

Abstract
Brain-derived neurotrophic factor (BDNF) is one of neurotrophins involved in the development and maintenance of both the peripheral nervous system and CNS. Although the expression of BDNF and its receptor TrkB still occurs in the adult stage, their physiological role in the mature CNS is not fully understood. In the present study we examined in detail the possibility that BDNF modulates synaptic neurotransmissions by using patch-clamp technique in rat hippocampal CA1 region. BDNF (20–100 ng/ml) did not show any appreciable effect on evoked EPSCs, but it markedly reduced both evoked and spontaneous IPSCs within 5 min, and the reduction persisted while BDNF was present. BDNF also attenuated GABAA receptor-mediated response to applied GABA. However, BDNF failed to attenuate IPSCs when the postsynaptic pyramidal neuron was loaded intracellularly with 200 nmK252a, an alkaloid that inhibits the kinase activity of Trk receptor family, through the patch pipette. Intracellular application of 200 nm K252b, a weaker inhibitor of Trk-type kinase, did not affect the inhibition. The attenuating effect also was prevented by postsynaptic injection of U73122 (5 μm), a broad-spectrum PLC inhibitor, and by strong chelation of intracellular Ca2+ with 10 mm BAPTA. These data suggest that BDNF modulates GABAA synaptic responses by postsynaptic activation of Trk-type receptor and subsequent Ca2+mobilization in the CNS.
Keywords: BDNF, GABAA receptor, disinhibition, plasticity, LTP, hippocampus
Brain-derived neurotrophic factor (BDNF) is one of the neurotrophins involved in the development and maintenance of both the peripheral nervous system and CNS. During brain development neurotrophins and their receptors display distinct stage- and tissue-specific patterns of expression (Ernfors et al., 1990a; Phillips et al., 1990; Merlio et al., 1992). BDNF mRNA is observed in the embryonic stage and is still present in the postnatal and adult stages (Ernfors et al., 1990b; Maisonpierre et al., 1990; Freidman et al., 1991). In the adult stage its expression level is modulated dramatically by neuronal activity (Falkenberg et al., 1992; Patterson et al., 1992; Rocamora et al., 1992; Bengzon et al., 1993; Kokaia et al., 1993). Moreover, expression of TrkB, a functional BDNF receptor, not only increases during embryonic development but also continues to increase until several weeks after birth in the hippocampus (Masana et al., 1993; Ringstedt et al., 1993). These observations suggest that in the adult CNS dynamic change in BDNF level still can trigger neuronal plasticity via activation of TrkB. Indeed, neurotrophins are secreted by neurons in both a constitutive and an activity-dependent manner (Blöchl and Thoenen, 1995, 1996; Griesbeck et al., 1995; Thoenen, 1995; Goodmann et al., 1996). Additionally, BDNF induces long-lasting enhancement of synaptic transmission (Kang and Schuman, 1995) and facilitates the induction of long-term potentiation (LTP) in the hippocampus (Figurov et al., 1996); however, the site and mechanism of action of BDNF remain unclear, because the effect of BDNF on synaptic transmission has been assessed by analyzing field (extracellular) potential changes in most cases. In the present study we examined in detail the possibility that BDNF regulates synaptic neurotransmissions by using the patch-clamp technique in rat hippocampal CA1 region and have presented evidence that BDNF directly modulates GABAAsynaptic responses by postsynaptic activation of Trk-type receptor and subsequent Ca2+ mobilization in the CNS.
MATERIALS AND METHODS
Slice preparation. Male Wistar rats, 12–18 d old, were used to prepare 250-μm-thick hippocampal slices in ice-cold artificial cerebrospinal fluid (ACSF). Rats were decapitated, and the brains were removed. Transverse hippocampal slices were cut with a vibratome. Slices were incubated at room temperature (22–25°C) in ACSF oxygenated with 95% O2 and 5% CO2 in a holding chamber at least for 1 hr. Then they were plated in the recording chamber under a platinum-supported nylon mesh and perfused at a rate of 2 ml/min with ACSF at room temperature. ACSF was composed of (in mm): NaCl 124, NaHCO3 26, KCl 2, KH2PO4 1.24, MgSO4 5, CaCl2 2, glucose 10, and ascorbic acid 0.4.
Electrophysiological recordings. Whole-cell patch-clamp recordings were performed with a blind approach. Patch electrodes (2–4 MΩ) were fabricated from borosilicate glass. The pipette solution contained (in mm): CsCl 140, CaCl2 0.2, EGTA 2, NaGTP 0.4, MgATP 4, and HEPES 10, pH 7.2 with CsOH. In some experiments 10 mm BAPTA and 125 mm CsCl were substituted for 2 mm EGTA and 140 mm CsCl, respectively. Chloride was used as a permeant anion in the internal solution to reverse the polarity and increase the amplitude of GABAA receptor-mediated IPSCs. LidocaineN-ethyl bromide (QX314, 5 mm; Research Biochemicals, Natick, MA) also was included in the recording electrode solution to prevent depolarizing IPSCs from triggering action potentials. Voltage-clamp (−60 mV) recordings from hippocampal CA1 pyramidal cells were obtained with an Axoclamp 1D amplifier, data were digitized by a TL-1 DMA interface, and acquisition and analysis were performed with the pCLAMP computer program (Axon Instruments, Foster City, CA). The series resistance (usually 10–25 MΩ) was monitored throughout the experiment; if it changed significantly, the experiment was rejected. Evoked EPSCs were obtained by stimulation of the Schaffer collateral–commissural afferents every 20 sec with bipolar tungsten electrodes in the presence of 20 μmpicrotoxin to block inhibitory inputs. In most cases evoked monosynaptic IPSCs were obtained by stratum pyramidal (SP) stimulation at 0.1 Hz in the presence of 20 μm6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 50 μm2-amino-5-phosphonovaleric acid (APV) to block excitatory inputs. Stable recordings of PSCs usually were obtained 20–30 min after the rupture of the patch and continued for at least 30 min. In gramicidin perforated-patch recordings another pair of stimulating electrodes was positioned within 500 μm from the recording electrode in the stratum lacunosum-moleculare (SL-M) to obtain another type of GABAA-mediated IPSCs (Pearce, 1993). IPSCs recorded for every 2 min were averaged for evaluation of their amplitude.
Gramicidin perforated-patch recording. In simultaneous recording of two types of IPSCs, we used the gramicidin perforated-patch method. The antibiotic gramicidin (Sigma, St. Louis, MO), when incorporated into lipid membranes, forms pores that are permeable to monovalent cations and small uncharged molecules. This method can avoid diffusion of cytoplasmic factors out of the recorded neuron and disturbance of intrinsic intracellular Cl−concentration. It is particularly important to avoid the latter problem to gain stable and long-lasting recordings of IPSCs generated at synapses distant from soma, such as IPSCs evoked by SL-M stimulation, because physiologically excess Cl− in the pipette does not distribute quickly to dendrites. The internal solution for gramicidin recordings was prepared according to the methods reported previously (Kyrozis and Reichling, 1995). Briefly, before each perforated-patch experiment gramicidin (2 mg/ml) was dissolved in dimethyl sulfoxide (DMSO). The gramicidin–DMSO solution was added to the electrode solution (composed of 150 mm KCl and 10 mmHEPES, pH 7.2 with KOH) to give a final concentration of 5 μg/ml. In filling the electrode with gramicidin-containing solution, we usually omitted the process of tip prefilling with gramicidin-free solution because in our cases we encountered no significant difficulty in seal formation.
