

Abstract
Activation of the amygdala in rats produces cardiovascular changes that include increases in heart rate and arterial pressure as well as behavioral changes characteristic of emotional arousal. The objective of the present study was to examine the interaction of GABA and excitatory amino acid (EAA) receptors in the basolateral amygdala (BLA) in regulating cardiovascular function. Microinjection of the GABAA receptor antagonist bicuculline methiodide (BMI) or the E A A receptor agonists NMDA or AMPA into the same region of the BLA of conscious rats produced dose-related increases in heart rate and arterial pressure. Injection of the nonselective EAA receptor antagonist kynurenic acid into the BLA prevented or reversed the cardiovascular changes caused by local injection of BMI or the noncompetitive GABA antagonist picrotoxin. Conversely, local pretreatment with the glutamate reuptake inhibitorl-trans-pyrrolidine-2,4-dicarboxylic acid enhanced the effects of intra-amygdalar injection of BMI. The cardiovascular effects of BMI were also attenuated by injection of either the NMDA antagonist 3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP) or the AMPA receptor antagonist 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide (NBQX). When these two EAA receptor antagonists were combined, their ability to suppress BMI-induced tachycardic and pressor responses was additive. These findings indicate that the cardiovascular effects caused by blockade of GABAergic inhibition in the BLA of the rat are dependent on activation of local NMDA and AMPA receptors.
Keywords: basolateral amygdala, excitatory amino acids, NMDA, AMPA, bicuculline, kynurenic acid, l-trans-PDC, heart rate, arterial pressure, cardiovascular control
The amygdala is thought to play a central role in a variety of behavioral, physiological, and endocrine responses. A significant body of evidence supports the view that the amygdala is an important component of central cardiovascular control mechanisms. Neuroanatomic studies indicate that the amygdala shares reciprocal connections to other areas of the CNS involved in cardiovascular control, including hypothalamic nuclei, the dorsomotor nucleus of the vagus, and the periaqueductal gray (Hopkins and Holstege, 1978; Krettek and Price, 1978; Beitz, 1982). Electrical stimulation of various sites in the amygdala evokes increases in heart rate and arterial pressure characteristic of the defense reaction (Hilton and Zbrozyna, 1963; Galeno and Brody, 1983; Gelsema et al., 1987, Maskati and Zbrozyna, 1989), whereas lesioning of this area can prevent stress-induced cardiovascular changes (Galeno et al., 1984;Sanders et al., 1994) and the development of hypertension (Galeno and Brody, 1982). More recently, it has been shown that injection of GABA antagonists into the anterior region of the basolateral amygdala (BLA) elicits increases in heart rate and arterial pressure (Sanders and Shekhar, 1991, 1995; Sanders et al., 1995). Thus, the BLA appears to contain a population of neurons that can elicit significant cardiovascular changes, and it appears that these neurons are under tonic GABAergic inhibition.
As yet, the excitatory input to neurons in the BLA that generate these cardiovascular changes has not been defined. GABA and EAA receptors mediate the majority of inhibitory and excitatory synaptic transmission in the mammalian CNS and interact to modulate neuronal activity in several brain nuclei (Jones, 1988; Steward et al., 1990; Soltis and DiMicco, 1991b), including the BLA (Rainnie et al., 1991a,b; Gean and Chang, 1992). Recent behavioral studies suggest a functional role for an interaction between GABA and EAA receptors in the BLA. Injection of GABA antagonists into the BLA can elicit anxiogenic behavior (Sanders and Shekhar, 1995) whereas, conversely, injection of EAA receptor antagonists into this same region can block the acquisition and expression of conditioned fear (Campeau et al., 1992; Falls et al., 1992; Kim et al., 1993; Campeau and Davis, 1995; Maren et al., 1996). If a similar relationship between GABA and EAA systems exists in the BLA with respect to cardiovascular function, then the cardiovascular changes caused by blockade of GABAergic inhibition in the BLA will depend on activation of local EAA receptors. To test this hypothesis, the EAA receptor agonists NMDA and AMPA and the postsynaptic GABAA antagonists bicuculline methiodide (BMI) and picrotoxin were injected into the region of the BLA of conscious rats while monitoring heart rate and arterial pressure. The effects of the GABA antagonists then were tested in the presence of the nonselective EAA receptor antagonist kynurenic acid (KYN) or the glutamate reuptake inhibitorl-trans-pyrrolidine-2,4-dicarboxylic acid (PDC). In addition, the role of specific subtypes of EAA receptors in generating the cardiovascular response to the GABA antagonist BMI was examined using the NMDA receptor antagonist 3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP) and the AMPA receptor antagonist 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide (NBQX).
MATERIALS AND METHODS
Male Sprague Dawley rats (270–350 gm, Harlan Sprague Dawley, Indianapolis, IN) were used in all experiments. Animals were housed individually under controlled temperature and light periodicity with free access to food and water. All procedures used were approved by Drake University Animal Care and Use Committee and followed guidelines set forth in the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Stereotaxic surgery. On day one of the surgical protocol, rats were anesthetized with Avertin (10 ml/kg, i.p.) and positioned in a stereotaxic frame (Kopf Instruments) with the incisor bar positioned at −3.3 mm below the horizontal plane. Rectal temperature was monitored and maintained at 36–37°C with a heating pad. Once the overlying skin and connective tissue were cleared from the skull, holes were drilled into the skull to allow access to the brain. Chronic guide cannulae (26 gauge, 11 mm length, Plastics One, Roanoke, VA) were directed bilaterally at the BLA (AP −2.1, RL ± 4.9, HD −8.7) and cemented in place with cranioplastic cement anchored to the skull with three jeweler’s screws. The guide cannulae were fitted with injector cannulae (33 gauge, 12 mm length) during the implantation procedure to prevent fluids and tissue from accumulating in the guide cannulae. After the cranioplastic cement had set, the injector cannulae were removed and the guide cannulae were sealed with wire dummy cannulae. Animals were removed from the stereotaxic frame and allowed to recover in individual cages.
Cardiovascular instrumentation. Five to seven days after implantation of the guide cannulae, animals were re-anesthetized with Avertin. The right femoral artery was cannulated with a 3.5 cm length of Microrenathane tubing (0.01 inner diameter, Braintree Scientific, Braintree, MA) attached to a length of Tygon tubing (0.02 inner diameter) filled with heparinized saline (100 U/ml), and the end was sealed with a stylet. The tubing was then routed subcutaneously to the nape of the neck, exteriorized, and secured to skin and underlying muscle with suture. The animals were allowed to recover in their individual cages for 24–48 hr before testing. By this time, the animals had resumed their regular eating, drinking, and grooming habits and exhibited no signs of pain or stress.
Microinjection procedure. Animals were tested while freely moving or resting in their home cage between 10:00 A.M. and 4:00 P.M. Arterial pressure was measured and recorded by connecting the arterial line in series to a pressure transducer, a MacLab/4 data acquisition system, and a Macintosh LCIII computer. Heart rate was derived from the arterial pulse pressure and was measured and recorded on a separate channel. Once a steady baseline of cardiovascular parameters was attained, the injector cannulae containing drug solution or vehicle were inserted into the guide cannulae. All injections were bilateral (250 nl/side, infused over 30 sec) and were made using two 5 μl Hamilton syringes mounted in a Harvard infusion pump. Injection cannulae remained in place for 1 min after ending the infusion and then removed. The order of injections was given in a staggered or varied sequence to control for order effects. Animals received no more than two injections per day and no more than five injections at one site.
