

Abstract
The intracellular balance between pro- and antiapoptotic members of the Bcl-2 gene family is thought to regulate cell death. Targeted disruption of bcl-x, a death repressing member, causes massive cell death of immature neurons in the developing mouse CNS, whereas targeted disruption of bax, a proapoptotic member, blocks the death of specific populations of sympathetic and motor neurons. In the present study, mice deficient in both Bcl-xL and Bax (bcl-x−/−/bax−/−) are used to examine the relative significance and potential interactions of Bcl-xL and Bax during early CNS development.bcl-x−/−/bax−/−mice demonstrate greatly reduced levels of apoptosis both in vivo and in vitro compared with the CNS of Bcl-xL-deficient mice, as assessed by histology and terminal deoxytransferase-mediated deoxyuridine triphosphate nick end-labeling. Bax-deficient mice, however, contain occasional apoptotic cells in the developing CNS, and cultures of bax-deficient telencephalic cells demonstrate similar levels of apoptosis as wild-type cultures. These results suggest that Bax critically interacts with Bcl-xL to regulate survival of immature neurons, but indicate that other cell death regulating proteins, in addition to Bcl-xL and Bax, also function during CNS development.
Keywords: apoptosis, programmed cell death, bcl-x, bax, bcl-2, development
Bcl-xL is a member of the Bcl-2 gene family. Members of this family regulate cell death by either promoting or reducing apoptosis in response to a variety of signals (Reed, 1994;Craig, 1995). Overexpression of Bcl-xL or Bcl-2 blocks apoptosis of lymphocytes (Vaux et al., 1988; Hockenbery et al., 1990;Boise et al., 1993) and sympathetic neurons (Garcia et al., 1992;Allsopp et al., 1993; Frankowski et al., 1995; Gonzalez-Garcia et al., 1995; Greenlund et al., 1995) after trophic factor withdrawal. Other family members, such as Bax, Bad, Bak, and Bcl-xS, can block the antiapo-ptotic effects of Bcl-xL or Bcl-2 (Boise et al., 1993; Oltvai et al., 1993; Chittenden et al., 1995b;Farrow et al., 1995; Kiefer et al., 1995; Yang et al., 1995; Minn et al., 1996). Bcl-2 family members contain three highly conserved homology regions (BH1, BH2, and BH3) that mediate protein–protein interactions, allowing various family members to form homo- and heterodimers (Yin et al., 1994; Chittenden et al., 1995a). This has led to the suggestion that the intracellular balance between proapoptotic and antiapoptotic members may serve as a rheostat to ultimately regulate whether a cell lives or dies in response to specific stimuli (Oltvai et al., 1993; Oltvai and Korsmeyer, 1994; Krajewski et al., 1995; Sedlak et al., 1995; Gillardon et al., 1996).
bcl-x is important for immature neuron survival.bcl-x is alternatively spliced intobcl-xL andbcl-xS, but onlybcl-xL is expressed in the mouse CNS (Boise et al., 1993; Gonzalez-Garcia et al., 1994; Krajewski et al., 1994b). bcl-xL expression is low in the ventricular zone and is upregulated in postmitotic cells of the intermediate zone (Motoyama et al., 1995). Targeted disruption ofbcl-x results in a massive increase in apoptosis in the intermediate zone of developing embryonic spinal cord and brainstem, and in dorsal root ganglia (DRG), whereas neuronal precursor cells in the ventricular zone are unaffected (Motoyama et al., 1995).bcl-x-deficient (bcl-x−/−) mice die around embryonic day (E) 13, and therefore, histological examination of the bcl-x−/− telencephalon, which consists largely of undifferentiated ventricular zone cells at E13, does not determine whether Bcl-xL regulates telencephalic neuron survival. bcl-x−/− E12 telencephalic cells grown 48 hr in low serum concentrations contain more apoptotic cells than wild-type cultures (Roth et al., 1996b), and adult chimeric mice demonstrate a reduced percentage of telencephalic neurons derived frombcl-x−/− embryonic stem (ES) cells compared with the ES cell contribution to non-neuronal tissues (Havlioglu et al., 1996), demonstrating that bcl-x plays a significant role in telencephalic development.
Based on the importance of a functional bcl-x gene and on potential interactions between members of this gene family, it can be hypothesized that proapoptotic members of the Bcl-2 gene family critically interact with Bcl-xL to regulate immature neuron survival. The proapoptotic Bax protein forms heterodimers with the largest number of other family members (Sato et al., 1994; Sedlak et al., 1995), and Bax dimerizes with Bcl-xL, blocking the ability of Bcl-xL to prevent apoptosis (Sedlak et al., 1995). Bax is expressed at high levels in adult CNS (Oltvai et al., 1993; Krajewski et al., 1994a) and is detected as early as E13 in rat brain (Zhang et al., 1995). Targeted disruption of baxprevents the death of sympathetic and motor neurons during development and after trophic factor deprivation (Deckwerth et al., 1996). These results demonstrate that Bax is present in the CNS, is capable of interacting with Bcl-xL, and does regulate survival of some neuronal populations, rendering it a likely candidate for the proapo-ptotic family member that regulates immature neuron survival in the CNS.
To determine whether Bax is critical during early CNS development,bax-deficient (bax−/−) embryos were examined. Neuronal apoptosis was assessed by histology and terminal deoxytransferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL) (Gavrieli et al., 1992) and was quantified further in vitro using a primary telencephalic cell culture system. Mice carrying disruptions of bcl-x or bax were interbred, and neuronal apoptosis was examined inbcl-x−/−/bax−/− mice. Results indicate that Bax interacts with Bcl-xL to regulate immature neuron survival, although Bax does not mediate all neuronal apoptosis in the developing CNS.
MATERIALS AND METHODS
Generation of mice carrying targeted gene disruptions.Generation of bcl-x-deficient mice by homologous recombination in ES cells has been described previously (Motoyama et al., 1995). Heterozygous bcl-x+/− male and female mice were bred to generate wild-type, heterozygous, andbcl-x-deficient embryos. bax-deficient mice have been generated by Dr. F. Wang in the laboratory of Dr. D. Y. Loh (Nippon Roche) by homologous recombination in ES cells. In these mice, a 5.6 kb BamHI–EcoRI segment of DNA containing exons 2–6 of bax was replaced with a neomycin expression cassette, and transfections of the construct, selections of E14 ES cells, and their injection into C57BL/6 blastocysts were done as described previously (Nakayama et al., 1993).bax+/− mice survived and bred normally.bax−/− mice survived normally, but malebax-deficient mice showed increased cell death in the testes (F. Wang, K. A. Roth, and D. Y. Loh, unpublished data) as has been reported previously for bax−/− mice (Knudson et al., 1995).
To generate mice deficient in both bcl-x and bax,bcl-x+/− males were first bred withbax+/− and bax−/−females to generate an F1 generation that included double heterozygous (bcl-x+/−/bax+/−) mice. Double heterozygotes were bred to produce embryos that contained nine different genotypic combinations, including 1 in 16bcl-x−/−/bax−/−embryos. To determine whether the distribution of generated genotypes followed the predicted Mendelian distribution, chi-square analysis of contingency tables was used.