Reagents. Human recombinant BDNF was dissolved (100 μg/ml) in phosphate buffer containing 0.1% BSA and stored at −30°C. Before every experiment the stock solution was diluted with ACSF to the final concentration (20–100 ng/ml). Therefore, all experiments were performed in ACSF containing 0.0001% BSA, which did not have any detectable effect on EPSCs or IPSCs. K252a, K252b (Kyowa Medex, Tokyo, Japan), and U73122 (Biomol Research Lab, Plymouth Meeting, PA) were dissolved in DMSO. The final concentration of DMSO, 0.1% in bath-applied ACSF or 0.05% in patch-pipette solution, did not have any significant effect on synaptic responses.
RESULTS
Modulative effect of BDNF on IPSCs without affecting EPSCs
CA1 pyramidal neurons are innervated by fast excitatory synaptic inputs from the CA3 region. Stimulation of Schaffer collaterals produced non-NMDA-type (CNQX-sensitive) glutamate receptor-mediated EPSCs in voltage-clamped (−60 mV) CA1 pyramidal cells in whole-cell recordings in the presence of a GABAAreceptor antagonist, bicuculline (20 μm; see Fig.2A). On the other hand, they are also under inhibitory innervation of neighboring interneurons. SP stimulation, in the presence of 50 μm APV and 20 μm CNQX to block glutamatergic inputs, produced bicuculline- and picrotoxin-sensitive GABAA receptor-mediated inward-directed IPSCs (Fig. 1A). BDNF (20–100 ng/ml) showed no apparent effects on the amplitude of evoked EPSCs (mean ± SEM; % of baseline, 90.5 ± 7.9;n = 6) (Fig. 2B,C). However, the amplitude of evoked IPSCs was reduced markedly within 5 min after the start of BDNF perfusion, and this reduction persisted while BDNF was present (Fig. 1B,C). The attenuating effect of BDNF was concentration-dependent [mean ± SEM; % of baseline: BDNF 20 ng/ml, 70.9 ± 6.9 (n = 6),p < 0.05; 100 ng/ml, 55.6 ± 9.9 (n = 6), p < 0.05]. Although we did not analyze the recovery of the amplitude of IPSCs systematically, the washout of BDNF resulted in the recovery of the amplitude (>80% of the baseline response) in 5 of 12 neurons within 30 min. The reduced IPSCs did not recover in the rest of the neurons. In several cases we observed transient increases in the amplitude of IPSCs as well as that of EPSCs within 2 min after the start of 100 ng/ml BDNF perfusion (Figs. 1C, 2C). This enhancement was accompanied by an increase in the frequency of spontaneous postsynaptic currents (data not shown), although it was not statistically significant.
Fig. 2.
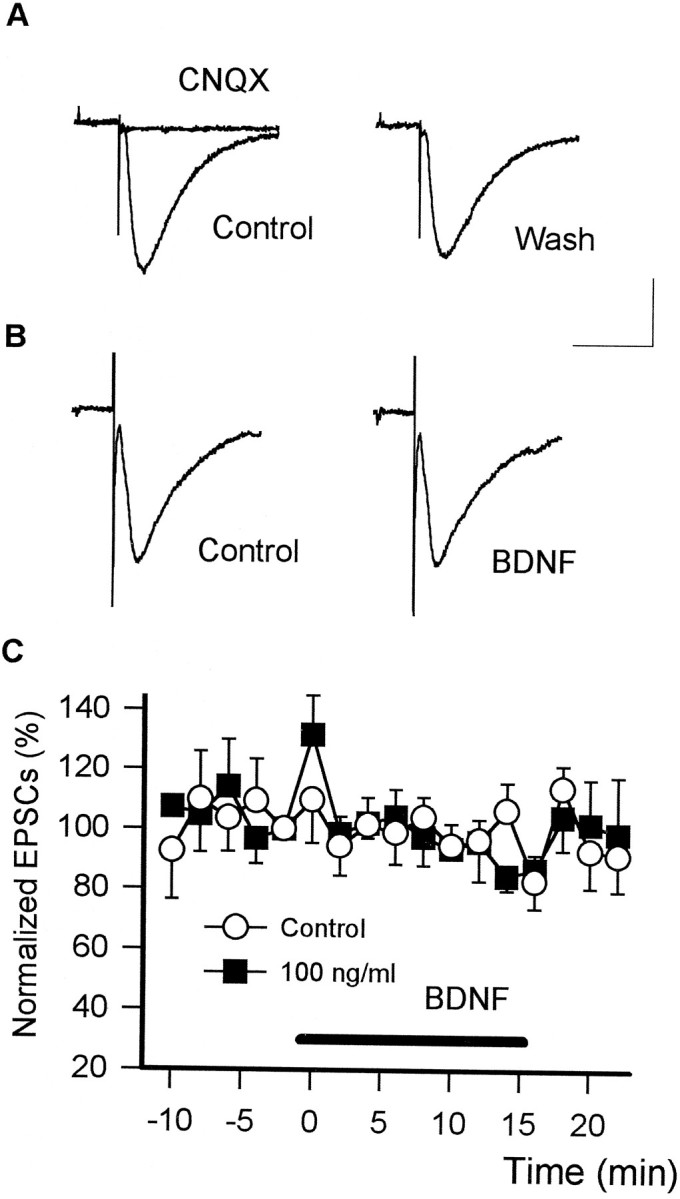
BDNF did not affect non-NMDA-type glutamate receptor-mediated EPSCs. A, Recording of the CNQX-sensitive EPSCs from voltage-clamped (−60 mV) CA1 pyramidal neurons. Stimulation of Schaffer collaterals produced the inward currents sensitive to 20 μm CNQX in the presence of 20 μm bicuculline. Representative traces are shown as the average of eight consecutive responses (calibration: 25 pA, 30 msec).B, Effect of BDNF on evoked EPSCs. BDNF (100 ng/ml, 15 min) did not significantly affect non-NMDA-type glutamate receptor-mediated EPSCs (right). Each trace represents the averaged EPSCs of eight consecutive sweeps (calibration: 40 pA, 30 msec). C, The time courses of changes in EPSCs. BDNF tended to increase the amplitude of EPSCs transiently (within 2 min) but had no significant effect on subsequent EPSCs. Data are indicated as mean ± SEM (n = 6).
Fig. 1.

