

Abstract
Bacterial infection causes fever, an adaptive but potentially self-destructive response, in the host. Also activated are counterregulatory systems such as the pituitary–adrenal axis. Antipyretic roles have also been postulated for certain endogenous central neuropeptides, including the melanocortins (α-MSH-related peptides). To test the hypothesis that endogenous central melanocortins have antipyretic effects mediated by central melanocortin receptors (MCRs), we determined the effect of intracerebroventricular injection of a synthetic MCR antagonist, Ac-Nle4,c-[Asp5,dNal(2′)7,Lys10]α-MSH(4–10)-NH2(SHU-9119) in endotoxin-challenged rats. The efficacy and specificity of SHU-9119 as an MCR antagonist in the rat was first validatedin vitro and in vivo. In vitro, in heterologous cells expressing either rat MC3-R or MC4-R, the major MCR subtypes expressed in brain, SHU-9119 showed no intrinsic agonism, but it inhibited α-MSH-induced cAMP accumulation (IC50 = 0.48 ± 0.19 and 0.41 ± 0.28 nm, respectively) and [125I]-[Nle4,dPhe7]-α-MSH binding (IC50 = 1.0 ± 0.1 and 0.9 ± 0.3 nm, respectively). In vivo, exogenous α-MSH (180 pmol) inhibited fever in rats when administered intracerebroventricularly 30 min after Escherichia colilipopolysaccharide (LPS) (25 μg/kg, i.p.). When co-injected with α-MSH, SHU-9119 (168 pmol, i.c.v.) prevented the antipyretic action of exogenous α-MSH. In contrast, neither α-MSH nor SHU-9119, alone or in combination, affected body temperatures in afebrile rats. In LPS-treated rats, intracerebroventricular injection of SHU-9119 significantly increased fever, whereas intravenous injection of the same dose of SHU-9119 had no effect. Neither intracerebroventricular nor intravenous SHU-9119 significantly affected LPS-stimulated plasma ACTH or corticosterone levels. The results indicate that endogenous central melanocortins exert an antipyretic influence during fever by acting on MCRs located within the brain, independent of any modulation of the activity of the pituitary–adrenal axis.
Keywords: fever, endotoxin, melanocortin receptor, pituitary–adrenal axis, neuroimmunomodulation, α-MSH, SHU-9119
Bacterial infection in vertebrates activates a potent array of adaptive but potentially destructive responses in the host, a hallmark of which is fever. Because survival of the host is threatened not only by insufficient antimicrobial responses but also by excessive responses, it is believed that robust intrinsic mechanisms for the self-limitation of systemic host defense reactions must exist, but these are poorly understood (Kluger, 1991; Catania and Lipton, 1993). Microbial infection activates the hypothalamic–pituitary–adrenal (HPA) axis (Reichlin, 1993), and the resulting rise in blood levels of adrenal glucocorticoids has a suppressive effect on fever (Coelho et al., 1992; McClellan et al., 1994) and various aspects of the immune response (Munck et al., 1984). Aside from glucocorticoids, fever-inhibitory roles have been proposed for certain cytokines and central neuropeptides, including the melanocortins (for review, see Kluger, 1991; Catania and Lipton, 1993).
Exogenous melanocortin (α-MSH-related) peptides are antipyretic when administered peripherally or centrally, and α-MSH antagonizes several actions of the proinflammatory cytokine interleukin-1 (IL-1), including fever (Catania and Lipton, 1993) and stimulation of pituitary ACTH release (Sundar et al., 1989; Shalts et al., 1992). In rabbits, intracerebroventricular administration of anti-α-MSH antiserum for several days before administration of leukocytic pyrogens prolonged the resulting fever, suggesting an antipyretic role of endogenous α-MSH (Shih et al., 1986). Furthermore, several brain regions important in thermoregulation are innervated by α-MSH-containing neurons (O’Donohue et al., 1979; Mezey et al., 1985) and contain melanocortin receptors (MCRs) (Tatro, 1990; Tatro and Entwistle, 1994a). Together, this evidence suggests that during fever, endogenous melanocortins may exert an antipyretic influence by acting on MCRs located within the brain; however, there is presently no direct evidence supporting this hypothesis.
Previously, the major obstacle to understanding the physiological roles of endogenous melanocortins has been a lack of availability of suitably potent and selective MCR antagonists. Recently, the synthetic α-MSH analog Ac-Nle4,c-[Asp5,dNal(2′)7,Lys10]α-MSH(4–10)-NH2(SHU-9119) was demonstrated (Hruby et al., 1995) to be a potent antagonist of the human homologs of the principal MCR isoforms expressed in rat brain (MC3-R and MC4-R) (for review, see Tatro, 1997), but not of the predominantly peripheral MCR isoforms MC1-R (human) and MC5-R (murine) (Hruby et al., 1995). It is unknown whether SHU-9119 antagonizes the rat MC3-R and MC4-R isoforms.
The present study was designed to determine whether MCRs in the brain mediate an antipyretic influence of endogenous melanocortins during fever. We first determined the abilities of SHU-9119 to antagonize the rat MC3-R and MC4-R in vitro, and its efficacy and specificity as an inhibitor of the antipyretic action of exogenous α-MSH in vivo. The role of central MCRs in fever was assessed by intracerebroventricular injection of SHU-9119 in endotoxin-challenged rats. The results indicate an antipyretic role of endogenous melanocortins, mediated via central MCRs.
A portion of the results have been published previously in abstract form (Tatro et al., 1996).
MATERIALS AND METHODS
Plasmid DNA, cell culture, and stable MCR expression.MCR-deficient B16-G4F mouse melanoma cells (Solca et al., 1993) were kindly provided by Drs. Alex Eberle and Johanna Chluba-de Tapia. Cells were maintained at 37°C in a humidified 95% air/5% CO2atmosphere using modified Eagle medium (MEM) supplemented with Earle’s salts, 10% heat-inactivated fetal calf serum, 2 mml-glutamine, 1% minimum nonessential amino acid solution, 50 U/ml penicillin, and 50 μg/ml streptomycin (Life Technologies, Gaithersburg, MD). The plasmid pcDNA Ineo (Invitrogen, San Diego, CA), containing cDNA encoding rat MC3-R (Roselli-Rehfuss et al., 1993), was kindly provided by Dr. Linda Roselli-Rehfuss. On the basis of our recent cloning of the full-length rat MC4-R cDNA (Alvaro et al., 1996), a new plasmid construct having enhanced expression in mammalian cells was produced for the present studies as follows. MC4-R-specific primers with flanking restriction sites were used in the PCR to amplify the full-length coding region of MC4-R (from bp −5 to +1001) using rat dorsal raphe single-stranded cDNA as a template. The PCR product was then directionally subcloned into the BamHI andXbaI sites of pcDNAIII (Invitrogen). Sequence analysis of both cDNA strands revealed that the PCR product had a predicted protein sequence identical to that of the published receptor (Alvaro et al., 1996).
B16-G4F cells were transfected with rat MC3-R- and MC4-R-encoding plasmids by a calcium phosphate method using a commercial kit (5Prime→3Prime, Inc.) according to the manufacturer’s instructions. Briefly, 5–7 × 105 cells were plated per 10 cm culture dish the day before they were transfected. Cells were fed with complete culture medium 3–4 hr before 1 ml of calcium phosphate-DNA precipitate containing ∼20 μg of DNA was added, and then were incubated for 4 hr with calcium phosphate, washed with serum-free medium, and shocked with 15% glycerol buffer, washed again with serum-free media, and incubated an additional 36–48 hr in complete medium. Cells were then selected for stable plasmid expression by passage into medium containing 1 mg/ml G418 (Geneticin; Life Technologies). Single colonies were isolated and subcultured 10–14 d later. Each stably transfected cell subline was maintained for >14 d in G418. MC3-R- and MC4-R-expressing sublines were identified by screening 8–12 colonies for specific binding of [125I]-NDP-MSH. In cell sublines selected for additional study, the presence of functional MCR having agonist binding profiles and coupling to adenylate cyclase consistent with those reported previously (Roselli-Rehfuss et al., 1993; Alvaro et al., 1996) was confirmed by assays of agonist-induced inhibition of [125I]-NDP-MSH binding and by demonstration of α-MSH-induced cAMP accumulation.
