

Abstract
We examined the cellular localization of nine different connexin32 (Cx32) mutants associated with X-linked Charcot–Marie–Tooth disease (CMTX) in communication-incompetent mammalian cells. Cx32 mRNA was made, but little or no protein was detected in one class of mutants. In another class of mutants, Cx32 protein was detectable in the cytoplasm and at the cell surface, where it appeared as plaques and punctate staining. Cx32 immunoreactivity in a third class of mutants was restricted to the cytoplasm, where it often colocalized with the Golgi apparatus. Our studies suggest that CMTX mutations have a predominant effect on the trafficking of Cx32 protein, resulting in a potentially toxic cytoplasmic accumulation of Cx32 in these cells. These results and evidence of cytoplasmic accumulation of other mutated myelin proteins suggest that diseases affecting myelinating cells may share a common pathophysiology.
Keywords: Cx32, gap junctions, X-linked Charcot–Marie–Tooth disease, protein trafficking, Golgi apparatus, myelin, Schwann cells, neuropathy
X-linked Charcot–Marie–Tooth disease (CMTX) is a form of hereditary motor and sensory neuropathy with clinical features that include progressive weakness and atrophy of distal limb muscles, loss of reflexes, sensory loss, and reduced nerve conduction velocity. In males affected with CMTX, the first clinical signs of the disease arise in late childhood or adolescence and lead to moderate disability by the third decade of life, whereas heterozygous women may be more mildly affected or asymptomatic (for review, seeDeschênes et al., 1996).
CMTX is caused by mutations in the gene for connexin32 (Cx32; also known as gap junction gene β1), a member of the family of gap junction proteins. Gap junctions are clusters of intercellular channels in plasma membranes that allow the passage of ions and small molecules between closely apposed cells (for review, see Bruzzone et al., 1996;Goodenough et al., 1996). Cx32 is expressed in many cell types (Bruzzone et al., 1996), including myelinating Schwann cells of peripheral nerve, where it localizes to noncompact myelin at the paranodal region and Schmidt–Lanterman incisures (Bergoffen et al., 1993; Scherer et al., 1995). In these structures, Cx32 is thought to form reflexive gap junctions between layers of the Schwann cell membrane (Bergoffen et al., 1993). The restriction of the CMTX phenotype to the peripheral nervous system implies that myelinating Schwann cells are particularly susceptible to Cx32 mutations.
Myelinating cells are also the targets of mutations in other myelin genes (Suter and Snipes, 1995; Scherer, 1997). For example, mutations in peripheral myelin protein 22 kDa (PMP-22) and myelin protein zero (P0) directly affect Schwann cells and are associated with inherited demyelinating neuropathies, such as CMT types 1A and 1B, hereditary neuropathy with liability to pressure palsies, and Dejerine–Sottas disease. Missense mutations in proteolipid protein (PLP) may kill oligodendrocytes, the myelin-producing cells of the CNS, and result in Pelizaeus–Merzbacher disease (PMD) (Nave and Boespflug-Tanguy, 1996). It is not yet known whether myelin gene mutants adversely affect myelinating Schwann cells via the same mechanisms.
To date, >90 different mutations in >120 CMTX families have been described (Scherer et al., 1997); their distribution throughout the coding region of Cx32 suggests that every domain of the protein is important for its normal function in peripheral nerve. Studies inXenopus oocytes (Bruzzone et al., 1994; Rabadan-Diehl et al., 1994) and in HeLa cells (Omori et al., 1996) have shown that some Cx32 mutants associated with CMTX are nonfunctional and may also exert dominant-negative effects on other connexins coexpressed in the same cell, whereas other mutants remain functional in these systems. Here, we report the effects of nine CMTX mutations on the trafficking and cellular localization of Cx32 stably expressed in a subclone of rat pheochromocytoma (PC12J) cells, a communication-incompetent mammalian cell line. We have defined three distinct effects of these CMTX mutations on Cx32 protein trafficking, suggesting that different defects in membrane protein transport in myelinating Schwann cells may have similar consequences.
MATERIALS AND METHODS
Cx32 mutations. The following abbreviations are used for Cx32 mutations studied in this paper: Gly12Ser (G12S), Arg15Gln (R15Q), Val63Ile (V63I), Val139Met (V139M), Arg142Trp (R142W), 175 frameshift (175fs), Glu186Lys (E186K), Glu208Lys (E208K), and Arg220stop codon (R220Stop). The CMTX families bearing these mutations have been described elsewhere [V139M (Heimler et al., 1978); G12S (Fryns and Van den Berghe, 1980); 175fs (Rozear et al., 1987); R142W and E186K (Bergoffen et al., 1993); and R15Q, V63I, E208K, and R220Stop (Fairweather et al., 1994)]. Table 1summarizes the phenotypes of these families; the severity of the clinical phenotype was based on a review of medical records and the published literature. Fifty percent of females at risk for inheriting the disease allele (symptomatic at-risk females, column 3) are expected to be symptomatic in families in which CMTX is dominant. The family with the 175fs mutation has the most severe phenotype, with affected men losing ambulation and many heterozygous women having symptoms nearly as severe as those of the men. Families with the G12S, R142W, E186K, and E208K mutations have moderate to severe symptoms, with reduced nerve conduction velocities in both males and heterozygous females. In families with the R15Q, V63I, V139M, and R220Stop mutations, the phenotype is mild to moderate in both men and women.
Table 1.
Comparison of CMTX phenotype with intracellular localization of Cx32
| Mutation | NCV (m/sec)1-a | Symptomatic at-risk female heterozygotes | Phenotype1-b | Cellular localization of Cx32 |
|---|---|---|---|---|
| G12S | Moderate (F) to marked (M) reductions | 1 /10 | Severe | Cytoplasm |
| R15Q | 20 (M); 30 (F) | 0 /3 | Moderate | Plasma membrane |
| V63I | 41 (M) | 2 /7 | Mild | Plasma membrane and cytoplasm |
| V139M | 28–32 (M) | 9 /15 | Moderate | Plasma membrane and cytoplasm |
| R142W | 27–35 (M) | 2 /5 | Moderate to severe | Cytoplasm |
| 175fs | 26–39 (M); 37–64 (F) | 9 /29 | Severe | No detectable protein |
| E186K | 39–40 (M); 35–48 (F) | 1 /12 | Moderate | Cytoplasm |
| E208K | Not known | 1 /3 | Moderate to severe | Cytoplasm |
| R220Stop | 48–50 (M) | 2 /2 | Moderate | Plasma membrane and cytoplasm |
Moderate indicates significant weakness and atrophy of distal muscles in all limbs, high stepping gait, and early onset, but not a serious impediment to a normal lifestyle in men and high-arched feet and mild weakness and atrophy in distal legs in women.Severe indicates marked weakness and atrophy of distal muscles in all limbs, sensory loss, and significant impediments to mobility that necessitate canes and wheelchairs in men and high-arched feet and sensory disturbances in women.