Genotyping mice. The endogenous and disrupted genes can be detected by PCR analysis of tail DNA extracts. Endogenousbcl-x was detected as an ∼400 bp product using primer sequences 5′-GTGCCATCAATGGCAACCCAT-3′ and 5′-CCGCCGTTCTCCTGGATCCAA-3′, and its targeted disruption was detected as a 1300 bp product using primers 5′-GCCTACCCGCTTCCATTGCTCAGC-3′ and 5′-GTAACAAACGCCTACCACGACAGC-3′. Cycling parameters included a 10 min hold at 94°C, then 94°C for 1 min, 66°C for 1.5 min, and 72°C for 2 min for 38 cycles, followed by a 10 min extension at 72°C. Endogenous bax amplification using primers 5′-GCTATCCAGTTCATCTCCAATTCGCC-3′ and 5′-GCTCTGAACAGATCATGAAGACAGGGG-3′ yielded a 120 bp product, and the disrupted gene generated a 110 bp product with primers 5′-ATGGACGGGTCCGGGGAGCAGCTT-3′ and 5′-GGGTGGGGTGGGATTAGATAAATG-3′. Cycling parameters were 10 min at 94°C, then 94°C for 1 min, 64°C for 1.5 min, and 72°C for 1.5 min for 30 cycles, followed by a 10 min extension at 72°C.
Histological preparation of tissue. Pregnant mice were killed on gestational day 12 by anesthetization with methoxyflurane followed by cervical dislocation, and whole embryos were removed. Samples of tail and limb tissue were taken from each embryo for DNA extraction. Embryos were then fixed in Bouin’s fixative overnight at 4°C and washed several times with 70% ethanol. Tissue was embedded in paraffin and cut in 4-μm-thick sagittal sections. Before staining, sections were deparaffinized by two washes in HemoDe (Fisher, Pittsburgh, PA), three washes in isopropanol, and rinsed with running tap water. Hematoxylin and eosin (H&E)-stained slides were treated as follows: 15 sec in hematoxylin solution, rinsed (with water), dipped in acid alcohol (2 ml of HCl/200 ml of 70% ethanol), rinsed, 15 sec in ammonia water (600 μl of ammonia/200 ml of distilled water), rinsed, 15 sec in eosin solution, rinsed, and then successively dipped in 70, 95, and 100% ethanol and two times in xylene.
TUNEL staining. TUNEL reactions were done with slight modifications of a method described previously (Tornusciolo et al., 1995). Briefly, deparaffinized tissue sections were permeabilized with 0.5% Triton X-100 in PBS (0.1 m PBS, pH 7.4) and then incubated with terminal deoxynucleotidyl transferase (TDT; 25 U/100 μl buffer; Boehringer Mannheim, Indianapolis, IN) and digoxygenin-conjugated deoxyuridine triphosphate (0.25 nmol/100 μl buffer; Boehringer Mannheim) for 60 min at 37°C in TDT buffer (30 mm Tris-base, pH 7.2, 140 mm sodium cacodylate, and 1 mm cobalt chloride). Reactions were stopped by a 15 min wash in a solution of 300 mm sodium chloride and 30 mm sodium citrate. TUNEL-labeled cells were visualized using tyramide signal amplification to increase the sensitivity of detection over that of previously described methods (Shindler and Roth, 1996a). TDT-reacted tissues were incubated overnight at 4°C with horseradish peroxidase-conjugated sheep antidigoxygenin antiserum (Boehringer Mannheim) diluted 1:1000 in PBS-blocking buffer (PBS with 1% bovine serum albumin, 0.2% powdered milk, and 0.3% Triton X-100). Three washes with Tris buffer (0.1 m Tris-HCl, pH 7.6, and 0.15 m NaCl) were followed by a 5 min incubation with SI-Red tyramide (Roth et al., 1996a; NEN Life Science Products, Boston, MA) diluted 1:1000. Tissue was counterstained for 10 min with a 0.04 μg/ml solution of bisbenzimide (Hoechst 33258; Sigma, St. Louis, MO). Staining was visualized on a Zeiss-Axioskop microscope equipped with epifluorescence.
Primary telencephalic cultures. E12 telencephalic cells were dissociated as described previously (Shindler and Roth, 1996b). Briefly, pregnant mice were killed on gestational day 12, the uterus was removed, and embryos were rapidly removed from the uterus and transferred to cold dissociation medium (DM) consisting of calcium- and magnesium-free HBSS (Life Technologies, Grand Island, NY) supplemented with 15 mm HEPES, 2.7 mm sodium bicarbonate, and 33.3 mm glucose. Separate samples of tail and limb tissue were taken from each embryo for DNA extraction. Whole brains were removed from the membranous skull and placed into a dish with cold DM, meninges were removed, and telencephalons were separated from the rest of the brain. Cells were dissociated with a solution of 0.01% trypsin with 0.004% EDTA (Sigma), and 0.001% deoxyribonuclease I (Sigma) in DM, followed by mild trituration with fire-polished Pasteur pipettes. Dissociated cells were washed twice with DM, resuspended in basal media [a 1:1 mix of DMEM and Ham’s F12 medium (Life Technologies) with 1.2 gm/l sodium bicarbonate and 15 mmHEPES, pH 7.4], and a small sample was stained with trypan blue and counted. Approximately 1 × 106 viable cells were obtained from each embryo.
A total of 20,000 cells diluted in basal media were plated per well of a 48-well tissue culture plate. Before plating, wells were precoated with successive overnight incubations in 0.1 mg/ml poly-l-lysine (Sigma) followed by 0.01 mg/ml laminin (Collaborative Biomedical Products, Bedford, MA). Cultures were incubated in 5% CO2 at 37°C for either 2 or 48 hr, as indicated. Cultures were fixed for 20 min at room temperature in PBS with 4% paraformaldehyde.
Immunostaining. Fixed cells were incubated overnight at 4°C with mouse anti-microtubule-associated protein (MAP) 2 antiserum (Sigma) diluted 1:10,000 in PBS-blocking buffer, then washed several times with PBS. Immunostaining was detected using a 1 hr room temperature incubation with Cy3-conjugated donkey anti-mouse secondary antiserum (Jackson ImmunoResearch, West Grove, PA) diluted 1:400. Cell nuclei were labeled with a 0.04 μg/ml solution of bisbenzimide for 10 min at room temperature.
Quantification of apoptosis. Apoptosis in E12 DRG was assessed by counting TUNEL-reactive cells in photomicrographs taken at 60× magnification. One field of cells was counted for each DRG examined. Six different DRG, from two to three different embryos and covering the same total area, were counted for each genotype. In telencephalic cultures, numbers of total nuclei and abnormally condensed, fragmented nuclei were counted. Typically, four randomly selected fields of nuclei were selected and counted at 40× magnification for each well (∼150–200 cells). Duplicate wells were set up and counted for each culture. The percentage of apoptotic cells was calculated as the number of abnormally bisbenzimide-labeled nuclei divided by the total number of nuclei. Two hour cultures were used to control for possible differences in initial plating density. Furthermore, data from cultures were compared as percentages to avoid possible differences introduced by differences in plating density. Significance was established using the nonparametric Kruskal–Wallis ANOVA on ranks.