Radioligand preparation and MCR binding assay. To measure MCR ligand binding radiometrically, a radiolabeled derivative of the superpotent MCR agonist and α-MSH analog [Nle4,dPhe7]-α-MSH (NDP-MSH) (Sawyer et al., 1980) was prepared by modifications of methods published previously (Tatro and Reichlin, 1987; Tatro, 1993), using a standard method (Thorell and Johansson, 1971) for lactoperoxidase-catalyzed radioiodination. Briefly, 4 μg of NDP-MSH in 100 μl of 10 mm sodium phosphate, pH 6.5, was added to 10 μl (1 mCi) of [125I]-Na (Amersham, Arlington Heights, IL) and 5 μl of lactoperoxidase (0.8 mg/ml) (Calbiochem, La Jolla, CA). The reaction was initiated by adding 5 μl of 0.003% H2O2 and was carried out for 4 min with the addition of three additional 5 μl aliquots of H2O2, once per minute, with continuous agitation. The reaction was stopped by adding 50 μl of 1 mm dithiothreitol (Boehringer Mannheim, Indianapolis, IN). The radiolabeled peptide was purified by reversed-phase HPLC using a 28–58% gradient of acetonitrile on a Beckman HPLC system equipped with gamma scintillation detector, UV spectrophotometer, and a Waters μBondapak C18 column. Fractions were pooled from the portion of the eluate peak showing the greatest immunoreactivity and specific activity. The biological activity and potency of this material, assessed by stimulation of melanogenesis in B16-F1C29 mouse melanoma cells as described (Tatro and Reichlin, 1987), in preliminary tests was determined to be similar to that of unlabeled peptide (data not shown).
Binding assays were performed using intact cells in monolayer cultures using modifications of published methods (Tatro et al., 1990;Roselli-Rehfuss et al., 1993). Cells were plated 48 hr before experiments in 24-well culture plates (Falcon Plastics) at a density of 5 × 104 per well and were 90–95% confluent on the day of assay. Cells were washed twice with wash buffer [10 mm sodium phosphate, 0.15 m NaCl, pH 7.2 (PBS), containing 0.2 mm CaCl2] and incubated with 8–12 × 104 cpm [125I]-NDP-MSH contained in 250 μl of a binding buffer consisting of Ham’s F-10 medium, 0.25% bovine serum albumin (BSA) (A-4503, Sigma), 10 mm HEPES (Life Technologies), 100 μg/ml bacitracin (Sigma), 500 KIU/ml aprotinin (Sigma), and 1 mm1,10-phenanthroline (Sigma), pH 7.2, for 30 min at room temperature, with gentle agitation on a rotary platform shaker. Cells were then washed and harvested by scraping in 1N NaOH.
cAMP accumulation assay. Agonist-induced intracellular accumulation of cAMP was assayed using a method modified from that described previously (Tatro et al., 1990), in cell monolayers plated 48 hr before assay as described for binding assays. Cells were washed twice with PBS and incubated in 0.25 ml assay buffer containing DMEM (Life Technologies), 0.25% BSA, and 100 μm isobutyl methyl xanthine (Sigma) for 5 min at 37°C in a humidified 95% air/5% CO2 atmosphere. Test solutions were then added for an additional 15 min, and incubations were stopped and intracellular cAMP was extracted as described (Tatro et al., 1990). The dried cell extracts were reconstituted in radioimmunoassay (RIA) buffer, and their cAMP contents were determined using a commercial cAMP RIA kit (DuPont NEN, Wilmington, DE).
Animals and surgical procedures. Adult male Sprague Dawley rats (Taconic, Germantown, NY), initially weighing 250–300 gm, were used. The rats were housed on a 12 hr light/dark cycle (lights on at 6 A.M.) and were provided standard laboratory rat chow and water ad libitum. All procedures were approved by the Animal Research Committee of Tufts University Medical School and New England Medical Center. In each rat, a miniature radio transmitter for telemetric monitoring of body temperatures (Tb) (Minimitter, Sunriver, OR) was implanted aseptically in the abdominal cavity under sodium pentobarbital anesthesia (50 mg/kg, i.p.), and a permanent cannula for intracerebroventricular drug administration was placed in the lateral cerebral ventricle as described (Huang et al., 1997). After surgery, the animals were housed in individual plastic cages and maintained in a separate room with temperature maintained at 25 ± 1°C, approximating the thermoneutral ambient temperature range for rats, by means of a convection heater with remote thermostat. Correct placement of intracerebroventricular cannulas was verified by injecting 10 μl of 0.1% cresyl violet through the cannula at the end of the experiment, followed by postmortem brain dissection; data obtained from improperly implanted animals were excluded from analysis. In experiments involving intravenous injection of drugs or blood sampling, rats were implanted aseptically with indwelling jugular catheters under pentobarbital anesthesia 2 d before the experiment. Animals showing weight loss or signs of wound infection after surgery were excluded from further study.
Animal handling procedures. Because of the sensitivity of thermoregulatory pathways and the pituitary–adrenal axis to stress, great care was taken to minimize the influence of nonspecific stress during experiments. The animals were allowed to recover from surgery for at least 7 d before experiments. For the five consecutive days preceding the study, each rat was conditioned to gentle handling for 3–5 min daily. This handling included a simulated intracerebroventricular injection performed by removing the dummy cannula and connecting the injection device to the guide cannula.
Intracerebroventricular injection. Drugs or injection vehicle was administered intracerebroventricularly via an internal cannula connected by flexible tubing to a 100 μl Hamilton syringe, allowing each rat to move about freely in its home cage during infusions, essentially as described (Huang et al., 1997). Drugs and vehicles were injected in a volume of 4 μl at a rate of 2 μl/min using a microinfusion pump (Bee Syringe Pump MF-9090, Bioanalytical Systems, West Lafayette, IN). After injection, the cannula was left in place for 2 min to prevent backflow of the injectate through the guide cannula.
Body temperature (Tb) measurements. Tb was monitored with implanted telemetry transmitters. The emitted frequencies were recorded at 1 hr intervals using a model RTA-500 receiver and a model SM-2372 frequency counter (Minimitter). Transmitters were calibrated before and after each experiment, and frequencies were converted toTb, according to the manufacturer’s instructions. Rats having baseline (time 0) Tb≥ 38.1°C were excluded from analysis.
Blood sampling and hormone measurements. Blood (0.5 ml) was collected via the indwelling jugular catheter into plastic tubes containing EDTA (2 mg) and aprotinin (500 KIU/ml) on ice. Immediately after the collection of each blood sample, 0.5 ml of sterile 0.9% saline was infused via the catheter to maintain blood volume. After centrifugation, plasma samples were harvested and stored at −20°C. ACTH was determined in unextracted plasma by RIA by modifications of a method reported previously (Takaki et al., 1994), using [125I]-human ACTH (ICN Pharmaceuticals, Costa Mesa, CA) and human ACTH antiserum provided by the National Hormone and Pituitary Program of the National Institute of Diabetes and Digestive and Kidney Diseases (lot AFP6328031). Synthetic human ACTH1–39(Sigma) was used as reference standard. Samples were incubated with first antibody overnight at 4°C, the tracer was then added, and the antibody-bound tracer was separated from unbound on the following day using a sheep anti-rabbit second antibody method. All samples were assayed in duplicate. The assay limit of detection was 32 pg/ml. The inter- and intra-assay coefficients of variation, each based on three determinations, were 7.2% and 8.7%, respectively.
Plasma corticosterone (CS) concentrations were measured by modifications of a method described previously (Takaki et al., 1994). Plasma samples diluted 1:100 in assay buffer (PBS containing 0.1% gelatin and 0.04% sodium azide, pH 7.4) were heat-denatured at 70°C for 30 min. Plasma samples or CS standard (Sigma) were incubated overnight at 4°C with [125I]-labeled CS and a rabbit anti-CS serum (ICN Pharmaceuticals) used at a final dilution of 1:17,500. Bound and unbound tracer were separated using a 4 hr incubation with sheep anti-rabbit second antibody (final dilution 1:120; diluted in 0.05 m sodium phosphate-EDTA buffer, pH 7.2), followed by the addition of 1 ml of 10% polyethylene glycol (Carbowax 8000, Fisher Scientific, Fairlawn, NJ) in PBS, centrifugation, and decanting of supernatants. Intra- and interassay coefficients of variation were 9.0% and 10.1%, respectively.