Histology. Microinjection sites were marked at the completion of each study. After induction of anesthesia (10 ml/kg Avertin, i.p.), 250 nl of a 5% solution of Alcian blue dye was infused at each site. The brain was perfused transcardially with 60 ml of heparinized saline followed by 150 ml of buffered 10% Formalin. The brain was removed and stored in buffered 10% Formalin. Coronal sections (60 μm) were cut on a microtome/cryostat, mounted on gelatin-coated slides, and stained with 1% neutral red solution. The locations of the sites of injection were determined according to the atlas of Paxinos and Watson (1986).
Chemicals. Drugs used in these experiments included BMI, NMDA, AMPA, PDC, CPP, NBQX (Research Biochemicals, Natick, MA), picrotoxin, KYN, and xanthurenic acid (Sigma, St. Louis, MO). All drugs were dissolved in saline, and the final pH was adjusted to 6–8. The anesthetic Avertin was prepared as follows: 10 gm of tribromoethanol (Aldrich, Milwaukee, WI), 5 gm tert-amyl alcohol (Sigma); 10 ml of this concentrate was dissolved in 40 ml of absolute ethanol and 450 ml of 0.9% saline/0.1 m phosphate buffer, pH 7.2 (Inglis et al., 1993).
Statistics. Results are expressed as mean ± SEM. In those experiments in which the data are expressed as a time course, the data were analyzed after calculation of the area under the curve (AUC). The AUC was determined for each experiment by plotting the data (change from baseline) over a grid (grid block = 5 min × mmHg or beats/min) and calculating the area using the trapezoidal method. All grids for a given experiment were summed (including both positive and negative areas, relative to baseline) to determine the total AUC (Feldman and Buccafusco, 1997). In those experiments in which the data are expressed as peak changes in heart rate or arterial pressure, the peak was defined as the highest value sustained for 1 min or longer. These values (AUCs and peak changes) were analyzed using one way ANOVA (with repeated measures where appropriate) using dose as the factor. Scheffé’s post hoc test was used to determine differences between groups; p < 0.01 was considered significant.
RESULTS
Bilateral injection of the GABA antagonist BMI (10–100 pmol) or the EAA agonists NMDA (10–100 pmol) or AMPA (3–30 pmol) into the BLA of conscious rats produced dose-related increases in heart rate and arterial pressure (Figs.1, 2, 3, 4). The time course of these changes was similar between the EAA agonists NMDA and AMPA in that the onset of effects occurred within 1–5 min of injection, peaked at 7–10 min, and returned to baseline by 25 min. Similarly, 100 pmol of NMDA produced maximal changes in heart rate (+80 ± 10 beats/min) and arterial pressure (+1 ± 2 mmHg) that were not significantly different from 30 pmol of AMPA (+70 ± 9 beats/min and +14 ± 1 mmHg, respectively). However, at a dose of 100 pmol, AMPA produced significantly smaller changes in heart rate (+34 ± 10, n = 6) and arterial pressure (+3 ± 4 mmHg) compared with the 30 pmol dose. Higher doses of NMDA were not systematically studied because of seizure-like activity in one rat after a 300 pmol injection of NMDA. The tachycardic and pressor responses to injection of BMI had a similar time to onset as the EAA agonists. However, the cardiovascular responses to BMI were of greater magnitude and of longer duration compared with the EAA agonists. The baseline heart rates (345 ± 11 beats/min) and mean arterial pressures (110 ± 4 mmHg) for this series of experiments were not significantly different among the groups of animals.
Fig. 1.
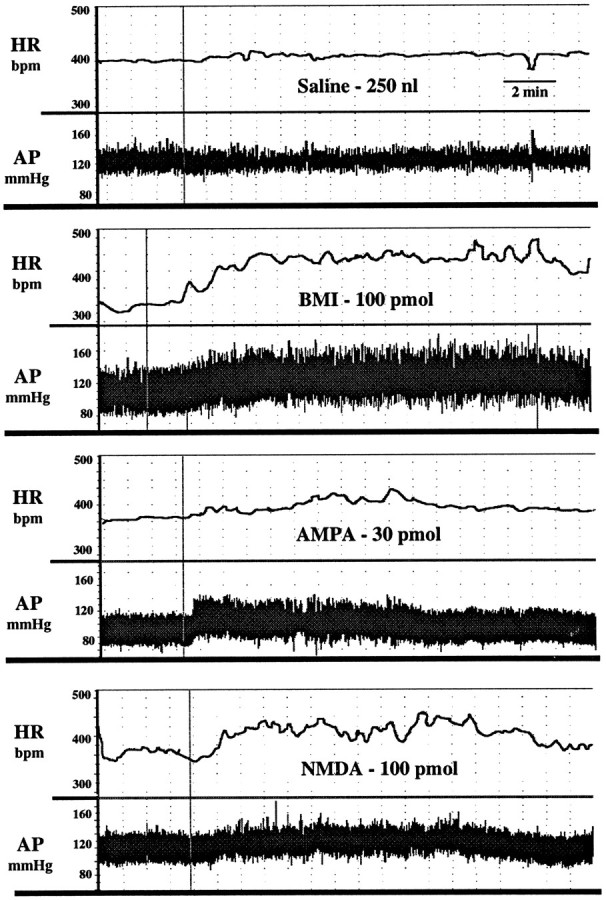
Tracings of heart rate (HR; beats/min) and arterial pressure (AP; mmHg) from four conscious, chronically instrumented rats depicting the effects of bilateral microinjection of Saline, BMI,AMPA, and NMDA into the basolateral amygdala. Drugs were injected in a volume of 250 nl/side over 30 sec.Vertical lines indicate the start of injection.
Fig. 2.
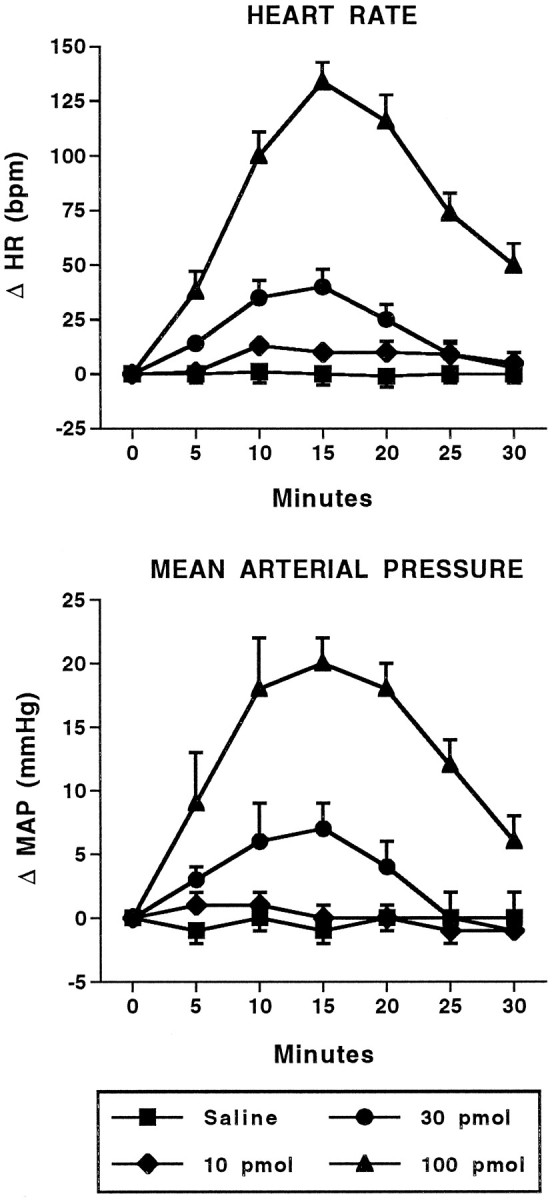
Cardiovascular effects of intra-amygdalar injection of bicuculline methiodide. Time course of changes in heart rate (HR; beats/min; top) and mean arterial pressure (MAP; mmHg; bottom) in conscious, chronically instrumented rats after bilateral injection of saline (250 nl/side) or various doses of bicuculline methiodide into the basolateral amygdala. The 30 pmol and 100 pmol doses for both HR and MAP are significantly different from saline. Each dose,n = 5. HR: F(3,16) = 66.23; MAP: F(3,16) = 34.99.
