

Abstract
The α7 subunit of the neuronal nicotinic acetylcholine receptor (nAChR) is abundantly expressed in hippocampus and is implicated in modulating neurotransmitter release and in binding α-bungarotoxin (α-BGT). A null mutation for the α7 subunit was prepared by deleting the last three exons of the gene. Mice homozygous for the null mutation lack detectable mRNA, but the mice are viable and anatomically normal. Neuropathological examination of the brain revealed normal structure and cell layering, including normal cortical barrel fields; histochemical assessment of the hippocampus was also normal. Autoradiography with [3H]nicotine revealed no detectable abnormalities of high-affinity nicotine binding sites, but there was an absence of high-affinity [125I]α-BGT sites. Null mice also lack rapidly desensitizing, methyllycaconitine-sensitive, nicotinic currents that are present in hippocampal neurons. The results of this study indicate that the α-BGT binding sites are equivalent to the α7-containing nAChRs that mediate fast, desensitizing nicotinic currents in the hippocampus. These mice demonstrate that the α7 subunit is not essential for normal development or for apparently normal neurological function, but the mice may prove to have subtle phenotypic abnormalities and will be valuable in defining the functional role of this gene product in vivo.
Keywords: acetylcholine receptor, α-bungarotoxin, gene targeting, hippocampus, mouse, nicotine, α7 subunit
Nicotinic acetylcholine receptors (nAChRs) are members of a superfamily of ligand-gated ion channels that include muscle and neuronal nAChRs and receptors for GABAA, glycine, and serotonin. Eleven genes have been identified that encode neuronal nAChR subunits: eight α subunits (α2–α9) and three β subunits (β2–β4) (Sargent, 1993;McGehee and Role, 1995). Transcripts for α8 have been found in avians but not in mammals (Schoepfer et al., 1990), and α9-containing nAChRs are expressed in auditory hair cells (Elgoyhen et al., 1994). In the vertebrate brain, two categories of nicotinic receptors are distinguishable based on high-affinity binding of the agonist nicotine or high-affinity binding of the antagonist α-bungarotoxin (α-BGT). The receptors from these two categories have distinct distributions (Wonnacott, 1986; Whiting and Lindstrom, 1988).
Biochemical and electrophysiological investigations have established that both neuronal and skeletal muscle nAChRs are heteromultimers consisting of five subunits each with four membrane-spanning segments (Sargent, 1993; McGehee and Role, 1995). Expression studies inXenopus oocytes demonstrated that functional neuronal nAChRs are comprised of two α subunits and three β subunits (Cooper et al., 1991; Bertrand and Changeux, 1995). The α7 subunit is an exception because it apparently cannot coassemble with other subunits when expressed in Xenopus oocytes. Thus, like α8 and α9, α7 forms a homo-oligomer receptor/channel that is inhibited with high affinity by α-BGT (Couturier et al., 1990; Séguéla et al., 1993). Because α7 is the only subunit of these three that is widely expressed in mammalian brain, it has been suspected that α7 contributes to the high-affinity α-BGT binding site, the structure and functional significance of which have been a long-standing source of controversy (Sargent, 1993).
Initially, it was thought that the α-BGT sites did not form functional ion channels, but more recent studies have demonstrated high-affinity inhibition by α-BGT and methyllycaconitine (MLA) of a rapidly desensitizing nicotinic current in hippocampal neurons (Albuquerque and Alkondon, 1991; Zorumski et al., 1992; Alkondon and Albuquerque, 1993; Gray et al., 1996) and in neurons of the peripheral nervous system (Zhang et al., 1994). Previous studies based on protein purification indicated that α-BGT sites were hetero-oligomers, but the copurified components may not have been integral subunits (Sargent, 1993). In chick brain and retina, α-BGT sites were found to be heterogeneous, containing α7, α8, or both, possibly in combination with another unknown component (Schoepfer et al., 1990; Gotti et al., 1995).
Despite morphological, biochemical, and electrophysiological analysis, the functions of neuronal α-BGT binding sites and nAChRs in the mammalian brain remain largely unknown. Various neuronal nAChRs are involved in nicotine addiction (Dani and Heinemann, 1996), and there is evidence that nicotine can improve attention, rapid information processing, and working memory (Levin, 1992; Ohno et al., 1993;Picciotto et al., 1995; McGehee and Role, 1996). Cholinergic mechanisms may be involved in neurobehavioral disorders including seizures and schizophrenia (Steinlein et al., 1995; Freedman et al., 1997). A locus for juvenile myoclonic epilepsy is mapped near the α7 gene in humans (Elmslie et al., 1997). Some functions attributed to the α-BGT sites and/or the α7 subunit include synapse formation (Fuchs, 1989; Pugh and Berg, 1994; Broide et al., 1995), mediation of nicotine-induced seizures (Miner and Collins, 1989; Stitzel et al., 1997), and presynaptic modulation of neurotransmitter release (McGehee et al., 1995; Gray et al., 1996; Zhang et al., 1996).
We prepared mice with a homozygous null mutation for the α7 subunit and found that the mice were viable and had no gross abnormalities in brain morphology, but the high-affinity α-BGT binding sites were not present. Furthermore, α7-deficient hippocampal neurons lack fast, rapidly desensitizing nicotinic currents, indicating that those currents in the hippocampus are derived from α7-containing nAChRs that are also likely to be the major high-affinity α-BGT binding sites in mouse brain.
MATERIALS AND METHODS
Acrα7 gene targeting in embryonic stem cells. A rat cDNA clone for the α7 subunit (Séguéla et al., 1993) was used to screen a mouse genomic DNA library prepared from the 129/SvJ strain (cat. #946305, Stratagene, La Jolla, CA). Detailed restriction maps were prepared for genomic clones, and the intron/exon boundaries of exons 5–10 and the 3′-untranslated region in exon 10 were sequenced (Fig.1a); sequences agreed with that published for the mouse α7 cDNA (Orr-Urtreger et al., 1995). Sequencing was performed using an Applied Biosystems (ABI) model 373-automated DNA sequencer and dye terminator protocols as provided by the manufacturer (ABI, Foster City, CA).