NCV (nerve conduction velocity) is based on studies in one or more affected individuals [M (male); F (female)]; normal NCV is >50 m/sec (Nicholson and Nash, 1993).
Mild indicates weakness and atrophy of distal muscles in all limbs but no difficulty walking in men and thin ankles and high-arched feet in women.
Cx32 mutants 175fs, R142W, and E186K were cloned previously byBruzzone et al. (1994). The coding regions were excised from theBglII site of pSP64T (Bruzzone et al., 1994) and cloned into the BamHI site of pREP9 (Invitrogen, San Diego, CA). The coding regions of wild-type Cx32 and CMTX mutants G12S, R15Q, V63I, V139M, E208K, and R220Stop were amplified from the genomic DNA of an unaffected man and male CMTX patients, respectively, by PCR (one cycle of 94°C for 10 min; 30 cycles of 94°C for 1 min, 59°C for 1 min, and 72°C for 50 sec) with Vent polymerase (New England Biolabs, Beverly, MA) in a PTC-100 thermal controller (MJ Research, Watertown, MA). The reaction buffer included the following reagents at their final concentrations: 400 μm dNTPs, 1 μm primers, 20% Luria–Bertani medium (Cease et al., 1994), 10% DMSO, and 0.5 U of Vent polymerase. The two pairs of PCR primers with restriction sites that were used to amplify a segment containing the translational start site and stop codon are listed as follows: for mutants G12S and V139M, sense Cx321.Bam 5′-CGC GGATCCGGCAGG ATGAACTGGACAGGT-3′ and antisense Cx32A1.Eco 5′-CCG GAATTCATGGCAGGTTGCCTGGTATG-3′, and for all other mutants, sense Cx32F.Kpn 5′-CGG GGTACCGGCAGG ATGAACTGGACAGG-3′ and antisense Cx32R.Bam 5′-GGC GGATCCATGGCAGGTTGCCTGGTATG-3′. The PCR products were electrophoresed on agarose gels, excised, purified by Geneclean (BIO 101, Vista, CA), and digested with BamHI andEcoRI (G12S and V139M) or KpnI andBamHI (all other mutants). The G12S and V139M inserts were ligated into the BamHI and EcoRI sites of pBS S/K and later excised from pBS S/K with NotI and XhoI for cloning into the same sites in pREP9. All other mutant inserts were ligated into the KpnI and BamHI sites of pREP9. The constructs were sequenced on both strands at least twice by automated sequencing (University of Pennsylvania Department of Genetics DNA Sequencing Facility) to verify the sequence.
Cell culture and stable transfection. The subclone of rat pheochromocytoma cells (PC12J; a gift of Dr. David Spray, Albert Einstein College of Medicine, Bronx, NY) that was used in this study expresses no endogenous Cx32 protein or mRNA (see Fig.2A,C,E) and very low amounts of Cx37 protein (data not shown). These cells are not electrically coupled, as determined by dual whole-cell patch-clamp analysis (D. Spray, personal communication). The cells were cultured in RPMI 1640 supplemented with 10% horse serum, 5% fetal bovine serum, and penicillin and streptomycin in a 37°C, 5% CO2 incubator. PC12J cells were stably transfected with 5–35 μg of plasmid DNA and Lipofectin reagent (Life Technologies, Gaithersburg, MD) according to the manufacturer’s protocol. For all transfections but those with G12S and V139M mutants, 35–55 independent clones were picked and expanded 2–3 weeks after selection in 400 μg/ml G418 (Life Technologies). For the G12S and V139M mutants, transfected cells were maintained in selective medium rather than isolating clones by limiting dilution. To test the effect of reduced growth temperatures on the expression of Cx32 protein, we also grew a subset of clones at 23–25°C on a bench top or in a 5% CO2 incubator.
Fig. 2.

Analysis of Cx32 expression in PC12J cells stably transfected with normal and mutated Cx32 cDNAs. A, As negative controls for B, immunoblots of the parental cells (P; lane 1) and two clones transfected with the vector alone (V; lanes 2–3, clones pREP9.6.1 and pREP9.6.4) were hybridized with a polyclonal antiserum (Lola) that recognizes an epitope in the cytoplasmic loop of Cx32. The bands seen across alllanes in A and B, including a high molecular weight band (asterisk), are proteins nonspecifically detected by Lola. B, Immunoblots of three clones transfected with 175fs(lanes 1–3, clones 5117.6.1, 5117.4.9, and 5117.4.3) were hybridized with Lola. C, Immunoblots of three clones expressing various amounts of R220Stop protein (lanes 1–3), the parental cells (P;lane 4), and two clones transfected with the vector alone (V; lanes 5–6) were performed with a mouse monoclonal antibody against the cytoplasmic loop (M12.13). An arrow indicates truncated Cx32. The two bands seen across all lanes (asterisks) are proteins nonspecifically detected by the M12.13 antibody.D, Immunoblots of rat liver (+;lane 1) and representative clones transfected with wild-type Cx32 (WT; lane 2, clone 517.4.7), R15Q (lanes 3–4, clones 15.23 and 15.57), V63I (lanes 5–6, clones 63.36 and 63.55), R142W (lanes 7–8, clones 116.41 and 116.38), E186K (lanes 9–10, clones 412.5 and 414.10), and E208K(lanes 11–12, clones 208.47 and 208.49) were performed with a mouse monoclonal antibody that also recognizes the C terminal (7C6.C7). Dashes indicate the positions of molecular weight markers of 46 and 30 kDa, respectively. E, Northern blots of four clones transfected with 175fs(lanes 1–4, 5117.2.3, 5117.4.9, 5117.2.10, and 5117.4.3), one clone expressing wild-type Cx32 (WT;lane 5, 517.4.7), and the parental cells (P; lane 6) were performed using a full-length, wild-type human Cx32 cDNA probe. The Cx32 transcripts are of the expected size (∼1.3 kb). The same blots were stripped and rehybridized with a full-length rat GAPDH cDNA probe to assess the relative quantity of RNA in each lane. Thedashes indicate the positions of 18S rRNA (∼2.4 kb). For immunoblots, the single and double arrowheads represent the positions of the monomeric and dimeric forms of rat liver Cx32 on the same gels. The migration of rat liver Cx32 is slower because the homogenate was prepared in a different lysis buffer relative to that used for the cell lysates (see Materials and Methods).
