

Abstract
Neuropeptide Y (NPY) inhibits excitatory synaptic transmission in the hippocampus and is implicated in control of limbic seizures. In the present study, we examined hippocampal function and the response to pharmacologically induced seizures in mutant mice lacking this peptide. In slice electrophysiology studies, no change in normal hippocampal function was observed in NPY-deficient mice compared with normal wild-type littermates. Kainic acid (KA) produced limbic seizures at a comparable latency and concentration in NPY-deficient mice compared with littermates. However, KA-induced seizures progressed uncontrollably and ultimately produced death in 93% of NPY-deficient mice, whereas death was rarely observed in wild-type littermates. Intracerebroventricular NPY infusion, before KA administration, prevented death in NPY-deficient mice. These results suggest a critical role for endogenous NPY in seizure control.
Keywords: anticonvulsant, homologous recombination, electrophysiology, epilepsy, mouse, hippocampus, neuropeptide Y
Neuropeptide Y (NPY), a 36-amino-acid member of the pancreatic polypeptide family, was first isolated from porcine brain in 1982 (Tatemoto, 1982). NPY is found only in neural tissue of the central and peripheral nervous systems, is one of the most abundant peptides in the nervous system of mammals, and is prominently expressed in the hippocampal formation (Allen et al., 1983;Chronwall et al., 1985). NPY expression patterns indicate an interneuron localization within the hippocampus (Morris, 1989; Gruber et al., 1994). The functional organization of the hippocampus has traditionally been described as a feed-forward trisynaptic circuit (Andersen et al., 1969). NPY-containing interneurons are thought to modulate excitability in this trisynaptic circuit by activating presynaptic Y2-type receptors at mossy fiber to CA3 pyramidal cell synapses and Schaffer collateral to CA1 pyramidal cell synapses (Fig. 1) (Colmers et al., 1988,1991; Martel et al., 1990; Bleakman et al., 1992; Larsen et al., 1993;Dumont et al., 1996). Exogenous application of NPY to hippocampal slices reduces the excitatory population spike evoked in CA1 pyramidal regions by electrical stimulation (Colmers et al., 1987). NPY application has also been shown to inhibit EPSPs at mossy fiber–CA3 and CA3–CA3 synapses within the hippocampus (Haas et al., 1987;Klapstein and Colmers, 1993).
Fig. 1.

Schematic representation of the hippocampal trisynaptic circuit. Feed-forward excitation enters the hippocampus from the entorhinal cortex (EC) via the perforant path (PP); glutamatergic granule cells (GC) in the dentate gyrus make excitatory synaptic connections ontoCA3 pyramidal neurons via mossy fibers (MF); and glutamatergic CA3pyramidal neurons make excitatory synaptic connections ontoCA1 pyramidal neurons via the Schaffer collaterals (SC). Interneurons containing NPY and GABA (filled triangles) are thought to inhibit excitation by acting at presynaptic sites to reduce excitatory neurotransmitter release at mossy fiber–CA3 and Schaffer collateral–CA1 synapses in the hippocampus.
Control of hippocampal excitability may be of particular significance with respect to epilepsy, because this region has long been implicated in the generation and modulation of seizure activity both clinically and experimentally (Schwartzkroin, 1994; Swanson, 1995; Mayanagi et al., 1996). Because NPY inhibits hippocampal excitability under normal conditions, it was hypothesized that this neuropeptide plays a critical role in modulation of hippocampal function during seizures. In support of this hypothesis, Woldbye at al. (1996) have shown that intracerebroventricular administration of NPY reduces both primary and secondary epileptiform afterdischarges elicited by electrical stimulation and inhibits kainic acid-induced motor seizures (Woldbye et al., 1997). Other studies have demonstrated that various types of acute experimentally induced seizures are associated with an increase in NPY gene expression in the hippocampus (Gruber et al., 1994; Kragh et al., 1994; Tønder et al., 1994). The correlation between seizures and NPY function suggests that NPY release may be a compensatory mechanism to reduce excitation via action at presynaptic sites in the hippocampus during a limbic seizure.
Erickson et al. (1996) have recently developed mutant mice in which theNPY gene was replaced with a lacZ reporter gene. These NPY-deficient mice exhibit mild, spontaneous seizure behaviors and a reduced threshold for pentylenetetrazol-induced seizures. However, it was not clear from this earlier study whether loss of NPY from CNS neurons resulted in alteration of synaptic function or whether NPY modulated limbic seizure activity. Because adequate pharmacological antagonists are not available for this neuropeptide, we used NPY-deficient mice to examine whether NPY plays a critical role in normal hippocampal function and/or the modulation of limbic seizure activity.
MATERIALS AND METHODS
Generation of mutant mice. Mice deficient for NPY were generated using homologous recombination techniques as described by Erickson et al. (1996). Briefly, a lacZ reporter gene with a nuclear localization signal replaced two exons and most of theNPY reading sequence. Mice heterozygous for NPY were inbred to produce mice of all possible genotypes. The genotypes were determined by hybridizing duplicate filters with tail DNA dot hybridization using probes for lacZ and a region of theNPY gene that had been deleted. All experiments were performed on F2 and F3 129SV/C57BL hybrid males. Mice were maintained under constant environmental conditions, a 12 hr:12 hr light/dark cycle, and given mouse chow and water ad libitum. All animal procedures complied with National Institutes of Health guidelines and were approved by the University of Washington Animal Care Facility Committee.
Hippocampal slice electrophysiology. Adult mice (8–12 weeks old) were anesthetized with Metofane and killed by decapitation. The top of the skull was removed, and the exposed brain was chilled with ice-cold, oxygenated slicing medium (Baraban and Schwartzkroin, 1997). The brain was then rapidly removed and divided in half, and one hemisphere was blocked and glued to the stage of a vibroslicer (Frederick Haer, Brunswick, ME). Slicing was performed in chilled (3–4°C), oxygenated sucrose-based artificial CSF consisting of (in mm): 220 sucrose, 3 KCl, 1.25 NaH2PO4, 2 MgSO4, 26 NaHCO3, 2 CaCl2, and 10 dextrose. The resulting transverse hippocampal slices (450 μm) were immediately transferred to a holding chamber in which they remained submerged in oxygenated recording medium consisting of (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 2 MgSO4, 26 NaHCO3, 2 CaCl2, and 10 dextrose. Slices were held at room temperature for at least 60 min before being transferred to a submersion recording chamber in which they were perfused with oxygenated recording medium at room temperature.
Extracellular recording electrodes, pulled from borosilicate glass and filled with 2 m NaCl (3–15 MΩ), were used to record field potentials (amplifier from Neurodata Instruments, New York, NY). Sharp intracellular recording electrodes, pulled from borosilicate glass and filled with 4 m potassium acetate (100–125 MΩ), were used to record intracellular potentials. For electrical stimulation of the tissue, a bipolar electrode (65 μm center to center) was placed on the surface of the slice. Stimuli consisted of 100–300 μsec constant current pulses of 50–750 μA. Spontaneous field activity and responses to stimulation were analyzed on-line and stored on videotape for further examination. Quantitative analyses of population spike amplitudes were performed on CA1 pyramidal cell and dentate granule cell responses.