BDNF attenuated GABAA-mediated IPSCs in CA1 pyramidal cells. A, Recording of the bicuculline-sensitive IPSCs from voltage-clamped (−60 mV) CA1 pyramidal neurons in the whole-cell mode. Electrical stimulation of the stratum pyramidal generates the inward currents in the presence of glutamate receptor blockers APV (50 μm) and CNQX (20 μm). The inward currents were blocked completely by a GABAA-receptor antagonist bicuculline (20 μm) (left traces). The right trace is the recovery after a 20 min wash of bicuculline (calibration: 100 pA, 50 msec). B, Effect of BDNF on evoked IPSCs. Bath-applied BDNF (100 ng/ml, 15 min) dramatically reduced the amplitude of IPSCs. Two representative IPSCs (left) are from the time points indicated in C. In this neuron IPSCs recovered almost completely after a 30 min washout of BDNF (right). Each trace represents the averaged IPSCs of eight consecutive sweeps (calibration: 100 pA, 50 msec). C, The time courses of inhibition of the amplitude of IPSCs. Perfusion of BDNF (indicated as ahorizontal solid bar) significantly reduced the amplitude of IPSCs (solid circles, 20 ng/ml;solid squares, 100 ng/ml;#p < 0.05, *p < 0.05, **p < 0.01, Dunnett test;n = 5–7). Data are indicated as mean ± SEM normalized to the amplitudes at the time point just before the application of BDNF.
Modulation of two types of inhibitory inputs
To confirm the suppressive effect of BDNF on IPSCs, we used the gramicidin-perforated patch-clamp technique (Kyrozis and Reichling, 1995), which avoids diffusion of cytoplasmic factors out of the recorded neuron and disturbance of intracellular Cl−concentration. CA1 pyramidal neurons receive inhibitory inputs from two sources, SP and SL-M. The two GABAA-mediated inhibitory responses are distinct in terms of physiological, pharmacological, and anatomical properties (Pearce, 1993; Pearce et al., 1995). Stimulation of either SP or SL-M evoked GABAA-mediated IPSCs in the perforated mode. The reversal potential of IPSCs evoked by SP or SL-M stimulation was 72.6 ± 4.6 or 75.8 ± 4.9 mV, respectively (n = 6). Therefore, each type of IPSCs was recorded as outward-directed currents when cells were voltage-clamped at −50 mV (Fig. 3A). In the perforated mode, once adequate access resistance (<30 MΩ) was achieved, we could obtain stable and long-lasting recordings of IPSCs for at least 1 hr. One of the typical simultaneous recordings of SP-evoked and SL-M-evoked IPSCs from the same neuron is shown in Figure 3. Application of 100 ng/ml BDNF silenced both types of IPSCs (Fig. 3B). In the perforated mode we could obtain similar results to those observed in the whole-cell mode. The reproducibility of the gramicidin-perforated mode confirmed that the attenuating effect of BDNF was not caused by experimental artifacts of the whole-cell mode.
Fig. 3.

Attenuation of two distinct types of inhibitory inputs to CA1 pyramidal neurons by BDNF in the gramicidin perforated-patch method. A, Stimulation of the stratum pyramidal (SP stim.) and the stratum lacunosum-moleculare (SL-M stim.) produced distinct types of outward-directed IPSCs in the same CA1 pyramidal cell voltage-clamped at −50 mV. In this mode BDNF (100 ng/ml) also reduced both types of IPSCs. The representative traces are from the time points indicated in B. B, The typical time courses of reduction of the amplitude of IPSCs generated by stimulation of two sources, SP (open circles) and SL-M (solid circles). BDNF at 100 ng/ml (horizontal solid bar) silenced both types of inhibitory inputs. Data are plotted as the mean of eight IPSCs evoked by alternative stimulation of SP or SL-M in the same neuron.
Involvement of a postsynaptic Trk-type receptor
BDNF specifically binds to TrkB, a neurotrophin receptor, including the catalytic domain of tyrosine kinase. When hippocampal slices were pretreated extracellularly with 200 nm K252a, an alkaloid that inhibits the kinase activity of the Trk receptor family (Knüsel and Hefti, 1992), BDNF failed to reduce the amplitude of IPSCs (Fig. 4A, solid squares), suggesting that BDNF requires Trk-type receptor activation for the depression of IPSCs. Attenuation of the amplitude of IPSCs by BDNF also was prevented when the postsynaptic pyramidal neuron was loaded intracellularly with K252a by use of a patch pipette containing 200 nm K252a (Fig. 4A, open squares), although it was not inhibited by intracellularly applied 200 nm K252b (Fig. 4C), which is a weaker kinase inhibitor of Trk-type [mean ± SEM; % of baseline: extracellular (ext) K252a, 90.2 ± 10.6 (n = 5); intracellular (int) K252a, 88.5 ± 13.0 (n = 5); int K252b, 61.6 ± 6.1 (n = 4), p< 0.05]. These data suggest that BDNF modulates IPSCs via activation of a postsynaptic Trk-type receptor.
Fig. 4.
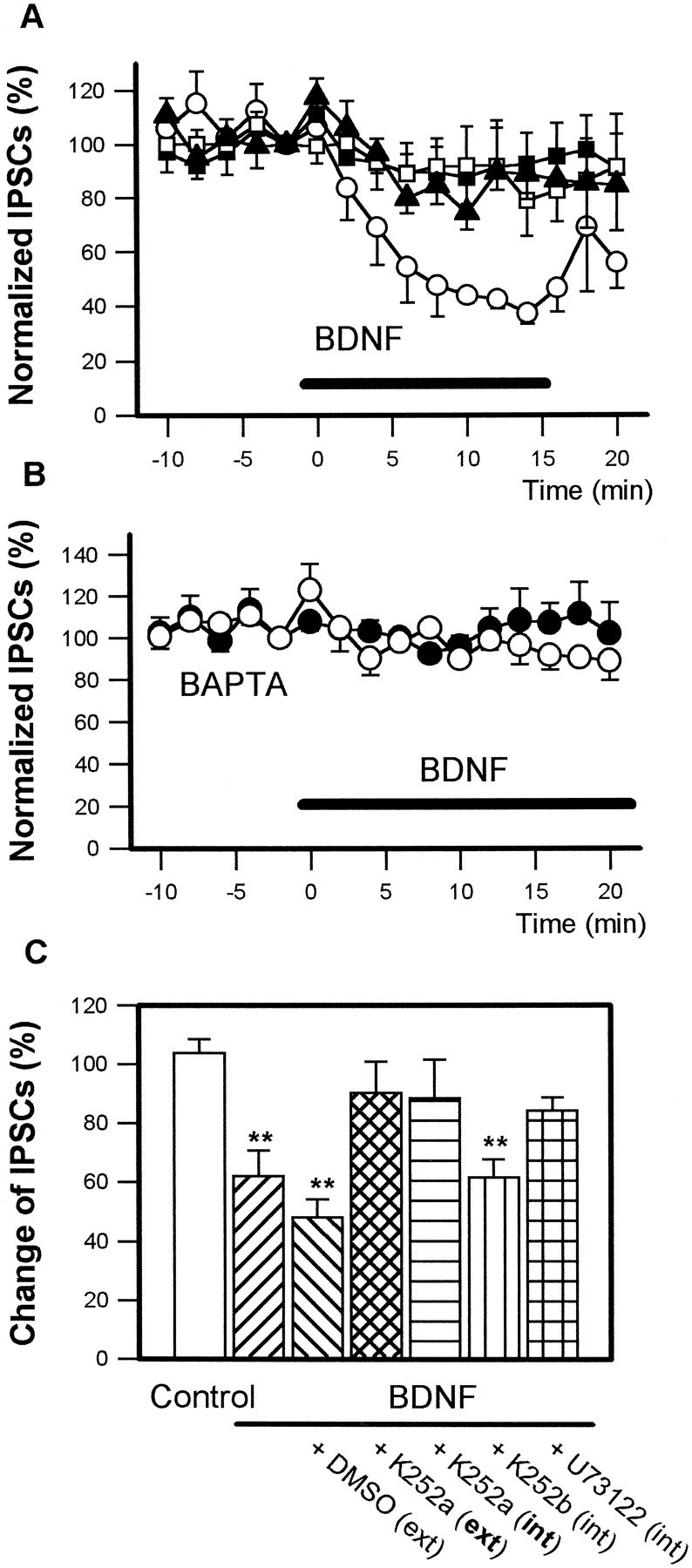
BDNF-induced inhibition of IPSCs requires postsynaptic activation of Trk-type receptor and subsequent Ca2+ mobilization. A, Perfusion of 100 ng/ml BDNF (horizontal solid bar) decreased the amplitude of IPSCs (open circles). Incubation of slices extracellularly with 200 nm K252a for 1 hr before the experiment prevented the 100 ng/ml BDNF-induced inhibition of IPSCs (solid squares). BDNF (100 ng/ml) also failed to attenuate IPSCs when CA1 pyramidal neurons were loaded intracellularly with 200 nm K252a (open squares) or 5 μm U73122 (solid triangles) in the whole-cell recording electrode. B, Intracellular BAPTA (10 mm) also blocked the 100 ng/ml BDNF-induced attenuation of IPSCs (open circles). Solid circlesindicate control responses in the absence of BDNF. C, Summarized graph from A. Data are ensemble averages measured between 8 and 14 min after the application of BDNF. Extracellular application (ext) of 200 nmK252a and intracellular application (int) of 200 nm K252a and 5 μm U73122 effectively prevented the attenuation by BDNF. Intracellular K252b (200 nm) was ineffective (**p < 0.01, Dunnett test; n = 5–6).