Fig. 3.
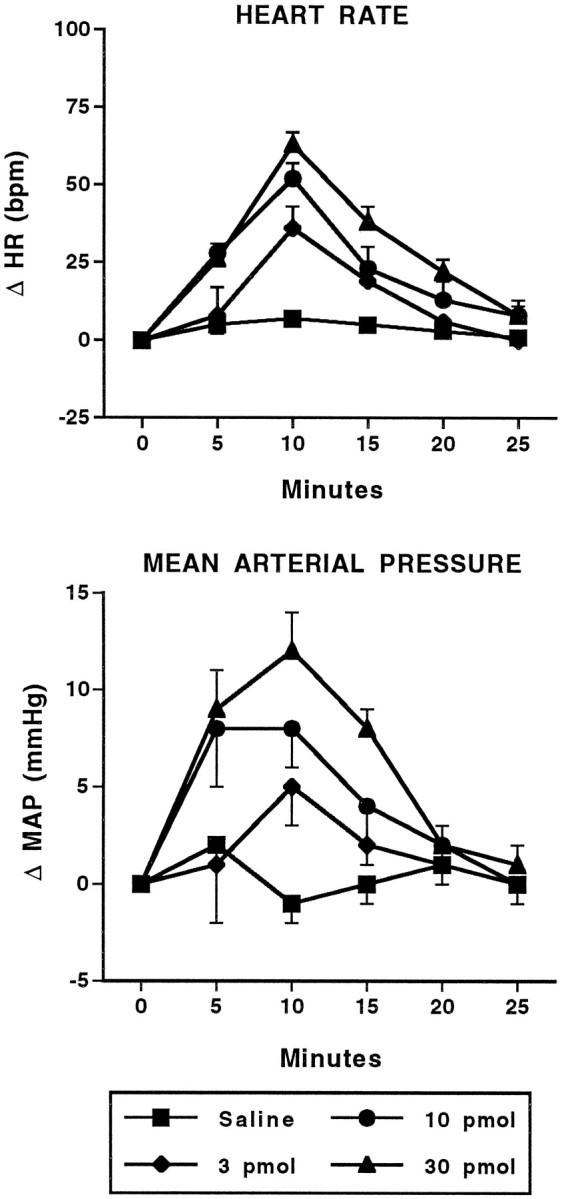
Cardiovascular effects of intra-amygdalar injection of AMPA. Time course of changes in heart rate (HR; beats/min; top) and mean arterial pressure (MAP; mmHg; bottom) in conscious, chronically instrumented rats after bilateral injection of saline (250 nl/side) or various doses of AMPA into the basolateral amygdala. The 10 pmol and 30 pmol doses for both HR and MAP are significantly different from saline. AMPA (30 pmol),n = 6; all other doses, n = 4. HR: F(3,14) = 20.19; MAP:F(3,14) = 5.77.
Fig. 4.
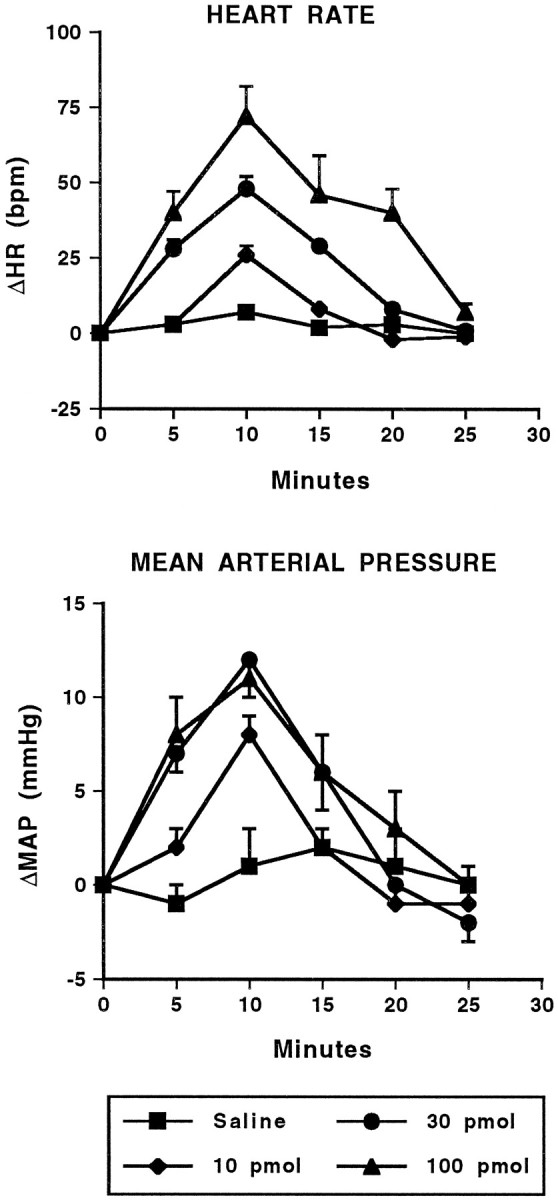
Cardiovascular effects of intra-amygdalar injection of NMDA. Time course of changes in heart rate (HR; beats/min; top) and mean arterial pressure (MAP; mmHg; bottom) in conscious, chronically instrumented rats after bilateral injection of saline (250 nl/side) or various doses of NMDA into the basolateral amygdala. The 30 pmol and 100 pmol doses for both HR and MAP are significantly different from saline. Each dose, n = 4. HR: F(3,12) = 58.54; MAP:F(3,12) = 21.14.
Microinjection of either EAA agonist or BMI also produced changes in locomotor activity. Although not quantitated, these changes included intermittent running and moving of bedding and appeared to follow the time course of the cardiovascular changes. However, in many animals, because the activity was intermittent, the heart rate and arterial pressure were consistently elevated during the active and quiescent periods in these rats.