X-gal staining. Brain slices (1000 μm) were prepared from 8-week-old heterozygote mice as described for slice electrophysiology. X-gal staining for β-galactosidase activity was performed as described previously by Mercer et al. (1991).
EEG implantation. Behavioral and electroencephalographic (EEG) observations were made using a time-locked, videodigital EEG monitoring system (Telefactor Corp., Conshocken, PA). For EEG recordings, mice (12 weeks old) were surgically implanted in the left and right frontoparietal cortex with electrodes. Each mouse was anesthetized with ketamine and xylazine (10 mg/kg and 1 mg/kg, i.p., respectively) so that there was no limb withdrawal response to a foot pinch. Animals were then placed in a stereotactic holder, and the scalp was opened with a sharp scalpel. Sterile stainless steel recording electrodes were placed epidurally through burr holes in the skull (one electrode on either side of the sagittal suture, approximately halfway between the bregma and lambdoid sutures and ∼1 mm from the midline). Electrodes were cemented in place with a fast-acting adhesive and dental acrylic, and electrode leads were attached to a microplug that was also cemented to the head of the animal. Animals were allowed to recover for 48 hr before kainic acid experiments were initiated. Simultaneous EEG and video recording sessions were made for each animal for 20 min before injection of kainic acid and continuously thereafter for a period of ∼2 hr.
Cannula implantation. Twelve-week-old male mice were anesthetized with an injection of Equithesin (34 mg/kg, i.p.) and atropine (0.35 mg/kg, i.p.). Animals were placed in a stereotactic device (Kopf Instruments), and cannulas were implanted into the lateral ventricle (0.6 mm posterior; 1.9 mm lateral; and 2.0 mm ventral to bregma). Mice were allowed to recover from surgery for 2 d. On the day of the experiment, intracerebroventricular injections of human NPY (American Peptide Company) were performed on awake, freely behaving animals. NPY (5 μg in 1 μl of neutral buffered solution) was administered using an infusion pump (kd Scientific) at a constant rate of 1 μl/min. Cannula placement was verified post hoc in all animals by injection of 1% cresyl violet solution before brain dissection.
Kainic acid studies. Kainic acid (stock solution, 4 mg/ml) was dissolved in neutral buffered saline. In initial experiments, intraperitoneal KA injections (20 mg/kg) were made every 20 min to determine the concentration of KA required to produce a full behavioral seizure in NPY−/− and NPY+/−mice (∼50 mg/kg; see Fig. 4). Subsequent seizure experiments (i.e., video-EEG, cannulation, and lacZ expression studies) began with a bolus injection of KA (40 mg/kg, i.p.) followed by a KA injection (20 mg/kg) every 20 min until a full behavioral seizure was elicited.
Fig. 4.

Effect of exogenous NPY on synaptic responses in mouse hippocampus. A, Representative CA1 field recording during stimulation of Schaffer collaterals in a hippocampal slice from an NPY+/+ mouse in normal recording medium (baseline) and ∼20 min after bath application of neuropeptide Y (0.5 μm NPY). B, Representative CA1 field recording during stimulation of Schaffer collaterals in a hippocampal slice from an NPY−/−mouse in normal recording medium (baseline) and ∼20 min after bath application of neuropeptide Y (0.5 μmNPY).
Statistics. Data were analyzed using the SigmaStat program (Jandel Scientific, Corte Madera, CA). Values represent the mean ± SEM. Significance was taken as p < 0.05 using Student’s t test.
RESULTS
Electrophysiological characterization of synaptic function in NPY-deficient mice
To determine whether NPY-deficient mice exhibit hyperexcitability at the cellular level, we examined electrophysiological properties of excitatory synaptic transmission in an acute hippocampal slice preparation. In previous electrophysiology studies, NPY was shown to modulate synaptic transmission at Schaffer collateral–CA1 in the hippocampus (Colmers et al., 1988, 1991; Klapstein and Colmers, 1993). In the present study, extracellular field potential recordings were obtained from the pyramidal cell body region of CA1 to evaluate the efficacy of the Schaffer collateral–CA1 synapse. Single-pulse stimulation (50–750 μA; 100–300 μsec) elicited an EPSP at low stimulus intensities and a sharp, negative population spike at higher stimulation intensities (Fig.2A). Input–output curves were constructed for slices from NPY-deficient (NPY−/−; n = 15 slices from five animals) and littermate wild-type control (NPY+/+;n = 15 slices from five animals) mice; no differences in Schaffer collateral–CA1 synapse function were observed (Fig.2C). A paired-pulse stimulation paradigm was also used to examine synaptic function at this synapse (King et al., 1985; Austin et al., 1989). Paired-pulse stimulation did not reveal differences in Schaffer collateral–CA1 synapse function between slices from NPY−/− mice and littermate wild-type controls (Fig. 2E). Consistent with the results from our extracellular recordings, intracellular synaptic responses in CA1 pyramidal neurons from NPY−/− mice seemed normal. EPSPs and IPSPs could be elicited by Schaffer collateral stimulation in cells from both NPY−/− (n = 9) and NPY+/+ (n = 2) mice with the amplitude of the EPSP (and the presence of an action potential) varying with stimulus intensity. In all CA1 cells, the EPSP was followed by an inhibitory postsynaptic response, presumably composed of a fast GABAA receptor-mediated and a slow GABABreceptor-mediated IPSP (Fig.3A). The amplitude and reversal potentials for fast and slow IPSPs evoked with Schaffer collateral stimulation in CA1 cells from NPY−/−mice (Fig. 3A–B) were within the range reported previously for rodents (Alger and Nicoll, 1982; Newberry and Nicoll, 1984; Biscoe and Duschen, 1985) and were comparable with those obtained in NPY+/+ mice (data not shown). Paired-pulse facilitation was also observed at interpulse intervals between 15 and 65 msec (Fig. 3C). To test the synaptic response to tetanic stimulation, we used a high frequency stimulation protocol (Figurov et al., 1996). Tetanic high frequency stimulation (100 pulses at 50 Hz; stimulation at 4× threshold) produces synaptic fatigue and a preferential release of synaptic vesicles containing neuropeptides (Zucker, 1989; Hökfelt, 1991; Vilim et al., 1996). High frequency stimulation produced a rapid reduction in population spike amplitude (∼75% reduction by the 10th pulse) at the Schaffer collateral–CA1 synapse; no significant differences in the response to high frequency stimulation were observed between slices from NPY-deficient mice and littermate wild-type controls (Fig.4).
Fig. 2.