Involvement of [Ca2+]i mobilization
In hippocampal pyramidal neurons phospholipase C-γ1 (PLC-γ1) is abundant in the cell soma as well as in dendrites (Yamada et al., 1991). This enzyme, activated by TrkB, generates inositol triphosphate (IP3), which mobilizes intracellular Ca2+ from endoplasmic reticulum (Widmer et al., 1992, 1993; Zirrgiebel et al., 1995). Thus, we investigated the possibility of involvement of intracellular Ca2+ mobilization. BDNF failed to attenuate IPSCs when U73122 (5 μm), a broad-spectrum PLC inhibitor (Chen et al., 1994), was injected intracellularly into pyramidal neurons (Fig. 4A, solid triangles) [mean ± SEM; % of baseline: int U73122, 84.3 ± 4.4 (n = 5)] or when intracellular Ca2+ concentration ([Ca2+]i) was reduced with 10 mmBAPTA, a potent chelator (Fig. 4B) [mean ± SEM; % of baseline: control, 101.8 ± 7.6 (n = 7); BDNF 20 ng/ml, 97.1 ± 8.4 (n = 5); BDNF 100 ng/ml, 97.7 ± 3.7 (n = 7)]. These data suggest that BDNF reduces the amplitude of IPSCs by postsynaptic elevation of [Ca2+]i via IP3 production.
Reduction of postsynaptic responsiveness to applied GABA
As mentioned above, we found that the second messenger system in postsynaptic neurons is necessary for the reduction of IPSCs. However, whether postsynaptic Trk activation results in a decrease of postsynaptic GABAA receptor responsiveness or in attenuation of presynaptic GABA release remains to be clarified. Metabotropic change in postsynaptic cells may influence presynaptic GABA release. Previous works (Pilter and Alger, 1992, 1994) indicate that an increase in postsynaptic [Ca2+]iinduced by spike firing or depolarization reduces GABAergic synaptic inhibition by decreasing the release of GABA presynaptically. Therefore, we analyzed the inhibitory effect of BDNF on the responsiveness to applied GABA.
GABA was applied to a hippocampal slice by gravity-driven flow through a multibarreled tube (diameter 1.5 mm) positioned <200 μm from the recorded CA1 pyramidal neuron. Introduction of 10 μm GABA for 2 sec at the CA1 region at 3 or 5 min intervals induced GABAA receptor-mediated inward current. Because GABA was applied for only 2 sec, no apparent desensitization or running-down of the responses was observed at these intervals for at least 30 min [96.4 ± 4.8% (n = 4) of the initial control response]. Bath application of 50 ng/ml BDNF for 15 min decreased GABA-induced current to 64.4 ± 8.8% (n = 4) of the control response before treatment (Fig. 5). This suggests that BDNF reduces postsynaptic responsiveness to GABA.
Fig. 5.

Reduction of postsynaptic responsiveness to GABA. Gravity-driven introduction of 10 μm GABA for 2 sec (horizontal solid bars) at 5 min intervals through a multibarreled tube positioned <200 μm from the CA1 region elicited inward-directed GABAA receptor-mediated current in voltage-clamped (−60 mV) CA1 pyramidal cells (left). This response was attenuated by a 15 min perfusion of 50 ng/ml BDNF (middle). More than 30 min of washout of BDNF was necessary for the recovery (right), but it was incomplete.
Effects on spontaneous IPSCs
Experiments on stimulus-evoked IPSCs were supplemented with data on spontaneous events that result from action potentials generated by interneurons, because these events probably reflect accurately the normal functioning of GABA synapses (Mody et al., 1994). Numerous spontaneously occurring currents were recorded from CA1 pyramidal neurons in the presence of 50 μm APV and 20 μm CNQX. They were blocked by 10 μmpicrotoxin or bicuculline, indicating that they are IPSCs generated by spontaneous GABA release from interneurons (Fig. 6). Perfusion of a hippocampal slice with BDNF (50 ng/ml) for 15 min markedly reduced the size of the events (Fig.7A) and caused a significant decrease in the average amplitude of IPSCs (control, 32.7 ± 10.2 pA; BDNF, 23.9 ± 9.9 pA; p < 0.01, paired ttest; n = 5), although the total frequency was not changed significantly (control, 2.85 ± 0.62 Hz; BDNF, 2.56 ± 0.77 Hz). This decrease of the mean amplitude resulted in a leftward shift in the amplitude distribution (Fig. 7B).
Fig. 6.