Northern blotting. RNA was isolated from confluent cultures of cells growing in 10 cm dishes using RNAzol B reagent (Tel-Test, Friends-wood, TX) according to the manufacturer’s directions. Ten microgram samples of total RNA were electrophoresed in 1% agarose gels containing 6.6% (v/v) formaldehyde and transferred and cross-linked as described previously (Scherer et al., 1995). Blots were prehybridized, hybridized, and washed using standard techniques (Sambrook, 1989), with a final wash in 2× SSC at 65°C. A full-length human Cx32 cDNA probe was prepared by isolating the 893 bp insert from the wild-type Cx32 construct described above. A full-length cDNA probe of rat glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Fort et al., 1985) was similarly prepared by restriction endonuclease digestion. Both inserts were electrophoresed on agarose gels, excised, purified by Geneclean (BIO 101), and labeled with 32P by random primer extension using the Rediprime kit (Amersham, Arlington Heights, IL).
Immunoblotting. Cells were grown to confluence in 100 mm plates and harvested in cold Dulbecco’s PBS lacking calcium and magnesium ions (Life Technologies). The cell pellet was either stored at −80°C or lysed directly in ice-cold 50 mm Tris, pH 7.0, 1% SDS, and 0.017 mg/ml phenylmethylsulfonyl fluoride (Sigma, St. Louis, MO), followed by a brief sonication on ice with a sonic dismembrator (Fisher Scientific, Pittsburgh, PA). Rat liver homogenate was prepared by pulverizing snap-frozen adult rat liver with a steel mortar and pestle on dry ice, homogenizing in 20 mm NaOH, and sonicating. Total protein concentrations were quantitated with a modified Lowry assay (DC Protein Assay Kit; Bio-Rad, Hercules, CA). One hundred microgram samples of protein lysate for each clone and 1 μl of the rat liver homogenate were loaded onto 12% SDS–polyacrylamide gels after a 5–15 min incubation in sample buffer at room temperature (RT), electrophoresed, and transferred to Immobilon–polyvinylidene fluoride membrane (Millipore, Bedford, MA) using a semidry blotting unit (Fisher) according to Millipore’s recommendations. The blots were blocked (5% powdered skim milk and 0.5% Tween-20 in Tris-buffered saline) 1 hr at RT and incubated overnight at 4°C in a polyclonal antiserum diluted 1:5000 (donated by David Paul, Harvard Medical School, Cambridge, MA) (Paul, 1986), monoclonal antibody M12.13 diluted 1:10 [supernatant donated by David Paul (Goodenough et al., 1988)], or monoclonal antibody 7C6.C7 (undiluted supernatant or ascites-diluted 1:1000; donated by Elliot Hertzberg, Albert Einstein College of Medicine, Bronx, NY) (Nagy et al., 1996). After washes in blocking solution, the blots were incubated in appropriate dilutions of peroxidase-coupled secondary antibodies against mouse or rabbit IgG (Jackson ImmunoResearch, West Grove, PA) for 1 hr at RT. After washes in blocking solution and Tris-buffered saline containing 0.5% Tween-20, blots were visualized by enhanced chemiluminescence (Amersham) according to the manufacturer’s protocols.
Immunocytochemistry. Eight-well plastic chamber slides (Lab-Tek Permanox-coated slides; Nunc, Naperville, IL) were coated with 10 μg/ml poly-l-lysine (Sigma), air dried, and seeded with clones expressing various Cx32 mutants. In some experiments, when cells reached 75% confluence, the medium was aspirated and replaced with medium containing 15 μg/ml brefeldin A (BFA, dissolved in 100% ethanol; Sigma) for 1 hr at 37°C in a 5% CO2 incubator. Control slides were incubated with medium containing the equivalent volume of ethanol. Cells were fixed in 95% ethanol/5% glacial acetic acid at −20°C for 10 min. After washes in PBS lacking calcium and magnesium ions, the slides were blocked in 5% bovine serum albumin, 10% fetal bovine serum, 0.1% Triton X-100, 0.1 mNaH2PO4, 0.1 mNa2HPO4, and 0.01% sodium azide at 37°C for 30–60 min. Incubations in the following antibodies were done for 24–72 hr at 4°C in a humidified chamber: M12.13 (undiluted supernatant), 7C6.C7 (undiluted supernatant or ascites-diluted 1:250), polyclonal antiserum B1J (diluted 1:100; donated by Norton Gilula, Scripps Research Institute, La Jolla, CA) (Milks et al., 1988), monoclonal antibody 10A8 (the supernatant was used either undiluted or diluted 1:150; donated by Nicholas Gonatas, University of Pennsylvania, Philadelphia, PA) (Gonatas et al., 1989), and a polyclonal antibody against rat liver Golgi α-mannosidase II (diluted 1:500; donated by Marilyn Farquhar, University of California, San Diego, CA) (Velasco et al., 1993). After washes in PBS, slides were incubated with 1:100 dilutions of rhodamine- or fluorescein-conjugated donkey anti-mouse or -rabbit IgG antibodies (Jackson ImmunoResearch). Slides were mounted in Vectashield (Vector Laboratories, Burlingame, CA) and visualized with a scanning laser confocal microscope (Leica, Nussloch, Germany), using ScanWare 5.10 software to generate ∼1-μm-thick optical sections.
RESULTS
Analysis of mutant Cx32 expression
The CMTX mutations shown in Figure 1are a subset of >90 different mutations that are distributed throughout the Cx32 protein (Scherer et al., 1997). They were chosen for this study because they occur in different domains of Cx32 and might be expected to have distinct effects on its localization and function. PC12J cells were stably transfected with each construct, and 41–80 clones were picked for each mutant. Clones were screened for Cx32 expression by immunoblotting and/or immunocytochemistry. Cx32 protein was not detected in parental cells (Fig.2A, lane 1) or in cells transfected with the vector alone (Fig.2A, lanes 2–3). Relative to other mutants and wild-type Cx32, few clones expressed detectable protein for mutants R142W and E186K (Table 2). The 175fs mutant is predicted to produce a truncated protein of 27 kDa, but no protein was detectable on immunoblots of 56 independent clones; three of these clones are shown in Figure 2B. On the other hand, cells transfected with R220Stop expressed a truncated protein at the expected size of 25 kDa (Fig. 2C, lanes 1–3) that was absent in parental cells and untransfected cells (Fig.2C, lanes 4–6). In clones expressing the other mutants, Cx32 migrated at the same position on denaturing polyacrylamide gels (Fig. 2D, lanes 3–12) as did Cx32 isolated from rat liver and cells transfected with wild-type Cx32 (Fig. 2D, lanes 1–2, respectively; V139M not shown). Cells transfected with G12S had no detectable Cx32 (data not shown). Northern blot analysis was performed to determine whether the absent or low-level Cx32 expression in G12S, R142W, and 175fs clones was because of mutational effects at the mRNA or protein levels. Transcripts of the appropriate size and amount relative to wild-type Cx32 transcripts (Fig. 2E,lane 5) were detected in four 175fs clones (Fig.2E, lanes 1–4) and two R142W clones (including 116.38; data not shown) but not in parental cells (Fig. 2E, lane 6) or cells transfected with G12S (data not shown). Immunocytochemical analysis of the four 175fs clones with an antibody that recognizes the cytoplasmic loop of Cx32 further confirmed the lack of Cx32 protein expression (data not shown). Except for the 175fs and G12S mutants, these results demonstrated that mutant Cx32 protein was expressed and migrated at the expected molecular weight.