Characterization of synaptic function in hippocampal slices. A, Representative population spike response in the CA1 pyramidal cell region in a hippocampal slice from an NPY−/− mouse. Schaffer collaterals were stimulated at 4 × threshold (T) for generation of a population spike. B, Representative population spike response in the GC region in a hippocampal slice from an NPY−/− mouse. The perforant path was stimulated at 4 × threshold for generation of a population spike. Stimulation protocols elicited typical paired-pulse facilitation of the second population spike response at both Schaffer collateral–CA1 (A) and perforant path–GC synapses (B). Stimulus artifacts are clipped in bothtraces. C, Input–output curves of population spike responses recorded in the CA1 pyramidal cell region (st. pyramidale) to stimulation of the Schaffer collaterals. Threshold for stimulation was defined for each slice as the minimum current required to elicit a detectable population spike (PS); the x-axis shows stimulus intensity in terms of threshold multiples. Responses are normalized with respect to maximumPS amplitude to allow averaging of responses from all slices from NPY-deficient animals (closed diamonds;n = 15) and all slices from littermate wild-type animals (open diamonds; n = 15). The values represent the mean ± SEM. D, Input–output curves of population spike responses recorded in the dentate GC body layer to stimulation of the perforant path. E, Plot of paired-pulse facilitation (amplitude of PS response to second stimulus divided by amplitude of population spike response to first stimulus) in the CA1 pyramidal cell region for hippocampal slices from NPY-deficient (closed diamonds;n = 11) and littermate wild-type control (open diamonds; n = 12) mice; stimulus intensity was at 4× threshold. F, Plot of paired-pulse facilitation in the GC region of hippocampal slices from NPY-deficient and littermate wild-type control mice.
Fig. 3.

Intracellular synaptic response in a CA1 pyramidal neuron from an NPY−/− mouse. A, Representative intracellular recording from a CA1 pyramidal neuron (resting membrane potential = −64 mV) during Schaffer collateral stimulation. Note the presence of an EPSP followed by a biphasic IPSP with fast (closed circle) and slow (open circle) components. B, Reversal potential plot of the fast and slow IPSPs for this CA1 pyramidal neuron. Reversal potentials were determined by systematically changing the membrane potential via intrasomatic current injection, evoking IPSPs at a stimulation intensity subthreshold for generation of an action potential and measuring the amplitude and polarity of the resulting IPSP at the time points indicated in A(open and closed circles). Vm, Membrane voltage. C, Intracellular response to paired-pulse stimulation of the Schaffer collaterals for this CA1 pyramidal neuron. Note the presence of paired-pulse facilitation of the EPSP at interpulse intervals between 15 and 65 msec.
Synaptic function was also examined in the perforant path–granule cell synapse. Recording electrodes were placed near dentate granule cells, and stimulation electrodes were placed on the perforant path (Fig.2B). At the perforant path–GC synapse, under normal conditions, paired-pulse inhibition is observed at short interpulse intervals (10–30 msec), whereas paired-pulse facilitation is seen at longer interpulse intervals (>30 msec). As seen with the Schaffer collateral–CA1 synapse, input–output curves and paired-pulse stimulation protocols in the perforant path–GC synapse did not reveal differences in synaptic function between slices from NPY-deficient mice (n = 11 slices from five animals) and littermate wild-type controls (n = 12 slices from five animals; Fig. 2D,F). The effect of tetanic stimulation on excitability in the perforant path–GC synapse was not quantitated because a >95% population spike amplitude reduction was observed by the fourth stimulation pulse in NPY-deficient and wild-type animals.
Neither multiple population spikes after single-pulse stimulation nor epileptiform-like afterdischarges after high frequency stimulation were observed in either the CA1 or GC recordings. Exogenous application of 0.5 μm NPY reduced the amplitude of the population spike elicited in CA1 during Schaffer collateral stimulation by 25% in hippocampal slices from NPY-deficient mice (n = 7) and by 33% in hippocampal slices from wild-type controls (n = 6; comparison of NPY−/− with NPY+/+, p = 0.58; Fig.5). These results suggest that NPY-deficient mice exhibit normal hippocampal synaptic function and have functional NPY receptors that can play a role in inhibition of excitatory transmission.
Fig. 5.

Effect of high frequency stimulation on synaptic responses. Plots of the response to high frequency tetanic stimulation (100 pulses at 50 Hz; stimulation at 4× threshold) in the CA1 pyramidal cell region of wild-type (NPY+/+) and NPY-deficient (NPY−/−) mice. They-axis shows population spike amplitude plotted as the percent of initial response (normalized to 100%). Representative extracellular field recordings are shown in insets. Calibration, 5 mV, 20 msec.
Seizure activity in NPY-deficient mice
To investigate whether NPY plays a role in the modulation of limbic seizure activity, we investigated the effect of pharmacologically induced seizures in awake, freely behaving mice. KA, an analog of the excitatory amino acid glutamate, is a potent convulsant agent and produces well characterized limbic motor seizures (Sperk, 1994). In initial experiments, KA (20 mg/kg, i.p.) was administered every 20 min until a full behavioral seizure was observed (e.g., bilateral forelimb clonus progressing to rearing, tonic-clonic extension and loss of posture). KA produced full behavioral seizure activity in all animals tested. The latency to first forelimb clonus and the concentration of KA required to produce this event were not different between NPY-deficient and littermate wild-type control mice (Fig. 6A–B). Strikingly, seizure activity was fatal within 127 ± 30 min of the first KA injection in almost all NPY-deficient mice (12/13), whereas only two of nine littermate wild-type control mice died as a result of seizure activity (Fig. 6C).
Fig. 6.
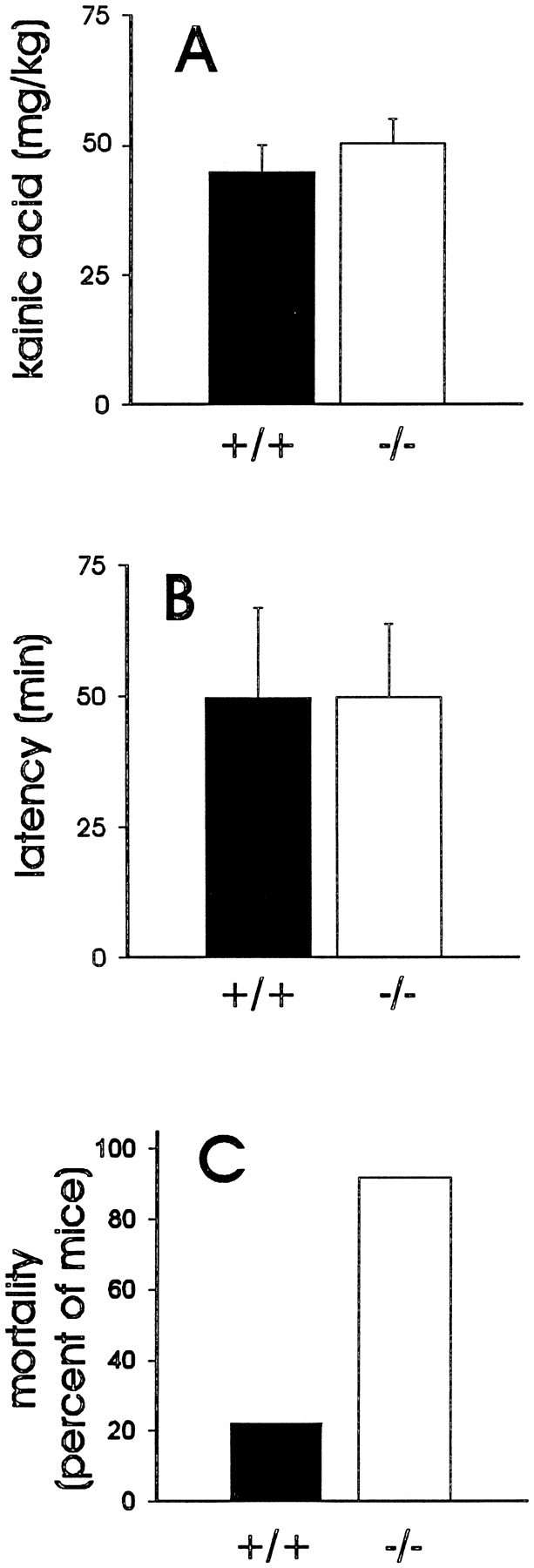
Response to pharmacologically induced seizure activity. A, Plot of the concentration of kainic acid required to elicit a full behavioral seizure in littermate wild-type control mice (closed bar;n = 9) or NPY-deficient mice (open bar; n = 13). B, Plot of the latencies to first forelimb clonus in wild-type and NPY-deficient mice. Bars in A andB show the mean ± SEM. C, Plot of the percent mortality in littermate wild-type and NPY-deficient mice after kainic acid administration (20–100 mg/kg, i.p.).