After histological examination of the brains from the above experiments, the injection sites were found to be within the coronal planes lying 1.8–2.6 mm posterior to bregma. The sites at which BMI, NMDA, and AMPA produced increases in heart rate and arterial pressure overlapped each other and were localized to areas within or 0.3 mm dorsal or lateral to the BLA (Fig. 5). To verify further that the BLA was the probable site of action of these agents, 10 animals were implanted with chronic guide cannulae directed at sites 0.5–1.5 mm medial or dorsal to the BLA (Fig. 5). Two of these sites are not identified in Figure 5 because the sites of injection were located in coronal planes anterior to those depicted in the figure. Each animal received injections of BMI (100 pmol), NMDA (100 pmol), and AMPA (30 pmol) at each of these sites. In all cases, injection of either of the agents failed to produce a tachycardic response greater than 25 beats/min or a pressor response greater than 5 mmHg. At three sites at which an increase in heart rate of 20–25 beats/min was observed, the onset was greater than 5 min.
Fig. 5.

Schematic representation of injection sites in coronal sections through the basolateral amygdala. Filled circles represent sites at which 100 pmol of BMI increased heart rate by 50 beats/min or greater. Filled trianglesrepresent sites at which 30 pmol of AMPA increased heart rate by 30 beats/min or greater. Filled squares represent sites at which 100 pmol of NMDA increased heart rate by 30 beats/min or greater.Open diamonds represent sites at which BMI, AMPA, and NMDA failed to increase heart rate by 25 beats/min or more.ACo, Anterior cortical amygdaloid nucleus;BLA, basolateral amygdaloid nucleus; BLV, ventral basolateral amygdaloid nucleus; BMA, anterior basomedial amygdaloid nucleus; CNA, central amygdaloid nucleus; DEn, dorsal endopiriform nucleus;IM, main intercalated amygdaloid nucleus;LaDL, dorsolateral lateral amygdaloid nucleus;OT; optic tract; Pir, piriform cortex;PLCo, posterolateral cortical amygdaloid nucleus;PVN, paraventricular nucleus of the hypothalamus;VMH, ventromedial hypothalamus; 3V, third ventricle. Adapted from the atlas of Paxinos and Watson (1986).
Given that stimulation of EAA receptors and blockade of GABA receptors at the same site in the BLA can produce similar changes in cardiovascular function, the next series of experiments examined the role of local EAA receptors in mediating the cardiovascular changes observed after injection of GABA antagonists into the BLA. Coinjection of the nonselective EAA receptor antagonist KYN (1–10 nmol) into the BLA blocked in a dose-dependent manner the increases in heart rate and arterial pressure seen with BMI 100 pmol (Fig.6). Coinjection of 100 pmol of BMI and 10 nmol of xanthurenic acid, a compound chemically similar to KYN with no activity at EAA receptors (Guyenet et al., 1987), produced increases in heart rate (124 ± 12 beats/min) and arterial pressure (18 ± 3 mmHg) similar to those produced by 100 pmol of BMI alone (134 ± 9 beats/min and 20 ± 1 mmHg, respectively; Fig. 2). Intra-amygdalar injection of KYN (10 nmol) alone produced no significant changes in baseline heart rate (+5 ± 8 beats/min) or arterial pressure (+2 ± 3 mmHg) (n = 4).
Fig. 6.
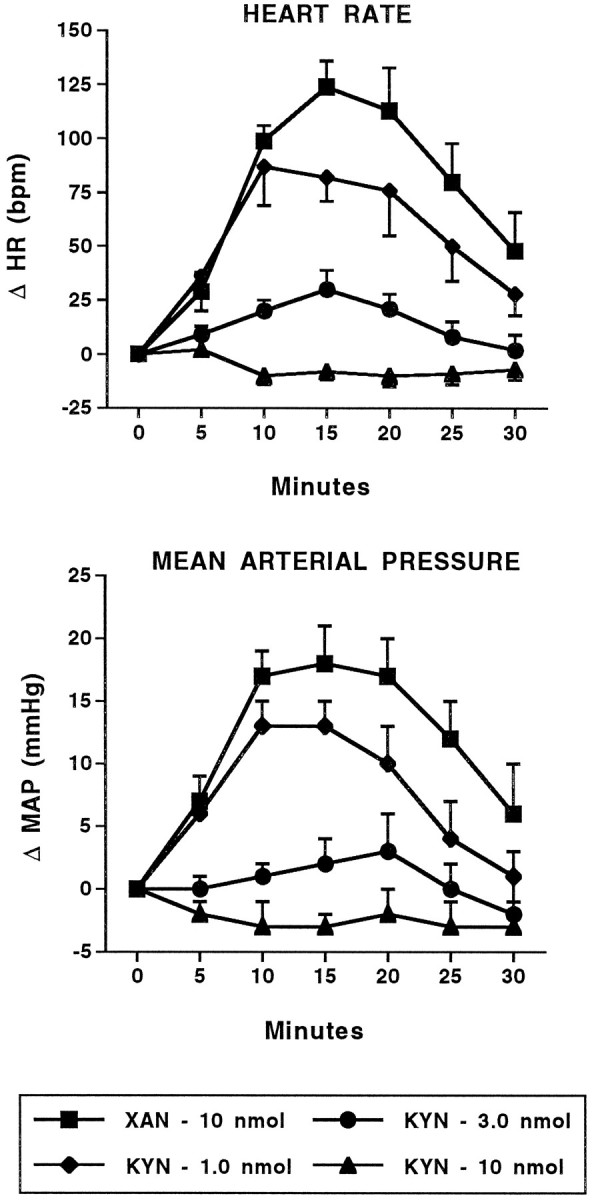
Kynurenic acid blocks the cardiovascular response to BMI. Time course of changes in heart rate (HR; beats/min; top) and mean arterial pressure (MAP; mmHg; bottom) in conscious, chronically instrumented rats after bilateral, intra-amygdalar coinjection of 100 pmol BMI with either xanthurenic acid (XAN) or kynurenic acid (KYN). The changes in HR and MAP for BMI + KYN (3.0 nmol) and for BMI + KYN (10 nmol) are significantly different compared with BMI + XAN and BMI + KYN (1.0 nmol). BMI + XAN and BMI + KYN (10 nmol), n = 5; all others,n = 4. HR: F(3,14) = 25.70; MAP: F(3,14) = 21.86.
In another series of experiments, we examined the effects of KYN using a different GABA antagonist and a different microinjection protocol. Microinjection of 100 pmol of picrotoxin, a noncompetitive, postsynaptic GABAA antagonist, produced significant increases in heart rate and arterial pressure similar to those seen with local injection of BMI (Fig. 7). Microinjection of 10 nmol of KYN into the BLA 5 min after local injection of picrotoxin reversed the picrotoxin-induced tachycardic and pressor responses. Injection of the inactive analog xanthurenic acid did not alter significantly the magnitude or time course of cardiovascular changes produced by picrotoxin.
Fig. 7.