RESULTS
Identification of genotypes
For each mouse, the presence of wild-type and disruptedbcl-x and bax was detected by PCR of tail DNA extracts. Endogenous bcl-x was amplified as a 400 bp product, and the disrupted bcl-x was amplified as a 1300 bp product (Fig. 1A). Endogenousbax was detected as a 120 bp product, and disruptedbax as a 110 bp product (Fig. 1B). Genotypes of E12 embryos were confirmed by PCR of a second DNA extract made from limb tissue.
Fig. 1.
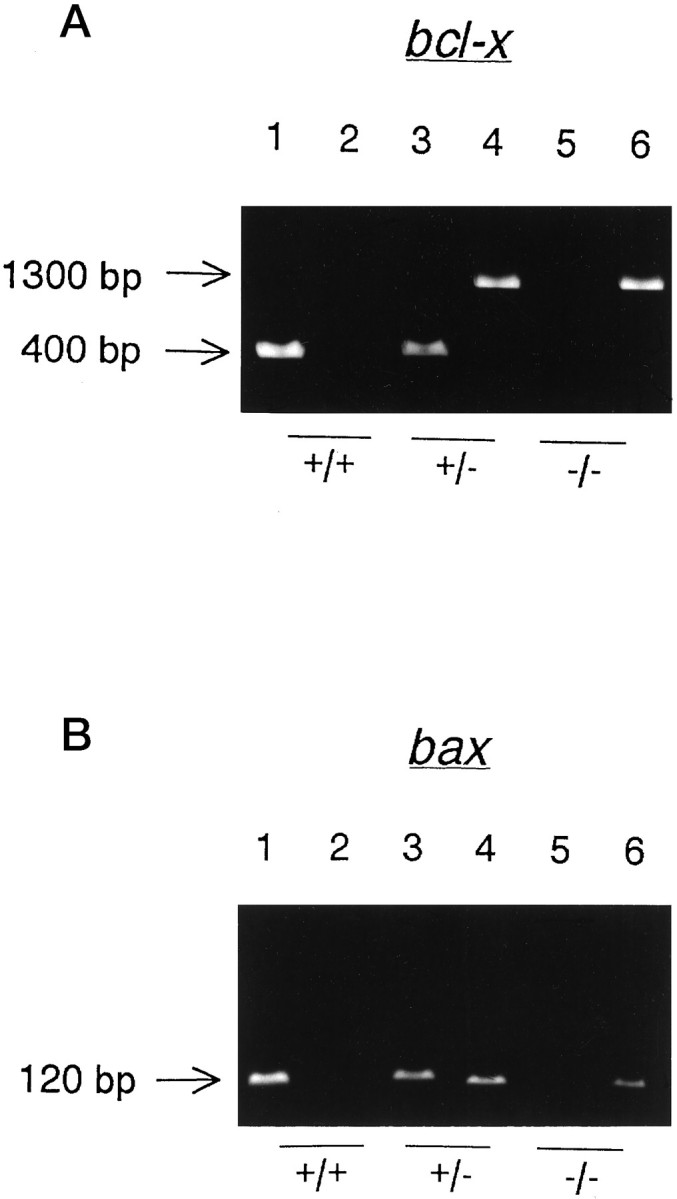
To determine the presence of endogenous and disrupted genes, separate PCR reactions of tail DNA extracts were set up and run in adjacent lanes of a 1.5% agarose gel for each mouse.A, Endogenous bcl-x was detected by the presence or absence of a 400 bp PCR product (lanes 1, 3,and 5), and disrupted bcl-x was detected as a 1300 bp product (lanes 2, 4, and6). Shown are results for three E12 embryos: onebcl-x+/+ (lanes 1 and2), one bcl-x+/−(lanes 3 and 4), and onebcl-x−/− (lanes 5 and6). B, Endogenousbax was detected by the presence or absence of a 120 bp PCR product (lanes 1, 3, and 5), and disrupted bax was detected as a 110 bp product (lanes 2, 4, and 6). Shown are results for the same three E12 embryos shown in A. Therefore, the embryo in lanes 1 and 2wasbcl-x+/+/bax+/+, in lanes 3 and 4 wasbcl-x+/−/bax+/−, and in lanes 5 and 6 wasbcl-x−/−/bax−/−.
Embryos generated from interbreedingbcl-x+/−/bax+/− mice should contain nine different genotypic combinations. All nine genotypes were found in the expected Mendelian frequency (p ≤ 0.05; Table 1). For example, 6.25% (1 of 16) of E12 embryos generated were predicted to bebcl-x−/−/bax−/−, and 6.45% (4 of 62) of embryos examined werebcl-x−/−/bax−/−.
Table 1.
Embryos and liveborn mice generated from interbreeding ofbcl-x+/−/bax+/−mice
| Genotype | E12 embryos | Liveborn mice |
|---|---|---|
| bcl-x+/+/bax+/+(1) | 4 | 7 |
| bcl-x+/+/bax+/−(2) | 7 | 10 |
| bcl-x+/+/bax−/−(1) | 5 | 5 |
| bcl-x+/−/bax+/+(2) | 4 | 8 |
| bcl-x+/−/bax+/−(4) | 20 | 15 |
| bcl-x+/−/bax−/−(2) | 6 | 9 |
| bcl-x−/−/bax+/+(1) | 4 | 0 |
| bcl-x−/−/bax+/−(2) | 8 | 0 |
| bcl-x−/−/bax−/−(1) | 4 | 0 |
Interbreeding ofbcl-x+/−/bax+/− mice resulted in nine different genotypic combinations shown in column 1. Numbers in parentheses indicate the predicted number of offspring out of every 16 mice generated. A total of 62 E12 embryos were generated in nine litters, and the number of embryos with each genotype is listed in column 2. All nine genotypes were found, and they followed the predicted Mendelian distribution (p ≤ 0.05). Fifty-four liveborn mice were generated in 11 litters, and the number of mice with each genotype is listed in column 3. Nobcl-x−/−/bax−/− mice were born, indicating that these mice were embryonic lethal (p ≤ 0.05). The numbers of liveborn mice containing the six nonlethal genotypes followed the predicted Mendelian distribution (p ≤ 0.05).
Bax deficiency does not prevent the embryonic lethality of Bcl-xL-deficient mice
Bcl-xL-deficient mice die around E13. To determine whether embryonic lethality is rescued by Bax deficiency,bcl-x+/−/bax+/− double heterozygote mice were interbred, and 11 litters containing 54 liveborn mice were generated. The 11 litters contained nobcl-x−/−/bax−/− mice, indicating that mice deficient in both genes were not viable (p ≤ 0.05). The frequency of liveborn mice followed the predicted Mendelian distribution of genotypes (p ≤ 0.05), allowing for the in utero death of all bcl-x−/− mice (Table1).
Bax deficiency prevents increased apoptosis in E12 brainstem and spinal cord of Bcl-xL-deficient mice
The wild-type E12 CNS, when visualized by H&E staining, contained only occasional cells with highly condensed, pyknotic nuclei in the brainstem and spinal cord (Fig. 2A). Occasional TUNEL-reactive cells were found primarily in the ventral spinal cord and intermediate zone of the developing brainstem (data not shown).
Fig. 2.