In a separate set of mice, a video-EEG system was used to monitor simultaneously cortical EEG activity and behavior in mice challenged with KA (40–60 mg/kg, i.p.). No qualitative differences in cortical EEG activity at rest were revealed between NPY-deficient (n = 3) and littermate wild-type control (n = 2) mice. In all animals, KA produced high voltage, synchronized epileptiform-like EEG activity (i.e., “EEG seizure”). Short duration EEG seizures that were accompanied by a staring episode were comparable between NPY-deficient (two to seven episodes per animal; duration, 18 ± 2 sec) and littermate wild-type control (three to seven episodes per animal; duration, 15 ± 1 sec) mice. However, full EEG seizures, which were accompanied by an episode of bilateral forelimb clonus, tonic-clonic extension, and a loss of posture, were different between NPY−/− mice and controls. Specifically, during a 75 min recording period, wild-type littermate control mice experienced two to six full EEG seizure episodes per animal (duration, 29 ± 2 sec; typical example in Fig. 7A). In NPY-deficient mice, full EEG seizure episodes occurred more frequently (6 to 10 episodes per animal), were longer in duration (80 ± 11 sec;p = 0.03), progressed to electrographic status epilepticus (duration of > 200 sec), and led to death within 50 ± 12 min of the first KA injection (typical example in Fig.7B).
Fig. 7.
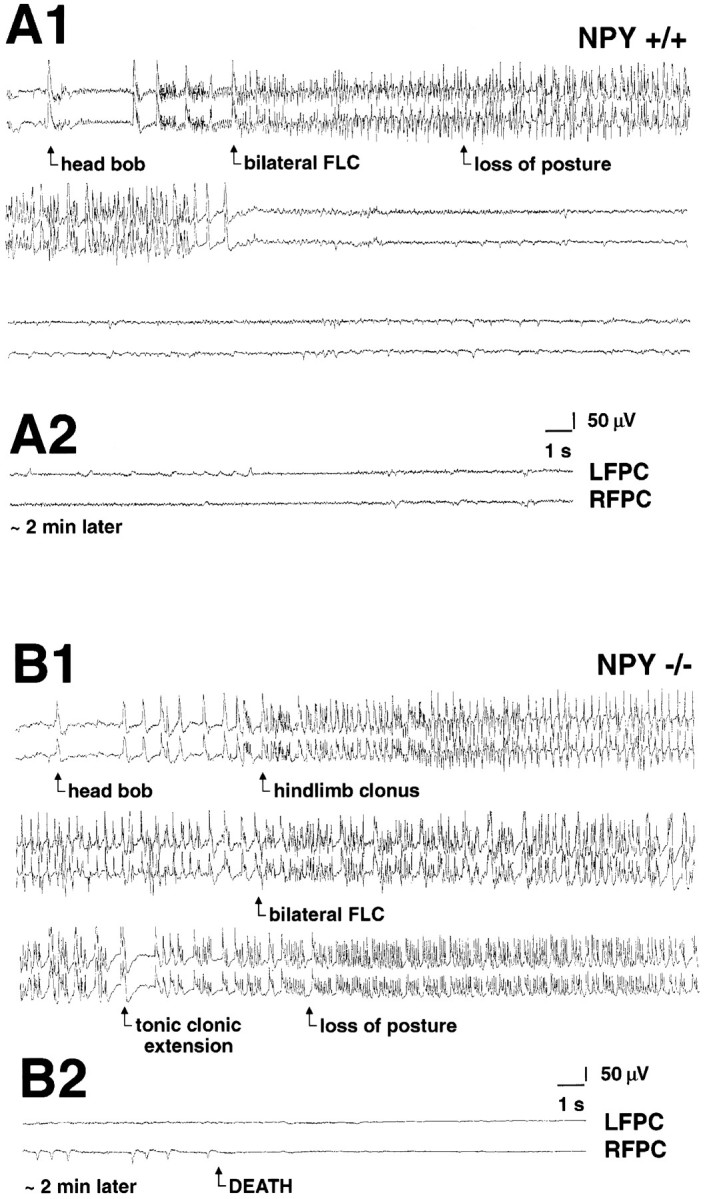
Electrographic activity during a kainate-induced seizure. A1, Representative electrographic (EEG) traces recorded from a wild-type mouse [top, left frontoparietal cortex (LFPC); bottom, right frontoparietal cortex (RFPC)] during a kainic acid-induced seizure (duration, 43 sec). EEG tracing was taken at ∼50 min after the first injection of kainic acid. A2, Same animal ∼2 min later (traces as described inA1). Note the termination of seizure activity and restoration of normal EEG activity. B1, Representative EEG traces recorded from a NPY-deficient mouse (traces as described in A1) during a kainic acid-induced seizure (duration, 220 sec). EEGtracing was taken at ∼50 min after the first injection of kainic acid. B2, Same animal ∼2 min later. Note the progression to a flat EEG associated with death of this animal.FLC, Forelimb clonus.
To investigate whether seizure-induced deaths could be prevented by replacing NPY, intracerebroventricular peptide infusion was performed in NPY−/− mice (n = 3). NPY infusion (5 μg) 20 min before KA administration (40–60 mg/kg, i.p.) prevented seizure-induced death in all NPY-deficient animals tested. The latencies to bilateral forelimb clonus with loss of posture were 22 and 112 min in two NPY-deficient animals; a third animal had no behavioral seizure activity during the 6 hr observation period. NPY infusion (5 μg) in wild-type littermates (n = 4) resulted in KA-induced seizure latencies between 11 and 97 min; for comparison, seizure latencies with this protocol were 5 and 10 min in wild-type mice tested in the video-EEG studies (without NPY infusion). These studies in awake, freely behaving animals reveal a critical antiepileptic role for NPY in the CNS.