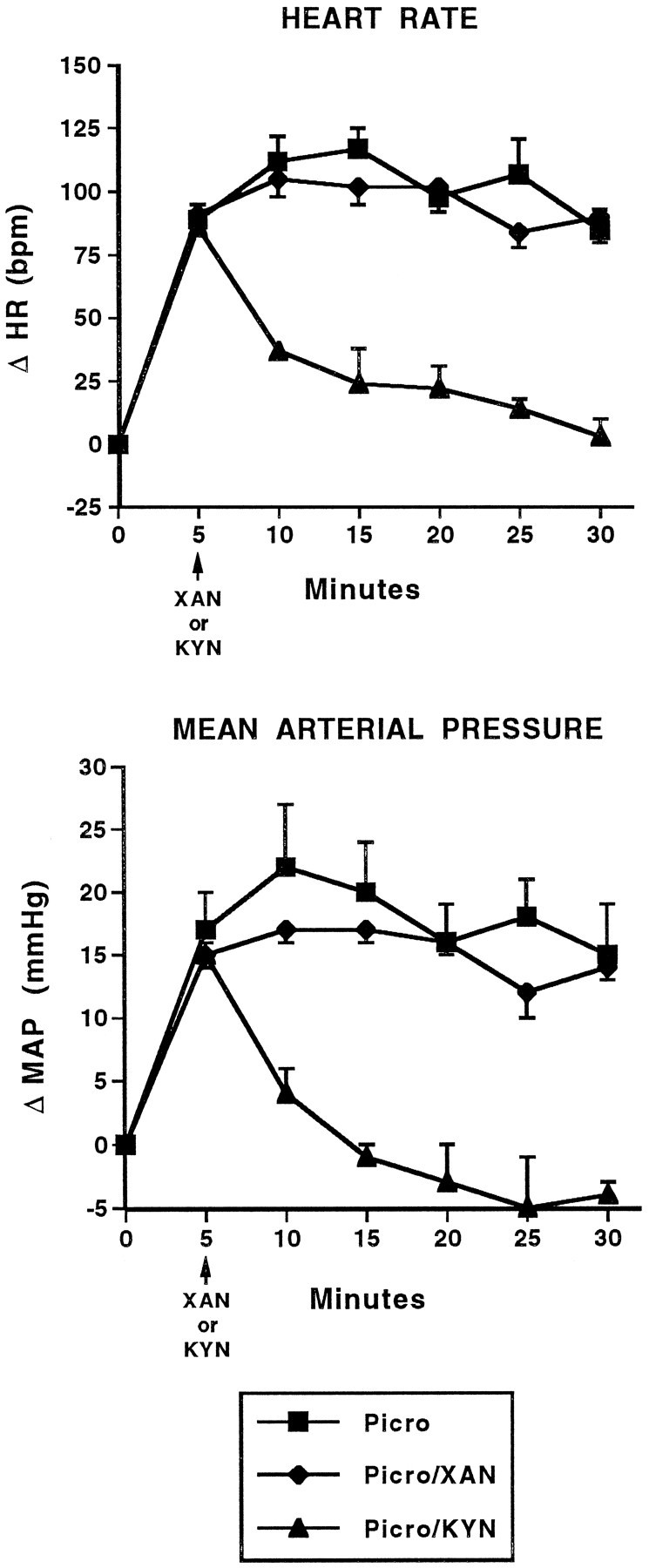
Kynurenic acid reverses the cardiovascular response to picrotoxin. Time course of changes in heart rate (HR; beats/min; top) and mean arterial pressure (MAP; mmHg; bottom) in conscious, chronically instrumented rats after bilateral, intra-amygdalar injection of picrotoxin (Picro; 100 pmol) without further treatment and picrotoxin followed by injection of xanthurenic acid (XAN; 10 nmol) or kynurenic acid (KYN; 10 nmol). The changes in HR and MAP for Picro + KYN are significantly different compared with Picro alone and Picro with XAN. Picro only, n = 4; all others,n = 3. HR: F(2,7) = 22.74; MAP: F(2,7) = 14.60.
Given that blockade of EAA receptors in the BLA can prevent or reverse the cardiovascular changes seen with removal of local GABAergic inhibition, the next series of experiments examined whether potentiation of local glutamatergic transmission would enhance the cardiovascular changes observed after injection of GABA antagonists into the BLA. Microinjection of the glutamate reuptake inhibitor PDC into the BLA (followed 5 min later with a local injection of saline) did not produce any significant changes in heart rate or arterial pressure (Fig. 8). Injection of an intermediate dose of BMI (30 pmol; preceded 5 min earlier with a local injection of saline) produced modest but significant increases in heart rate (+64 ± 9 beats/min) and arterial pressure (+14 ± 2 mmHg). However, when this intermediate dose of BMI was preceded by a local injection of the glutamate reuptake inhibitor PDC, the response to BMI was significantly potentiated (+130 ± 15 beats/min and +24 ± 5 mmHg, respectively).
Fig. 8.
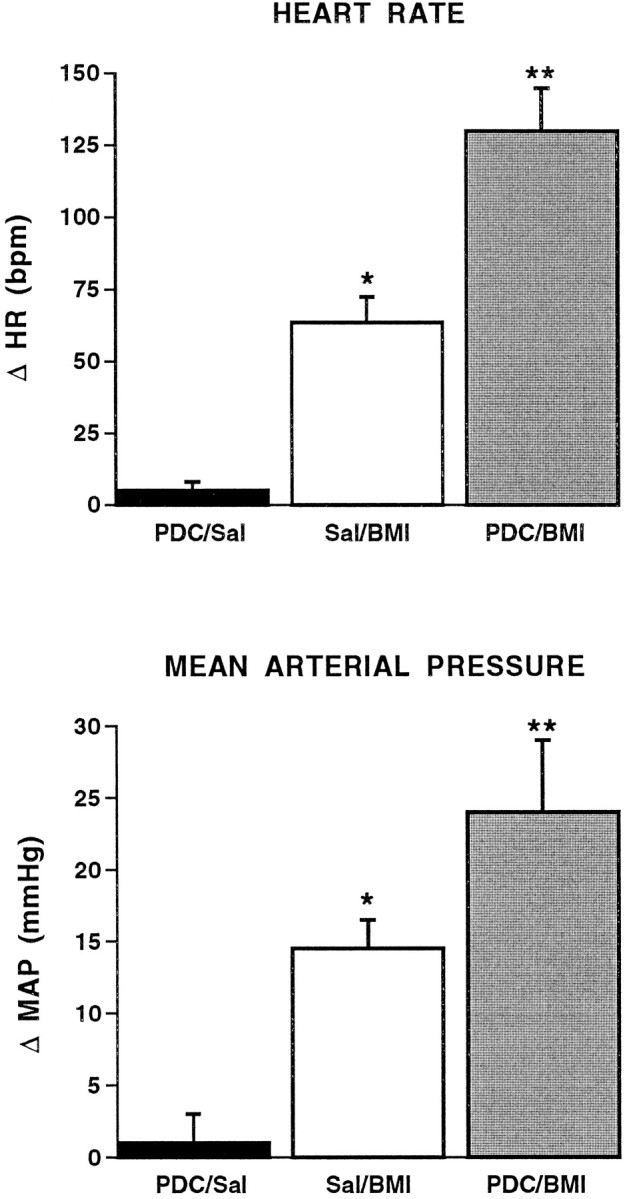
PDC potentiates the cardiovascular response to BMI. Maximal changes in heart rate (HR; beats/min;top) and mean arterial pressure (MAP; mmHg; bottom) in conscious, chronically instrumented rats after bilateral, intra-amygdalar injection ofl-trans-pyrrolidine-2,4-dicarboxylic acid (PDC; 1 nmol) and saline (Sal; 250 nl), saline and BMI (30 pmol), or PDC and BMI. Single asterisks denote significant differences from PDC/saline.Double asterisks denote significant differences from saline/BMI. Each group, n = 4. HR:F(2,9) = 42.88; MAP:F(2,9) = 33.71.