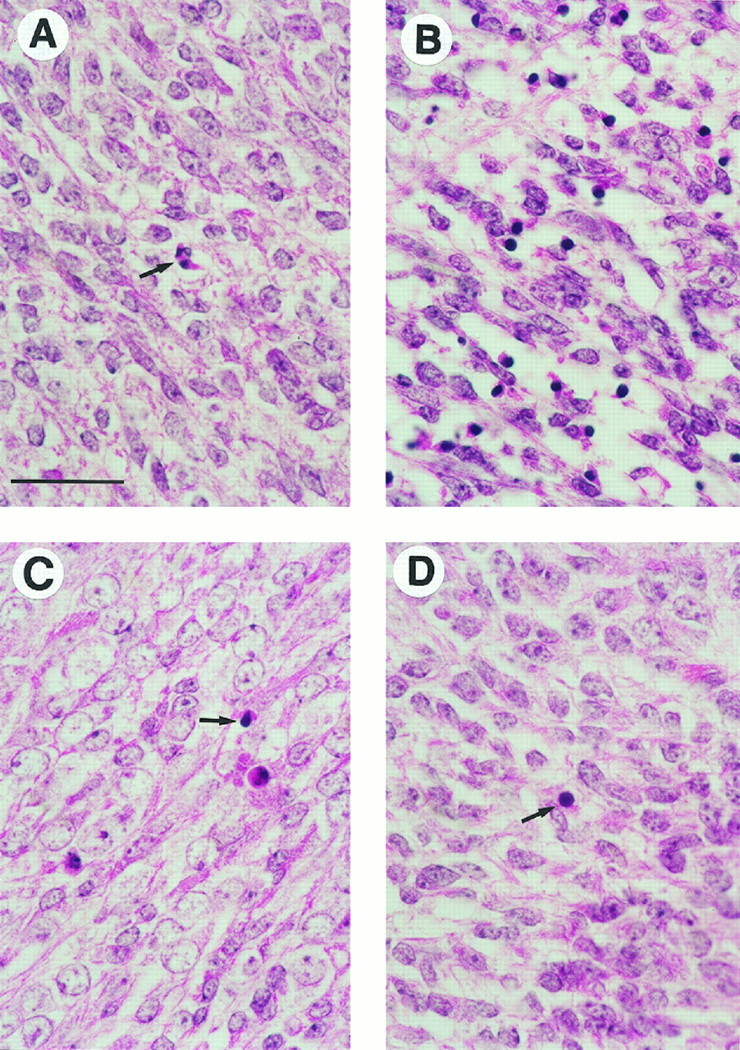
E12 embryos were fixed in Bouin’s solution, embedded in paraffin, and cut into 4-μm-thick sagittal sections. H&E staining of spinal cords from four embryos is shown. A, The wild-type spinal cord showed only occasional condensed pyknotic cells (arrow) indicative of ongoing apoptosis.B, The bcl-x−/− spinal cord was filled with numerous pyknotic, apoptotic cells. The photomicrograph shown is from abcl-x−/−/bax+/−embryo. C, Thebcl-x−/−/bax−/−spinal cord contained only occasional pyknotic cells (arrow). Note that the general appearance and amount of apoptosis is similar to the wild-type embryo shown in A, and remarkably different from the bcl-x−/−embryo shown in B. D, The spinal cord ofbax−/− mice appeared to be normal, with only a rare pyknotic cell identified (arrow). Scale bar, 35 μm.
As has been reported previously, mice carrying a targeted disruption of the bcl-x gene (bcl-x−/−) contained a large increase in the number of apoptotic cells, defined by histological criteria and TUNEL staining, in the intermediate zone of E12 brainstem and spinal cord. This phenotype was seen inbcl-x-deficient mice containing either one (bcl-x−/−/bax+/−) or two (bcl-x−/−/bax+/+) copies of endogenous bax (Figs. 2B,3A,B). The CNS ofbcl-x−/−/bax−/− mice, however, contained few apoptotic cells (Figs. 2C,3C,D) compared with bcl-x−/− mice, and were similar to wild-type littermates.
Fig. 3.

Apoptotic cells in Bouin’s fixed sagittal sections of E12 embryos were identified by TUNEL. Cell nuclei were counterstained with bisbenzimide (Hoechst 33258). A, The spinal cord of abcl-x−/−/bax+/−embryo demonstrates a tremendous number of TUNEL-positive cells (red). B, Dual-label TUNEL (red) and bisbenzimide (blue) of the same field shown in A. Many of the TUNEL-labeled cells contained highly condensed chromatin, observed as bright blue bisbenzimide-stained nuclei, demonstrating the correlation between these two measures of apoptosis. C, The spinal cord of abcl-x−/−/bax−/−embryo showed a greatly reduced number of apoptotic cells compared with the bcl-x−/− spinal cord shown inA. Occasional apoptotic cells are present, illustrated by two red TUNEL-positive cells in thecenter of the field. D, Dual-label TUNEL and bisbenzimide of the same field shown in Cdemonstrates the normal chromatin pattern of most cells. Scale bar, 25 μm.
The microscopic appearance of brainstem and spinal cord inbax−/− mice was similar to wild-type littermates (Fig. 2D), with no obvious difference in the size of the ventricular or intermediate zones. However, only rare TUNEL-labeled cells were detected in the ventral spinal cord and brainstem (data not shown). In contrast, and similar to wild-type embryos, numerous TUNEL-positive cells were viewed in the dorsal midline of the spinal cord (data not shown), where cell death related to neural tube closure occurs (Geelan and Langman, 1977).
Quantification of apoptosis in E12 DRG
TUNEL staining of E12 DRG was examined to quantitatively compare in vivo levels of apoptosis in a defined population of cells. At E12, normal programmed cell death was observed in wild-type DRG, whereas the number of apoptotic cells detected was more than doubled in DRG of bcl-x−/− embryos (Table2). This increased apoptosis was reduced by 54% in DRG of bcl-x−/−/bax−/−mice in which apoptosis was not significantly different from wild type (a small 5% increase was observed). Apoptosis was further reduced by 80% in bax−/− DRG.
Table 2.
Apoptosis in E12 DRG
| Genotype | Number of TUNEL+ cells | TUNEL+ cells/field |
|---|---|---|
| Wild type (3) | 100 (6) | 16.7 ± 2.0 |
| bcl-x−/− (3) | 228 (6) | 38.0 ± 3.4 |
| bcl-x−/−/bax−/−(3) | 105 (6) | 17.5 ± 4.1 |
| bax−/−(2) | 19 (6) | 3.2 ± 0.7 |
Sagittal sections of Bouin’s fixed, paraffin-embedded E12 wild-type, bcl-x−/−, bcl-x−/−/bax−/−, andbax−/− mice were labeled by TUNEL, and photomicrographs of DRG were taken at 60× magnification. The total number of TUNEL-labeled cells counted from DRG of each genotype is shown in column 2. The number of different DRG examined is shown in parentheses in column 2, and the number of different embryos these DRG came from is shown in parenthesis in column 1. One photomicrograph was counted from each DRG. Column 3 shows the mean ± SE number of TUNEL-labeled cells per 60× field. The difference in number of TUNEL+ cells/field between any two genotypes was significant (p ≤ 0.05), except for between wild-type andbcl-x−/−/bax−/−DRG.