Fig. 1.
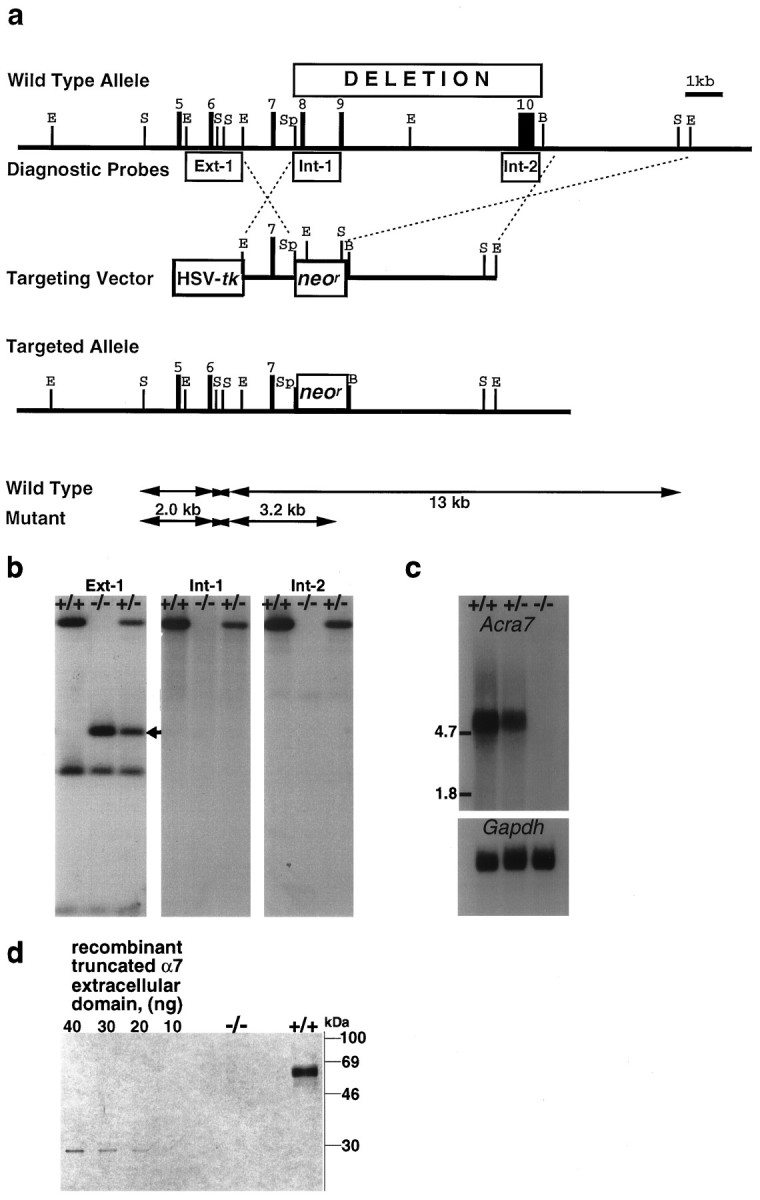
Gene targeting of the neuronal nAChR α7 subunit. a, Partial genomic structure of the murine α7 subunit gene including exons 5–10 is shown. The homologous recombination event generated a 7 kb genomic deletion that removes exons 8–10. Restriction enzyme sites are as follows:E, EcoRI;S, SacI; Sp,SpeI; B, BamHI. The diagnostic probes include a flanking probe to genotype ES cells and animals (external probe 1, Ext-1) and two internal probes (Int-1 andInt-2) to confirm the deletion. The targeting vector was used to obtain a replacement mutation and contains a neomycin resistance gene (neor) as a positive selectable marker and the herpes simplex thymidine kinase gene (HSV-tk) as a negative selectable marker. The sites of predicted homologous recombination are shown. The expected wild-type and mutant restriction fragments, after SacI enzyme digest and hybridization with Ext-1 probe, are shownbelow the targeting vector. b, Southern blot analysis identifies the α7 homozygous null (−/−), heterozygous (+/−), and littermate control (+/+) animals using each of the three probes as indicated. The small arrow indicates the mutant band with the Ext-1 probe. Constant fragments of 2.0 and 0.3 kb are seen with the Ext-1 probe. c, Northern blot analysis of α7 gene expression in brains of (+/+), (+/−), and (−/−) animals, using the Acrα7 cDNA (Acra7) and control (Gapdh) probes. d, Western blot analysis of brains from +/+ and −/− animals. As a positive control, 10, 20, 30, and 40 ng of recombinant extracellular domain of the rat α7 protein (Chen and Patrick, 1997) were used.
The deletion mutation was introduced into the ABI 2.1 embryonic stem (ES) cell line (Soriano et al., 1991) and transmitted to the germline as described previously (Bullard et al., 1996). Chimeric mice were bred with C57BL/6J mice, and the mice used in these studies were maintained on a mixed 129/SvEv and C57BL/6J background. The mutation is being back-crossed on the C57BL/6J background for future work.
Southern, Northern, and Western analyses. Southern blot hybridization was performed according to standard methods (Sambrook et al., 1989) using a hybridization solution of 0.125m NaPO4, pH 7.0, 0.25 mNaCl, 1 mm EDTA, 10% polyethylene glycol (PEG-8000), 7% SDS, and 1% bovine serum albumin (BSA) at 65°C overnight followed by washing to a final stringency of 0.2× SSC/0.1% SDS at 65°C and autoradiography at −80°C.
Total RNA was isolated from brain after homogenization in Ultraspec II (Tel Test, Houston, TX). Total RNA was resolved on a 1.2% agarose gel in 10 mm NaPO4 buffer, pH 6.8, after glyoxal/dimethylsulfoxide denaturation according to standard methods (Sambrook et al., 1989). RNA was visualized by ethidium bromide staining, transferred to Hybond N+ membrane, and hybridized with full-length (mouse) Acrα7 cDNA as probe. Hybridization and washing conditions were identical to those used for Southern hybridization.