Fig. 1.

Diagram of Cx32, with the mutations studied in this paper indicated by arrows. Asterisks indicate conserved cysteine residues.
Table 2.
List of clones picked under selective pressure after stable transfection with mutants R15Q, V63I, R142W, 175fs, E186K, E208K, and R220Stop
| Construct | # Clones picked | Surviving clones (%) | Expressors2-a |
|---|---|---|---|
| pREP9 | 41 | 33 (80) | 0 |
| Wild-type | 54 | 41 (76) | 16 |
| R15Q | 68 | 41 (60) | 20 |
| V63I | 72 | 38 (53) | 23 |
| R142W | 80 | 51 (64) | 6 (2 low-moderate)b,c |
| 175fs | 62 | 56 (90) | 0 |
| E186K | 78 | 54 (69) | 9 (4 low-moderate)2-b |
| E208K | 72 | 45 (62) | 26 |
| R220Stop | 52 | 41 (78) | 16 |
Assessment of expression level was based on signal intensity in immunocytochemical analyses and/or on band intensity on immunoblots, relative to that seen with the wild-type clone 517.4.7. As determined through immunocytochemistry, Cx32 expression was heterogeneous in all clones; not all cells in a given population expressed Cx32 protein, even though they were maintained under selective pressure.
Relative to other constructs, few clones expressing these Cx32 mutants were isolated, and the Cx32 expression level in these clones was generally lower and more heterogeneous than the level in clones expressing wild-type or mutant Cx32.
The extent of Cx32 expression in this clone was especially low, accounting for the weak Cx32 bands on immunoblots (see Figure 2).
Cellular localization
We examined the localization of wild-type and mutant Cx32 by indirect immunofluorescence using scanning laser confocal microscopy. Cells expressing wild-type Cx32 had plaques and punctate staining, especially in regions of cell–cell contact, as well as punctate cytoplasmic staining (Fig.3B). Clones expressing R15Q had a staining pattern that was indistinguishable from that of cells expressing wild-type Cx32 (Fig. 3C). The localization of Cx32 in the plasma membrane of clones expressing either wild-type Cx32 or R15Q was independent of the level of protein expression (data not shown).
Fig. 3.
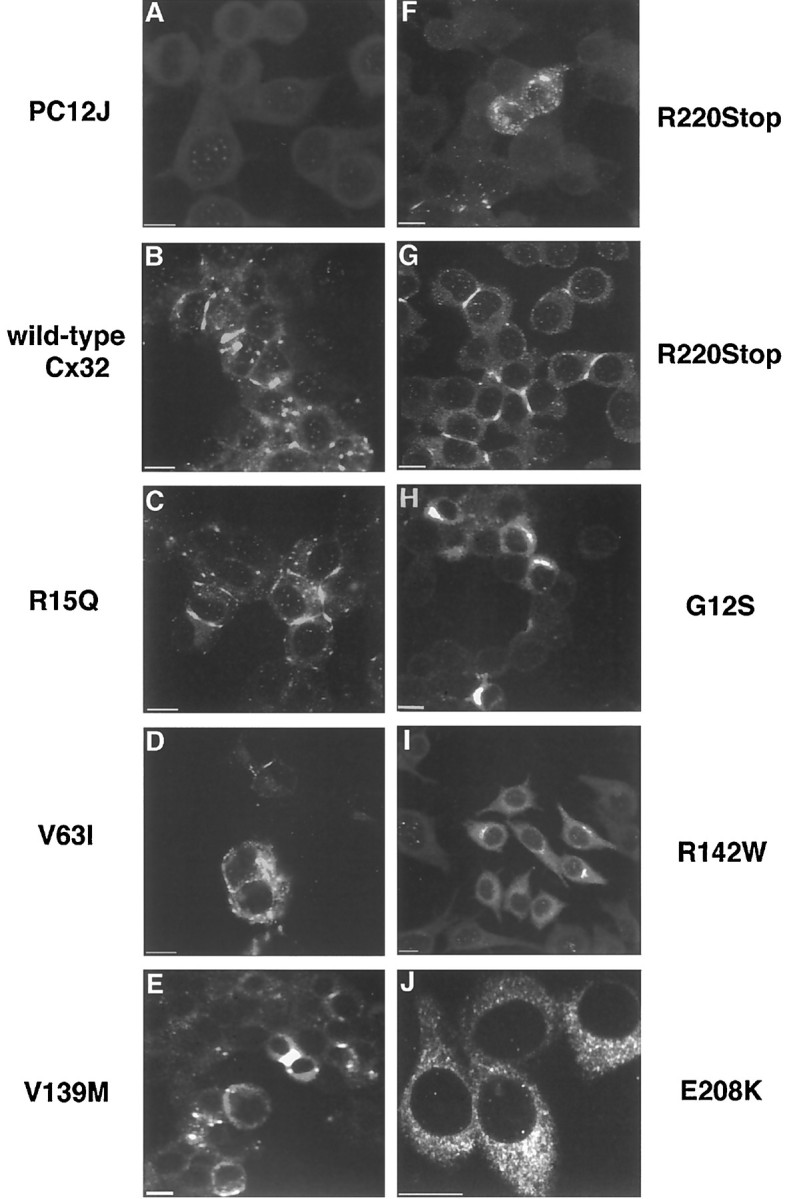
Localization of wild-type Cx32 and CMTX mutants in PC12J cells by indirect immunofluorescence using scanning laser confocal microscopy. Immunocytochemistry was performed with 7C6.C7 for all clones but R220Stop, for which M12.13 was used. A, Untransfected cells, PC12J. B,Wild-type Cx32, clone 517.4.7. C,R15Q, clone 15.38. D,V63I, clone 63.55. E,V139M, nonclonal cells. F,R220Stop, clone 220.37. G,R220Stop, clone 220.37. H,G12S, nonclonal cells. I,R142W, clone 116.38. J,E208K, clone 208.37. The magnification ofJ is higher to show greater cytoplasmic detail. Note that the diffuse staining seen in clone 208.37 is the predominant pattern observed in all clones expressing E208K. Scale bars, 10 μm.