Drugs. SHU-9119 (molecular weight 1188) was prepared as described (Hruby et al., 1995) and dissolved in sterile pyrogen-free 0.9% NaCl (saline) containing 0.1% low-endotoxin BSA (Sigma, A-3675) at a concentration of 1 μg/μl and stored at −70°C. Immediately before experiments, the stock SHU-9119 was diluted further with saline to a final concentration of 50 ng/μl and was injected intracerebroventricularly at a dose of 200 ng. α-MSH (Peninsula Laboratories, Belmont, CA) was dissolved in saline and injected intracerebroventricularly at a volume of 4 μl and dose of 300 ng per rat. Lipopolysaccharide (LPS) (Escherichia coli serotype 055:B5, L-4005, Sigma) was dissolved in saline and injected at a dose of 25 μg/kg, i.p.
Statistics. All data are represented as mean ± SEM unless indicated otherwise. Half-maximal inhibitory concentrations (IC50) for inhibition of [125I]-NDP-MSH binding and for inhibition of α-MSH-induced cAMP accumulationin vitro were estimated by a curve-fitting method as described (Tatro and Entwistle, 1994b). Tb and plasma concentrations of ACTH and CS immediately before intraperitoneal injection of LPS or saline, respectively, were defined as basal levels. The areas under the temperature-, ACTH-, or CS-time response curves (AUC) for each rat were determined by trapezoidal integration after subtracting the respective basal levels from each subsequent value, and were used for tests of statistical significance of treatments. The AUC data for each group were analyzed by one-way ANOVA (Figs. 2, 3, 5) or by two-way ANOVA in studies incorporating a 2 × 2 design (Figs.4, 6), followed by t tests corrected for multiple comparisons by the method of Scheffé (Scheffé, 1959).
Fig. 2.
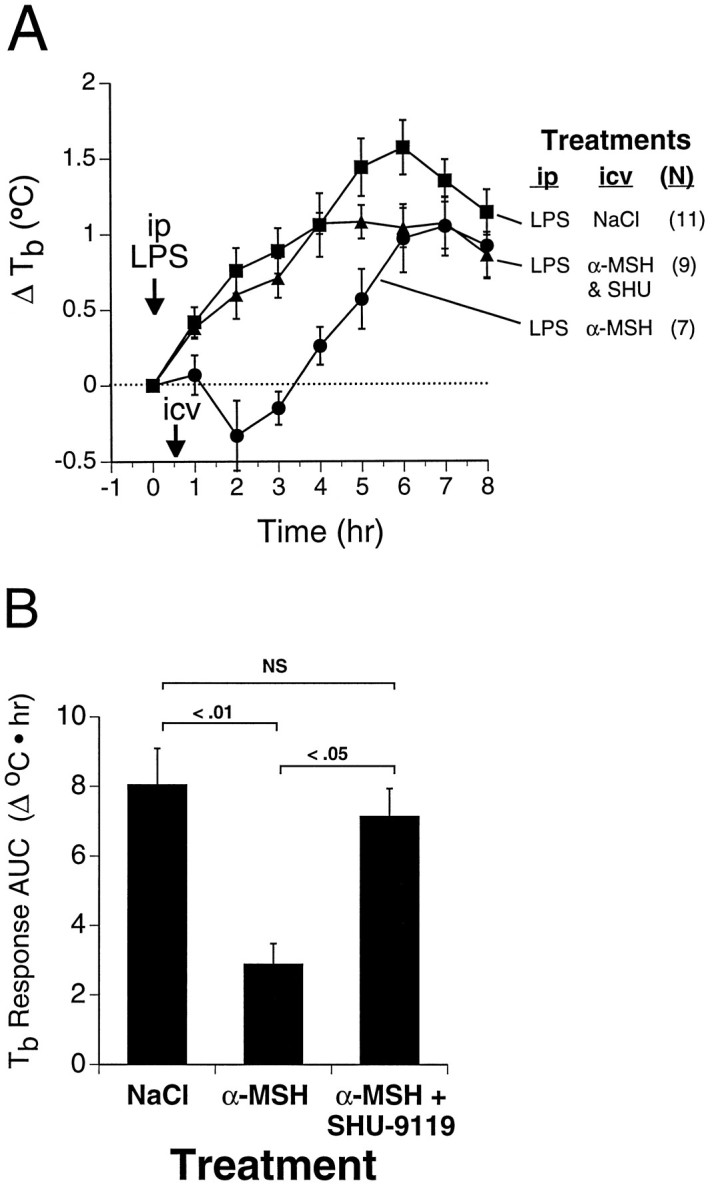
Inhibition of antipyretic effect of α-MSHin vivo by SHU-9119. Rats were treated with LPS (25 μg/kg, i.p.) at time 0 (arrow) and received the indicated injectates intracerebroventricularly 30 min later (arrow). A, Time course of LPS-induced fever in the presence and absence of intracerebroventricular α-MSH (300 ng; 180 pmol) or α-MSH plus SHU-9119 (200 ng; 168 pmol). Numbers of animals in each group are indicated in parentheses.Symbols represent mean ± SEM. B, Data from A represented as area under the temperature-time response curves (mean ± SEM). Overall statistical significance of treatment effects isF(2,24) = 8, p = 0.002; statistical significance of between-group comparisons is indicated in the figure. Baseline (time 0) Tb values for the respective groups were (°C; mean ± SEM) LPS/NaCl (▪), 37.6 ± 0.1; LPS/α-MSH (•), 37.7 ± 0.1; LPS/(α-MSH+SHU-9119) (▴), 37.6 ± 0.1.
Fig. 3.

Lack of thermoregulatory effects of α-MSH (300 ng) and SHU-9119 (200 ng) in afebrile rats. Rats received saline injections intraperitoneally at time 0 (arrow), and the indicated injectates intracerebroventricularly 30 min later (arrow). Numbers of animals in each group are indicated in parentheses. Symbols represent mean ± SEM. Tb profiles and areas under temperature-time response curves (not shown) were not significantly different. Baseline Tb values for the respective groups were (°C) NaCl (▪), 37.4 ± 0.1; α-MSH (•), 37.5 ± 0.1; α-MSH/SHU-9119 (▴), 37.4 ± 0.2.
Fig. 5.

Lack of effect on LPS-induced fever of SHU-9119 administered intravenously. Rats were treated with LPS (25 μg/kg, i.p.) at time 0 (arrow), and received the indicated injectates via indwelling jugular catheter 30 min later (arrow). Numbers of animals in each group are indicated in parentheses. By two-way repeated measures ANOVA,Tb response after LPS is highly significant (F(8,56) = 30, p < 0.0001), but there was no significant effect of SHU-9119 treatment. Areas under the temperature-time response curves (not shown) were not significantly different. Baseline Tb values for the respective groups were (°C) LPS/SHU-9119 (•), 37.7 ± 0.1; LPS/NaCl (▪), 37.5 ± 0.03.
Fig. 4.