For immunoblotting, whole brain from three homozygous α7 null (−/−) mice and three littermate control (+/+) mice was homogenized in 10 ml of ice-cold buffer containing 10 mm HEPES, pH 7.4, 5 mm EDTA, 5 mm EGTA, and 1 mmphenylmethylsulfonylfluoride (PMSF). Total membranes were pelleted by centrifugation at 100,000 × g for 60 min at 4°C. Membranes were resuspended in 10 ml of solubilization buffer containing 10 mm HEPES, pH 7.4, 5 mm EDTA, 5 mm EGTA, 1% Triton X-100, and 1 mm PMSF and shaken for 2 hr at 4°C. The detergent-solubilized membrane protein supernatant was recovered after centrifugation at 100,000 ×g for 60 min. Protein concentration was determined by the BCA method (Pierce, Rockford, IL). Solubilized membrane proteins (36 mg) were incubated overnight with 25 μl of α-cobratoxin matrix (0.5 mg toxin/ml matrix, Sigma, St. Louis, MO), which was preequilibrated with the solubilization buffer. The matrix was recovered by brief centrifugation in a benchtop microcentrifuge and washed four times with 1 ml of solubilization buffer. Bound receptors were eluted with 30 μl of SDS/sample buffer and separated by SDS-PAGE followed by transfer to a nitrocellulose membrane. The membrane was blocked for 30 min with 5% dry milk and 0.1% Tween 20 in PBS. A polyclonal sheep antibody (Chen and Patrick, 1997) against a bacterial expressed extracellular domain of the rat α7 protein was diluted 1:1000 in the above blocking solution and incubated with the membrane overnight at 4°C. The membrane was washed and probed with a rabbit anti-sheep IgG antibody coupled to peroxidase (Cappel, Durham, NC) at a dilution of 1:10,000. After we washed the membranes, we detected peroxidase activity using enhanced chemiluminescence (ECL; Amersham, Arlington Heights, IL).
Nicotine and α-BGT autoradiography. For nicotine autoradiography, coronal sections (10 μm) of fresh frozen mouse brain were prepared and dried onto slides. [3H]nicotine (75 Ci/mmol, DuPont NEN, Boston, MA) was diluted in 50 mmTris, pH 7.4, and 1% BSA to a final concentration of 2 nm. Slides were incubated with the solution for 1 hr at room temperature. Nonspecific binding was assessed by incubating sections with an excess (10 μm) of unlabeled ligand nicotine. Slides were washed twice in ice-cold 50 mm Tris, pH 7.4, for 2 min each, dried, and exposed to tritium-sensitive film (3H Hyperfilm, Amersham, Arlington Heights, IL) for 12 weeks.
For α-BGT autoradiography, unfixed, frozen brain was sectioned at 20 μm, thaw-mounted onto slides, and dried. Sections were preincubated for 30 min at room temperature in 1% BSA and 50 mm Tris, pH 7.4, either with or without 10 μmunlabeled α-BGT as a competitor. [125I]α-BGT (initial specific activity of 4.8 × 108cpm/nmol and used within 1 half-life; Amersham) was diluted in the above blocking solution to a final concentration of 5 nmand added to the sections for 1 hr at room temperature. Sections were washed four times for 5 min in ice-cold 50 mm Tris, pH 7.4, after which they were dried and exposed to Kodak BioMax MR film for 1–3 d.
Histological analysis. Brain tissue for Nissl stain, immunohistochemistry, and acetylcholinesterase histochemistry was prepared by perfusion with PBS/4% paraformaldehyde in PBS, pH 7.4, and post-fixed overnight at 4°C in the same fixative. Tissue was equilibrated in 30% sucrose, frozen in prechilled isopentane, and sectioned at 30–50 μm.
For Nissl stains, cleared and rehydrated sections were incubated with 0.5% cresyl violet acetate, washed, rapidly dehydrated, cleared, and mounted on coverslips. Immunohistochemistry for glial fibrillary acidic protein (GFAP) was performed with antibody against GFAP (clone G-A-C, mouse IgG1, Boehringer Mannheim, Indianapolis, IN); free-floating sections were stained according to a standard ABC-protocol (Vector Laboratories, Burlingame, CA). Staining for acetylcholine esterase was performed according to a published protocol (Geneser-Jensen and Blackstad, 1971). A modified Timm stain protocol was used to demonstrate mossy fibers in paraffin-embedded, 8-μm-thick sections of the hippocampus (Sloviter, 1982).
Labeling for cytochrome oxidase was performed on mounted sections using PBS containing 4% sucrose, 25 μm cytochrome c(type III, Sigma), and 1 mm diaminobenzidine tetrahydrochloride at 37°C until staining of the desired intensity could be detected, after which the reaction was stopped by washing in PBS.
Electrophysiology. Hippocampal neurons were obtained from newborn mice (12–36 hr postnatal). The brain was removed immediately and kept throughout the dissection in a cold solution containing (in mm): 137 NaCl, 5.3 KCl, 0.2 Na2HPO4, 0.2 KH2PO4, and 10 HEPES, pH 7.4. Hippocampal tissue from both hemispheres was gently removed and cut into small pieces. The tissue was digested for 40 min at 37°C in a dissecting solution containing 20 U/ml papain (Worthington, Freehold, NJ). After digestion, the tissue was triturated with fire-polished Pasteur pipettes of decreasing diameter. Hippocampal cells were plated on collagen/poly-d-lysine-coated coverslips (Fisher, Pittsburgh, PA). A separate cell culture was prepared for each mouse in litters born to heterozygote parents, with the expectation that +/+, +/−, and −/− pups would occur in a ratio of 1:2:1. The genotype of each mouse was determined after electrophysiological studies were completed and interpreted. Cells were kept in minimum essential medium (MEM) containing 5% fetal bovine serum (HyClone, Logan, UT), 1 μl/ml Serum Extender (Collaborative Research, Bedford, MA), 0.5 μm tetrodotoxin (Calbiochem, Pasadena, CA), 20 mm glucose, and 2.5 mm MgCl2. Hippocampal neurons were studied from day 15 to day 25 in culture because nicotinic currents are larger and more commonly expressed after day 10 in culture (Alkondon and Albuquerque, 1993; Gray et al., 1996).