Bax deficiency prevents increased apoptosis of Bcl-xL-deficient telencephalic cells in vitro
Primary cultures of undifferentiated ventricular zone cells from E12 telencephalon of mice carrying targeted gene disruptions can be used to determine the effects of Bcl-xL and Bax on immature telencephalic neuronal death, and to quantitate apoptosis of cells with different genotypes. Two hours after plating on laminin-coated wells, cultures consisted mainly of small, round cells, and 24.8 ± 1.2% cells expressed MAP2 immunoreactivity (data not shown). Less than 3% of cells, regardless of genotype, contained highly condensed, fragmented chromatin such as that found in apoptotic cells labeled with bisbenzimide (Fig. 5A). After 48 hr in serum-free, unsupplemented DMEM/F12, wild-type cells sprouted neurites and 50.9 ± 1.4% expressed MAP2 immunoreactivity (Fig.4B). A total of 25–30% of cells in wild-type cultures were apoptotic, as determined by the presence of highly condensed, fragmented chromatin visualized by bisbenzimide staining (Figs. 4A, 5B–D).
Fig. 5.

Primary dissociated E12 telencephalic cells were grown for either 2 or 48 hr in unsupplemented DMEM/F12, fixed in 4% paraformaldehyde, and stained with bisbenzimide. Total nuclei and condensed, fragmented apoptotic nuclei were counted, and the percentage of apoptotic cells in each culture was calculated. Data represent the mean ± SEM for all cultures generated from embryos with an indicated genotype. A, After 2 hr in vitro, <3% of telencephalic cells were apoptotic, with no significant differences observed between cultures generated from mice with different genotypes. Shown are data from wild-type (n = 13 embryos),bcl-x−/− (n = 11),bax−/− (n = 9), andbcl-x−/−/bax−/−(n = 4) cultures. B, Embryos generated from interbreeding of bcl-x+/−mice were used to examine apoptosis of bcl-x-deficient and heterozygous cells grown 48 hr in vitro. No significant difference in apoptosis was found betweenbcl-x+/+ (n = 13) andbcl-x+/− (n = 21) cells, whereas bcl-x−/−(n = 11) cultures contained significantly more apoptotic cells than wild-type or heterozygote cultures (*p ≤ 0.05). C, Embryos generated from interbreeding of bax+/− mice were used to examine apoptosis of bax-deficient and heterozygous cells grown 48 hr in vitro. No significant differences were found between bax+/+(n = 9), bax+/−(n = 17), or bax−/−(n = 9) cultures. D, Embryos generated from interbreeding ofbcl-x+/−/bax+/−mice were used to examine apoptosis of cells deficient in both genes grown 48 hr in vitro. Because no difference in the amount of apoptosis was found between wild-type andbcl-x+/− cultures, or betweenbax-deficient, heterozygote, and wild-type cultures, data from telencephalic cultures of these mice were pooled together.o indicates presence of either the endogenous (+) or disrupted (−) gene. Cultures of bcl-x-deficient cells that contained at least one endogenous bax gene (bcl-x−/−/bax+/o;n = 6) contained significantly more apoptosis than cultures of non-bcl-x-deficient (bcl-x+/o/baxo/o;n = 20) cells (*p ≤ 0.05). Cultures of cells deficient in both genes (bcl-x−/−/bax−/−;n = 4) contained significantly less apoptosis thanbcl-x−/−/bax+/ocultures and significantly more apoptosis thanbcl-x+/o/baxo/ocultures (**p ≤ 0.05).
Fig. 4.
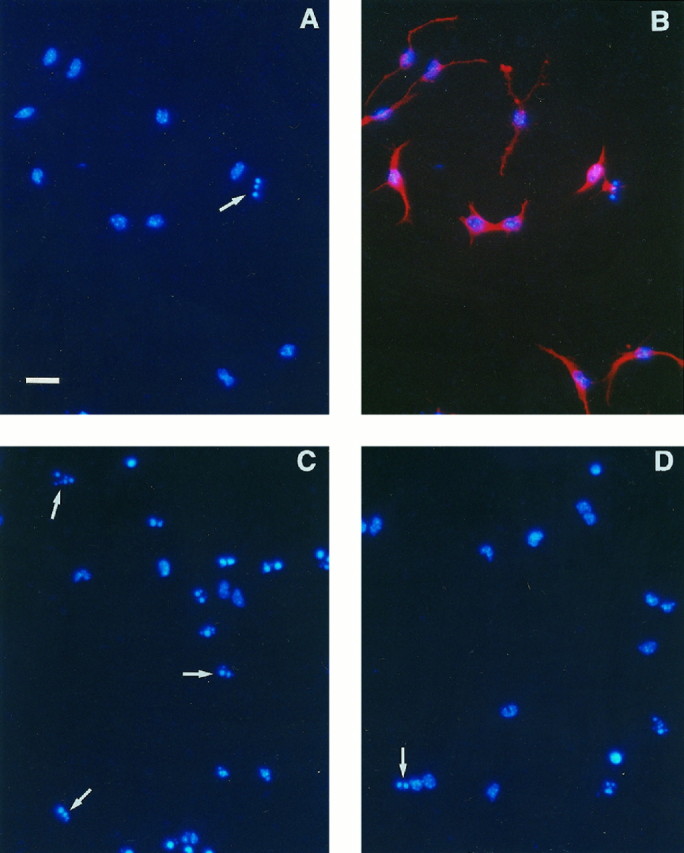
Primary dissociated cells from telencephalons of individual E12 embryos were grown for 48 hr in unsupplemented DMEM/F12 media and fixed in 4% paraformaldehyde. A, Cells from a wild-type embryo stained with bisbenzimide demonstrate the normal chromatin staining of many cells and the highly condensed, fragmented staining pattern used to identify apoptotic cells (arrow). B, The same cells as shown inA were dual-labeled with bisbenzimide (blue) and anti-MAP2 antibodies (red). The presence of MAP2-immunoreactive neuritic processes shows that telencephalic cells differentiated into neurons in culture.C, Cells from abcl-x−/−/bax+/−embryo stained with bisbenzimide demonstrate a large increase in apoptotic cells (arrows) compared with the wild-type culture shown in A. D, Cells from abcl-x−/−/bax−/−embryo show a decreased number of apoptotic cells (arrow) compared with otherbcl-x−/− embryos such as that shown inC. Scale bar, 25 μm.
Comparison of cultures generated from bcl-x+/+,bcl-x+/−, and bcl-x−/−mice revealed no significant difference in the amount of apoptosis between wild-type (27.3 ± 2.3%, n = 13) and heterozygote (30.2 ± 2.1%, n = 21) cultures after 48 hr in vitro. Cultures of bcl-x-deficient cells, however, contained significantly more apoptotic cells than wild-type or heterozygote cultures, with 78.9 ± 2.8% (n = 11) cells containing abnormally bisbenzimide-stained nuclei (p ≤ 0.05; Figs.4C, 5B).
Comparison of cultures generated from bax+/+,bax+/−, and bax−/−mice grown 48 hr in vitro revealed no significant differences in the percentage of apoptotic cells, as measured by bisbenzimide staining, from wild-type (26.5 ± 2.6%,n = 9), heterozygote (25.8 ± 1.7%,n = 17), or bax-deficient (22.8 ± 1.9%, n = 9) littermates (Fig. 5C).