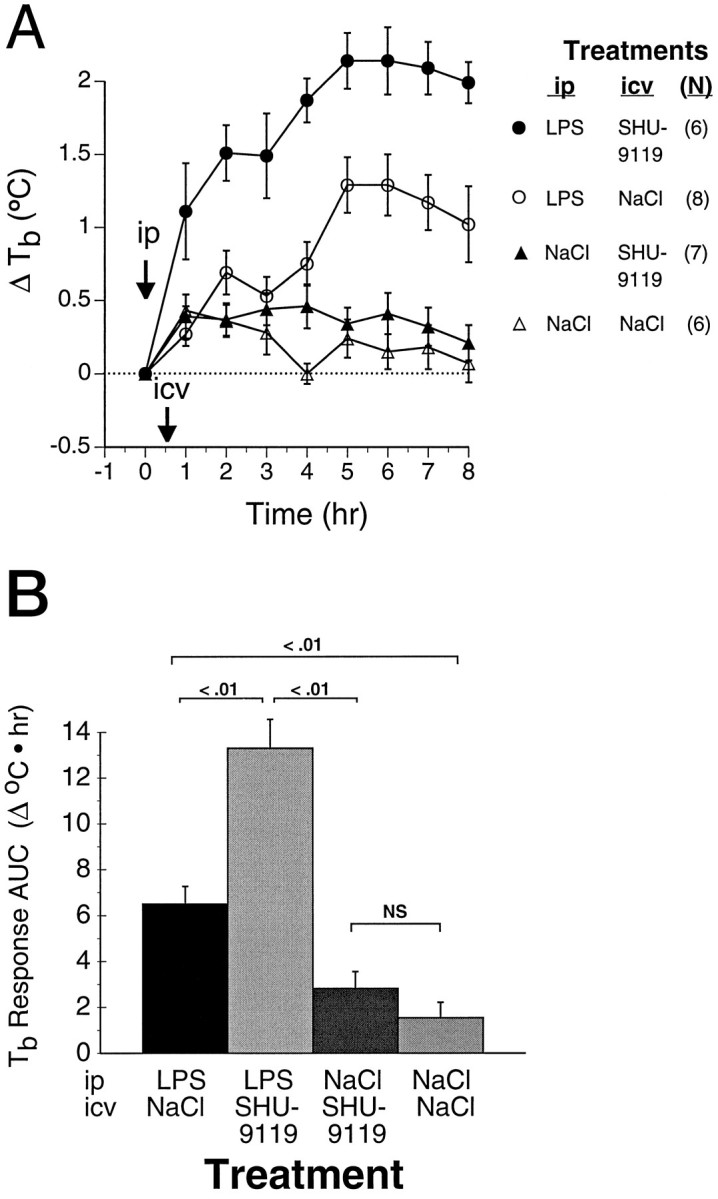
Exacerbation of LPS-induced fever by central MCR blockade. Rats were treated with LPS (25 μg/kg, i.p.) or with saline at time 0 (arrow), and with the indicated intracerebroventricular injectates 30 min later (arrow).A, Time course of LPS-induced fever in the presence and absence of intracerebroventricular SHU-9119 (200 ng). Also shown areTb profiles of rats receiving comparable intracerebroventricular infusions after intraperitoneal injection of saline rather than LPS. Numbers of animals in each group are indicated in parentheses. Baseline Tbvalues for the four respective treatment groups were (°C) LPS/SHU-9119 (•), 37.5 ± 0.1; LPS/NaCl (○), 37.7 ± 0.1; NaCl/SHU-9119 (▴), 37.5 ± 0.2; NaCl/NaCl (▵), 37.6 ± 0.1. B, Data from A represented as areas under the temperature-time response curves. By two-way ANOVA, significance of main effects for LPS and for SHU-9119, respectively, are F(1,23) = 79, p < 0.0001, and F(1,23) = 22,p < 0.0001; F(1,23) = 10, p = 0.004 for interaction. Statistical significance of between-group comparisons is indicated in the figure.
Fig. 6.

Lack of effect of centrally administered SHU-9119 on LPS-induced ACTH and CS secretion. Rats were treated with LPS (25 μg/kg, i.p.) or with saline injection vehicle at time 0, and received the indicated injectates intracerebroventricularly 30 min later.A, B, Time course of LPS-induced plasma ACTH (A) and CS (B) levels in the presence and absence of intracerebroventricular SHU-9119 (200 ng). Data fromA and B, respectively, were represented as area under the plasma hormone-time response curves (not shown) for statistical analysis by two-way ANOVA. For ACTH (A), significance of the main effect of LPS isF(1,31) = 64, p < 0.0001. For CS (B), significance of the main effect of LPS is F(1,31) = 30, p< 0.0001. Effect of SHU-9119 was not significant for either ACTH or CS. Data from a subset of the rats used in this experiment are also included in the study shown in Figure 4.
RESULTS
Validation of SHU-9119 as a potent antagonist of rat MC3-R and MC4-R in vitro
The ability of SHU-9119 to antagonize α-MSH-induced activation of the rat MC3 and MC4 receptor subtypes was assessed in heterologous B16-G4F mouse melanoma cells transfected with plasmid vectors encoding the corresponding receptor proteins. Untransfected B16-G4F cells, which are deficient in expression of the native melanocytic MC1 receptor (Solca et al., 1993), accordingly failed to exhibit detectable α-MSH-induced cAMP accumulation or specific [125I]-NDP-MSH binding (data not shown). In contrast, the B16-G4F sublines stably transfected with either rat MC3-R (B16-G4F-rMC3) or MC4-R (B16-G4F-rMC4) showed robust specific [125I]-NDP-MSH binding and α-MSH-induced cAMP accumulation. Preliminary concentration–response experiments (not shown) indicated that 10 nm α-MSH was very close to the half-maximal stimulating concentration for induction of cAMP accumulation in both the B16-G4F-rMC3 and B16-G4F-rMC4 sublines.In vitro, SHU-9119 inhibited α-MSH-induced cAMP accumulation in B16-G4F-rMC3 and B16-G4F-rMC4 cells in a concentration-dependent manner (IC50 = 0.48 ± 0.19 nm and 0.41 ± 0.28 nm, respectively) (Fig. 1). In contrast, in the absence of α-MSH, SHU-9119 failed to show any intrinsic agonism in B16-G4F-rMC3 or B16-G4F-rMC4 cells (legend, Fig. 1). SHU-9119 also inhibited binding of [125I]-NDP-MSH with a concentration dependence both similar to that for its inhibition of α-MSH-induced cAMP accumulation and parallel to that of α-MSH (Fig. 1), suggesting that it antagonizes activation of the rat MC3-R and MC4-R by inhibiting agonist binding (IC50 for rMC3-R = 1.0 ± 0.1 nm; IC50 for rMC4-R = 0.9 ± 0.3 nm). The relative binding affinity of SHU-9119 was approximately 60-fold greater than that of α-MSH on both rMC3-R and rMC4-R (Fig. 1).
Fig. 1.