Whole-cell currents were elicited with 500 μm nicotine and measured with standard patch-clamp techniques (Zarei and Dani, 1995). The external bath solution contained (in mm): 150 NaCl, 2.5 KCl, 5 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, pH 7.4, 0.5 μm tetrodotoxin, and 10 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX). The pipette solution contained (in mm): 150 CsCH3SO3, 5 NaCl, 0.2 EGTA, 2 Na2ATP, 2 MgATP, 0.3 Na3GTP, and 10 HEPES, pH 7.3. The holding potential was −60 mV. Currents were amplified and filtered on line (1 kHz) using an Axopatch 1D voltage clamp with a 4-pole Bessel filter and were digitally sampled to exceed the Nyquist criterion. No series resistance compensation or leak subtraction was performed. Drugs were applied through flow pipes mounted on a motorized manipulator (Newport, Fountain Valley, CA) and connected to computer-driven valves (General Valve Corporation, Fairfield, NJ). Nicotine and MLA were applied for 600 msec and 90 sec, respectively. Solution changes were complete in ∼30 msec (Gray et al., 1996). All experiments were conducted at room temperature.
RESULTS
Preparation of a null mutation for the α7 subunit
Mice deficient in the α7 subunit were generated by introducing a 7 kb deletion into ES cells followed by transmission to the germline. The mutation deletes the last three exons (8–10) of the α7 locus (Acrα7); these exons encode the second transmembrane domain (MII), forming the putative ion channel and the third and fourth transmembrane domains and the cytoplasmic loop (Fig.1a). Southern blot analysis using a flanking genomic probe detected a new 3.2 kb mutant fragment in the heterozygote (+/−) and homozygote (−/−) mice (Fig. 1b); adjacent smaller DNA fragments were unchanged. Analysis with genomic probes from the regions of the deleted exons confirmed the absence of these exons (Fig.1b). The effect of the mutation on mRNA transcripts was examined using Northern blotting, and no detectable transcripts were found in homozygous mutant mice, indicating that any altered transcripts were unstable (Fig. 1c). Because the deletion leaves intact the coding sequences for the extracellular, ligand-binding, and toxin-binding domains of the α7 subunit, a partially functional, truncated protein could occur; this possibility is unlikely, however, considering the results of Northern blotting. Immunoblots (Fig. 1d) of protein extracts prepared by α-BGT affinity chromatography from wild-type and mutant mice were probed with antibodies to the N-terminal portion of the α7 subunit, demonstrating absence of toxin-binding α7 protein in homozygous mice. The results of Northern analysis and immunoblotting confirm the generation of a null mutation for the α7 subunit.
Normal growth, viability, and neuroanatomy in α7 null mice
The α7 null mice are viable, are present in the expected proportion in matings of heterozygote mice, grow to normal size, and show no obvious physical or neurological deficit. Homozygous male and female mice are fertile, although there may be some reduction in fertility.
The overall brain structure and organization in homozygous null mice appear normal as assessed by comparable Nissl-stained sections in +/+ and −/− mice (Fig. 2a). All major neuronal structures are intact, and there are no apparent abnormalities in cell density or layering of cortical structures. Because α7 is expressed most abundantly in the hippocampus, a more detailed anatomical analysis was performed (Fig. 2b). Nissl staining revealed that the major classes of neurons (i.e., granule cells, pyramidal cells, and hilar interneurons) are present in their appropriate location and, therefore, do not appear to depend on the presence of α7 for their formation or migration within the network. Histochemical staining for acetylcholinesterase (AChE) was performed to evaluate the hippocampus, and no abnormalities were found. Because a mutation in an α7-like gene in C. elegans is associated with neurodegeneration (Treinin and Chalfie, 1995), we performed immunohistochemical staining for GFAP, a marker for astrocytes, to search for glial scars representing neuronal injury or cell death. No glial scars were found in the hippocampus of mutant mice, suggesting that deficiency of α7 does not lead to abnormal neuronal degeneration in this region. The status of intrahippocampal connections, particularly the mossy fiber projections, was evaluated by Timm staining, and no differences were found between mutants and controls.
Fig. 2.

Neuroanatomy and histochemistry of mutant mice. Panels with +/+ and −/− indicated are as follows: (a) coronal sections of brain with Nissl stain; (b) sagittal sections of hippocampus with Nissl stain (Nissl), AChE-acetylcholinesterase (AChE), glial fibrillary acidic protein (GFAP), and Timm stain (Timm) as indicated; and (c) sections through the barrels (arrows) in the primary somatosensory cortex stained with cytochrome oxidase.
The primary somatosensory cortex of rodents is characterized by anatomically defined barrel structures that reflect projections from the whiskers. Because staining with α-BGT outlines barrels at the time of their appearance, it was suggested that α7 may be important for barrel formation (Fuchs, 1989; Broide et al., 1995). Mutant animals were analyzed for the presence of barrels using histochemical staining for cytochrome oxidase (Fig. 2c). Sections through the somatosensory cortex of mutant animals show characteristic barrel-like structures that were indistinguishable from those in wild-type littermates, indicating that structures with the general appearance of barrels develop in the absence of α7.
Null mice lack α-BGT binding sites
The distribution of high-affinity nicotine binding sites and α-BGT binding sites in the brain of mutant (−/−) and control (+/+) littermates was examined (Fig. 3). The pattern of distribution of binding sites for these two ligands is significantly different, with the greatest abundance of high-affinity nicotine binding sites in thalamic structures and the predominance of α-BGT binding sites in the hippocampus (Clarke et al., 1985). The use of [3H]nicotine at low nanomolar concentrations detects only high-affinity binding sites and does not detect low-affinity nicotine binding sites such as those present in hippocampus (Clarke et al., 1985; Picciotto et al., 1995). The pattern of high-affinity nicotine binding sites in mutant and control animals was indistinguishable, and there was no evidence for significant upregulation of nicotine binding sites in α7-deficient mice (Fig. 3). The distribution of α-BGT binding sites in the α7-deficient mice is of particular interest because of the possibility that α7 represents the major or sole α-BGT binding protein in rodent brain. Brain sections of homozygous null mice and littermate controls were analyzed using autoradiography with [125I]α-BGT (Fig. 3). The characteristic distribution of α-BGT binding sites was found in normal mice with a prominent pattern in telencephalic structures such as the hippocampus, amygdala, and neocortex. In contrast, there was no significant α-BGT binding above background levels in the null mice, suggesting that α7 is required for high-affinity α-BGT binding sites in the mouse brain.