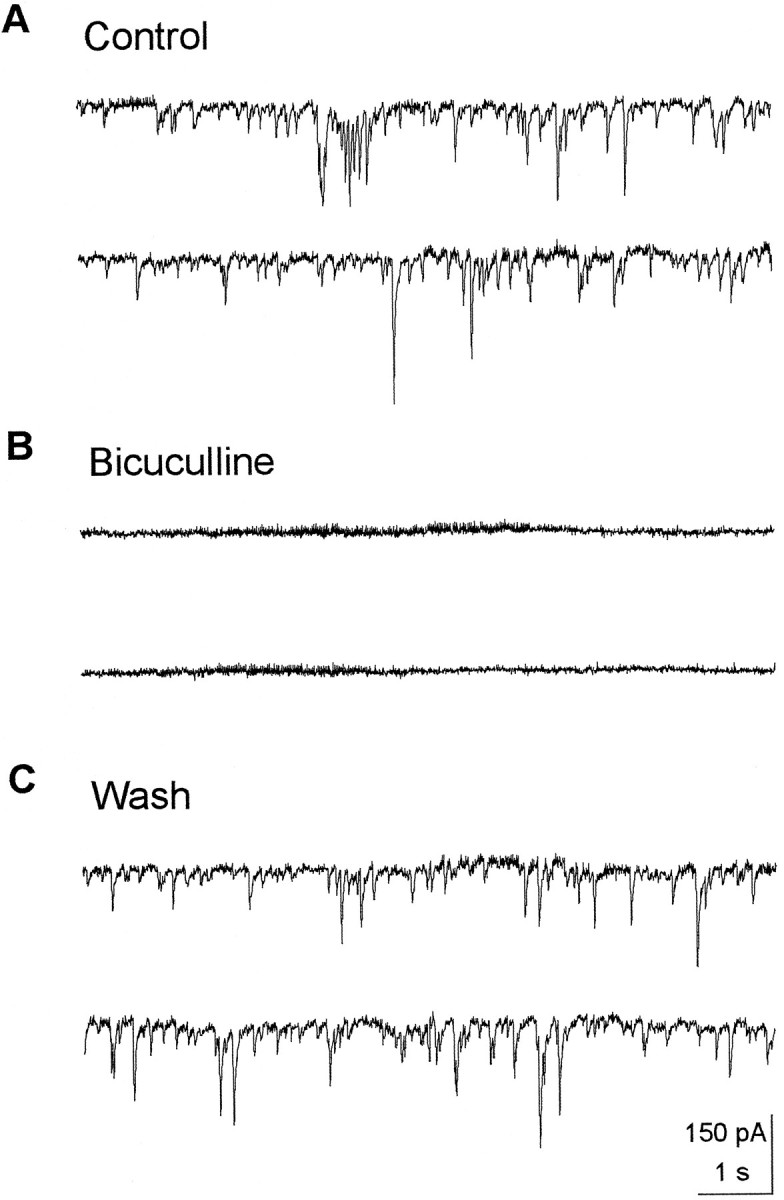
Recording of spontaneous IPSCs from CA1 pyramidal cells. A, Numerous spontaneous events were recorded from voltage-clamped (−60 mV) CA1 pyramidal cells in the presence of 50 μm APV and 20 μm CNQX. B, Bicuculline (20 μm), a GABAA receptor antagonist, completely blocked the spontaneous events.C, Shown is the recovery of the spontaneous events after the washout of bicuculline.
Fig. 7.
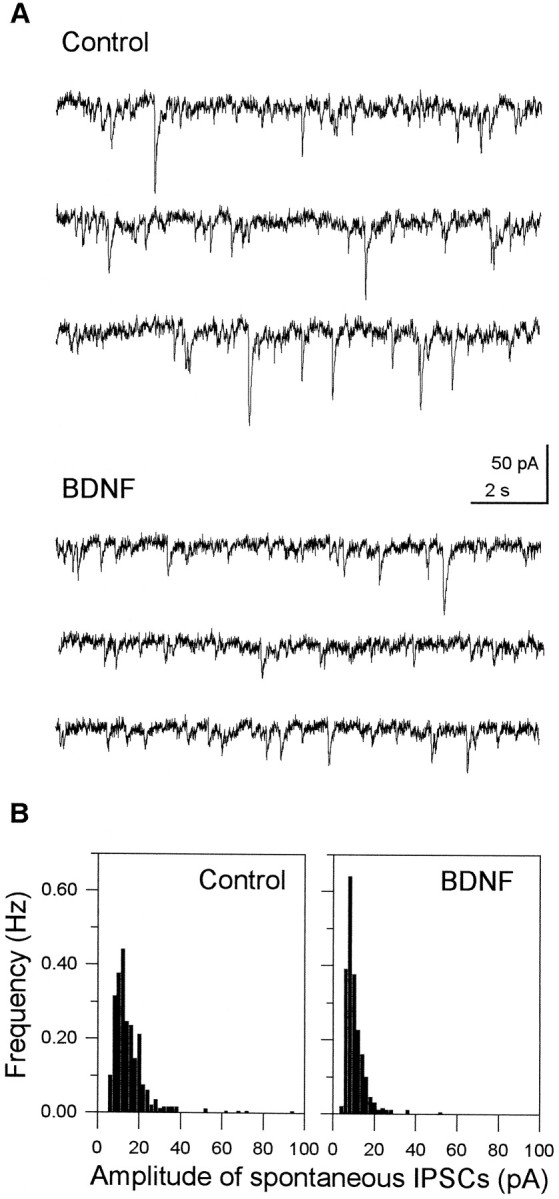
Reduction of the amplitude, but not the frequency, of spontaneous IPSCs by BDNF. BDNF (50 ng/ml, 15 min) reduced the amplitude, but not the frequency, of spontaneous IPSCs (lower traces). B, Summarized histograms from the data shown in A. BDNF caused a slight leftward shift in the amplitude distribution, reflecting a decrease in the mean size of these events. Data were sampled for 200 sec in the absence (left) or presence (right) of 50 ng/ml BDNF.
DISCUSSION
Inhibition of GABAA receptor-mediated IPSCs by BDNF
The present experiments demonstrated that BDNF regulates GABAA receptor responses via activation of a Trk-type receptor located in postsynaptic neurons. In the hippocampus excitatory neuronal networks are controlled by inhibitory innervation. An anatomical and electrophysiological approach identified three distinct types of GABAergic neurons (axo-axonic, basket, and bistratified cells) in the hippocampus. Each type of inhibitory neuron innervates a different domain of the surface of the principal neurons by activation of postsynaptic GABAA receptor. The axo-axonic cell primarily forms synapses on the axon initial segment of the principal cell, the basket cell primarily on the soma, and the bistratified cell on the apical and basal dendrites (Buhl et al., 1994). GABAergic interneurons mediate different inhibitory effects, such as recurrent inhibition, shunting of dendritic excitatory inputs, or governing of the firing threshold. Therefore, suppression of postsynaptic GABAA receptor by BDNF will facilitate neural excitation and generation of action potential firing. Indeed, exogenous BDNF promotes the induction of LTP in the young hippocampus; in turn, endogenous BDNF is suggested to be involved in the triggering mechanism of LTP in the adult (Figurov et al., 1996). Disinhibition induced by BDNF will participate in the induction of LTP, because elimination of GABAergic hyperpolarizing influence relieves voltage-dependent Mg2+ block of NMDA receptor-channel and facilitates the induction of LTP (Davies et al., 1991; Mott and Lewis, 1991). This also is supported by the results of mutant mice lacking BDNF (Korte et al., 1995), which showed significantly reduced LTP in the hippocampus. Moreover, judging from the figure, field EPSP of the mutant mice clearly was enhanced more by GABAA-receptor blocker than that of control mice, although the authors did not mention it. This result agrees with our data that BDNF suppresses GABAAsynaptic responses. GABAA-mediated synaptic transmission in the normal hippocampus may be regulated adequately by endogenous BDNF.