In cells expressing the V63I, V139M, and R220Stop mutants, Cx32 was localized in the plasma membrane in a punctate or plaque-like staining pattern (Fig. 3D,E,G) and in the cytoplasm as a focal immunoreactivity (Fig. 3D–F). The predominant pattern in many V63I and R220Stop clones expressing lesser amounts of Cx32, however, was a diffuse cytoplasmic immunoreactivity (data not shown). Because cytoplasmic immunoreactivity was consistently seen in multiple clones expressing different amounts of mutant Cx32 (as determined by immunoblot), overexpression of Cx32 can be eliminated as the cause of cytoplasmic accumulation in the V63I and R220Stop clones.
Cx32 immunoreactivity was entirely cytoplasmic in clones expressing G12S (Fig. 3H), R142W (Fig.3I), E186K (Fig.4A), and E208K (Figs.3J, 4D), regardless of the level of protein expression (data not shown). Via immunocytochemistry, few cells transfected with G12S and R142W were found to express Cx32 protein, accounting for its complete (G12S) or near absence (R142W) on immunoblots. To determine the intracellular compartment in which mutant Cx32 was located, we performed double immunolabeling with antibodies against resident proteins of the Golgi apparatus (Gonatas et al., 1989;Velasco et al., 1993). In cells expressing E186K, Cx32 appeared to be exclusively localized in the Golgi apparatus (Fig.4A–C). Although the E208K mutant sometimes colocalized with the Golgi apparatus (Fig.4D–F), it was predominantly found in a diffuse cytoplasmic pattern that overlapped but was not limited to the Golgi apparatus (Fig. 3J). In clones expressing lower amounts of E208K protein, Cx32 staining appeared entirely diffuse. Although double immunolabeling was not performed for G12S and R142W, the identical cytoplasmic immunoreactivities of G12S, R142W, and E186K (Figs. 3H,I, 4A) suggest that G12S and R142W were also localized in the Golgi apparatus in the few cells expressing the protein.
Fig. 4.

Localization of Cx32 mutantsE186K and E208K in PC12J cells by indirect immunofluorescence using scanning laser confocal microscopy.A–C, Cells expressing E186K (clone 414.10) were double-stained with the mouse monoclonal antibody 7C6.C7 against Cx32 (A; rhodamine) and a polyclonal antiserum against rat α-mannosidase II (MnII) (B; fluorescein).A and B are superimposed inC. D–F, Cells expressingE208K (clone 208.49) were double-stained with the polyclonal antiserum B1J against Cx32(D; rhodamine) and the monoclonal antibody 10A8 againstMG160 (E; fluorescein). Dand E are superimposed in F. The characteristically punctate staining of E186K obtained with 7C6.C7 (see Fig. 5; data not shown) is difficult to see because of high background staining contributed by the α-mannosidase II antibody; arrowheads are used to indicateCx32 immunoreactivity in the cytoplasm of these cells. Note the absence of Cx32 at the cell surface in these two mutants. Scale bars, 10 μm.
To confirm further the localization of E186K and E208K in the Golgi apparatus, we treated cells expressing each mutant with brefeldin A (BFA), which blocks anterograde transport into the Golgi compartment, thereby causing a redistribution of Golgi resident proteins to the endoplasmic reticulum (ER) (for review, see Hunziker et al., 1992;Klausner et al., 1992). BFA did not dramatically alter the localization of Cx32 protein in clones expressing the E208K mutant (data not shown). BFA did, however, change Cx32 immunoreactivity in cells expressing the E186K mutant from a concise localization (Fig.5A) to a diffuse, weaker-staining pattern (Fig. 5B) that paralleled changes in the localization of MG160 (Fig. 5C,D, respectively), a resident protein of the medial cisternae of the Golgi apparatus (Gonatas et al., 1989). Taken together, these data demonstrate that substitutions at residues 12, 142, 186, and 208 may block the proper trafficking of Cx32 to the plasma membrane.
Fig. 5.

Effect of BFA on intracellular localization of E186K in clone 414.10. Immunocytochemistry was performed on cells treated with ethanol or 15 μg/ml BFA for 1 hr, and the cells were visualized with scanning laser confocal microscopy. A, Cx32 (antibody 7C6.C7); ethanol; 520×. B, Cx32; BFA; 760×. C, MG160 (antibody 10A8); ethanol; 760×.D, MG160; BFA; 760×. Arrows indicate the compact Cx32 or Golgi staining in control cells. Regardless of BFA concentration (data not shown), residual bright MG160 immunoreactivity on one side of the nucleus (arrowheads) was always observed in BFA-treated cells, including wild-type Cx32-expressing clones and vector-alone clones.
DISCUSSION
Three classes of CMTX mutations
Via immunocytochemical localization of Cx32, we have defined three different effects of CMTX mutations, which represent different classes of mutants. One class is represented by Cx32 mutant 175fs, a frameshift that truncates Cx32 by 43 amino acids and changes the last 67 residues, including the fourth transmembrane domain. Although clones expressing this mutant had the same amount of transcript as a wild-type Cx32 clone (Fig. 2E), little or no Cx32 protein was detected, suggesting that this CMTX mutation interferes with translation or results in the rapid degradation of Cx32 protein. This finding contrasts with the expression and cell surface localization of the same mutant in Xenopus oocytes (Bruzzone et al., 1994), indicating that protein stability and/or trafficking differ between the two cell types. Because our results were obtained from cultured mammalian cells, they are probably more relevant to the disease mechanism. The family bearing this mutation has a particularly severe phenotype, in which older male patients are often wheelchair-bound and many females at risk for carrying the disease allele are symptomatic (Table 1; Rozear et al., 1987; Bergoffen et al., 1993).
Another class of CMTX mutations is defined by Cx32 mutants R15Q, V63I, V139M, and R220Stop, in which at least some Cx32 is properly routed to the cell surface. It may be that the mutant Cx32 that arrives at the plasma membrane is not capable of forming functional gap junctions or that a fraction of the protein is misrouted, with dominant-negative or toxic effects on Schwann cells. The V139M and R220Stop mutants were studied previously by Omori et al. (1996). Our findings with V139M are consistent with their report, but the localization of R220Stop differed in that it was also in the cytoplasm. It may be that the amount of R220Stop in our cells is higher than the amount was in this previous study, resulting in the formation of intracellular gap junctions, as shown by Kumar et al. (1995). Nonetheless, the identification of the specific effects of R15Q, V63I, V139M, and R220Stop on the function of gap junctions awaits electrophysiological and dye-coupling studies and examination of the mutant proteins in the myelin sheath.