Fig. 3.
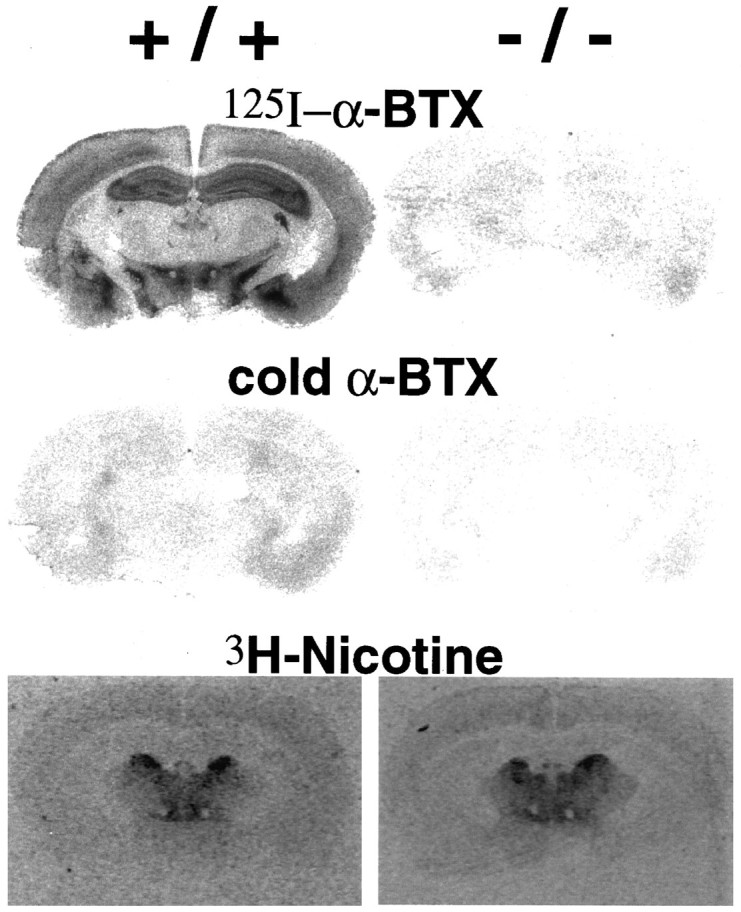
Nicotine and α-BGT binding in mutant mice. Autoradiography was performed with [3H]nicotine and [125I]α-BGT as described in Materials and Methods with +/+ mice shown on the left and −/− shown on the right. The panels marked coldα-BGT included excess nonradioactive α-BGT as competitor.
Null mice lack hippocampal fast nicotinic currents
Electrophysiological evaluation of the mice was important, because rapidly desensitizing nicotinic currents in hippocampal neurons are attributed to expression of α7 (Alkondon and Albuquerque, 1993; Gray et al., 1996). Neonatal littermates from matings of heterozygotes were examined, and genotypes were determined after collection and interpretation of the electrophysiological data. Hippocampal neurons were cultured from neonatal mice, and rapid application of 500 μm nicotine evoked a fast, desensitizing current that could be blocked by 5 nm MLA, a potent and selective antagonist of α7-containing nAChRs (Alkondon and Albuquerque, 1993;Gray et al., 1996) (Fig. 4a). The block by MLA was reversible after 2 min of washout with bath solution. When neurons from α7-deficient mice were exposed to 500 μm nicotine, no currents could be recorded; none of the 35 cells from the four null mice examined revealed any current (Fig.4c). Neurons from both wild-type and heterozygous mice showed bimodal response distributions to nicotine; some cells responded by giving measurable currents, and other cells had no response. The lack of nicotine-induced currents in the α7 null mice indicates that the predominant currents found in cultured hippocampal neurons from control mice are dependent on expression of the α7 nAChR.
Fig. 4.

Nicotine induces currents in control but not in α7 null mice. a, Nicotine (0.5 mm) evoked a fast, desensitizing current in hippocampal neurons from control (+/+) mice. The currents were completely blocked by 5 nm MLA (90 sec application) and recovered to 89% of the original peak current after washout (recovery at 150 sec). Thesolid black lines indicate the duration of the nicotine applications. b, Nicotine (0.5 mm) failed to induce ionic currents in all of the hippocampal cells studied from α7 null mice. c, The majority (72%) of hippocampal cells from wild-type (+/+) mice (3 animals) showed currents in response to nicotine application, and 28% did not show any response. Approximately half (54%) of hippocampal cells from α7 heterozygous (+/−) mice (8 animals) responded to nicotine, whereas 46% did not show any current. No nicotine-induced current could be recorded in any of the cells from α7 null (−/−) mice (4 animals, 35 cells). n, Number of cells recorded; I, current in pA.
DISCUSSION
A mutation deleting numerous exons was introduced into the gene for the α7 subunit, completely eliminating its potential for participation in an ion channel. Although this mutation might allow for the synthesis of a truncated protein with ligand-binding capacity, Northern blotting and immunoblotting studies demonstrate the absence of detectable mRNA or protein and ensure that this mutation produces a null allele. The function of the α7 subunit is largely unknown, although many possible roles have been discussed (Sargent, 1993;McGehee and Role, 1995). Homozygous mutant mice demonstrate normal general appearance, growth, survival, gait, and anatomy. Histological evaluation of the nervous system did not reveal any developmental abnormalities. Thus, the phenotypic consequences of deficiency of the α7 nAChR are not immediately obvious.
