronal migration defects in human brain. Hum Mol Genet. 1997;6:555–562. doi: 10.1093/hmg/6.4.555. [DOI] [PubMed] [Google Scholar]
- 37.Sheldon M, Rice DS, D’Arcangelo G, Yoneshima H, Nakajima K, Mikoshiba K, Howell BW, Cooper JA, Goldowitz D, Curran T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature. 1997;389:730–733. doi: 10.1038/39601. [DOI] [PubMed] [Google Scholar]
- 38.Sheppard AM, Pearlman AL. Abnormal reorganization of preplate neurons and their associated extracellular matrix: an early manifestation of altered neocortical development in the reeler mutant mouse. J Comp Neurol. 1997;378:173–179. doi: 10.1002/(sici)1096-9861(19970210)378:2<173::aid-cne2>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 39.Soriano E, Del Rio JA, Martinez A, Super H. Organization of the embryonic and early postnatal murine hippocampus. I. Immunocytochemical characterization of neuronal populations in the subplate and marginal zone. J Comp Neurol. 1994;342:571–595. doi: 10.1002/cne.903420406. [DOI] [PubMed] [Google Scholar]
- 40.Stanfield BB, Cowan WM. The morphology of the hippocampus and dentate gyrus in normal and reeler mice. J Comp Neurol. 1979a;185:393–422. doi: 10.1002/cne.901850302. [DOI] [PubMed] [Google Scholar]
- 41.Stanfield BB, Cowan WM. The development of the hippocampus and dentate gyrus in normal and reeler mice. J Comp Neurol. 1979b;185:423–459. doi: 10.1002/cne.901850303. [DOI] [PubMed] [Google Scholar]
- 42.Sweet HO, Bronson RT, Johnson KR, Cook SA, Davisson MT. Scrambler, a new neurological mutation of the mouse with abnormalities of neuronal migration. Mamm Genome. 1996;7:798–802. doi: 10.1007/s003359900240. [DOI] [PubMed] [Google Scholar]
- 43.Takahashi T, Nowakowski TRS, Caviness VS., Jr BUdR as an S-phase marker for quantitative studies of cytokinetic behaviour in the murine cerebral ventricular zone. J Neurocytol. 1992;21:185–197. doi: 10.1007/BF01194977. [DOI] [PubMed] [Google Scholar]
- 44.Tsai LH, Delalle I, Caviness VS, Jr, Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- 45.Ware ML, Fox JW, Gonzalez JL, Davis NM, Lambert de Rouvroit C, Russo CJ, Chua SC, Goffinet AM, Walsh CA. Aberrant splicing of a mouse disabled homologue, mdab1, in the scrambler mouse. Neuron. 1997;19:239–249. doi: 10.1016/s0896-6273(00)80936-8. [DOI] [PubMed] [Google Scholar]