Increased apoptosis of bcl-x-deficient telencephalic cells was reduced by disruption of bax.bcl-x−/−/bax−/−cultures contained 39.3 ± 1.3% (n = 4) apoptotic cells, whereas cultures of their bcl-x-deficient littermates (bcl-x−/−/bax+/+ andbcl-x−/−/bax+/−) had significantly more (71.8 ± 2.0%, n = 6) apoptotic cells (p ≤ 0.05), as seen previously in bcl-x-deficient cultures (Figs. 4D,5D). Data frombcl-x−/−/bax+/+ andbcl-x−/−/bax+/−cultures were pooled together because no difference in amount of apoptosis was found between these cells (data not shown). Similarly, telencephalic cultures generated from mice containing at least one functional bcl-x gene revealed no differences in the amount of apoptosis, regardless of genotype (data not shown). Pooled data from these cultures showed significantly fewer apoptotic cells (28.3 ± 0.9%, n = 20) than eitherbcl-x−/− orbcl-x−/−/bax−/−cultures (p ≤ 0.05).
DISCUSSION
Targeted disruption of the bax gene prevents the massive cell death of immature neurons in the developingbcl-x-deficient CNS. The spinal cord and brainstem ofbcl-x−/−/bax−/− mice are similar to wild-type mice both histologically and by the pattern of TUNEL staining, and there is little difference in levels of apo-ptosis between wild-type andbcl-x−/−/bax−/− DRG. This is in stark contrast to bcl-x-deficient mice containing a functional bax gene, in which the DRG and intermediate zone of postmitotic immature neurons contain extensive numbers of pyknotic cells that are TUNEL-labeled, measures that have been used previously to indicate that cells are undergoing apoptosis (Gavrieli et al., 1992; Motoyama et al., 1995). Similarly, bax deficiency results in a large reduction of the number of abnormally bisbenzimide-labeled nuclei in cultures of telencephalic cells frombcl-x-deficient mice. Abnormally condensed and fragmented bisbenzimide staining has also been used previously to identify apoptotic cells, and is correlated with TUNEL-reactive cells (Deckwerth and Johnson, 1993; Roth et al., 1996b). Together, these results suggest that Bax interacts with Bcl-xL to regulate survival of immature neurons throughout the CNS.
Interestingly, bax deficiency does not eliminate cell death in the developing CNS. Occasional pyknotic, TUNEL-labeled cells are found in the intermediate zone of E12 brainstem and spinal cord ofbcl-x−/−/bax−/− andbax−/− mice, and cell death related to neural tube closure appears unaffected, indicating that neuronal apoptosis can occur in the absence of Bax. Similar to previous observations (Deckwerth et al., 1996), apoptosis is significantly reduced in DRG ofbax−/− mice, although a small number of TUNEL-labeled cells can be detected. A primary telencephalic culture system was used to further examine whether bax deficiency leads to a reduction in immature neuron apoptosis. Cells were grown for 48 hr in unsupplemented basal medium on laminin-coated plates. This system allows undifferentiated E12 telencephalic cells to begin to differentiate into neurons, as determined by sprouting of neurites and immunoreactivity for MAP2, a neuron-specific protein restricted to dendrites of mature neurons but also found in cell bodies and axons early in neuronal development (Papandrikopoulou et al., 1989; Tucker, 1990). Plating in basal medium also establishes a sizable and reproducible baseline level of apoptosis in wild-type cultures, with 25–30% of cells containing condensed, fragmented chromatin after 48 hr in vitro. Cultures of bax-deficient telencephalic cells show no significant reduction in this baseline level of apoptosis, suggesting that cell death in this population of cells is not dependent on Bax. In addition, cultures ofbcl-x−/−/bax−/− cells show a small but significant increase in levels of apoptosis (40% vs 25–30%) compared with wild-type cells. Therefore, although Bax critically interacts with Bcl-xL in many immature neurons, there also must be other pathways of cell death that are active during this early developmental period and that are not dependent on Bcl-xL or Bax. Such pathways may be regulated by other members of the Bcl-2 gene family, or may act independently of this family.
Cell death in the bcl-x-deficient CNS occurs relatively early in neuronal development. Although cell death has long been recognized as a normal process in nervous system development, with up to 50% of all neurons generated in some regions ultimately undergoing apoptosis (Oppenheim, 1991), much of this work has focused on apoptosis that occurs after synapse formation and the development of target-derived trophic factor dependency. However, an earlier period of cell death, before synapse formation, has been described in the retina, spinal cord, sensory ganglia, and telencephalon (Maruyama and D’Agostino, 1967; Lance-Jones, 1982; Acklin and van der Kooy, 1993;Homma et al., 1994; Blaschke et al., 1996; Galli-Resta and Ensini, 1996), and recent reports indicate that the amount of death occurring at these early periods is extensive. In the telencephalon, 80% of precursors in the E17 rat gives rise to at least one daughter cell that dies within the next 48 hr (Acklin and van der Kooy, 1993). In the E14 mouse cortex, 70% of all cortical cells, including those found in proliferative zones, contain fragmented DNA characteristic of apo-ptotic cells (Blaschke et al., 1996). The increased apoptosis seen in bcl-x-deficient mice also occurs early in development, and therefore it was thought that death during this period may normally result from the failure of a cell to upregulatebcl-x. Although this may account for some of the normal cell death during this period, this is clearly not the case for all death, because bcl-x- and bax-independent pathways of death are present. This does suggest that although apoptosis of immature neurons is regulated by Bcl-xL and Bax, the death of cells within proliferative precursor regions may be subject to different regulation. Other antiapo-ptotic molecules, such as Bcl-2 or Bcl-y/w (Guastella et al., 1995; Gibson et al., 1996), may contribute to the survival of precursor cells or early postmitotic neurons within these regions. Preliminary investigations support this possibility, because mice carrying targeted disruptions of bothbcl-x and bcl-2(bcl-x−/−/bcl-2−/−) contain more apoptotic cells in the embryonic CNS thanbcl-x-deficient mice (K. A. Roth, unpublished observations).
In addition to identifying Bax as the proapoptotic protein that interacts with Bcl-xL in the CNS, the ability ofbax deficiency to rescue bcl-x-deficient immature neurons from death provides in vivo evidence to support the hypothesis that the balance between anti- and proapoptotic Bcl-2 family members regulates apoptosis. This was suggested previously based on the ability of these proteins to form heterodimers and on the fact that the ratio of Bcl-xL to Bax, or Bcl-2 to Bax, in transfected cells determines the susceptibility of the cell to undergo apoptosis (Oltvai et al., 1993; Sedlak et al., 1995). In the developing CNS, Bcl-xL and Bax expression overlap in many regions (Mai et al., 1996). Targeted disruption of bcl-x presumably upsets the normal Bcl-xL to Bax ratio, resulting in apoptosis. Subsequent disruption of bax eliminates the imbalance between Bcl-xL and Bax, and restores the viability of immature neurons. This result also provides insight into the question of which Bcl-2 family members inhibit the function of other members. It has been suggested that antiapoptotic Bcl-2 family members regulate apoptosis by acting as death repressors, and proapoptotic members inhibit this action by heterodimerizing with the antiapoptotic members; or, alternatively, proapoptotic members could function as death effectors inhibited by antiapoptotic members. In the case of immature neurons of the CNS, it appears that Bax functions as a death effector and is inhibited by Bcl-xL. This assessment is based on the fact that in the absence of Bcl-xL, these cells undergo apoptosis presumably because of the increased numbers of Bax homodimers that can form, whereas in the absence of both genes, these immature neurons are able to survive normally.