There is evidence that expression of the α7 subunit may be correlated with differences in nicotine binding, nicotine-induced seizures, nicotine preference, and effect of nicotine on body temperature in various strains of mice (Miner and Collins, 1989;Stitzel et al., 1997), and it will be of interest to analyze the mutant mice for these traits. In addition, α7-deficient mice might be expected to show resistance to α-conotoxin (Johnson et al., 1995). There is also a report suggesting that differences in response to auditory stimuli that are associated with schizophrenia show linkage to human chromosome 15 in a region near the α7 locus (Freedman et al., 1997). There is evidence of increased smoking in schizophrenics, and α7 has been proposed as having a role in the pathophysiology of schizophrenia (Freedman et al., 1994, 1997). It will be important to evaluate learning and behavior in the mutant mice, and the mutation is being back-crossed to C57BL/6J background, because nicotine-induced seizures, auditory gaiting, and learning are all known to be variable among inbred strains of mice (Miner and Collins, 1989; Stitzel et al., 1997). Behavioral studies seeking to identify schizophrenia-like behavior in mice (Dains et al., 1996; Kafka and Corbett, 1996) will also be of interest.
The hippocampus is a center for learning and memory and receives cholinergic innervation mainly from the medial septum and diagonal band (Woolf, 1991). Presynaptic terminals containing choline acetyltranferase (the enzyme that catalyzes the synthesis of acetylcholine) have been found to synapse directly onto pyramidal and granule cells and their dendrites (Alonso and Amaral, 1995). Nicotine and cytisine autoradiography and in situ hybridization using probes for various subunits indicate that nAChRs are expressed throughout the hippocampus and that α7 and β2 are the most abundant subunits (Deneris et al., 1988; Wada et al., 1990; Dineley-Miller and Patrick, 1992; Perry et al., 1993; Séguéla et al., 1993). The hippocampus also is known to possess a high density of α-BGT binding sites. Our results with α7 null mice indicate that the α-BGT sites are not detected when the α7 gene is disrupted, but the high-affinity nicotine sites in the brain are not detectably different. Although the results do not eliminate the possibility of other low-affinity sites or of another very minor component of α-BGT sites, the major α-BGT sites that have been at the center of attention and controversy are absent in α7 null mice. These results indicate that, unlike chick, in which multiple forms of α-BGT sites are seen based on the presence of either the α7 or the α8 subunit (Schoepfer et al., 1990; Gotti et al., 1995), the α-BGT sites require the α7 subunit in mice.
In rat hippocampal slices and cultures, it was found that nicotinic agonists can enhance glutamate release by acting through presynaptic nAChRs (Gray et al., 1996). This enhanced release of glutamate and the rapidly desensitizing nicotinic currents were inhibited by α-BGT and MLA, indicating that those nAChRs contain the α7 subunit. Although other types of nicotinic currents occasionally could be seen, the predominant current displayed fast activation and rapid desensitization (Alkondon and Albuquerque, 1993; Gray et al., 1996). Similarly for mice, we found nicotine-activated currents in the majority (72%) of hippocampal neurons, and in all cases those currents were rapid and inhibited by MLA. In cultures from four α7 null mice, however, none of the 35 hippocampal neurons that were studied displayed nicotinic currents. These results suggest that the rapid nicotinic currents in hippocampal neurons are mediated by the α-BGT binding sites and that those sites require the α7 subunit for their formation into a receptor/ion channel complex.
Whether homo-oligomer α7 receptors exist in the brain is still open to question, but evidence is mounting that α7 might form a receptor without requiring any of the other presently known nicotinic subunits. The α7 null mice lack the α-BGT sites but have the high-affinity nicotine sites, and β2 null mice lack the nicotine sites but still have the α-BGT sites (Picciotto et al., 1995). These results indicate that β2 is not required for the α-BGT site and that α7 is not required for the high-affinity nicotine site. Because α7 and β2 are by far the predominant subunits in the hippocampus, it is unlikely that any other known subunit is abundant enough to be present in all of the α-BGT sites in the hippocampus. Thus, it is unlikely that all of the α-BGT sites contain a known subunit other than α7. In addition, α-BGT sites from PC12 cells showed size and pharmacological similarities to homo-oligomeric chimeric α7 receptors expressed in tsA201 cells (Rakhilin et al., 1996), further supporting the hypothesis that homo-oligomeric α7 receptors may exist naturally as well as in heterologous expression systems.
Nicotine obtained from tobacco has complex psychopharmacological effects (Dani and Heinemann, 1996), but it is reasonable to hypothesize that nicotine acts, in part, on the hippocampus (Gray et al., 1996) to enhance learning and memory on various tasks (Levin, 1992; Ohno et al., 1993). The results of the present work suggest that the actions of nicotine on the hippocampus are likely to arise largely from activation, desensitization, or modification of α-BGT binding sites that contain the α7 subunit.
Although it may be surprising that mice with complete deficiency of the α7 subunit do not have gross or obvious abnormalities, these mice do demonstrate that the α7 subunit is not essential for normal development or for superficially normal neurological function. These mice may prove to have subtle phenotypic abnormalities, and they will be valuable in defining the functional role of the α7 subunitin vivo.
Footnotes
This work was supported by National Institutes of Health Grants NS-21229, DA-09411, DA-04077, and TW-04861.
Correspondence should be addressed to Dr. Arthur L. Beaudet, Department of Molecular and Human Genetics, Baylor College of Medicine, One Baylor Plaza, Room T619, Houston, TX 77030.
Dr. Orr-Urtreger’s present address: Tel-Aviv Sourasky Medical Center, Tel-Aviv, Israel.