The ability of a glutamate receptor antagonist to block or reverse the cardiovascular effects of GABA antagonists and the ability of a glutamate reuptake inhibitor to enhance these same effects suggest that local EAA receptors play a role in this response. To examine the role of EAA receptor subtypes in these changes, the NMDA receptor antagonist CPP and the AMPA receptor antagonist NBQX were used. Coinjection of either 100 pmol of CPP or 100 pmol of NBQX with 50 pmol of BMI into the BLA attenuated, to a similar extent, the tachycardic and pressor responses associated with BMI (Fig. 9). Furthermore, in the presence of the two EAA receptor antagonists, the changes in heart rate and arterial pressure observed with BMI were reduced further compared with BMI in the presence of either antagonist alone, suggesting that the effects of the two EAA antagonists were additive.
Fig. 9.
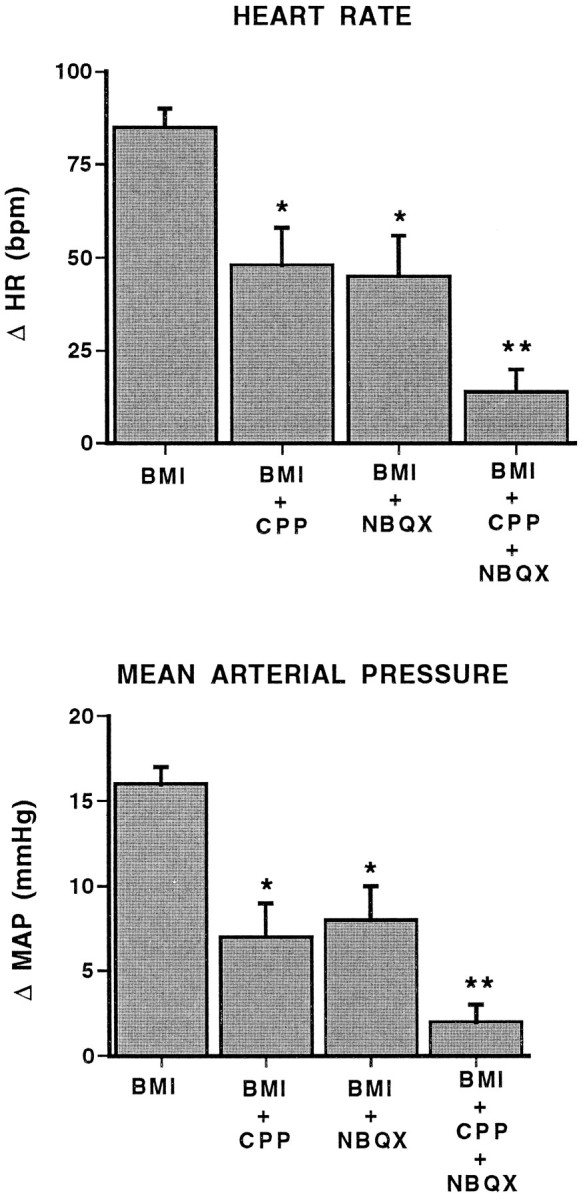
CPP and NBQX block the cardiovascular response to BMI. Maximal changes in heart rate (HR; beats/min;top) and mean arterial pressure (MAP; mmHg; bottom) in conscious, chronically instrumented rats after bilateral injection of 50 pmol of BMI alone or coinjection of BMI with CPP (BMI +CPP; 100 pmol), NBQX (BMI +NBQX; 100 pmol), or CPP and NBQX (BMI +CPP + NBQX). Single asterisks denote significant differences from BMI alone.Double asterisks denote significant differences from BMI alone, BMI + CPP, and BMI + NBQX. BMI alone, n = 8; all others, n = 5. HR:F(3,19) = 15.30; MAP:F(3,19) = 23.22.
DISCUSSION
Data presented in this study provide evidence that the cardiovascular effects resulting from blockade of GABAergic inhibition in the BLA of conscious rats are dependent on activation of local EAA receptors. This study is consistent with and expands on previous reports indicating that GABA and EAA receptors in the amygdala play an important role in anxiogenic behavior and, additionally, provides functional support of previous work describing the GABA/EAA receptor interactions in the amygdala at the cellular level.
Microinjection of the EAA receptor agonists NMDA and AMPA into the BLA produced dose-related increases in heart rate and arterial pressure similar to those produced by injection of the GABAAreceptor antagonist BMI at the same site. This pattern of cardiovascular changes is consistent with anxiety-like or defense-like reactions in this species (Hilton and Zbrozyna, 1963; Hilton and Redfern, 1986). Interestingly, the dose–response curve to AMPA was bell-shaped in that at higher doses the tachycardic and pressor responses were significantly diminished. This observation is consistent with other reports demonstrating that higher doses or concentrations of ionotropic EAA agonists can produce changes that are opposite or inhibitory to those seen at lower concentrations (Lipski et al., 1988;Soltis and DiMicco, 1991a). It has been postulated that the mechanism of this diminished response is associated with either receptor desensitization or depolarization blockade (Lipski et al., 1988; Otis et al., 1996).
The sites from which injection of BMI, NMDA, and AMPA elicited the greatest increases in heart rate and arterial pressure were localized to the anterior portions of the BLA. It can be argued that the volume of injection (250 nl) used in the present study does not restrict the drug to the BLA exclusively. Indeed, histological examination of the injection sites after infusion of the dye Alcian blue indicates that the dye, a crude estimator of drug diffusion, does spread, in some cases, to nuclei adjacent to the BLA. Although there are no definitive methods by which to determine the exact site of action of a drug after its injection into the brain, we attempted to establish the boundaries of the reactive area by implanting cannulae at sites near the proposed active site. Areas dorsal to the active site were examined based on the cannula tract (located dorsally) representing the path of least resistance for drug diffusion and, therefore, a likely site of action. Areas medial to the BLA were examined because this area includes the central nucleus of the amygdala (CNA), a site where electrical stimulation produces cardiovascular and behavioral changes that replicate the defense reaction (Hilton and Zbrozyna, 1963; Galeno and Brody, 1983; Gelsema et al., 1987). In this study, the areas medial and dorsal to the BLA were clearly nonreactive to BMI, NMDA, or AMPA at the doses tested. It is possible that the CNA is the active site and that the doses used in the present study were too high to elicit a response when injected directly at the active site. That is, the cardiovascular responses attributed to injection of these agents into the BLA may actually be a result of the drugs diffusing to the CNA at a lesser but pharmacologically active concentration. However, if the CNA were the site of action and the doses used were too high to produce an effect when injected directly into the CNA, then those injections that were medial to the CNA (see Fig. 5) should have produced tachycardic and pressor responses similar to those seen after injections at sites an equal distance lateral to the CNA, that is, in the BLA. Given that this did not occur, the histological data suggest that the anterior region of the BLA is the site of action of these agents. This pattern of localization and the description of cardiovascular changes associated with injection of these agents are consistent with previous reports that used similar volumes of injections to demonstrate that intra-amygdalar injections of GABAA antagonists and glutamate produce stress-like cardiovascular changes (Maskati and Zbrozyna, 1989; Sanders and Shekhar, 1991, 1995).