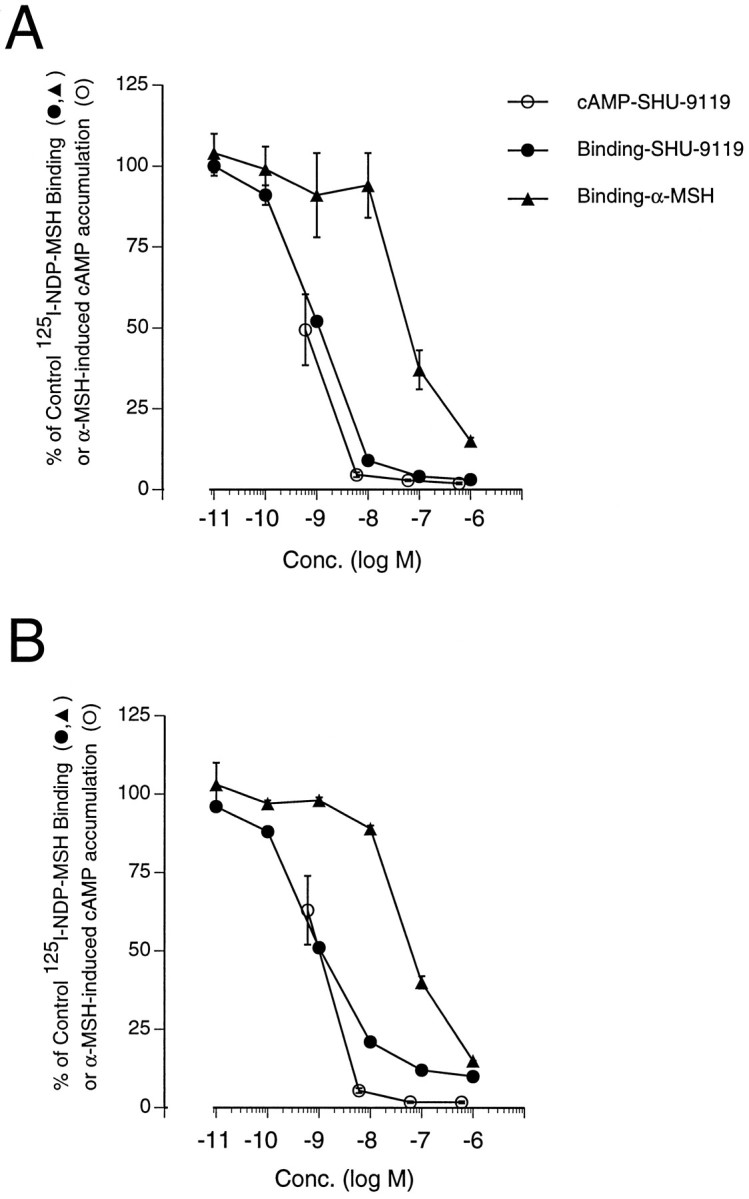
SHU-9119-mediated inhibition of α-MSH (10 nm)-induced cAMP accumulation (○) and specific [125I]-NDP-MSH binding (•) in B16-G4F cells stably expressing the rat MC3-R (A) or MC4-R (B). The potency of α-MSH in inhibiting [125I]-NDP-MSH binding is shown for comparison (▴). For cAMP, the stimulation by 10 nm α-MSH is defined as 100%, and the symbols indicate mean ± SEM of three independent experiments. For [125I]-NDP-MSH binding, control binding in the absence of SHU-9119 or α-MSH was defined as 100%, and the symbols indicate means of two to three separate experiments for SHU-9119 and of one experiment for α-MSH. Because of the different numbers of experiments shown in the various binding curves, their respective error terms are indicated separately.A, B16-G4F-rMC3. cAMP levels under basal and α-MSH-stimulated conditions were 2.2 ± 0.4 and 93.9 ± 36.8 pmol/well (mean ± SEM), respectively. SHU-9119 was tested alone for intrinsic agonism in one experiment at concentrations ranging between 6 × 10−11m and 6 × 10−7m, and all values were ≤ basal levels. Mean control [125I]-NDP-MSH binding ranged between 7142 ± 351 and 1680 ± 147 cpm/well (mean ± SD); the curve represents data pooled from three experiments.B, B16-G4F-rMC4. cAMP levels under basal and α-MSH-stimulated conditions were 0.9 ± 0.4 and 93.9 ± 24.1 pmol/well (mean ± SEM), respectively. SHU-9119 was tested alone for intrinsic agonism in one experiment at concentrations ranging between 6 × 10−11m and 6 × 10−7m, and all values were ≤ 2.8 ± 0.8 pmol/well (mean ± SD). Mean control [125I]-NDP-MSH binding ranged between 901 ± 51 and 1068 ± 169 cpm/well (mean ± SD); the curve represents means of data pooled from two experiments.
Ability of SHU-9119 to block antipyretic action of exogenous α-MSH
To validate the use of SHU-9119 as an antagonist of central MCRin vivo, we tested the ability of intracerebroventricular SHU-9119 to block the antipyretic action of exogenous intracerebroventricular α-MSH in LPS-treated rats. Intracerebroventricular injection of α-MSH 30 min after a moderate dose of LPS suppressed the LPS-induced fever (Fig. 2). When a roughly equimolar dose of SHU-9119 was co-administered intracerebroventricularly with α-MSH, the antipyretic effect of α-MSH was prevented (Fig. 2), demonstrating its efficacy as anin vivo antagonist of central melanocortin action. In contrast, intracerebroventricular administration of α-MSH, or of α-MSH plus SHU-9119, at the same respective doses in the absence of previous LPS treatment had no significant effect onTb (Fig. 3).
MCR-mediated antipyretic role of endogenous central melanocortins
To determine whether endogenous central melanocortins act via central MCR to modulate fever, rats were administered LPS (25 μg/kg, i.p.) or saline, followed 30 min later by intracerebroventricularly administered SHU-9119 (200 ng) or saline. Tbvalues were monitored hourly for 8 hr. LPS-induced fevers in the SHU-9119-injected rats were approximately twice as great, as assessed by Tb AUC (Fig. 4). The increasedTb response of rats treated with LPS followed by intracerebroventricular SHU-9119, relative to that of controls treated with LPS followed by intracerebroventricular saline, was evident within 30 min, and persisted for ≥8 hr (Fig. 4A). The effect of intracerebroventricular SHU-9119 on Tbwas negligible in control rats receiving saline vehicle intraperitoneally (Fig. 4).
To determine whether the observed increase in fever after intracerebroventricular SHU-9119 could be attributed to peripheral actions of SHU-9119 entering the peripheral blood from cerebrospinal fluid, SHU-9119 (200 ng) was administered via intrajugular catheters 30 min after LPS administration. In these experiments, intravenous SHU-9119 had no significant effect on LPS-inducedTb responses (Fig. 5), indicating that the effects of the same dose of SHU-9119 given intracerebroventricularly were not mediated peripherally.
Lack of SHU-9119 effect on LPS activation of the pituitary–adrenal axis
Because LPS activates the pituitary–adrenal axis, and because exogenous α-MSH reportedly inhibits LPS-induced stimulation of ACTH and CS secretion in mice (Rivier et al., 1989), we assessed the effects of central MCR blockade on LPS-induced ACTH and CS levels in rats. LPS, SHU-9119, and saline vehicle were administered as described for the experiment shown in Figure 4. As expected, LPS treatment led to increased plasma levels of ACTH (maximal at 1 hr) and CS (maximal at 2 hr) (Fig. 6). No significant differences in plasma ACTH or CS levels, however, were detected between intraperitoneal LPS/intracerebroventricular SHU-9119-treated and intraperitoneal LPS/intracerebroventricular saline-treated rats (Fig. 6), indicating that central MCR blockade did not affect the pituitary–adrenal axis under the same conditions in which it produced augmented fevers. Likewise, intravenous injection of SHU-9119 had no effect on LPS-induced plasma ACTH and CS levels, as determined in the same rats whose Tb profiles are shown in Figure 5 (data not shown).
DISCUSSION
These results provide significant new insights into several aspects of melanocortin pharmacology and the antipyretic roles of melanocortins. An initial series of studies validated the experimental model used. First, the efficacy and potency of SHU-9119 as a blocker of agonist-induced activation of rat MC3 and MC4 receptors in vitro was demonstrated. This was important because the potent antagonism of human MC3-R and MC4-R by SHU-9119 reported earlier (Hruby et al., 1995) does not necessarily predict its effects on the brain-associated MC3-R and MC4-R homologs of the rat, because differences of even a single amino acid in primary structure can dramatically alter the pharmacological profile of MCR isoforms (Robbins et al., 1993; Doré et al., 1996). Second, because previous information on antipyretic actions of α-MSH in the rat has been limited (Bull et al., 1990; Villar et al., 1991), the antipyretic effect of intracerebroventricular α-MSH in rats was confirmed. Third, the ability of intracerebroventricular SHU-9119 to block the antipyretic effect of exogenous intracerebroventricular α-MSH was demonstrated. Furthermore, the same dose of SHU-9119, administered either in the absence or presence of co-administered α-MSH, had no significant effect on Tb of afebrile rats, indicating its specificity. This is consistent with the recent finding that cardiovascular effects of α-MSH microinjected into the dorsal vagal complex, but not baseline cardiovascular parameters, were selectively blocked by co-administration of SHU-9119 (Li et al., 1996).
With these key technical points established, the core finding of the present study is that central administration of SHU-9119 to febrile rats exacerbated fever. In contrast, intravenous administration of the same dose of SHU-9119 had no effect on LPS-induced fever. Therefore, these findings indicate that endogenous melanocortins act on MCR within the brain during fever to exert an antipyretic effect. It is reasonable to hypothesize that the MCRs involved may be those known to be present in certain brain regions believed to be involved in thermoregulation and fever, including the preoptic region, anterior hypothalamus and paraventricular nucleus, ventral and lateral septal regions, and autonomic nuclei of the hindbrain (Tatro, 1990; Tatro and Entwistle, 1994a). These findings also raise the question of whether the antipyretic effects of peripherally administered melanocortins (Bull et al., 1990; Catania and Lipton, 1993) are likewise mediated by direct actions on MCRs located within the brain.