A third class of CMTX mutations, which includes G12S, R142W, E186K, and E208K, is characterized by the cytoplasmic accumulation of Cx32 and the absence of cell surface expression. Our findings indicate that these CMTX mutations alter Cx32 trafficking so that the protein accumulates in intracellular compartments such as the Golgi apparatus (e.g., G12S, R142W, and E186K) or in other sites such as the ER or lysosomes (e.g., E208K). We postulate that transmembrane domains 3 (TM3) and 4 (TM4) play important roles in Cx32 trafficking. A similar role for Cx32 transmembrane domains has been proposed by Leube, who showed that swapping the transmembrane domains of Cx32 and synaptophysin altered the routing of the hybrid proteins (Leube, 1995). In a previous study of the R142W and E186K mutants in Xenopus oocytes, Cx32 was shown to localize to the plasma membrane (Bruzzone et al., 1994). Again, the disparities in protein targeting between Xenopusoocytes and PC12J cells likely relate to cell-type and species differences in connexin processing. Finally, because the N terminal has been implicated in the insertion of connexins into membranes (Bennett et al., 1991; Falk et al., 1994), the cytoplasmic retention of the G12S mutant may result from improper membrane insertion.
The nature of the particular changes in the third class of CMTX mutations may provide additional clues to their adverse effects. Two of the mutations, E186K and E208K, change the polarity of a charged residue (− to +) that flanks TM4, whereas the third mutation R142W replaces a charged residue with an uncharged amino acid in TM3. Because charged residues in transmembrane domains are important for retention of proteins in the ER and the Golgi apparatus (Bonifacino et al., 1991;Machamer et al., 1993), these CMTX mutations might cause conformational changes within the Cx32 molecule or affect interactions between Cx32 monomers forming a hemichannel. The relationship between hemichannel assembly and the site of mutant Cx32 accumulation may be complex, considering the different paradigms for connexin oligomerization (Musil and Goodenough, 1993; Kumar et al., 1995). Because the substitution at residue 139 is conservative, few if any effects on membrane insertion or protein trafficking would be expected. Further mutational analysis will be necessary to determine which domains of Cx32 are most important for its proper oligomerization and transport in the cell.
Cx32 mutations and the CMTX phenotype
What is the effect of trafficking defects on the CMTX disease phenotype and its pattern of inheritance? An association between Cx32 mutations and disease severity has been suggested by Ionasescu et al. (1996), but it is difficult to draw strict correlations between genotype and clinical phenotype because of the variable expressivity of the disease (e.g., Table 1). An additional complication is the incomplete penetrance of the disease in women. For example, 50% of women at risk for inheriting the disease allele (Table 1, symptomatic at-risk female heterozygotes) should be symptomatic when CMTX is dominantly inherited. Although cellular localization does not correlate with disease penetrance in females, it may parallel the clinical variability observed in CMTX patients. At least some mutant Cx32 from families with mild to moderate symptoms (Table 1) reaches the cell surface in transfected cells (e.g., R15Q, V63I, V139M, and R220Stop). The most severe neuropathy is associated with 175fs (Table 1), for which little or no protein is detectable in transfected cells; the disease mechanism may be a simple loss of function. Although Cx32-null mice do not have an overt phenotype, clear evidence of demyelination is manifest by 4 months of age (Anzini et al., 1997), indicating that loss of Cx32 is associated with changes in the myelin sheath. It may be that mice are less sensitive than are humans to Cx32 loss. Investigations of additional loss-of-function mutations in Cx32 will be necessary to resolve this apparent difference.
In the families with moderate to severe clinical phenotypes (e.g., G12S, R142W, and E208K), Cx32 protein trafficking is defective in vitro. This association suggests that these Cx32 mutants may have dominant-negative effects in peripheral nerve. Toxic interactions with other proteins such as chaperones or proteins of the trafficking machinery might also occur in compartments in which mutant Cx32 accumulates. Other studies have focused on interactions between normal and mutant Cx32 (Rabadan-Diehl et al., 1994; Omori et al., 1996), but these are not likely to exist in myelinating Schwann cells of women because Cx32 is subject to X-inactivation (Scherer et al., 1997).
The possibility of dominant-negative interactions with other connexins in the same cell, however, is supported by at least two lines of evidence. First, Bruzzone et al. (1994) showed that mutant R142W can exert dominant-negative effects on Cx26 in Xenopus oocytes, which is coexpressed with Cx32 in many tissues (Bruzzone et al., 1996). Second, the expression of unidentified connexins in cultured Schwann cells has been reported (Chanson et al., 1993; Chandross et al., 1995,1996); mutant Cx32 may abrogate the normal function of these other connexins in myelinating Schwann cells.
Dominant effects of mutations in myelin proteins have been noted in other inherited diseases of myelin (Suter and Snipes, 1995; Scherer, 1997). In the Trembler mouse, for example, a point mutation in PMP-22 causes a severe dysmyelinating phenotype (Suter et al., 1992) and results in the accumulation of P0 in the Golgi apparatus (Heath et al., 1991). An accumulation of mutant PLP in the ER was observed in oligodendrocytes of jimpy mice (Roussel et al., 1987), which also have a severe dysmyelinating phenotype (for review, see Nave and Boespflug-Tanguy, 1996). Gow et al. (1994)transfected the jimpy allele and human PLP missense mutants into Cos cells and found that they, too, caused defects in protein trafficking. A more detailed study of PLP alleles involved in different subtypes of PMD (for review, see Nave and Boespflug-Tanguy, 1996) revealed that connatal (more severe) PMD correlated with ER retention of both mutant PLP and its alternatively spliced product DM-20, whereas classical (less severe) PMD was associated with ER accumulation of full-length PLP but not of DM-20 (Gow and Lazzarini, 1996). Unlike their milder counterparts, DM-20 alleles associated with connatal PMD could not facilitate transport of PLP to the cell surface, indicating a dominant-negative effect of these mutations on PLP (Gow and Lazzarini, 1996). Taken together, these findings and our results suggest that inherited diseases of myelin share a common pathophysiology in which mutant protein retained in intracellular compartments has deleterious effects on the protein folding, degradation, or trafficking machineries. Myelin-producing cells may be particularly susceptible to damage caused by misrouted protein, perhaps accounting in part for the tissue specificity of these diseases.
As with all model systems, it is difficult to know for certain whether our results reflect the mechanism of disease in vivo. The myelinating Schwann cells that are affected by Cx32 mutations have a phenotype and architecture that are different from PC12J cells and a more complex pattern of expression of plasma membrane proteins. Analysis of mutant Cx32 in in vivo models, such as transgenic and knock-out mice, will shed light on these issues and help to define further the normal role of gap junctions in peripheral nerve.