The coincident site of injection and the similar pattern and onset of cardiovascular changes seen with injection of NMDA, AMPA, and BMI suggest that these agents are activating the same population of neurons to generate the cardiovascular changes. To determine whether the cardiovascular responses to intra-amygdalar injection of GABA antagonists are mediated by local EAA receptors, the nonselective EAA receptor antagonist KYN was used. When coinjected with BMI, KYN attenuated in a dose-dependent manner the tachycardic and pressor responses. Similarly, microinjection of KYN 5 min after injection of the GABA antagonist picrotoxin reversed the tachycardic and pressor effects caused by this agent. Conversely, xanthurenic acid, a structural analog of KYN without significant effects on EAA receptors (Guyenet et al., 1987), failed to alter the response when coinjected with BMI or locally injected 5 min after picrotoxin. Therefore, taking together the staggered order of treatments within the series of experiments, the negative control using xanthurenic acid, and the different injection protocols (coinjection versus reversal with a separate injection), it is likely that the reduced response observed in the presence of KYN can be attributed to blockade of EAA receptors and not to changes in the responsiveness of the preparation or to nonspecific actions of KYN.
To characterize further the interaction of GABA- and EAA-mediated synaptic transmission in the BLA in regulating cardiovascular function, we used the glutamate reuptake inhibitor PDC (Bridges et al., 1991). We hypothesized that if antagonism of local EAA receptors can block the effects of a GABA antagonist, then enhancing activity at local EAA receptors should potentiate the effects of a GABA antagonist. To demonstrate an enhanced effect of BMI, we used an intermediate dose of BMI (30 pmol) as determined in the dose–response experiments. Injection of PDC at a dose (1 nmol) shown to be capable of producing cardiorespiratory changes after microinjection into the medulla (McManigle et al., 1995) did not produce significant changes in resting heart rate or arterial pressure. However, when injected before BMI, PDC potentiated the response to BMI to a magnitude otherwise seen only with the 100 pmol dose of BMI. Thus, the effects of PDC in combination with BMI are consistent with a role for EAA receptor-mediated transmission in BMI-induced cardiovascular changes. However, EAA receptor-mediated input does not appear to play an important role in regulating cardiovascular function under basal conditions, as evidenced by the lack of effect after injection of PDC or KYN alone. Rather, it appears that the predominate tone in the BLA under basal conditions in conscious rats is GABA-mediated inhibition.
In the final series of experiments, we examined the role of specific EAA receptor subtypes in mediating the cardiovascular responses after blockade of GABAergic inhibition in the BLA. Injection of either the NMDA receptor antagonist CPP or the AMPA receptor antagonist NBQX attenuated the tachycardic and pressor responses to a similar degree. When the two EAA antagonists were combined and coinjected with BMI, the effects were additive. Therefore, there appear to be two distinct components mediating the cardiovascular response to injection of BMI. One component relies on activity at NMDA receptors, whereas the other is mediated through AMPA receptors. The notion that neuronal excitability in the BLA can be modulated by a balance between EAA receptor-mediated excitation and GABA receptor-mediated inhibition has been investigated using other preparations. Intracellular recordings from brain slices have shown that stimulation of afferent pathways to the BLA elicits EPSPs consisting of fast and slow components that are blocked by AMPA/kainate receptor antagonists and NMDA receptor antagonists, respectively (Rainnie et al., 1991a; Gean and Chang, 1992). Furthermore, in the presence of bicuculline, epileptiform burst discharges occur in normally quiescent BLA neurons, suggesting that the overriding input to the neurons is GABAA receptor-mediated inhibition (Rainnie et al., 1991b). In the present study, we have provided in vivo evidence that a similar interaction between GABA and EAA systems in the BLA plays a role in cardiovascular regulation.
The present study is also important in light of recent studies examining the role of GABA and EAA receptors in the BLA with respect to anxiogenic behavior. Injection of NMDA and non-NMDA EAA receptor antagonists into the BLA blocks in a dose-dependent manner the acquisition and the expression, respectively, of conditioned fear (Campeau et al., 1992; Falls et al., 1992; Kim et al., 1993; Campeau and Davis, 1995; Maren et al., 1996). Furthermore, injection of GABA antagonists into the BLA elicits anxiogenic behavior as measured in various behavioral paradigms (Sanders and Shekhar, 1995). Therefore, these studies and the data presented here suggest that an interaction between GABA and EAA receptors in the BLA may control the cardiovascular responses associated with conditioned rewarding or aversive stimuli.
In summary, the present study provides evidence that the cardiovascular response caused by removal of GABAergic inhibition in the BLA is dependent on activation of local EAA receptors. Therefore, the neurons mediating this response are regulated by a balance of GABA receptor- and EAA receptor-mediated synaptic transmission.
Footnotes
This work was supported by a grant from the American Heart Association, Iowa Affiliate.
Correspondence should be addressed to Robert P. Soltis, Department of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, Drake University, 2507 University Avenue, Des Moines, IA 50311.