NPY gene expression in the mouse hippocampus after a limbic seizure
In situ hybridization and immunocytochemistry techniques have been used previously to identify the cellular localization of NPY in the CNS (Allen et al., 1983; Morris, 1989). In our studies, a novel molecular approach was used to identify NPY localization in the CNS. In mutant mice that we generated, theNPY gene was replaced with a lacZ reporter gene with a nuclear localization signal (Erickson et al., 1996). In these animals, lacZ is expressed in neurons that would normally transcribe NPY mRNA. In brain slices from heterozygous (NPY+/−) mice, X-gal staining for β-galactosidase activity was used to identify these “NPY-containing” neurons. The pattern of β-galactosidase activity that we observed in these slices was similar to those obtained for NPY expression by the other techniques (Allen et al., 1983; Morris, 1989). Specifically, prominent nuclear X-gal staining was observed in the cerebral cortex, hippocampus, striatum, brainstem, and hypothalamus. In the hippocampus, nuclear staining was observed in the stratum oriens and alveus regions of CA1–CA3 and the dentate gyrus (Fig.8A,C). The greatest density of lacZ positive neurons was observed in the hilar and granule cell regions of the dentate gyrus (Fig.8A,E)
Fig. 8.

Expression of lacZ in the hippocampus of heterozygous mice; X-gal staining of tissue from NPY+/− mice. A, X-gal staining in a whole-brain section through the hippocampal formation from a normal heterozygous mouse. B, X-gal staining in a whole-brain section through the hippocampal formation 24 hr after a kainic-acid induced seizure (60 mg/kg, i.p.). Note the increased X-gal staining in the stratum oriens region of CA3 (arrow) and particularly in the granule cell layer of the dentate gyrus (arrowhead) in this thick tissue section.C, High magnification of X-gal staining in the stratum oriens region of CA3 (1000-μm-thick tissue section) from a different heterozygous mouse. D, High magnification of X-gal staining in the stratum oriens region of CA3 (1000-μm-thick tissue section) 24 hr after a kainic acid-induced seizure. E, High magnification of X-gal staining in the dentate gyrus (1000-μm-thick tissue section) from a different heterozygous mouse.F, High magnification of X-gal staining in the dentate gyrus (1000-μm-thick tissue section) 24 hr after a kainic acid-induced seizure.
NPY gene expression in the hippocampus is upregulated after seizure induction (Sloviter, 1989; Gruber et al., 1994; Kragh et al., 1994;Tønder et al., 1994). Because NPY−/− mice did not survive limbic seizures, we could not determine whether NPY expression is upregulated in these animals. However, we did investigate whether X-gal staining could be used to monitor seizure-induced changes in gene expression in NPY+/− mice (Fig.8B,D,F). At 24 hr after a KA-induced seizure, increased X-gal staining was observed most dramatically in the hippocampus, consistent with the idea that KA produces limbic seizures. In particular, increased X-gal staining was observed in stratum oriens of CA1–CA3 regions (Fig. 8D) and most prominently in the granule cell layer of the dentate gyrus (Fig.8B). These data indicate that limbic seizures induce NPY gene transcription.
DISCUSSION
Previous studies have shown a correlation between hippocampal NPY expression and seizures, suggesting a critical role for NPY in modulation of excitability (Gruber et al., 1994; Kragh et al., 1994). Given that NPY inhibits excitatory neurotransmission in normal hippocampus (Colmers et al., 1988, 1991), one possibility is that by dampening the excess excitation associated with seizures, NPY acts as an endogenous anticonvulsant agent. Our data from mutant mice lacking NPY are consistent with this hypothesis.
NPY may not be required for hippocampal function under normal conditions
Our studies provide the first electrophysiological assessment of hippocampal function in mice lacking NPY. Although exogenous NPY application modulates synaptic activity in vitro (Colmers et al., 1988, 1991; Klapstein and Colmers, 1993), no clear electrophysiological deficits were observed in our studies on acute hippocampal slices from NPY-deficient animals. Standard protocols designed to assess synaptic function (e.g., input–output curves, paired-pulse stimulation, and EPSP/IPSP sequence) failed to reveal any deficits in the Schaffer collateral to CA1 pyramidal cell and perforant path to granule cell synapses in tissue from NPY-deficient animals. A high frequency stimulation protocol, which preferentially releases neuropeptides at the synaptic terminal (Hökfelt, 1991; Vilim et al., 1996), also failed to uncover any differences between NPY-deficient and littermate wild-type mice. In addition, activation of NPY receptors by exogenous peptide application produced levels of inhibition in NPY-deficient mice similar to that seen in wild-type littermate controls. These studies indicate that under the standard recording conditions used in these experiments (and within the limits of extracellular and intracellular recording protocols), the excitatory and inhibitory postsynaptic responses in the hippocampus are not mediated by endogenous NPY release.
NPY plays an important role in regulation of hippocampal function during a condition of hyperexcitability
Increased expression of NPY, as seen with immunocytochemistry andin situ hybridization, has been demonstrated in the hippocampal formation after experimentally induced seizures initiated by electroconvulsive stimulation, electrical kindling, kainic acid, and pentylenetetrazol (Marksteiner et al., 1989; Pitkänen et al., 1989; Gruber et al., 1994; Kragh et al., 1994; Tønder et al., 1994). Sloviter observed changes in NPY expression associated with perforant path stimulation (Sloviter, 1989) and demonstrated that changes in NPY expression only occurred in cocaine-treated rats that exhibited behavioral seizure activity (Goodman and Sloviter, 1993). Moreover,Vezzani et al. (1994) have demonstrated a twofold increase in NPY release in hippocampal slices from KA-treated rats. Sadamatsu et al. (1995) observed increased levels of NPY immunoreactivity in the hippocampus of spontaneously epileptic rats, suggesting that the accumulation of NPY is not an artifact of drug administration in the kainate model. In the present study, expression of a lacZreporter gene inserted downstream from the NPY promoter in mutant mice showed a consistent seizure-induced upregulation in the hippocampus. Although it is unclear from our thick tissue sections which cell types upregulate NPY–lacZ expression (see Fig. 8), previous studies demonstrated that NPY expression in the dentate granule cells and hippocampal interneurons is increased after a limbic seizure (Sloviter, 1989; Gruber et al., 1994; Kragh et al., 1994). These observations suggest that the seizure-induced accumulation of NPY in the hippocampus is the result of transcriptional activation and may serve a self-limiting role to reduce excitatory neurotransmission in the hippocampus during a condition of hyperexcitability.