Footnotes
This study was supported by grants from the Muscular Dystrophy Association and National Institutes of Health Grant NS08075. We thank Dr. David Spray for the gift of PC12J cells, Dr. Neva Haites for genomic DNA from CMTX patients, Dr. Phillip Chance for clinical information from the CMTX family with the E186K mutation, Drs. Roberto Bruzzone and David Paul for Cx32 clones, and Drs. Elliot Hertzberg, David Paul, Norton Gilula, Nicholas Gonatas, and Marilyn Farquhar for antibodies. We are grateful to Tracey Oliver, Melissa Davey, Susan Shumas, and Melanie Hartman for technical assistance and to Linda Jo Bone and Drs. Cecilia Lo and David Spray for helpful discussion.
Correspondence should be addressed to Dr. Kenneth H. Fischbeck, Department of Neurology, University of Pennsylvania Medical Center, 415 Curie Boulevard, CRB 247, Philadelphia, PA 19104.
REFERENCES
- 1.Anzini P, Neuberg DH, Schachner M, Nelles E, Willecke K, Zielasek J, Toyka KV, Suter U, Martini R. Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin32. J Neurosci. 1997;17:4545–4551. doi: 10.1523/JNEUROSCI.17-12-04545.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett MVL, Barrio LC, Bargiello TA, Spray DC, Hertzberg E, Saez JC. Gap junctions: new tools, new answers, new questions. Neuron. 1991;6:305–320. doi: 10.1016/0896-6273(91)90241-q. [DOI] [PubMed] [Google Scholar]
- 3.Bergoffen J, Scherer SS, Wang S, Scott MO, Bone LJ, Paul DL, Chen K, Lensch MW, Chance PF, Fischbeck KH. Connexin mutations in X-linked Charcot–Marie–Tooth disease. Science. 1993;262:2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- 4.Bonifacino JS, Cosson P, Shah N, Klausner RD. Role of potentially charged transmembrane residues in targeting proteins for retention and degradation within the endoplasmic reticulum. EMBO J. 1991;10:2783–2793. doi: 10.1002/j.1460-2075.1991.tb07827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruzzone R, White TW, Scherer SS, Fischbeck KH, Paul DL. Null mutations of connexin32 in patients with X-linked Charcot–Marie–Tooth disease. Neuron. 1994;13:1253–1260. doi: 10.1016/0896-6273(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 6.Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- 7.Cease KB, Potcova CA, Lohff CJ, Zeigler ME. Optimized PCR using Vent polymerase. PCR Methods Applic. 1994;3:298–300. doi: 10.1101/gr.3.5.298. [DOI] [PubMed] [Google Scholar]
- 8.Chandross KJ, Chanson M, Spray DC, Kessler JA. Transforming growth factor-β1 and forskolin modulate gap junctional communication and cellular phenotype of cultured Schwann cells. J Neurosci. 1995;15:262–273. doi: 10.1523/JNEUROSCI.15-01-00262.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chandross KJ, Spray DC, Cohen RI, Kumar NM, Kremer M, Dermietzel R, Kessler JA. TNFα inhibits Schwann cell proliferation, connexin46 expression, and gap junctional communication. Mol Cell Neurosci. 1996;7:479–500. doi: 10.1006/mcne.1996.0035. [DOI] [PubMed] [Google Scholar]
- 10.Chanson M, Chandross KJ, Rook MB, Kessler JA, Spray DC. Gating characteristics of a steeply voltage-dependent gap junction channel in rat Schwann cells. J Gen Physiol. 1993;102:925–946. doi: 10.1085/jgp.102.5.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deschênes SM, Bone LJ, Fischbeck KH, Scherer SS. Connexin32 and X-linked Charcot–Marie–Tooth disease. In: Spray DC, Dermietzel R, editors. Gap junctions in the nervous system. Landes; Austin, TX: 1996. pp. 213–227. [Google Scholar]
- 12.Fairweather N, Bell C, Cochrane S, Chelly J, Wang S, Mostacciuolo ML, Monaco AP, Haites NE. Mutations in the connexin 32 gene in X-linked dominant Charcot–Marie–Tooth disease (CMTX1). Hum Mol Genet. 1994;3:29–34. doi: 10.1093/hmg/3.1.29. [DOI] [PubMed] [Google Scholar]
- 13.Falk MM, Kumar NM, Gilula NB. Membrane insertion of gap junction connexins: polytopic channel forming membrane proteins. J Cell Biol. 1994;127:343–355. doi: 10.1083/jcb.127.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fort P, Marty L, Piechaczyk M, Sabrouty SE, Dani C, Jeanteur P, Blanchard JM. Various rat adult tissues express only one major mRNA species from the glyceraldehyde-3-phosphate-dehydrogenase multigenic family. Nucleic Acids Res. 1985;13:1431–1442. doi: 10.1093/nar/13.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fryns JP, Van den Berghe H. Sex-linked recessive inheritance in Charcot–Marie–Tooth disease with partial clinical manifestations in female carriers. Hum Genet. 1980;55:413–415. doi: 10.1007/BF00290228. [DOI] [PubMed] [Google Scholar]
- 16.Gonatas JO, Mezitis SGE, Stieber A, Fleischer B, Gonatas NK. MG160, a novel sialoglycoprotein of the medial cisternae of the Golgi apparatus. J Biol Chem. 1989;264:646–653. [PubMed] [Google Scholar]
- 17.Goodenough DA, Paul DL, Jesaitis L. Topological distribution of two connexin32 antigenic sites in intact and split rodent hepatocyte gap junctions. J Cell Biol. 1988;107:1817–1824. doi: 10.1083/jcb.107.5.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodenough DA, Goliger JA, Paul DL. Connexins, connexons, and intercellular communication. Annu Rev Biochem. 1996;65:475–502. doi: 10.1146/annurev.bi.65.070196.002355. [DOI] [PubMed] [Google Scholar]
- 19.Gow A, Lazzarini RA. A cellular mechanism governing the severity of Pelizaeus–Merzbacher disease. Nat Genet. 1996;13:422–428. doi: 10.1038/ng0896-422. [DOI] [PubMed] [Google Scholar]
- 20.Gow A, Friedrich VL, Lazzarini RA. Many naturally occurring mutations of myelin proteolipid protein impair its intracellular transport. J Neurosci Res. 1994;37:574–583. doi: 10.1002/jnr.490370504. [DOI] [PubMed] [Google Scholar]
- 21.Heath JW, Inuzuka T, Quarles RH, Trapp BD. Distribution of P0 protein and the myelin-associated glycoprotein in peripheral nerves from Trembler mice. J Neurocytol. 1991;20:439–449. doi: 10.1007/BF01252272. [DOI] [PubMed] [Google Scholar]