REFERENCES
- 1.Albuquerque EX, Alkondon M. Initial characterization of the nicotinic acetylcholine receptors in rat hippocampal neurons. J Recept Res. 1991;11:1001–1021. doi: 10.3109/10799899109064693. [DOI] [PubMed] [Google Scholar]
- 2.Alkondon M, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. J Pharmacol Exp Ther. 1993;265:1455–1473. [PubMed] [Google Scholar]
- 3.Alonso JR, Amaral DG. Cholinergic innervation of the primate hippocampal formation. I. Distribution of choline acetyltransferase immunoreactivity in the Macaca fascicularis and Macaca mulatta monkeys. J Comp Neurol. 1995;355:135–170. doi: 10.1002/cne.903550202. [DOI] [PubMed] [Google Scholar]
- 4.Bertrand D, Changeux J-P. Nicotinic receptor: an allosteric protein specialized for intercellular communication. Neuroscience. 1995;7:75–90. [Google Scholar]
- 5.Broide RS, O’Connor LT, Smith MA, Smith JAM, Leslie I. Developmental expression of α7 neuronal nicotinic receptor messenger RNA in rat sensory cortex and thalamus. Neuroscience. 1995;67:83–94. doi: 10.1016/0306-4522(94)00623-d. [DOI] [PubMed] [Google Scholar]
- 6.Bullard DC, Kunkel EJ, Kubo H, Hicks MJ, Lorenzo I, Doyle NA, Doerschuk CM, Ley K, Beaudet AL. Infectious susceptibility and severe deficiency of leukocyte rolling and recruitment in E-selectin and P-selectin double mutant mice. J Exp Med. 1996;183:2329–2336. doi: 10.1084/jem.183.5.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen D, Patrick JW. The alpha-bungarotoxin-binding nicotinic acetylcholine receptor from rat brain contains only the alpha7 subunit. J Biol Chem. 1997;272:24024–24029. doi: 10.1074/jbc.272.38.24024. [DOI] [PubMed] [Google Scholar]
- 8.Clarke PBS, Schwartz RD, Paul SM, Pert CB, Pert A. Nicotinic binding in rat brain: autoradiographic comparison of [3H]acetylcholine, [3H]nicotine, and [125I]-α-bungarotoxin. J Neurosci. 1985;5:1307–1315. doi: 10.1523/JNEUROSCI.05-05-01307.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooper E, Couturier S, Ballivet M. Pentameric structure and subunit stoichiometry of a neuronal nicotinic acetylcholine receptor. Nature. 1991;350:235–238. doi: 10.1038/350235a0. [DOI] [PubMed] [Google Scholar]
- 10.Couturier S, Bertrand D, Matter J-M, Hernandez M-C, Bertrand S, Millar N, Valera S, Barkas T, Ballivet M. A neuronal nicotinic acetylcholine receptor subunit (α7) is developmentally regulated and forms a homo-oligomeric channel blocked by α-BTX. Neuron. 1990;5:847–856. doi: 10.1016/0896-6273(90)90344-f. [DOI] [PubMed] [Google Scholar]
- 11.Dains K, Hitzemann B, Hitzemann R. Genetics, neuroleptic response and the organization of cholinergic neurons in the mouse striatum. J Pharmacol Exp Ther. 1996;279:1430–1438. [PubMed] [Google Scholar]
- 12.Dani JA, Heinemann S. Molecular and cellular aspects of nicotine abuse. Neuron. 1996;16:905–908. doi: 10.1016/s0896-6273(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 13.Dineley-Miller K, Patrick J. Gene transcripts for the nicotinic acetylcholine receptor subunit, β4, are distributed in multiple areas of the rat central nervous system. Mol Brain Res. 1992;16:339–344. doi: 10.1016/0169-328x(92)90244-6. [DOI] [PubMed] [Google Scholar]
- 14.Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S. α9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell. 1994;79:705–715. doi: 10.1016/0092-8674(94)90555-x. [DOI] [PubMed] [Google Scholar]
- 15.Elmslie FV, Rees M, Williamson MP, Kerr M, Kjeldsen MJ, Pang KA, Sundqvist A, Friis ML, Chadwick D, Richens A, Covanis A, Santos M, Arzimanoglou A, Panayiotopoulos CP, Curtis D, Whitehouse WP, Gardiner RM. Genetic mapping of a major susceptibility locus for juvenile myoclonic epilepsy on chromosome 15q. Hum Mol Genet. 1997;6:1329–1334. doi: 10.1093/hmg/6.8.1329. [DOI] [PubMed] [Google Scholar]
- 16.Freedman R, Adler LE, Bickford P, Byerley W, Coon H, Cullum CM, Griffith JM, Harris JG, Leonard S, Miller C, Myles-Worsley M, Nagamoto HT, Rose G, Waldo M. Schizophrenia, nicotinic receptors, and cigarette smoking. Harvard Rev Psychiatry. 1994;2:179–192. doi: 10.3109/10673229409017136. [DOI] [PubMed] [Google Scholar]
- 17.Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, Polymeropoulos M, Holik J, Hopkins J, Hoff M, Rosenthal J, Waldo MC, Reimherr F, Wender P, Yaw J, Young DA, Breese CR, Adams C, Patterson D, Adler LE, Kruglyak L, Leonard S, Byerley W. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Natl Acad Sci USA. 1997;94:587–592. doi: 10.1073/pnas.94.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuchs JL. [125I]α-bungarotoxin binding marks primary sensory areas of developing rat neocortex. Brain Res. 1989;501:223–234. doi: 10.1016/0006-8993(89)90640-9. [DOI] [PubMed] [Google Scholar]
- 19.Geneser-Jensen FA, Blackstad TW. Distribution of acetylcholinesterase in the hippocampal region of the guinea pig. I. Entorhinal area, parasubiculum, and presubiculum. Z Zellforsch. 1971;114:460–481. doi: 10.1007/BF00325634. [DOI] [PubMed] [Google Scholar]
- 20.Gotti C, Hanke W, Moretti M, Longhi R, Balestra B, Briscini L, Clementi F. α-Bungarotoxin receptor subtypes. In: Clarke PBS, Quik M, Adlkofer F, Thurau K, editors. Effects of nicotine on biological systems, Pt II. Birkhauser Verlag; Boston: 1995. pp. 37–44. [Google Scholar]
- 21.Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- 22.Johnson DS, Martinez J, Elgoyhen AB, Heinemann SF, McIntosh JM. α-Conotoxin Imi exhibits subtype-specific nicotinic acetylcholine receptor blockade: preferential inhibition of homomeric α7 and α9 receptors. Mol Pharmacol. 1995;48:194–199. [PubMed] [Google Scholar]