Kim et al. (1994) reported previously that neurotrophin-3 (NT-3), another member of neurotrophins, inhibits GABAA synaptic transmission in cultured embryonic cortical neurons. NT-3 primarily binds to TrkC but also can activate TrkB (Soppet et al., 1991; Squinto et al., 1991). Therefore, it was not clear whether TrkB participates in the regulation of GABAA receptor function. The present study using BDNF, a specific ligand for TrkB, indicates that TrkB participates in the modulation of GABAA receptor function, although TrkC also may regulate it. However, in contrast to BDNF expression, NT-3 expression level is downregulated in the adult brain by neuronal activity (Lindvall et al., 1992; Rocamora et al., 1992,1994; Bengzon et al., 1993) (but also see Patterson et al., 1992). In this respect, it is difficult to regard NT-3 as a signal that is secreted activity dependently, which triggers neural plasticity. BDNF seems to be a more promising candidate for the signal because of its mRNA upregulation (Falkenberg et al., 1992; Patterson et al., 1992;Rocamora et al., 1992; Bengzon et al., 1993; Kokaia et al., 1993) and secretion by neuronal activity (Griesbeck et al., 1995; Goodmann et al., 1996). Generally, expression of NT-3 is highest in immature CNS and dramatically decreases with maturation. On the contrary, expression of BDNF is low in developing regions of the CNS and increases as these regions mature (Maisonpierre et al., 1990). Therefore, TrkC activated by NT-3 may modulate synaptic transmission in the developing synapses, but not in the matured synapses.
BDNF did not affect non-NMDA-type glutamate receptor-mediated EPSCs in the present study. However, Levine et al. (1995) reported that BDNF increased both the frequency and amplitude of excitatory transmission in cultured embryonic neurons. Two contradictory results were reported from the analysis of field potentials in hippocampal slices. Kang and Schuman (1995) showed long-lasting BDNF-induced enhancement of synaptic transmission, whereas Figurov et al. (1996) recently reported no change of baseline response. We do not have a reasonable explanation for the discrepancy; however, Kang et al. (1996) reported that the perfusion rate is critical for the penetration of BDNF into the hippocampal slices.
In several cases we observed transient increases in the amplitude of EPSCs as well as that of IPSCs within 2 min after the start of BDNF perfusion (Fig. 1B,D), although it was not statistically significant. This enhancement was accompanied by an increase in frequency of spontaneous postsynaptic currents, which was similar to the results in cultured embryonic neurons (Leβmann et al., 1994). The transient effect is probably attributable to the presynaptic enhancement, because the intracellular application of K252a or BAPTA into postsynaptic neurons did not affect it.
Modulation of two types of inhibitory inputs
CA1 pyramidal neurons receive GABAA receptor-mediated inhibitory inputs from two sources, SP and SL-M. The two GABAA-mediated inhibitory responses are distinct in term of physiological, pharmacological, and anatomical properties (Pearce, 1993; Pearce et al., 1995). However, BDNF inhibited both IPSCs evoked by SP and SL-M stimulations, suggesting the colocalization of GABAA receptor and BDNF receptor TrkB. TrkB immunoreactivity is distributed widely to the pyramidal cell soma and dendrites (Zhou et al., 1993). Additionally, PLC-γ1 also is distributed widely and abundantly in the pyramidal cell (Yamada et al., 1991) in correspondence to the wide distribution of TrkB, suggesting the functional coupling between GABAA receptor and TrkB signaling cascade.
Involvement of postsynaptic TrkB activation
BDNF specifically binds to TrkB, a neurotrophin receptor, including the catalytic domain of tyrosine kinase (Soppet et al., 1991;Squinto et al., 1991). BDNF failed to reduce the amplitude of IPSCs when the recorded neuron was loaded intracellularly with K252a as well as when hippocampal slices were pretreated with K252a. K252a is an alkaloid that inhibits biological activities of neurotrophins and kinase activity of the Trk receptor family (Knüsel and Hefti, 1992). Therefore, these data suggest that BDNF modulates IPSCs via activation of a postsynaptic Trk-type receptor. K252b (200 nm) could not inhibit BDNF-induced reduction in the amplitude of IPSCs. It is a weaker inhibitor and is usually effective at concentrations higher than 1 μm (Widmer et al., 1992).
Involvement of intracellular Ca2+ mobilization
The binding of neurotrophins to the Trk receptor family initiates a signaling cascade involving phosphorylation of intracellular proteins on tyrosine residues. BDNF-activated TrkB rapidly associates with and phosphorylates the cytoplasmic proteins, PLC-γ1, SUS-associated neurotrophic factor-induced tyrosine-phosphor-ylated target (SNT), and Erk1 (Knüsel et al., 1994). In hippocampal pyramidal neurons PLC-γ1 is abundant in the cell soma as well as in the dendrites (Yamada et al., 1991). This enzyme, activated by TrkB, generates inositol triphosphate (IP3), which mobilizes intracellular Ca2+ from endoplasmic reticulum (Widmer et al., 1992, 1993;Zirrgiebel et al., 1995). BDNF failed to attenuate IPSCs when U73122, a broad-spectrum PLC inhibitor (Chen et al., 1994), was injected intracellularly into pyramidal neurons (Fig. 3A, solid triangles) or when intracellular Ca2+ concentration ([Ca2+]i) was reduced with 10 mmBAPTA, a potent chelator (Fig. 3B). These data suggest that BDNF reduces the amplitude of IPSCs by postsynaptic elevation of [Ca2+]i via IP3 production. BDNF stimulates PLC-γ1 phosphorylation within 20 sec (Widmer et al., 1993) and mobilizes intracellular Ca2+ within 1 min (Berninger et al., 1993). In this respect, there seems to be a time lag between elevation of [Ca2+]i and BDNF-induced reduction of the amplitude of IPSCs, suggesting the involvement of activation of some signal cascade after the rise in [Ca2+]i. GABAA receptor function is maintained by phosphorylation and disrupted by [Ca2+]i elevation (Stelzer et al., 1988;Marchenko, 1991; Gyenes et al., 1994; Moss et al., 1995). In this respect, dephosphorylation induced by a Ca2+-dependent phosphatase may be involved in the downstream of BDNF-activated signal transduction and lead to the attenuation of GABAA response (Stelzer and Shi, 1994; Chen and Wong, 1995).
Reduction in the amplitude of spontaneous IPSCs
BDNF reduced the amplitude of spontaneous IPSCs (sIPSCs), but not total frequency (Fig. 7). The spontaneous events that result from action potentials generated by interneurons probably reflect accurately the normal functioning of GABA synapse (Mody et al., 1994). These sIPSCs result from the highly synchronous release of GABA at several boutons, although stimulus-evoked IPSCs tends to reflect asynchronous release. Therefore, analysis of sIPSCs attributes the decrease of amplitude by BDNF to the reduction of postsynaptic receptor responsiveness to released GABA. This result coincides with the postsynaptic reduction of responses to exogenously applied GABA (Fig.5) and also is supported by the pharmacological attempts to block postsynaptic Trk-type receptor kinase activity (Fig. 4).
Conclusion
The present study demonstrates that BDNF directly modulates GABAA function, but not glutamate receptor function, via postsynaptic activation of Trk-type receptors. This disinhibition would enhance synaptic responses and facilitate the induction of LTP by eliminating dendritic shunting. BDNF not only enhances synaptic efficacy but also may promote the transduction of output signal.