Bcl-x plays a critical role in regulating survival not only of immature neurons, but also of hematopoietic precursors and hepatic cells in the liver, and the embryonic lethality of bcl-x-deficient mice is thought to be secondary to hematopoietic and/or hepatic cell death (Motoyama et al., 1995). Although bax deficiency prevented the increased neuronal death of bcl-x-deficient mice, the embryonic lethality was not altered inbcl-x−/−/bax−/− mice. This suggests that Bax is not the proapoptotic Bcl-2 family member that interacts with Bcl-x in the developing liver. Cell death in the developing liver may, for example, be mediated by another proapoptotic member, such as Bak or Bad, thus accounting for the continued embryonic lethality ofbcl-x−/−/bax−/− mice. These results demonstrate that although many Bcl-2 family members are expressed in a wide range of tissues, the relative importance of any one member can vary in specific tissues. Similar results have been observed in mice carrying targeted disruptions of bcl-2 andbax. bcl-2-deficient mice show increased apoptosis of mature lymphocytes, as well as kidney cells, resulting in polycystic kidney disease (Veis et al., 1993; Nakayama et al., 1994).bax deficiency rescues the increased death ofbcl-2-deficient lymphocytes, but not the death seen in the kidneys (S. J. Korsmeyer, personal communication).
bax deficiency prevents the increased apoptosis ofbcl-x-deficient immature neurons throughout the CNS, although bax deficiency alone does not eliminate all apoptosis normally present during early CNS development. These results indicate that Bax interacts with Bcl-xL to regulate immature neuron survival, suggest that Bax acts as a dominant death effector molecule, and provide in vivo evidence consistent with the hypothesis that the balance between pro- and antiapoptotic Bcl-2 gene family members ultimately determines the susceptibility of a cell to apoptosis. Although this regulation appears to be the case for a large percentage of immature neurons, cell death that is not dependent on Bax or Bcl-xL is also important. The relative role of other Bcl-2 family members, as well as Bcl-2 family-independent mechanisms, in regulating immature neuron and neuronal precursor cell survival needs to be examined.
Footnotes
This work was supported by National Institutes of Health Grant NS35107. We thank Dr. Dennis Y. Loh (Nippon Roche) for the generous gift ofbcl-x-deficient and bax-deficient mice and Dr. Mark N. Bobrow (NEN Life Science Products) for tyramide signal amplification reagents. We also thank Drs. Anise A. Ardelt and Anne Marie Yunker for valuable discussions and reviewing of this manuscript.
Correspondence should be addressed to Kevin A. Roth, Department of Pathology, Washington University School of Medicine, 660 South Euclid Avenue, Box 8118, St. Louis, MO 63110.
REFERENCES
- 1.Acklin SE, van der Kooy D. Clonal heterogeneity in the germinal zone of the developing rat telencephalon. Development. 1993;118:175–192. doi: 10.1242/dev.118.1.175. [DOI] [PubMed] [Google Scholar]
- 2.Allsopp TE, Wyatt S, Paterson HF, Davies AM. The proto-oncogene bcl-2 can selectively rescue neurotrophic factor-dependent neurons from apoptosis. Cell. 1993;73:295–307. doi: 10.1016/0092-8674(93)90230-n. [DOI] [PubMed] [Google Scholar]
- 3.Blaschke AJ, Staley K, Chun J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development. 1996;122:1165–1174. doi: 10.1242/dev.122.4.1165. [DOI] [PubMed] [Google Scholar]
- 4.Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 5.Chittenden T, Flemington C, Houghton AB, Ebb RG, Gallo GJ, Elangovan B, Chinnadurai G, Lutz RJ. A conserved domain in Bak, distinct from BH1 and BH2, mediates cell death and protein binding functions. EMBO J. 1995a;14:5589–5596. doi: 10.1002/j.1460-2075.1995.tb00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chittenden T, Harrington EA, O’Connor R, Flemington C, Lutz RJ, Evan GI, Guild BC. Induction of apoptosis by the Bcl-2 homologue Bak. Nature. 1995b;374:733–736. doi: 10.1038/374733a0. [DOI] [PubMed] [Google Scholar]
- 7.Craig RW. The BCL-2 gene family. Semin Cancer Biol. 1995;6:35–43. doi: 10.1006/scbi.1995.0005. [DOI] [PubMed] [Google Scholar]
- 8.Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 10.Farrow SN, White JHM, Martinou I, Raven T, Pun K-T, Grinham CJ, Martinou J-C, Brown R. Cloning of a bcl-2 homologue by interaction with adenovirus E1B 19K. Nature. 1995;374:731–733. doi: 10.1038/374731a0. [DOI] [PubMed] [Google Scholar]
- 11.Frankowski H, Missotten M, Fernandez P-A, Martinou I, Michel P, Sadoul R, Martinou J-C. Function and expression of the Bcl-x gene in the developing and adult nervous system. NeuroReport. 1995;6:1917–1921. doi: 10.1097/00001756-199510020-00023. [DOI] [PubMed] [Google Scholar]
- 12.Galli-Resta L, Ensini M. An intrinsic time limit between genesis and death of individual neurons in the developing retinal ganglion cell layer. J Neurosci. 1996;16:2318–2324. doi: 10.1523/JNEUROSCI.16-07-02318.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia I, Martinou I, Tsujimoto Y, Martinou J-C. Prevention of programmed cell death of sympathetic neurons by the bcl-2 proto-oncogene. Science. 1992;258:302–304. doi: 10.1126/science.1411528. [DOI] [PubMed] [Google Scholar]
- 14.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geelan JAG, Langman J. Closure of the neural tube in the cephalic region of the mouse embryo. Anat Rec. 1977;189:625–640. doi: 10.1002/ar.1091890407. [DOI] [PubMed] [Google Scholar]
- 16.Gibson L, Holmgreen SP, Huang DCS, Bernard O, Copeland NG, Jenkins NA, Sutherland GR, Baker E, Adams JM, Cory S. bcl-w, a novel member of the bcl-2 family, promotes cell survival. Oncogene. 1996;13:665–675. [PubMed] [Google Scholar]
- 17.Gillardon F, Zimmermann M, Uhlmann E, Krajewski S, Reed JC, Klimaschewski L. Antisense oligodeoxynucleotides to bax mRNA promote survival of rat sympathetic neurons in culture. J Neurosci Res. 1996;43:726–734. doi: 10.1002/(SICI)1097-4547(19960315)43:6<726::AID-JNR9>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez-Garcia M, Perez-Ballestero R, Ding L, Duan L, Boise LH, Thompson CB, Nunez G. bcl-xL is the major bcl-x mRNA form expressed during murine development and its product localizes to mitochondria. Development. 1994;120:3033–3042. doi: 10.1242/dev.120.10.3033. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez-Garcia M, Garcia I, Ding L, O’Shea S, Boise LH, Thompson CB, Nunez G. bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc Natl Acad Sci USA. 1995;92:4304–4308. doi: 10.1073/pnas.92.10.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenlund LJS, Korsmeyer SJ, Johnson EM., Jr Role of BCL-2 in the survival and function of developing and mature sympathetic neurons. Neuron. 1995;15:649–661. doi: 10.1016/0896-6273(95)90153-1. [DOI] [PubMed] [Google Scholar]