The present results confirm and extend those of an earlier study in which endogenous α-MSH was depleted by passive immunization using anti-α-MSH antibodies (Shih et al., 1986). In that study, the resulting prolongation of fever was quite long lasting, similar to the effect of intracerebroventricular SHU-9119 (Fig. 4A), and suggestive of a continuous damping effect of endogenous melanocortins throughout the course of fever. The anti-α-MSH antibodies were administered intracerebroventricularly, but they were given daily for 3 d. Because blood antibody titers were not determined, the possibility could not be ruled out that the effects were exerted in the periphery. Furthermore, the potential role of central MCRs in mediating the antipyretic effects of melanocortins could not be tested directly by the earlier passive immunization approach (Shih et al., 1986).
The source of endogenous α-MSH whose antipyretic action is blocked by intracerebroventricular SHU 9119 is probably central rather than peripheral, because the access of blood-borne melanocortins to brain parenchyma is severely restricted by the blood-brain barrier (Wilson, 1988). Accordingly, MCR-bearing brain regions, including the preoptic region, anterior hypothalamus and paraventricular nucleus, ventral and lateral septal regions, and autonomic nuclei of the hindbrain, are innervated by melanocortinergic neurons projecting from the hypothalamic arcuate nucleus (O’Donohue et al., 1979; Mezey et al., 1985; Palkovits et al., 1987). Furthermore, several lines of evidence suggest that neuronal release of melanocortins is activated within MCR-bearing brain regions during fever. First, arcuate neurons of unknown identity but having a distribution and morphology similar to pro-opiomelanocortin neurons are activated, as indicated by increased Fos expression, after intraperitoneal injection of LPS (Sagar, 1994) or intracerebroventricular injection of IL-1 (Rivest et al., 1992). The latter finding is relevant because intraparenchymal IL-1 is believed to be an important mediator of LPS-induced fever (Klir et al., 1994) and HPA activation (Kakucska et al., 1993), based on studies involving intracranial IL-1 immunoneutralization or IL-1 receptor blockade. Second, the α-MSH content of arcuate neurons, which comprise the sole source of melanocortin peptides in the forebrain, decreased within 2 hr after peripheral IL-1 treatment of rats as indicated by semiquantitative immunostaining (Vriend et al., 1994), suggestive of acute α-MSH export. Third, during fever in rabbits, immunoreactive α-MSH levels increased in the septum (Samson et al., 1981; Holdeman et al., 1985), a putative site of α-MSH-mediated antipyretic action based on microinjection studies (Glyn-Ballinger et al., 1983). A push–pull perfusion study from the same laboratory suggested increased septal neurosecretion of α-MSH during fever (Bell and Lipton, 1987).
Regardless of potential changes in α-MSH neurosecretion, the present results clearly indicate that a febrile state-dependent increase in central responsiveness to the antipyretic action of melanocortins occurs during LPS-induced fever, because the same doses of SHU-9119 and of α-MSH that modulated fever failed to significantly affect Tb of afebrile rats. This conclusion is consistent with the results of earlier studies (Lipton et al., 1981;Shih et al., 1986; Catania and Lipton, 1993). The mechanisms potentially accounting for this phenomenon, such as MCR upregulation during fever, remain to be elucidated.
The fever-augmenting effect of central MCR blockade appeared within the first hour after LPS treatment, indicating that the onset of antipyretic activity of endogenous melanocortins occurs early during fever. Furthermore, the effect persisted for at least 8 hr. One hypothesis to explain this long-lasting effect is that SHU-9119 may dissociate very slowly from its target MCRs. Other Nle4-containing α-MSH analogs dissociate very slowly after binding to MCRs in vitro (Haskell-Leuvano et al., 1996). In particular, the structurally similar cyclic lactam α-MSH analog Ac-Nle4-c[Asp5,dPhe7,Lys10]α-MSH(4–10)-NH2had the slowest dissociation rate, with 86% of tracer remaining bound after 6 hr (Haskell-Leuvano et al., 1996). In any event, the present results indicate that the physiological response to endogenous melanocortins is triggered early during fever and persists throughout its course.
The specific MCR subtype(s) that mediate the antipyretic effects of endogenous central melanocortins in the rat cannot be determined from these studies, because SHU-9119 does not discriminate between rat MC3-R and MC4-R (Fig. 1). Nevertheless, it is likely that one or both of these MCR subtypes contributes to the antipyretic central action of endogenous melanocortins, because the mRNAs encoding rat MC3-R and MC4-R are distributed among ventral forebrain structures involved in thermoregulation (Roselli-Rehfuss et al., 1993; Mountjoy et al., 1994) and are the predominant MCR genes known to be expressed in rat brain. In contrast, mRNA encoding the other MCR subtype reportedly expressed in brain, MC5-R, is of very low abundance in the rat brain, because it is not detectable by sensitive RNase protection assay or in situ hybridization (Griffon et al., 1994; Alvaro et al., 1996) but only by the ultrasensitive PCR (Griffon et al., 1994). Most importantly, like α-MSH, SHU-9119 acts as an agonist on the mouse MC5-R as well as on the peripherally expressed MC1-R (Hruby et al., 1995), whereas in the present studies SHU-9119 blocked the antipyretic action of exogenous α-MSH. Furthermore, neither α-MSH nor SHU-9119 administered intracerebroventricularly significantly affectedTb of afebrile rats. Taken together, the present results therefore do not support a role of central MC5-R or other identified MCR subtypes in modulating fever in the rat.
The present results clearly demonstrate that central melanocortins can exert a suppressive influence on fever in rats, consistent with earlier studies in several species (Catania and Lipton, 1993; Bock et al., 1994). Nevertheless, the spectrum of central thermoregulatory actions of exogenous melanocortins seems to be quite broad. High doses of centrally administered α-MSH or ACTH were hypothermic in afebrile rabbits (Lipton et al., 1981) and guinea pigs (Kandasamy and Williams, 1984). In contrast, other groups have observed hyperthermic effects of intraparenchymal central α-MSH microinjection in rats (Raible and Knickerbocker, 1993; Resch and Millington, 1993). These hyperthermic effects (Raible and Knickerbocker, 1993) exhibited biphasic, bell-shaped dose–response relationships, as commonly observed for both thermoregulatory and nonthermoregulatory actions of exogenous melanocortins (Cannon et al., 1986; Hiltz and Lipton, 1990; Catania and Lipton, 1993). Furthermore, Rothwell and colleagues concluded that γ1-MSH contributes to the thermogenic effect of centrally administered CRF in rats, based on studies using centrally administered anti-γ-MSH antibodies (Rothwell et al., 1991). Rather than contradicting the present results, these earlier findings probably reflect the complex neuropharmacology of the thermoregulatory actions of melanocortins.
In fact, a combination of neuroanatomic and pharmacological factors may potentially contribute to the apparent complexity of thermoregulatory actions of melanocortins. Whereas intraseptal infusion of α-MSH or γ2-MSH, respectively, was antipyretic in rabbits (Lipton et al., 1981) or rats (Bock et al., 1994), intrapreoptic α-MSH microinjections in afebrile rats were hyperthermic (Raible and Knickerbocker, 1993; Resch and Millington, 1993). In rats, mRNA encoding multiple MCR isoforms (primarily MC3-R and MC4-R) are differentially distributed among thermosensitive areas and autonomic control centers of the ventral forebrain, including the septal area, preoptic region, anterior hypothalamus, paraventricular nucleus, and arcuate nucleus, as well as in autonomic nuclei of the hindbrain (Roselli-Rehfuss et al., 1993; Mountjoy et al., 1994). These MCR isoforms exhibit distinct agonist profiles. For instance, the MC4-R, mRNA for which is expressed widely in neuroendocrine- and autonomic-related structures in the ventral forebrain and hindbrain of the rat (Mountjoy et al., 1994), preferentially binds α-MSH (Gantz et al., 1993b; Alvaro et al., 1996). In contrast, the MC3-R, for which mRNA expression in relevant brain structures in the rat is largely restricted to hypothalamus and preoptic and septal regions, is at least as sensitive to γ-MSH as to α-MSH (Gantz et al., 1993a;Roselli-Rehfuss et al., 1993). The heterogeneity of MCR populations expressed in different brain regions is supported further by the demonstration of regional differences in ligand binding affinity profiles (Tatro and Entwistle, 1994b). Thus the differential distribution of MCR subtypes in different brain regions may contribute to the qualitative differences in thermoregulatory responses to the melanocortins observed, depending on the site, dose, and form of melanocortin administered centrally. Taken together, the available evidence thus suggests that exogenous melanocortins have multiple, dose-dependent, site-dependent, and physiological state-dependent thermoregulatory central actions. Determining which of these effects truly reflects a role of endogenous melanocortins requires precise definition of experimental conditions, as in the present study.