Footnotes
We thank Sumitomo Pharmaceutical Company for providing human recombinant BDNF and Dr. Hiroshi Katsuki for excellent technical advice.
Correspondence should be addressed to Dr. Norio Matsuki at the above address.
REFERENCES
- 1.Bengzon J, Kokaia Z, Ernfors P, Kokaia M, Leanza G, Nilsson OG, Persson H, Lindvall O. Regulation of neurotrophin and trkA, trkB, and trkC tyrosine kinase receptor messenger RNA expression in kindling. Neuroscience. 1993;53:433–446. doi: 10.1016/0306-4522(93)90207-v. [DOI] [PubMed] [Google Scholar]
- 2.Berninger B, García DE, Inagaki N, Hahnel C, Lindoholm D. BDNF and NT-3 induce intracellular Ca2+ elevation in hippocampal neurons. NeuroReport. 1993;4:1303–1306. doi: 10.1097/00001756-199309150-00004. [DOI] [PubMed] [Google Scholar]
- 3.Blöchl A, Thoenen H. Characterization of nerve growth factor (NGF) release from hippocampal neurons: evidence for a constitutive and an unconventional sodium-dependent regulated pathway. Eur J Neurosci. 1995;7:1220–1228. doi: 10.1111/j.1460-9568.1995.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 4.Blöchl A, Thoenen H. Localization of cellular storage compartments and sites of constitutive and activity-dependent release of nerve growth factor (NGF) in primary cultures of hippocampal neurons. Mol Cell Neurosci. 1996;7:173–190. doi: 10.1006/mcne.1996.0014. [DOI] [PubMed] [Google Scholar]
- 5.Buhl EH, Halasy K, Somogyi P. Diverse sources of hippocampal unitary inhibitory postsynaptic potentials and the number of synaptic release sites. Nature. 1994;368:823–828. doi: 10.1038/368823a0. [DOI] [PubMed] [Google Scholar]
- 6.Chen P, Xie H, Sekar C, Gupta K, Wells A. Epidermal growth factor receptor-mediated cell motility: phospholipase C activity is required, but mitogen-activated protein kinase activity is not sufficient for induced cell movement. J Cell Biol. 1994;127:847–857. doi: 10.1083/jcb.127.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen QX, Wong RKS. Suppression of GABAA receptor responses by NMDA application in hippocampal neurons acutely isolated from the adult guinea-pig. J Physiol (Lond) 1995;482:353–362. doi: 10.1113/jphysiol.1995.sp020522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies CH, Starkey SJ, Pozza MF, Collingridge GL. GABAB autoreceptors regulate the induction of LTP. Nature. 1991;349:609–611. doi: 10.1038/349609a0. [DOI] [PubMed] [Google Scholar]
- 9.Ernfors P, Ibáñez CF, Ebendal T, Olson L, Persson H. Molecular cloning and neurotrophic activities of a protein with structural similarities to nerve growth factor: developmental and topographical expression in the brain. Proc Natl Acad Sci USA. 1990a;87:5454–5458. doi: 10.1073/pnas.87.14.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ernfors P, Wetmore C, Olson L, Persson H. Identification of cells in rat brain and peripheral tissues expressing mRNA for members of the nerve growth factor family. Neuron. 1990b;5:511–526. doi: 10.1016/0896-6273(90)90090-3. [DOI] [PubMed] [Google Scholar]
- 11.Falkenberg T, Ernfors P, Persson H, Lindefors N. Cortical trans-synaptic activation of tyrosine kinase receptor trkB messenger RNA expression in rat hippocampus. Neuroscience. 1992;51:883–889. doi: 10.1016/0306-4522(92)90527-9. [DOI] [PubMed] [Google Scholar]
- 12.Figurov A, Pozzo-Miller L, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 13.Friedman WJ, Ernfors P, Persson H. Transient and persistent expression of NT-3/HDNF mRNA in the rat brain during postnatal development. J Neurosci. 1991;11:1577–1584. doi: 10.1523/JNEUROSCI.11-06-01577.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodmann LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996;7:222–238. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- 15.Griesbeck O, Blöchl A, Carnahan JF, Nawa H, Thoenen H (1995) Characterization of brain-derived neurotrophic factor (BDNF) secretion from hippocampal neurons. Soc Neurosci Abstr 21[part 2]:1046.
- 16.Gyenes M, Wang Q, Gibbs TT, Farb DH. Phosphorylation factors control neurotransmitter and neuromodulator actions at the γ-aminobutyric acid type A receptor. Mol Pharmacol. 1994;46:542–549. [PubMed] [Google Scholar]
- 17.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;264:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 18.Kang H, Jia LZ, Suh KY, Tang L, Schuman EM. Determination of BDNF-induced hippocampal synaptic plasticity: role of the TrkB receptor and the kinetics of neurotrophin delivery. Learn Mem. 1996;3:188–196. doi: 10.1101/lm.3.2-3.188. [DOI] [PubMed] [Google Scholar]
- 19.Kim HG, Wang T, Olafsson P, Lu B. Neurotrophin 3 potentiates neuronal activity and inhibits γ-aminobutyratergic synaptic transmission in cortical neurons. Proc Natl Acad Sci USA. 1994;91:12341–12345. doi: 10.1073/pnas.91.25.12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knüsel B, Hefti F. K252 compounds: modulators of neurotrophin signal transduction. J Neurochem. 1992;59:1987–1996. doi: 10.1111/j.1471-4159.1992.tb10085.x. [DOI] [PubMed] [Google Scholar]
- 21.Knüsel B, Rabin SJ, Hefti F, Kaplan DR. Regulated neurotrophin receptor responsiveness during neural migration and early differentiation. J Neurosci. 1994;14:1542–1554. doi: 10.1523/JNEUROSCI.14-03-01542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kokaia Z, Bengzon J, Metsis M, Kokaia M, Persson H, Lindvall O. Coexpression of neurotrophins and their receptors in neurons of the central nervous system. Proc Natl Acad Sci USA. 1993;90:6711–6715. doi: 10.1073/pnas.90.14.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kyrozis A, Reichling DB. Perforated-patch recording with gramicidin avoids artifactual changes in intracellular chloride concentration. J Neurosci Methods. 1995;57:27–35. doi: 10.1016/0165-0270(94)00116-x. [DOI] [PubMed] [Google Scholar]
- 25.Leβmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurons. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- 26.Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindvall O, Ernfors P, Bengzon J, Kokaia Z, Smith ML, Siesjö BK, Persson H. Differential regulation of mRNAs for nerve growth factor, brain-derived neurotrophic factor, and neurotrophin 3 in the adult rat brain following cerebral ischemia and hypoglycemic coma. Proc Natl Acad Sci USA. 1992;89:648–652. doi: 10.1073/pnas.89.2.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 29.Marchenko SM. Mechanism of modulation of GABA-activated current by internal calcium in rat central neurons. Brain Res. 1991;546:355–357. doi: 10.1016/0006-8993(91)91502-r. [DOI] [PubMed] [Google Scholar]