REFERENCES
- 1.Beitz AJ. The organization of afferent projections to the midbrain periaqueductal gray of the rat. Neuroscience. 1982;7:133–159. doi: 10.1016/0306-4522(82)90157-9. [DOI] [PubMed] [Google Scholar]
- 2.Bridges RJ, Stanley MS, Anderson MW, Cotman CW, Chamberlin AR. Conformationally defined neurotransmitter analogues. Selective inhibition of glutamate uptake by one pyrrolidine-2,4-dicarboxylic diastereomer. J Med Chem. 1991;34:717–723. doi: 10.1021/jm00106a037. [DOI] [PubMed] [Google Scholar]
- 3.Campeau S, Davis M. Involvement of the central nucleus and the basolateral complex of the amygdala in fear conditioning measured with fear-potentiated startle in rats trained concurrently with auditory and visual conditioned stimuli. J Neurosci. 1995;15:2301–2311. doi: 10.1523/JNEUROSCI.15-03-02301.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campeau S, Miserendino MJD, Davis M. Intra-amygdala infusion of the N-methyl-d-aspartate receptor antagonist AP5 blocks acquisition but not expression of fear potentiated startle to an auditory conditioned stimulus. Behav Neurosci. 1992;106:569–574. doi: 10.1037//0735-7044.106.3.569. [DOI] [PubMed] [Google Scholar]
- 5.Falls WA, Miserendino MJD, Davis M. Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feldman DS, Buccafusco JJ. Spinal muscarinic, glutamatergic and GABAergic receptor systems in cardiovascular regulation. J Pharmacol Exp Ther. 1997;281:274–283. [PubMed] [Google Scholar]
- 7.Galeno TM, Brody MJ. Hemodynamic responses to electrical amygdaloid stimulation in spontaneously hypertensive rats. Am J Physiol. 1983;245:281–286. doi: 10.1152/ajpregu.1983.245.2.R281. [DOI] [PubMed] [Google Scholar]
- 8.Galeno TM, Van Hoesen GW, Brody MJ. Central amygdaloid lesion attenuates exaggerated hemodynamic responses to noise stress in spontaneously hypertensive rats. Brain Res. 1984;219:249–259. doi: 10.1016/0006-8993(84)91257-5. [DOI] [PubMed] [Google Scholar]
- 9.Gean PW, Chang FC. Pharmacological characterization of excitatory synaptic potentials in the rat basolateral amygdaloid neurons. Synapse. 1992;11:1–9. doi: 10.1002/syn.890110102. [DOI] [PubMed] [Google Scholar]
- 10.Gelsema AJ, McKitrick DJ, Calaresu FR. Cardiovascular response to chemical and electrical stimulation of the amygdala in rats. Am J Physiol. 1987;253:R712–R718. doi: 10.1152/ajpregu.1987.253.5.R712. [DOI] [PubMed] [Google Scholar]
- 11.Guyenet PG, Filtz TM, Donaldson SR. Role of excitatory amino acids in rat vagal and sympathetic baroreflexes. Brain Res. 1987;407:272–284. doi: 10.1016/0006-8993(87)91105-x. [DOI] [PubMed] [Google Scholar]
- 12.Hilton SM, Redfern WS. A search for brainstem groups integrating the defence reaction in the rat. J Physiol (Lond) 1986;378:213–228. doi: 10.1113/jphysiol.1986.sp016215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hilton SM, Zbrozyna AW. Amygdaloid region for defence reaction and its efferent pathway to the brainstem. J Physiol (Lond) 1963;165:160–173. doi: 10.1113/jphysiol.1963.sp007049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hopkins DA, Holstege G. Amygdaloid projections to the mesencephalon, pons and medulla oblongata in the cat. Exp Brain Res. 1978;32:529–547. doi: 10.1007/BF00239551. [DOI] [PubMed] [Google Scholar]
- 15.Inglis WL, Dunbar JS, Winn P. Barbiturate anesthesia reduces the neurotoxic effects of quinolinate but not ibotenate in the rat pedunculopontine tegmental nucleus. Neurosci Lett. 1993;156:78–82. doi: 10.1016/0304-3940(93)90444-p. [DOI] [PubMed] [Google Scholar]
- 16.Jones RSG. Epileptiform events induced by GABA antagonists in entorhinal cortical cells in vitro are partly mediated by N-methyl-d-aspartate receptors. Brain Res. 1988;457:113–121. doi: 10.1016/0006-8993(88)90062-5. [DOI] [PubMed] [Google Scholar]
- 17.Kim M, Campeau S, Falls WA, Davis M. Infusion of the non-NMDA receptor antagonist CNQX into amygdala blocks the expression of fear potentiated startle. Behav Neural Biol. 1993;59:5–8. doi: 10.1016/0163-1047(93)91075-x. [DOI] [PubMed] [Google Scholar]
- 18.Krettek JE, Price JL. Amygdaloid projections to subcortical structures within the basal forebrain and brainstem in the rat and cat. J Comp Neurol. 1978;178:225–254. doi: 10.1002/cne.901780204. [DOI] [PubMed] [Google Scholar]
- 19.Lipski J, Bellingham MC, West MJ, Pilowsky J. Limitations of the technique of pressure microinjection of excitatory amino acids for evoking responses from localized regions of the CNS. J Neurosci Methods. 1988;126:169–176. doi: 10.1016/0165-0270(88)90166-5. [DOI] [PubMed] [Google Scholar]
- 20.Maren S, Aharonov G, Stote DL, Fanselow MS. N-methyl-d-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of conditional fear in rats. Behav Neurosci. 1996;110:1365–1374. doi: 10.1037//0735-7044.110.6.1365. [DOI] [PubMed] [Google Scholar]
- 21.Maskati HAA, Zbrozyna AW. Cardiovascular and motor components of the defence reaction elicited in rats by electrical and chemical stimulation in amygdala. J Auton Nerv Syst. 1989;28:127–132. doi: 10.1016/0165-1838(89)90085-4. [DOI] [PubMed] [Google Scholar]
- 22.McManigle JE, Panico WH, DaSilva AMT, Gillis RA. Respiratory effects produced by microinjection of l-glutamate and an uptake inhibitor of l-glutamate into the caudal subretrofacial area of the medulla. Eur J Pharmacol. 1995;280:257–275. doi: 10.1016/0014-2999(95)00203-w. [DOI] [PubMed] [Google Scholar]
- 23.Otis T, Zhang S, Trussell LO. Direct measurement of AMPA receptor desensitization induced by glutamatergic synaptic transmission. J Neurosci. 1996;16:7496–7504. doi: 10.1523/JNEUROSCI.16-23-07496.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; New York: 1986. [DOI] [PubMed] [Google Scholar]
- 25.Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Excitatory transmission in the basolateral amygdala. J Neurophysiol. 1991a;66:986–998. doi: 10.1152/jn.1991.66.3.986. [DOI] [PubMed] [Google Scholar]
- 26.Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Inhibitory transmission in the basolateral amygdala. J Neurophysiol. 1991b;66:999–1009. doi: 10.1152/jn.1991.66.3.999. [DOI] [PubMed] [Google Scholar]
- 27.Sanders BJ, Wirtz-Nole CS, DeFord M, Erling BF. Central amygdaloid lesions attenuate cardiovascular responses to acute stress in borderline hypertensive rats. Physiol Behav. 1994;56:709–713. doi: 10.1016/0031-9384(94)90231-3. [DOI] [PubMed] [Google Scholar]
- 28.Sanders SK, Shekhar A. Blockade of GABAA receptors in the region of the anterior basolateral amygdala of rats elicits increases in heart rate and blood pressure. Brain Res. 1991;576:101–110. doi: 10.1016/0006-8993(91)91441-3. [DOI] [PubMed] [Google Scholar]
- 29.Sanders SK, Shekhar A. Regulation of anxiety by GABAA receptors in the rat amygdala. Pharmacol Biochem Behav. 1995;52:701–706. doi: 10.1016/0091-3057(95)00153-n. [DOI] [PubMed] [Google Scholar]
- 30.Sanders SK, Morzorati SL, Shekhar A. Priming of experimental anxiety by repeated subthreshold GABA blockade in the rat amygdala. Brain Res. 1995;699:250–259. doi: 10.1016/0006-8993(95)00915-d. [DOI] [PubMed] [Google Scholar]
- 31.Soltis RP, DiMicco JA. GABAA and excitatory amino acid receptors in dorsomedial hypothalamus and heart rate in rats. Am J Physiol. 1991a;260:R13–R20. doi: 10.1152/ajpregu.1991.260.1.R13. [DOI] [PubMed] [Google Scholar]
- 32.Soltis RP, DiMicco JA. Interaction of hypothalamic GABAA and excitatory amino acid receptors controlling heart rate in rats. Am J Physiol. 1991b;261:R427–R433. doi: 10.1152/ajpregu.1991.261.2.R427. [DOI] [PubMed] [Google Scholar]
- 33.Steward O, Tomasulo R, Levy WB. Blockade of inhibition in a pathway with dual excitatory and inhibitory action unmasks a capability for LTP that is otherwise not expressed. Brain Res. 1990;516:292–300. doi: 10.1016/0006-8993(90)90930-a. [DOI] [PubMed] [Google Scholar]