In these studies, intracerebroventricular administration of a fever-enhancing dose of SHU-9119 did not significantly affect LPS-induced ACTH and CS secretion. These findings indicate that the observed effect of central MCR blockade on fever cannot be attributed to interference with activation of the pituitary–adrenal axis, a major cytokine-counterregulatory feedback pathway (Reichlin, 1993). Furthermore, because ACTH and CS levels provide a sensitive index of stress, the lack of HPA activation in control (non-LPS-injected) rats receiving intracerebroventricular SHU-9119, as well as in rats receiving intracerebroventricular saline, provide important negative controls in these studies. In fact, even such seemingly minor stressors as exposure to a novel environment or “open field” can cause fever in rats (Kluger, 1991), but no such effect was observed in the present studies. Therefore, taken together, the present findings rule out nonspecific stress as a causative or confounding factor in their interpretation.
In summary, the present studies clearly demonstrate an antipyretic role of endogenous melanocortins acting on MCRs within the brain during LPS-induced fever. These findings indicate that central mechanisms operate during fever to mitigate as well as to promote fever, illustrating the intricate balance of central pathways through which the CNS exerts fine control and coordination of host responses to systemic infection.
Footnotes
This work was supported by National Institutes of Health Grants MH44694 (J.B.T.), DA08227 (R.S.D), and DK17420 (V.J.H.). We thank Dr. W. Yuan for preparing the α-MSH analog SHU-9119.
Correspondence should be addressed to Dr. Jeffrey B. Tatro, Division of Endocrinology, Diabetes, Metabolism and Molecular Medicine, Box 268, New England Medical Center, 750 Washington Street, Boston, MA 02111.
REFERENCES
- 1.Alvaro JD, Tatro JB, Quillan JM, Fogliano M, Eisenhard M, Lerner MR, Nestler EJ, Duman RS. Morphine down-regulates melanocortin-4 receptor expression in brain regions that mediate opiate addiction. Mol Pharmacol. 1996;50:583–591. [PubMed] [Google Scholar]
- 2.Bell RC, Lipton JM. Pulsatile release of antipyretic neuropeptide α-MSH from septum of rabbit during fever. Am J Physiol. 1987;252:R1152–R1157. doi: 10.1152/ajpregu.1987.252.6.R1152. [DOI] [PubMed] [Google Scholar]
- 3.Bock M, Roth J, Kluger MJ, Zeisberger E. Antipyresis caused by stimulation of vasopressinergic neurons and intraseptal or systemic infusions of γ-MSH. Am J Physiol. 1994;266:R614–R621. doi: 10.1152/ajpregu.1994.266.2.R614. [DOI] [PubMed] [Google Scholar]
- 4.Bull DF, King MG, Pfister HP, Singer G. Alpha-melanocyte-stimulating hormone conditioned suppression of a lipopolysaccharide-induced fever. Peptides. 1990;11:1027–1031. doi: 10.1016/0196-9781(90)90028-4. [DOI] [PubMed] [Google Scholar]
- 5.Cannon JG, Tatro JB, Reichlin SR, Dinarello CA. α-Melanocyte stimulating hormone inhibits immunostimulatory and inflammatory actions of interleukin-1. J Immunol. 1986;137:2232–2236. [PubMed] [Google Scholar]
- 6.Catania A, Lipton JM. α-Melanocyte stimulating hormone in the modulation of host reactions. Endocr Rev. 1993;14:564–576. doi: 10.1210/edrv-14-5-564. [DOI] [PubMed] [Google Scholar]
- 7.Coelho MM, Souza GE, Pela IR. Endotoxin-induced fever is modulated by endogenous glucocorticoids in rats. Am J Physiol. 1992;263:R423–R427. doi: 10.1152/ajpregu.1992.263.2.R423. [DOI] [PubMed] [Google Scholar]
- 8.Doré B, Entwistle ML, Tatro JB, Drouin J, Roselli-Rehfuss L (1996) Mutations in three amino acids of the rat melanocortin 3 receptor result in altered receptor activity. Proceedings of the 10th International Congress of Endocrinology, San Francisco, CA, June 1996, p 794.
- 9.Gantz I, Kondaf Y, Tashiro T, Shimoto Y, Miwa H, Munzert G, Watson SJ, DelValle J, Yamada T. Molecular cloning of a novel melanocortin receptor. J Biol Chem. 1993a;268:8248–8250. [PubMed] [Google Scholar]
- 10.Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, DelValle J, Yamada T. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J Biol Chem. 1993b;268:15174–15179. [PubMed] [Google Scholar]
- 11.Glyn-Ballinger JR, Bernardini GL, Lipton JM. α-MSH injected into the septal region reduces fever in rabbits. Peptides. 1983;4:199–203. doi: 10.1016/0196-9781(83)90114-6. [DOI] [PubMed] [Google Scholar]
- 12.Griffon N, Mignon V, Facchinetti P, Diaz J, Schwartz J, Sokoloff P. Molecular cloning and characterization of the rat fifth melanocortin receptor. Biochem Biophys Res Commun. 1994;200:1007–1014. doi: 10.1006/bbrc.1994.1550. [DOI] [PubMed] [Google Scholar]
- 13.Haskell-Leuvano C, Miwa H, Dickinson C, Hadley ME, Hruby VJ, Yamada T, Gantz I. Characterizations of the unusual dissociation properties of melanotropin peptides from the melanocortin receptor, hMC1R. J Med Chem. 1996;39:432–435. doi: 10.1021/jm950407s. [DOI] [PubMed] [Google Scholar]
- 14.Hiltz ME, Lipton JM. Alpha-MSH peptides inhibit acute inflammation and contact sensitivity. Peptides. 1990;11:979–982. doi: 10.1016/0196-9781(90)90020-6. [DOI] [PubMed] [Google Scholar]
- 15.Holdeman M, Khorram O, Samson WK, Lipton JM. Fever-specific changes in central MSH and CRF concentrations. Am J Physiol. 1985;248:R125–R129. doi: 10.1152/ajpregu.1985.248.1.R125. [DOI] [PubMed] [Google Scholar]
- 16.Hruby VJ, Lu D, Sharma SD, Castrucci AL, Kesterson RA, Al-Obeidi FA, Hadley ME, Cone RD. Cyclic lactam α-melanotropin analogues of Ac-Nle4-cyclo[Asp5, D-Phe7, Lys10]-α-melanocyte-stimulating hormone(4–10)-NH2 with bulky aromatic amino acids at position 7 show high antagonist potency and selectivity at specific melanocortin receptors. J Med Chem. 1995;38:3454–3461. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- 17.Huang Q-H, Takaki A, Arimura A (1997) Central noradrenergic system modulates plasma interleukin-6 production by peripheral interleukin-1β. Am J Physiol, in press. [DOI] [PubMed]
- 18.Kakucska I, Qi Y-P, Clark BD, Lechan RM. Endotoxin-induced corticotropin-releasing hormone gene expression in the hypothalamic paraventricular nucleus is mediated centrally by interleukin-1. Endocrinology. 1993;113:815. doi: 10.1210/endo.133.2.8344218. [DOI] [PubMed] [Google Scholar]
- 19.Kandasamy SB, Williams BA. Hypothermic and antipyretic effects of ACTH (1–24) and alpha-melanotropin in guinea-pigs. Neuropharmacology. 1984;23:49–53. doi: 10.1016/0028-3908(84)90216-8. [DOI] [PubMed] [Google Scholar]