Recent work by Woldbye and colleagues provides additional evidence of a compensatory, antiepileptic role for NPY in the CNS. They showed that intracerebroventricular administration of NPY in rats reduced epileptiform-like afterdischarge activity and stimulation-induced wet dog shakes (Woldbye et al., 1996) and inhibited kainic acid-induced motor seizures (Woldbye et al., 1997). In vitro experiments indicate that NPY is also effective in reducing picrotoxin-induced epileptiform activity (Smialowska et al., 1996) and spontaneous population bursts induced by a Mg-free solution (Bijak, 1995). Because Y2 receptors are present in high density in the hippocampal formation (Dumont et al., 1996) and have been shown to mediate presynaptic glutamate release in hippocampal slices (Haas et al., 1987;Colmers et al., 1991), it is possible that the anticonvulsant action of NPY is mediated by this receptor subtype. However, Woldbye et al. (1997) found that an intracerebroventricular injection of 6 nmol of the Y1/Y4/Y5 agonist 3–36, [Leu31, Pro34]NPY, and the Y4/Y5 agonist human pancreatic polypeptide but not the Y2 selective agonist NPY 13–36 inhibited KA-induced seizures, perhaps indicating a role for the Y5 receptor subtype. Although further studies are required to determine precisely which receptor subtype is responsible for the anticonvulsant actions of NPY, these intriguing results are consistent with the hypothesis that NPY plays a critical role in modulation of limbic seizures.
We have provided additional evidence of this hypothesis by examining seizure activity in animals lacking NPY. Specifically, seizures induced by excess excitation (e.g., kainic acid) were not controlled in a “normal” manner, resulting in prolonged seizure activity and death in most NPY-deficient mice. It is possible that lack of NPY expression in peripheral nervous system structures regulating autonomic function contributed to the observed seizure-related deaths. However, because intracerebroventricular infusion of NPY was effective in preventing death in mice lacking NPY, a critical CNS mechanism is likely. It is also possible that intracerebroventricular NPY is an effective anticonvulsant in these mice because the absence of NPY during development leads to receptor compensation (indicated by an increased responsiveness to exogenous NPY). To address whether there were compensatory changes in NPY−/− mice, we examined the feeding response elicited by intracerebroventricular injection of NPY in separate studies (Palmiter et al., 1997). NPY-stimulated food intake was measured for NPY+/+ and NPY−/− mice, and the dose–response curves were identical. We also tested the response to exogenous NPY application in hippocampal slices from NPY+/+ and NPY−/− mice and found no significant difference in the magnitude of inhibition achieved (see Fig. 5). These data argue against a general increase in NPY receptor sensitivity in NPY−/− animals. Finally, it is possible that the absence of NPY during gestation results in a subtle change in physiology that makes NPY−/− mice more susceptible to the induction of status epilepticus and death. We have found no evidence of hyperexcitable changes in hippocampal physiology using standard extracellular and intracellular recording techniques in tissue from NPY−/− mice. Additional whole-cell voltage-clamp and single-channel studies will be necessary to establish clearly that physiological function is normal in these animals. Nonetheless, our studies in NPY−/− mice suggest that normal mechanisms required to modulate limbic seizures involve the release of endogenous pools of NPY at the presynaptic terminal.
NPY may be an endogenous anticonvulsant in the hippocampus
Neuronal synchronization is required for the generation and spread of epileptiform activity in the CNS. It is thought that control of synchronization and modulation of seizures can be achieved by directly influencing the firing activity of burst generator (i.e., “pacemaker”) neurons and/or by changing the efficacy of synaptic coupling (Connors and Amitai, 1993). Kainic acid-induced seizures arise by excess excitation of neurons, leading to synchronized epileptiform discharge in the dentate gyrus, followed by feed-forward excitatory propagation of epileptiform discharge through the trisynaptic hippocampal circuit (Sperk, 1994). We suggest that NPY controls seizures by modulating synaptic coupling in the hippocampal formation because (1) NPY is present in interneurons at critical sites within the trisynaptic circuit (Fig. 1; Morris, 1989; Dumont et al., 1996), (2) exogenous NPY application modulates excitatory neurotransmission in the hippocampus (Fig. 5; Haas et al., 1987; Colmers et al., 1991; Bleakman et al., 1992), and (3) seizure activity is fatal in mice lacking NPY but can be prevented by intracerebroventricular administration of the peptide (this study). Furthermore, the expression of NPY and of Y2 receptors is increased in the granule cell–mossy fiber pathway (a critical synapse in the spread of synchronized epileptiform activity through the hippocampus) in rats with experimentally induced epilepsy (Marksteiner et al., 1990; Sperk et al., 1992; Causing et al., 1996; Röder et al., 1996).
An important antiepileptic role for neuropeptide Y could lead to development of novel anticonvulsant agents. Established antiepileptic drugs (1) modulate the activity of voltage-dependent ion channels critical for control of neuronal firing (Kelly et al., 1990; Ragsdale et al., 1991), (2) potentiate the inhibitory effects of GABA at postsynaptic GABAA receptors (Sieghart, 1995), or (3) block the excitatory effects of glutamate at postsynaptic AMPA-type glutamate receptors (Macdonald and Barker, 1978). Recently developed anticonvulsant drugs such as vigabatrin, lamotrigine, and topiramate have similar mechanisms of action (Meldrum, 1996). Neuropeptide Y, presumably modulating synaptic efficacy via a presynaptic action, has now been shown to be an effective anticonvulsant in rats (Woldbye et al., 1996, 1997) and mice (see Results). Taken together, these studies suggest that rational drug design might profitably focus on developing a novel class of anticonvulsants that could reduce excitation via action at NPY receptors in the CNS.
Footnotes
This work was supported in part by a Predoctoral Merck Fellowship (J.C.E.), National Institutes of Health Grants NS-07144 (S.C.B.) and NS-18895 (P.A.S.), and the Howard Hughes Medical Institute (R.D.P.). We express our gratitude to Carol R. Robbins and Dr. Jong Rho for assistance with the video-EEG system.
Correspondence should be addressed to Dr. Scott C. Baraban, Department of Pediatrics, MS 6003, Case Western Reserve University, 11100 Euclid Avenue, Cleveland, OH 44106-6003.