- 22.Heimler A, Friedman E, Rosenthal AD. Naevoid basal cell carcinoma syndrome and Charcot–Marie–Tooth disease. J Med Genet. 1978;15:288–291. doi: 10.1136/jmg.15.4.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hunziker W, Whitney JA, Mellman I. Brefeldin A and the endocytic pathway. FEBS Lett. 1992;307:93–96. doi: 10.1016/0014-5793(92)80908-y. [DOI] [PubMed] [Google Scholar]
- 24.Ionasescu V, Ionasescu R, Searby C. Correlation between connexin 32 gene mutations and clinical phenotype in X-linked dominant Charcot–Marie–Tooth neuropathy. Am J Med Genet. 1996;63:486–491. doi: 10.1002/(SICI)1096-8628(19960614)63:3<486::AID-AJMG14>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 25.Klausner RD, Donaldson JG, Lippincott-Schwartz J. Brefeldin A: insight into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar NM, Friend DS, Gilula NB. Synthesis and assembly of human β1 gap junctions in BHK cells by DNA transfection with the human β1 cDNA. J Cell Sci. 1995;108:3725–3734. doi: 10.1242/jcs.108.12.3725. [DOI] [PubMed] [Google Scholar]
- 27.Leube RE. The topogenic fate of the polytopic transmembrane proteins, synaptophysin and connexin, is determined by their membrane-spanning domains. J Cell Sci. 1995;108:883–894. doi: 10.1242/jcs.108.3.883. [DOI] [PubMed] [Google Scholar]
- 28.Machamer CE, Grim MG, Esquela A, Chung SW, Rolls M, Ryan K, Swift AM. Retention of a cis Golgi protein requires polar residues on one face of a predicted α-helix in the transmembrane domain. Mol Biol Cell. 1993;4:695–704. doi: 10.1091/mbc.4.7.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milks LC, Kumar NM, Houghten R, Unwin N, Gilula NB. Topology of the 32-kd liver gap junction protein determined by site-directed antibody localizations. EMBO J. 1988;7:2967–2975. doi: 10.1002/j.1460-2075.1988.tb03159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Musil LS, Goodenough DA. Multisubunit assembly of an integral plasma-membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell. 1993;74:1065–1077. doi: 10.1016/0092-8674(93)90728-9. [DOI] [PubMed] [Google Scholar]
- 31.Nagy JI, Hossain MZ, Hertzberg EL, Marotta CA. Induction of connexin43 and gap junctional communication in PC12 cells overexpressing the carboxy terminal region of amyloid precursor protein. J Neurosci Res. 1996;44:124–132. doi: 10.1002/(SICI)1097-4547(19960415)44:2<124::AID-JNR4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 32.Nave K-A, Boespflug-Tanguy O. X-linked developmental defects of myelination: from mouse mutants to human genetic diseases. Neuroscientist. 1996;2:33–43. [Google Scholar]
- 33.Nicholson GA, Nash J. Intermediate nerve conduction velocities define X-linked Charcot–Marie–Tooth neuropathy families. Neurology. 1993;43:2558–2564. doi: 10.1212/wnl.43.12.2558. [DOI] [PubMed] [Google Scholar]
- 34.Omori Y, Mesnil M, Mironov N, Yamasaki H. Connexin32 mutations from X-linked Charcot–Marie–Tooth disease patients: functional defects and dominant-negative effects. Mol Biol Cell. 1996;7:907–916. doi: 10.1091/mbc.7.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paul DL. Molecular cloning of cDNA for rat liver gap junction protein. J Cell Biol. 1986;103:123–134. doi: 10.1083/jcb.103.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rabadan-Diehl C, Dahl G, Werner R. A connexin-32 mutation associated with Charcot–Marie–Tooth disease does not affect channel formation in oocytes. FEBS Lett. 1994;351:90–94. doi: 10.1016/0014-5793(94)00819-1. [DOI] [PubMed] [Google Scholar]
- 37.Roussel G, Neskovic NM, Trifilieff E, Artault J-C, Nussbaum J-L. Arrest of proteolipid transport through the Golgi apparatus in jimpy brain. J Neurocytol. 1987;16:195–204. doi: 10.1007/BF01795303. [DOI] [PubMed] [Google Scholar]
- 38.Rozear MP, Pericak-Vance MA, Fischbeck K, Stajich JM, Gaskell PC, Krendel DA, Graham DG, Dawson DV, Roses AD. Hereditary motor and sensory neuropathy, X-linked: a half century follow-up. Neurology. 1987;37:1460–1465. doi: 10.1212/wnl.37.9.1460. [DOI] [PubMed] [Google Scholar]
- 39.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 40.Scherer SS. Molecular genetics of demyelination: new wrinkles on an old membrane. Neuron. 1997;18:13–16. doi: 10.1016/s0896-6273(01)80042-8. [DOI] [PubMed] [Google Scholar]
- 41.Scherer SS, Deschênes SM, Xu Y-T, Grinspan JG, Fischbeck KH, Paul DL. Connexin32 is a myelin-related protein in the PNS and CNS. J Neurosci. 1995;15:8281–8294. doi: 10.1523/JNEUROSCI.15-12-08281.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scherer SS, Bone LJ, Deschênes SM, Fischbeck KH, Balice-Gordon R. The role of the gap junction protein connexin32 in the myelin sheath. In: Devon RM, Doucette JR, Juurlink BHJ, Nazarali AJ, Schreyer DJ, Verge VMK, editors. Cell biology and pathology of myelin: evolving biological concepts and therapeutic approaches. Plenum; New York: 1997. pp. 83–102. [Google Scholar]
- 43.Scherer SS, Xu Y-T, Fischbeck KH, Willecke K, Bone LJ (1997) Connexin 32-null mice develop demyelinating peripheral neuropathy. Glia, in press. [DOI] [PubMed]
- 44.Suter U, Snipes GJ. Biology and genetics of hereditary motor and sensory neuropathies. Annu Rev Neurosci. 1995;18:45–75. doi: 10.1146/annurev.ne.18.030195.000401. [DOI] [PubMed] [Google Scholar]
- 45.Suter U, Welcher AA, Ozcelik T, Snipes GJ, Kosaras B, Francke U, Billings-Gagliardi S, Sidman RL, Shooter EM. The Trembler mouse carries a point mutation in a myelin gene. Nature. 1992;356:241–244. doi: 10.1038/356241a0. [DOI] [PubMed] [Google Scholar]
- 46.Velasco A, Hendricks L, Moremen KW, Tulsiani DRP, Touster O, Farquhar MG. Cell-type dependent variations in the subcellular distribution of alpha-mannosidase-I and alpha-mannosidase-II. J Cell Biol. 1993;122:39–51. doi: 10.1083/jcb.122.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]