- 21.Guastella J, Weber E, Zhang L. Isolation of bcl-y, a novel bcl-2 homologue, from rat and human brain. Soc Neurosci Abstr. 1995;21:1067. [Google Scholar]
- 22.Havlioglu N, Motoyama N, Loh DY, Roth KA. Analysis of the bcl-x deficient adult chimeric mouse nervous system. J Neuropathol Exp Neurol. 1996;55:613. [Google Scholar]
- 23.Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 24.Homma S, Yaginuma H, Oppenheim RW. Programmed cell death during the earliest stages of spinal cord development in the chick embryo: a possible means of early phenotypic selection. J Comp Neurol. 1994;345:377–395. doi: 10.1002/cne.903450305. [DOI] [PubMed] [Google Scholar]
- 25.Kiefer MC, Brauer MJ, Powers VC, Wu JJ, Umansky SR, Tomei LD, Barr PJ. Modulation of apoptosis by the widely distributed Bcl-2 homologue Bak. Nature. 1995;374:736–739. doi: 10.1038/374736a0. [DOI] [PubMed] [Google Scholar]
- 26.Knudson CM, Tung KSK, Tourtellotte WG, Brown GAJ, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- 27.Krajewski S, Krajewska M, Shabaik A, Miyashita T, Wang HG, Reed JC. Immunohistochemical determination of in vivo distribution of Bax, a dominant inhibitor of Bcl2. Am J Pathol. 1994a;145:1323–1336. [PMC free article] [PubMed] [Google Scholar]
- 28.Krajewski S, Krajewska M, Shabaik A, Wang H-G, Irie S, Fong L, Reed JC. Immunohistochemical analysis of in vivo patterns of bcl-x expression. Cancer Res. 1994b;54:5501–5507. [PubMed] [Google Scholar]
- 29.Krajewski S, Mai JK, Krajewska M, Sikorska M, Mossakowski MJ, Reed JC. Upregulation of bax protein levels in neurons following cerebral ischemia. J Neurosci. 1995;15:6364–6376. doi: 10.1523/JNEUROSCI.15-10-06364.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lance-Jones C. Motoneuron cell death in the developing lumbar spinal cord of the mouse. Dev Brain Res. 1982;4:473–479. doi: 10.1016/0165-3806(82)90192-4. [DOI] [PubMed] [Google Scholar]
- 31.Mai JK, Krajewska M, Reed JC, Krajewski S. Expression pattern of bcl-2, bcl-X and bax genes in the developing mouse nervous system. Soc Neurosci Abstr. 1996;22:1216. [Google Scholar]
- 32.Maruyama S, D’Agostino AN. Cell necrosis in the central nervous system of normal rat fetuses. Neurology. 1967;17:550–558. doi: 10.1212/wnl.17.6.550. [DOI] [PubMed] [Google Scholar]
- 33.Minn AJ, Boise LH, Thompson CB. Bcl-xS antagonizes the protective effects of Bcl-xL. J Biol Chem. 1996;271:6306–6312. doi: 10.1074/jbc.271.11.6306. [DOI] [PubMed] [Google Scholar]
- 34.Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K-i, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, Loh DY. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- 35.Nakayama K, Nakayama K-i, Negishi I, Kuida K, Sawa H, Loh DY. Targeted disruption of Bcl-2αβ in mice: occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proc Natl Acad Sci USA. 1994;91:3700–3704. doi: 10.1073/pnas.91.9.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakayama K-i, Nakayama K, Negishi I, Kuida K, Shinkai Y, Louie MC, Fields LE, Lucas PJ, Stewart V, Alt FW, Loh DY. Disappearance of the lymphoid system in Bcl-2 homozygous mutant chimeric mice. Science. 1993;261:1584–1588. doi: 10.1126/science.8372353. [DOI] [PubMed] [Google Scholar]
- 37.Oltvai ZN, Korsmeyer SJ. Checkpoints of dueling dimers foil death wishes. Cell. 1994;79:189–192. doi: 10.1016/0092-8674(94)90188-0. [DOI] [PubMed] [Google Scholar]
- 38.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 39.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 40.Papandrikopoulou A, Doll T, Tucker RP, Garner CC, Matus A. Embryonic MAP2 lacks the cross-linking sidearm sequences and dendritic targeting signal of adult MAP2. Nature. 1989;340:650–652. doi: 10.1038/340650a0. [DOI] [PubMed] [Google Scholar]
- 41.Reed JC. Bcl-2 and the regulation of programmed cell death. J Cell Biol. 1994;124:1–6. doi: 10.1083/jcb.124.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roth KA, Bobrow MN, Erickson TJ. Immunohistochemical detection using SI-Red tyramide signal amplification. J NIH Res. 1996a;8:68. [Google Scholar]
- 43.Roth KA, Motoyama N, Loh DY. Apoptosis of bcl-x-deficient telencephalic cells in vitro. J Neurosci. 1996b;16:1753–1758. doi: 10.1523/JNEUROSCI.16-05-01753.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sato T, Hanada M, Bodrug S, Irie S, Iwama M, Boise LH, Thompson CB, Golemis E, Fong L, Wang H-G, Reed JC. Interactions among members of the Bcl-2 family analyzed with a yeast two-hybrid system. Proc Natl Acad Sci USA. 1994;91:9238–9242. doi: 10.1073/pnas.91.20.9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sedlak TW, Oltvai ZN, Yang E, Wang K, Boise LH, Thompson CB, Korsmeyer SJ. Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc Natl Acad Sci USA. 1995;92:7834–7838. doi: 10.1073/pnas.92.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shindler KS, Roth KA. Double immunofluorescent staining using two unconjugated primary antisera raised in the same species. J Histochem Cytochem. 1996a;44:1331–1335. doi: 10.1177/44.11.8918908. [DOI] [PubMed] [Google Scholar]
- 47.Shindler KS, Roth KA. Cholera toxin binds to differentiating neurons in the developing murine basal ganglia. Dev Brain Res. 1996b;92:199–210. doi: 10.1016/0165-3806(95)00215-4. [DOI] [PubMed] [Google Scholar]
- 48.Tornusciolo DRZ, Schmidt RE, Roth KA. Simultaneous detection of TDT-mediated dUTP-biotin nick end labeling (TUNEL)-positive cells and multiple immunohistochemical markers in single tissue sections. Biotechniques. 1995;19:800–805. [PubMed] [Google Scholar]
- 49.Tucker RP. The roles of microtubule-associated proteins in brain morphogenesis: a review. Brain Res Rev. 1990;15:101–120. doi: 10.1016/0165-0173(90)90013-e. [DOI] [PubMed] [Google Scholar]
- 50.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 51.Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75:229–240. doi: 10.1016/0092-8674(93)80065-m. [DOI] [PubMed] [Google Scholar]
- 52.Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-xL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 53.Yin X-M, Oltvai ZN, Korsmeyer SJ. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature. 1994;369:321–323. doi: 10.1038/369321a0. [DOI] [PubMed] [Google Scholar]
- 54.Zhang LX, Zhang L, Smith MA, Reed JC, Clark M, Feldman AN, Rubinow DR, Post RM. Ratio of bcl-2 and bax expression during development and apoptosis of neurons in the CNS. Soc Neurosci Abstr. 1995;21:559. [Google Scholar]