REFERENCES
- 1.Alger BE, Nicoll RA. Feedforward dendritic inhibition in rat hippocampal pyramidal cells studied in vitro. J Physiol (Lond) 1982;328:105–123. doi: 10.1113/jphysiol.1982.sp014255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen YS, Adrian TE, Allen JM, Tatemoto K, Crow TJ, Bloom SR, Polak JM. Neuropeptide Y distribution in the rat brain. Science. 1983;221:877–879. doi: 10.1126/science.6136091. [DOI] [PubMed] [Google Scholar]
- 3.Andersen P, Bliss TVP, Lømo T, Olsen LI, Skrede KK. Lamellar organization of hippocampal excitatory pathways. Acta Physiol Scand. 1969;76:4A–5A. doi: 10.1111/j.1748-1716.1969.tb04499.x. [DOI] [PubMed] [Google Scholar]
- 4.Austin KB, Bronzino JD, Morgane PJ. Paired-pulse facilitation and inhibition in the dentate gyrus is dependent on behavioral state. Exp Brain Res. 1989;77:594–604. doi: 10.1007/BF00249612. [DOI] [PubMed] [Google Scholar]
- 5.Baraban SC, Schwartzkroin PA. Effects of prenatal cocaine exposure on the developing hippocampus: intrinsic and synaptic physiology. J Neurophysiol. 1997;77:126–136. doi: 10.1152/jn.1997.77.1.126. [DOI] [PubMed] [Google Scholar]
- 6.Bijak M. Inhibitory effect of neuropeptide Y on epileptiform activity in the frontal cortex and hippocampus in vitro. Pol J Pharmacol. 1995;47:461–463. [PubMed] [Google Scholar]
- 7.Biscoe TJ, Duschen MR. An intracellular study of dentate, CA1 and CA3 neurones in the mouse hippocampal slice. Q J Exp Physiol. 1985;70:189–202. doi: 10.1113/expphysiol.1985.sp002902. [DOI] [PubMed] [Google Scholar]
- 8.Bleakman D, Harrison NL, Colmers WF, Miller RJ. Investigations into neuropeptide Y-mediated presynaptic inhibition in cultured hippocampal neurones of the rat. Br J Pharmacol. 1992;107:334–340. doi: 10.1111/j.1476-5381.1992.tb12747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Causing CG, Makus KD, Ma Y, Miller FD, Colmers WF. Selective upregulation of T alpha 1 alpha-tubulin and neuropeptide Y mRNAs after intermittent excitatory stimulation in adult rat hippocampus in vivo. J Comp Neurol. 1996;367:132–146. doi: 10.1002/cne.903670102. [DOI] [PubMed] [Google Scholar]
- 10.Chronwall BM, DiMaggio DA, Massari VJ, Pickel VM, Ruggiero DA, O’Donohue TL. The anatomy of neuropeptide-Y-containing neurons in rat brain. Neuroscience. 1985;15:1159–1181. doi: 10.1016/0306-4522(85)90260-x. [DOI] [PubMed] [Google Scholar]
- 11.Colmers WF, Lukowiak K, Pittman QJ. Presynaptic action of neuropeptide Y in area CA1 of the rat hippocampal slice. J Physiol (Lond) 1987;383:285–299. doi: 10.1113/jphysiol.1987.sp016409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colmers WF, Lukowiak K, Pittman QJ. Neuropeptide Y action in the rat hippocampal slice: site and mechanism of presynaptic inhibition. J Neurosci. 1988;8:3827–3837. doi: 10.1523/JNEUROSCI.08-10-03827.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colmers WF, Klapstein GJ, Fournier A, St-Pierre S, Treherne KA. Presynaptic inhibition by neuropeptide Y in rat hippocampal slice in vitro is mediated by a Y2 receptor. Br J Pharmacol. 1991;102:41–44. doi: 10.1111/j.1476-5381.1991.tb12129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Connors BW, Amitai Y. Generation of epileptiform discharge by local circuits of neocortex. In: Schwartzkroin PA, editor. Epilepsy. Models, mechanisms, and concepts. Cambridge UP; Cambridge, U.K.: 1993. pp. 388–423. [Google Scholar]
- 15.Dumont Y, Fournier A, St-Pierre S, Quirion R. Autoradiographic distribution of [125I]Leu31, Pro34-PYY and [125I]PYY3–36 binding sites in the rat brain evaluated with two newly developed Y1 and Y2 receptor radioligands. Synapse. 1996;22:139–158. doi: 10.1002/(SICI)1098-2396(199602)22:2<139::AID-SYN7>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 16.Erickson JC, Clegg KE, Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature. 1996;381:415–418. doi: 10.1038/381415a0. [DOI] [PubMed] [Google Scholar]
- 17.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 18.Goodman JH, Sloviter RS. Cocaine neurotoxicity and altered neuropeptide Y immunoreactivity in the rat hippocampus; a silver degeneration and immunocytochemical study. Brain Res. 1993;616:263–272. doi: 10.1016/0006-8993(93)90217-b. [DOI] [PubMed] [Google Scholar]
- 19.Gruber B, Greber S, Rupp E, Sperk G. Differential NPY mRNA expression in granule cells and interneurons of the rat dentate gyrus after kainic acid injection. Hippocampus. 1994;4:474–482. doi: 10.1002/hipo.450040409. [DOI] [PubMed] [Google Scholar]
- 20.Haas HL, Hermann A, Greene RW, Chan-Palay V. Action and location of neuropeptide tyrosine (Y) on hippocampal neurons of the rat in slice preparations. J Comp Neurol. 1987;257:208–215. doi: 10.1002/cne.902570207. [DOI] [PubMed] [Google Scholar]
- 21.Hökfelt T. Neuropeptides in perspective: the last ten years. Neuron. 1991;7:867–879. doi: 10.1016/0896-6273(91)90333-u. [DOI] [PubMed] [Google Scholar]
- 22.Kelly KM, Gross RA, Macdonald RL. Valproic acid selectively reduces the low-threshold (T) calcium current in rat nodose neurons. Neurosci Lett. 1990;116:233–238. doi: 10.1016/0304-3940(90)90416-7. [DOI] [PubMed] [Google Scholar]
- 23.King GL, Dingledine R, Giacchino JL, McNamara JO. Abnormal neuronal excitability in hippocampal slices from kindled rats. J Neurophysiol. 1985;54:1295–1304. doi: 10.1152/jn.1985.54.5.1295. [DOI] [PubMed] [Google Scholar]
- 24.Klapstein GJ, Colmers WF. On the sites of presynaptic inhibition by neuropeptide Y in rat hippocampus in vitro. Hippocampus. 1993;3:103–112. doi: 10.1002/hipo.450030111. [DOI] [PubMed] [Google Scholar]
- 25.Kragh J, Tønder N, Finsen BR, Zimmer J, Bolwig TG. Repeated electroconvulsive shocks cause transient changes in rat hippocampal somatostatin and neuropeptide Y immunoreactivity and mRNA in situ hybridization signals. Exp Brain Res. 1994;98:305–313. doi: 10.1007/BF00228418. [DOI] [PubMed] [Google Scholar]
- 26.Larsen PJ, Sheikh SP, Jakobsen CR, Schwartz TW, Mikkelsen JD. Regional distribution of putative NPY Y1 receptors and neurons expressing Y1 mRNA in forebrain areas of the rat central nervous system. Eur J Neurosci. 1993;5:1622–1637. doi: 10.1111/j.1460-9568.1993.tb00231.x. [DOI] [PubMed] [Google Scholar]