- 30.Masana Y, Wanaka A, Kato H, Asai T, Tohyama M. Localization of trkB mRNA in postnatal development. J Neurosci Res. 1993;35:468–479. doi: 10.1002/jnr.490350503. [DOI] [PubMed] [Google Scholar]
- 31.Merlio JP, Ernfors MJ, Persson H. Molecular cloning of rat trkC and distribution of cells expressing messenger RNAs for members of the trk family in the rat central nervous system. Neuroscience. 1992;51:513–532. doi: 10.1016/0306-4522(92)90292-a. [DOI] [PubMed] [Google Scholar]
- 32.Mody I, Koninck D, Otis TS, Sotesz I. Bridging the cleft at GABA synapses in the brain. Trends Neurosci. 1994;17:517–525. doi: 10.1016/0166-2236(94)90155-4. [DOI] [PubMed] [Google Scholar]
- 33.Moss SJ, Gorrie GH, Amato A, Smart TG. Modulation of GABAA receptors by tyrosine phosphorylation. Nature. 1995;377:344–348. doi: 10.1038/377344a0. [DOI] [PubMed] [Google Scholar]
- 34.Mott DD, Lewis DV. Facilitation of the induction of long-term potentiation by GABAB receptors. Science. 1991;252:1718–1720. doi: 10.1126/science.1675489. [DOI] [PubMed] [Google Scholar]
- 35.Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNA. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- 36.Pearce RA. Physiological evidence for two distinct GABAA responses in rat hippocampus. Neuron. 1993;10:189–200. doi: 10.1016/0896-6273(93)90310-n. [DOI] [PubMed] [Google Scholar]
- 37.Pearce RA, Grunder SD, Faucher LD. Different mechanisms for use-dependent depression of two GABAA-mediated IPSCs in rat hippocampus. J Physiol (Lond) 1995;484:425–435. doi: 10.1113/jphysiol.1995.sp020675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Phillips HS, Hains HM, Laramee GR, Rosenthal A, Winslow A. Widespread expression of BDNF but not NT3 by target areas of basal forebrain cholinergic neurons. Science. 1990;250:290–294. doi: 10.1126/science.1688328. [DOI] [PubMed] [Google Scholar]
- 39.Pilter TA, Alger BE. Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci. 1992;12:4122–4132. doi: 10.1523/JNEUROSCI.12-10-04122.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pilter TA, Alger BE. Depolarization-induced suppression of GABAergic inhibition in rat hippocampal pyramidal cells: G-protein involvement in a presynaptic mechanism. Neuron. 1994;13:1447–1455. doi: 10.1016/0896-6273(94)90430-8. [DOI] [PubMed] [Google Scholar]
- 41.Ringstedt T, Lagercrantz H, Persson H. Expression of members of the trk family in the developing postnatal rat brain. Dev Brain Res. 1993;72:119–131. doi: 10.1016/0165-3806(93)90165-7. [DOI] [PubMed] [Google Scholar]
- 42.Rocamora N, Palacios JM, Mengod G. Limbic seizures induce a differential regulation of the expression of nerve growth factor, brain-derived neurotrophic factor, and neurotrophin-3 in the rat hippocampus. Mol Brain Res. 1992;13:27–33. doi: 10.1016/0169-328x(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 43.Rocamora N, Massieu L, Boddeke HWGM, Palacios JM, Mengod G. Differential regulation of the expression of nerve growth factor, brain-derived neurotrophic factor, and neurotrophin-3 mRNAs in adult rat brain after intrahippocampal injection of quinolinic acid. Mol Brain Res. 1994;26:89–98. doi: 10.1016/0169-328x(94)90078-7. [DOI] [PubMed] [Google Scholar]
- 44.Soppet D, Escandon D, Maragos J, Middlemas DS, Reid SW, Blair J, Burton LE, Stanton BR, Kaplan DR, Hunter T, Nikolics K, Parada LF. The neurotrophic factors, brain-derived neurotrophic factor and neurotrophin-3, are ligands for the trkB tyrosine kinase receptor. Cell. 1991;65:895–903. doi: 10.1016/0092-8674(91)90396-g. [DOI] [PubMed] [Google Scholar]
- 45.Squinto SP, Stitt TN, Aldrich TH, Davis S, Bianco SM, Radziejewski C, Glass DJ, Masiakowski P, Furth ME, Valenzuela DM, DiStefano PS, Yancopoulos GD. TrkB encodes a functional receptor for brain-derived neurotrophic factor and neurotrophin-3, but not nerve growth factor. Cell. 1991;65:885–893. doi: 10.1016/0092-8674(91)90395-F. [DOI] [PubMed] [Google Scholar]
- 46.Stelzer A, Shi H. Impairment of GABAA receptor function by N-methyl-d-aspartate-mediated calcium influx in isolated CA1 pyramidal cells. Neuroscience. 1994;62:813–828. doi: 10.1016/0306-4522(94)90479-0. [DOI] [PubMed] [Google Scholar]
- 47.Stelzer A, Kay AR, Wong RKS. GABAA-receptor function in hippocampal cells is maintained by phosphorylation factors. Science. 1988;241:339–341. doi: 10.1126/science.2455347. [DOI] [PubMed] [Google Scholar]
- 48.Stuart GJ, Sakmann B. Active propagation of somatic action potentials into neocortical pyramidal cell dendrites. Nature. 1994;367:69–72. doi: 10.1038/367069a0. [DOI] [PubMed] [Google Scholar]
- 49.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 50.Widmer HR, Knüsel B, Hefti F. Stimulation of phosphatidylinositol hydrolysis by brain-derived neurotrophic factor and neurotrophin-3 in rat cerebral cortical neurons developing in culture. J Neurochem. 1992;59:2113–2124. doi: 10.1111/j.1471-4159.1992.tb10102.x. [DOI] [PubMed] [Google Scholar]
- 51.Widmer HR, Kaplan DR, Rabin SJ, Beck KD, Hefti F, Knüsel B. Rapid phosphorylation of phospholipase Cγ1 by brain-derived neurotrophic factor and neurotrophin-3 in cultures of embryonic rat cortical neurons. J Neurochem. 1993;60:2111–2123. doi: 10.1111/j.1471-4159.1993.tb03496.x. [DOI] [PubMed] [Google Scholar]
- 52.Yamada M, Mizuguchi M, Rhee SG, Kim SU. Developmental changes of three phosphoinositide-specific phospholipase C isozymes in the rat nervous system. Dev Brain Res. 1991;59:7–16. doi: 10.1016/0165-3806(91)90023-c. [DOI] [PubMed] [Google Scholar]
- 53.Zhou XF, Parada LF, Soppet D, Rush RA. Distribution of TrkB tyrosine kinase immunoreactivity in the rat central nervous system. Brain Res. 1993;622:63–70. doi: 10.1016/0006-8993(93)90802-t. [DOI] [PubMed] [Google Scholar]
- 54.Zirrgiebel U, Ohga Y, Carter B, Berninger B, Inagaki N, Thoenen H, Lindoholm D. Characterization of TrkB receptor-mediated signaling pathways in rat cerebellar granule neurons: involvement of protein kinase C in neuronal survival. J Neurochem. 1995;65:2241–2250. doi: 10.1046/j.1471-4159.1995.65052241.x. [DOI] [PubMed] [Google Scholar]