- 23.Kafka SH, Corbett R. Selective adenosine A2A receptor/dopamine D2 receptor interactions in animal models of schizophrenia. Eur J Pharmacol. 1996;295:147–154. doi: 10.1016/0014-2999(95)00668-0. [DOI] [PubMed] [Google Scholar]
- 24.Levin DL. Nicotinic systems and cognitive function. Psychopharmacology. 1992;108:417–431. doi: 10.1007/BF02247415. [DOI] [PubMed] [Google Scholar]
- 25.McGehee DS, Heath MJS, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- 26.McGehee DS, Role LW. Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu Rev Physiol. 1995;57:521–546. doi: 10.1146/annurev.ph.57.030195.002513. [DOI] [PubMed] [Google Scholar]
- 27.McGehee DS, Role LW. Memories of nicotine. Nature. 1996;383:670–671. doi: 10.1038/383670a0. [DOI] [PubMed] [Google Scholar]
- 28.Miner LL, Collins AC. Strain comparison of nicotine-induced seizure sensitivity and nicotinic receptors. Pharmacol Biochem Behav. 1989;33:469–475. doi: 10.1016/0091-3057(89)90532-7. [DOI] [PubMed] [Google Scholar]
- 29.Ohno M, Yamamoto T, Watanabe S. Blockade of hippocampal nicotinic receptors impairs working memory but not reference memory in rats. Pharmacol Biochem Behav. 1993;45:89–93. doi: 10.1016/0091-3057(93)90091-7. [DOI] [PubMed] [Google Scholar]
- 30.Orr-Urtreger A, Seldin MF, Baldini A, Beaudet AL. Cloning and mapping of the mouse α7-neuronal acetylcholine receptor. Genomics. 1995;26:399–402. doi: 10.1016/0888-7543(95)80228-e. [DOI] [PubMed] [Google Scholar]
- 31.Picciotto MR, Zoll M, Lena C, Bessis A, Lallemand Y, LeNovere N, Vincent P, Pich EM, Brulet P, Changeux J-P. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature. 1995;374:65–67. doi: 10.1038/374065a0. [DOI] [PubMed] [Google Scholar]
- 32.Pugh PC, Berg DK. Neuronal acetylcholine receptors that bind α-bungarotoxin mediate neurite retraction in a calcium-dependent manner. J Neurosci. 1994;14:889–896. doi: 10.1523/JNEUROSCI.14-02-00889.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rakhilin SV, Atluri P, Drisdel RC, Ko E, Rangwala F, Salman WN, Green WN. α7/5HT3 chimeric homomers: similarity to PC12 α-bungarotoxin receptors. Neuroscience. 1996;22:1521. [Google Scholar]
- 34.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. Cold Spring Harbor Laboratory; A laboratory manual. Cold Spring Harbor, NY: 1989. [Google Scholar]
- 35.Sargent PB. The diversity of neuronal nicotinic acetylcholine receptors. Annu Rev Neurosci. 1993;16:403–443. doi: 10.1146/annurev.ne.16.030193.002155. [DOI] [PubMed] [Google Scholar]
- 36.Schoepfer R, Conroy WG, Whiting P, Gore M, Lindstrom J. Brain α-bungarotoxin binding protein cDNAs and mAbs reveal subtypes of this branch of the ligand-gated ion channel gene superfamily. Neuron. 1990;5:35–48. doi: 10.1016/0896-6273(90)90031-a. [DOI] [PubMed] [Google Scholar]
- 37.Séguéla P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sloviter RS. A simplified Timm stain procedure compatible with formaldehyde fixation and routine paraffin embedding of rat brain. Brain Res Bull. 1982;8:771–774. doi: 10.1016/0361-9230(82)90104-6. [DOI] [PubMed] [Google Scholar]
- 39.Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 40.Steinlein OK, Mulley JC, Propping P, Wallace RH, Phillips HA, Sutherland GR, Scheffer IE, Berkovic SF. A missense mutation in the neuroinal nicotinic acetylcholine receptor α4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 1995;11:201–203. doi: 10.1038/ng1095-201. [DOI] [PubMed] [Google Scholar]
- 41.Stitzel JA, Robinson SF, Marks MJ, Collins AC (1997) Differences in response to nicotine are determined by genetic factors. Adv Pharmacol Sci 279–284.
- 42.Treinin M, Chalfie M. A mutated acetylcholine receptor subunit causes neuronal degeneration in C. elegans. Neuron. 1995;14:871–877. doi: 10.1016/0896-6273(95)90231-7. [DOI] [PubMed] [Google Scholar]
- 43.Whiting PJ, Lindstrom JM. Characterization of bovine and human neuronal nicotinic acetylcholine receptors using monoclonal antibodies. J Neurosci. 1988;8:3395–3404. doi: 10.1523/JNEUROSCI.08-09-03395.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wonnacott S. α-Bungarotoxin binds to low-affinity nicotine binding sites in rat brain. J Neurochem. 1986;47:1706–1712. doi: 10.1111/j.1471-4159.1986.tb13078.x. [DOI] [PubMed] [Google Scholar]
- 45.Woolf NJ. Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol. 1991;37:475–524. doi: 10.1016/0301-0082(91)90006-m. [DOI] [PubMed] [Google Scholar]
- 46.Zarei MM, Dani JA. Structural basis for explaining open-channel blockage of the NMDA receptor. J Neurosci. 1995;15:1446–1454. doi: 10.1523/JNEUROSCI.15-02-01446.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Z-W, Coggan JS, Berg DK. Synaptic currents generated by neuronal acetylcholine receptors sensitive to α-bungarotoxin. Neuron. 1996;17:1231–1240. doi: 10.1016/s0896-6273(00)80253-6. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Z, Vijayaraghavan S, Berg D. Neuronal acetylcholine receptors that bind α-bungarotoxin with high affinity function as ligand-gated ion channels. Neuron. 1994;12:167–177. doi: 10.1016/0896-6273(94)90161-9. [DOI] [PubMed] [Google Scholar]
- 49.Zorumski CF, Thio LL, Isenberg KE, Clifford DB. Nicotinic acetylcholine currents in cultured postnatal rat hippocampal neurons. Mol Pharmacol. 1992;41:931–936. [PubMed] [Google Scholar]