- 27.Macdonald RL, Barker JL. Different actions of anticonvulsant anesthetic barbiturates revealed by use of cultured mammalian neurons. Science. 1978;200:775–777. doi: 10.1126/science.205953. [DOI] [PubMed] [Google Scholar]
- 28.Marksteiner J, Sperk G, Maas D. Differential increases in brain levels of neuropeptide Y and vasoactive intestinal polypeptide after kainic acid-induced seizures in the rat. Naunyn Schmiedebergs Arch Pharmacol. 1989;339:173–177. doi: 10.1007/BF00165140. [DOI] [PubMed] [Google Scholar]
- 29.Marksteiner J, Ortler M, Bellman R, Sperk G. Neuropeptide Y biosynthesis is markedly induced in mossy fibers during temporal lobe epilepsy of the rat. Neurosci Lett. 1990;112:143–148. doi: 10.1016/0304-3940(90)90193-d. [DOI] [PubMed] [Google Scholar]
- 30.Martel J-C, Fournier A, St Pierre S, Quirion R. Quantitative autoradiographic distribution of [125I]Bolton–Hunter neuropeptide Y receptor binding sites in rat brain. Comparison with [125I]peptide YY receptor sites. Neuroscience. 1990;36:255–283. doi: 10.1016/0306-4522(90)90367-d. [DOI] [PubMed] [Google Scholar]
- 31.Mayanagi Y, Watanabe E, Kaneko Y (1996) Mesial temporal lobe epilepsy: clinical features and seizure mechanism. Epilepsia 37[Suppl 3]:57–60. [DOI] [PubMed]
- 32.Meldrum BS (1996) Update on the mechanism of action of antiepileptic drugs. Epilepsia 37[Suppl 6]:4–11. [DOI] [PubMed]
- 33.Mercer EH, Hoyle GW, Kapur RP, Brinster RL, Palmiter RD. The dopamine β-hydroxylase gene promoter directs expression of E. coli lacZ to sympathetic and other neurons in adult transgenic mice. Neuron. 1991;7:703–716. doi: 10.1016/0896-6273(91)90274-4. [DOI] [PubMed] [Google Scholar]
- 34.Morris BJ. Neuronal localization of neuropeptide Y gene expression in rat brain. J Comp Neurol. 1989;290:358–368. doi: 10.1002/cne.902900305. [DOI] [PubMed] [Google Scholar]
- 35.Newberry NR, Nicoll RA. A bicuculline-resistant inhibitory post-synaptic potential in rat hippocampal pyramidal cells in vitro. J Physiol (Lond) 1984;348:239–254. doi: 10.1113/jphysiol.1984.sp015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palmiter RD, Erickson JC, Hollopeter G, Baraban SC, Schwartz MW (1997) Life without neuropeptide Y. Recent Prog Horm Res, in press. [PubMed]
- 37.Pitkänen A, Beal MF, Sirviö J, Swartz KJ, Männistö PT, Riekkinen PJ. Somatostatin, neuropeptide Y, GABA and cholinergic enzymes in brain of pentylenetetrazol-kindled rats. Neuropeptides. 1989;14:197–207. doi: 10.1016/0143-4179(89)90045-0. [DOI] [PubMed] [Google Scholar]
- 38.Ragsdale DS, Scheuer T, Catterall WA. Frequency and voltage-dependent inhibition of type IIA Na+ channels, expressed in a mammalian cell line, by local anesthetic, antiarrhythmic, and anticonvulsant drugs. Mol Pharmacol. 1991;40:756–765. [PubMed] [Google Scholar]
- 39.Röder C, Schwarzer C, Vezzani A, Gobbi M, Mennini T, Sperk G. Autoradiographic analysis of neuropeptide Y receptor binding sites in the rat hippocampus after kainic acid-induced limbic seizure. Neuroscience. 1996;70:47–55. doi: 10.1016/0306-4522(95)00332-d. [DOI] [PubMed] [Google Scholar]
- 40.Sadamatsu M, Kanai H, Masui A, Serikawa T, Yamada J, Sasa M, Kato N. Altered brain contents of neuropeptides in spontaneously epileptic rats (SER) and tremor rats with absence seizures. Life Sci. 1995;57:523–531. doi: 10.1016/0024-3205(95)00302-m. [DOI] [PubMed] [Google Scholar]
- 41.Schwartzkroin PA. Role of the hippocampus in epilepsy. Hippocampus. 1994;4:239–242. doi: 10.1002/hipo.450040302. [DOI] [PubMed] [Google Scholar]
- 42.Sieghart W. Structure and pharmacology of γ-aminobutyric acidA receptor subtypes. Pharmacol Rev. 1995;47:181–234. [PubMed] [Google Scholar]
- 43.Sloviter RS. Chemically defined hippocampal interneurons and their possible relationship to seizure mechanisms. In: Chan-Palay V, Kohler C, editors. The hippocampus–new vistas. Liss; New York: 1989. pp. 443–461. [Google Scholar]
- 44.Smialowska M, Bijak M, Sopala M, Tokarski K. Inhibitory effect of NPY on the picrotoxin-induced activity in the hippocampus: a behavioral and electrophysiological study. Neuropeptides. 1996;30:7–12. doi: 10.1016/s0143-4179(96)90048-7. [DOI] [PubMed] [Google Scholar]
- 45.Sperk G. Kainic acid seizures in the rat. Prog Neurobiol. 1994;42:1–32. doi: 10.1016/0301-0082(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 46.Sperk G, Marksteiner J, Gruber B, Bellmann R, Mahata M, Ortler M. Functional changes in neuropeptide Y- and somatostatin-containing neurons induced by limbic seizures in the rat. Neuroscience. 1992;50:831–846. doi: 10.1016/0306-4522(92)90207-i. [DOI] [PubMed] [Google Scholar]
- 47.Swanson TH. The pathophysiology of human mesial temporal lobe epilepsy. J Clin Neurophysiol. 1995;12:2–22. [PubMed] [Google Scholar]
- 48.Tatemoto K. Neuropeptide Y: complete amino acid sequence of the brain peptide. Proc Natl Acad Sci USA. 1982;79:5485–5489. doi: 10.1073/pnas.79.18.5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tønder N, Kragh J, Finsen BR, Bolwig TG, Zimmer J. Kindling induces transient changes in neuronal expression of somatostatin, neuropeptide Y, and calbindin in adult rat hippocampus and fascia dentata. Epilepsia. 1994;35:1299–1308. doi: 10.1111/j.1528-1157.1994.tb01802.x. [DOI] [PubMed] [Google Scholar]
- 50.Vezzani A, Civenni G, Rizzi M, Monno A, Messali S, Samanin R. Enhanced neuropeptide Y release in the hippocampus is associated with chronic seizure susceptibility in kainic acid treated rats. Brain Res. 1994;660:138–143. doi: 10.1016/0006-8993(94)90847-8. [DOI] [PubMed] [Google Scholar]
- 51.Vilim FS, Cropper EC, Price DA, Kupfermann I, Weiss KR. Release of peptide cotransmitters in Aplysia: regulation and functional implications. J Neurosci. 1996;16:8105–8114. doi: 10.1523/JNEUROSCI.16-24-08105.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Woldbye DPD, Madsen TM, Larsen PJ, Mikkelsen JD, Bolwig TG. Neuropeptide Y inhibits hippocampal seizures and wet dog shakes. Brain Res. 1996;737:162–168. doi: 10.1016/0006-8993(96)00730-5. [DOI] [PubMed] [Google Scholar]
- 53.Woldbye DPD, Larsen PJ, Mikkelsen JD, Klemp K, Madsen TM, Bolwig TG. Powerful inhibition of kainic acid seizures by neuropeptide Y via Y5-like receptors. Nat Med. 1997;3:761–764. doi: 10.1038/nm0797-761. [DOI] [PubMed] [Google Scholar]
- 54.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]