- 20.Klir JJ, McClellan JL, Kluger MJ. Interleukin-1β causes the increase in anterior hypothalamic interleukin-6 during LPS-induced fever in rats. Am J Physiol. 1994;266:R1845–R1848. doi: 10.1152/ajpregu.1994.266.6.R1845. [DOI] [PubMed] [Google Scholar]
- 21.Kluger MJ. Fever: role of pyrogens and cryogens. Physiol Rev. 1991;71:93–127. doi: 10.1152/physrev.1991.71.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li S-J, Varga K, Archer P, Hruby VJ, Sharma SD, Kesterson RA, Cone RD, Kunos G. Melanocortin antagonists define two distinct pathways of cardiovascular control by α- and γ-melanocyte-stimulating hormones. J Neurosci. 1996;16:5182–5188. doi: 10.1523/JNEUROSCI.16-16-05182.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lipton JM, Glyn JR, Zimmer JA. ACTH and alpha-melanotropin in central temperature control. Fed Proc. 1981;40:2760–2764. [PubMed] [Google Scholar]
- 24.McClellan JL, Klir JJ, Morrow LE, Kluger MJ. Central effects of glucocorticoid receptor antagonist RU-38486 on lipopolysaccharide and stress-induced fever. Am J Physiol. 1994;267:R705–R711. doi: 10.1152/ajpregu.1994.267.3.R705. [DOI] [PubMed] [Google Scholar]
- 25.Mezey E, Kiss JZ, Mueller GP, Eskay R, O’Donohue TL, Palkovits M. Distribution of the pro-opiomelanocortin derived peptides, adrenocorticotropic hormone, alpha-melanocyte-stimulating hormone and beta-endorphin (ACTH, α-MSH, β-END) in the rat hypothalamus. Brain Res. 1985;328:341–347. doi: 10.1016/0006-8993(85)91046-7. [DOI] [PubMed] [Google Scholar]
- 26.Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol. 1994;8:1298–1308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 27.Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev. 1984;5:25–44. doi: 10.1210/edrv-5-1-25. [DOI] [PubMed] [Google Scholar]
- 28.O’Donohue TL, Miller RL, Jacobowitz DM. Identification, characterization and stereotaxic mapping of intraneuronal α-melanocyte stimulating hormone-like immunoreactive peptides in discrete regions of the rat brain. Brain Res. 1979;176:101–123. doi: 10.1016/0006-8993(79)90873-4. [DOI] [PubMed] [Google Scholar]
- 29.Palkovits M, Mezey E, Eskay RL. Pro-opiomelanocortin-derived peptides (ACTH/β-endorphin/α-MSH) in brainstem baroreceptor areas of the rat. Brain Res. 1987;436:323–328. doi: 10.1016/0006-8993(87)91676-3. [DOI] [PubMed] [Google Scholar]
- 30.Raible LH, Knickerbocker D. Alpha-melanocyte-stimulating hormone (MSH) and [Nle4, D-Phe7]-alpha-MSH: effects on core temperature in rats. Pharmacol Biochem Behav. 1993;44:533–538. doi: 10.1016/0091-3057(93)90163-n. [DOI] [PubMed] [Google Scholar]
- 31.Reichlin S. Neuroendocrine-immune interactions. New Engl J Med. 1993;329:1246–1253. doi: 10.1056/NEJM199310213291708. [DOI] [PubMed] [Google Scholar]
- 32.Resch GE, Millington WR. Glycyl-l-glutamine antagonizes α-MSH-elicited thermogenesis. Peptides. 1993;14:971–975. doi: 10.1016/0196-9781(93)90073-p. [DOI] [PubMed] [Google Scholar]
- 33.Rivest S, Torres G, Rivier C. Differential effects of central and peripheral injection of interleukin-1β on brain c-fos expression and neuroendocrine functions. Brain Res. 1992;587:13–23. doi: 10.1016/0006-8993(92)91424-d. [DOI] [PubMed] [Google Scholar]
- 34.Rivier C, Chizzonite R, Vale W. In the mouse, the activation of the hypothalamic-pituitary-adrenal axis by a lipopolysaccharide (endotoxin) is mediated through interleukin-1. Endocrinology. 1989;125:2800–2805. doi: 10.1210/endo-125-6-2800. [DOI] [PubMed] [Google Scholar]
- 35.Robbins LS, Nadeau JH, Johnson KR, Kelly MA, Roselli-Rehfuss L, Baack E, Mountjoy KG, Cone RD. Pigmentation phenotypes of variant extension locus alleles result from point mutations that alter MSH receptor function. Cell. 1993;72:827–834. doi: 10.1016/0092-8674(93)90572-8. [DOI] [PubMed] [Google Scholar]
- 36.Roselli-Rehfuss L, Mountjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro JB, Entwistle ML, Simerly RB, Cone RD. Identification of a receptor for POMC peptides in the medial basal hypothalamus and limbic system. Proc Natl Acad Sci USA. 1993;90:8856–8860. doi: 10.1073/pnas.90.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rothwell NJ, Hardwick A, LeFeuvre RA, Crosby SR, White A. Central actions of CRF on thermogenesis are mediated by pro-opiomelanocortin products. Brain Res. 1991;541:89–92. doi: 10.1016/0006-8993(91)91078-f. [DOI] [PubMed] [Google Scholar]
- 38.Sagar SM. Functional neuroanatomy of the acute phase response. Ann NY Acad Sci. 1994;739:282–291. doi: 10.1111/j.1749-6632.1994.tb19830.x. [DOI] [PubMed] [Google Scholar]
- 39.Samson WK, Lipton JM, Zimmer JA, Glyn JR. The effect of fever on central alpha-MSH concentrations in the rabbit. Peptides. 1981;2:419–423. doi: 10.1016/s0196-9781(81)80098-8. [DOI] [PubMed] [Google Scholar]
- 40.Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. [Nle4,D-Phe7]α-melanocyte stimulating hormone: a highly potent α-melanotropin with ultralong biological activity. Proc Natl Acad Sci USA. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheffé H. The analysis of variance. Wiley; New York: 1959. [Google Scholar]
- 42.Shalts E, Feng Y-J, Ferin M, Wardlaw SL. alpha-Melanocyte-stimulating hormone antagonizes the neuroendocrine effects of corticotropin-releasing factor and interleukin-1-alpha in the primate. Endocrinology. 1992;131:132–138. doi: 10.1210/endo.131.1.1319315. [DOI] [PubMed] [Google Scholar]
- 43.Shih ST, Khorram O, Lipton JM, McCann SM. Central administration of alpha-MSH antiserum augments fever in the rabbit. Am J Physiol. 1986;250:803–806. doi: 10.1152/ajpregu.1986.250.5.R803. [DOI] [PubMed] [Google Scholar]
- 44.Solca FF, Chluba-de Tapia J, Iwata K, Eberle AN. B16–G4F mouse melanoma cells: an MSH receptor-deficient cell clone. FEBS Lett. 1993;322:177–180. doi: 10.1016/0014-5793(93)81563-f. [DOI] [PubMed] [Google Scholar]
- 45.Sundar SK, Becker KJ, Cierpial MA, Carpenter MD, Rankin LA, Fleener SL, Ritchie JC, Simson PE, Weiss JM. Intracerebroventricular infusion of interleukin 1 rapidly decreases peripheral cellular immune responses. Proc Natl Acad Sci USA. 1989;86:6398–6402. doi: 10.1073/pnas.86.16.6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takaki A, Huang QH, Somogyvari VA, Arimura A. Immobilization stress may increase plasma interleukin-6 via central and peripheral catecholamines. Neuroimmunomodulation. 1994;1:335–342. doi: 10.1159/000097185. [DOI] [PubMed] [Google Scholar]
- 47.Tatro JB. Melanotropin receptors in the brain are differentially distributed and recognize both corticotropin and α-melanocyte stimulating hormone. Brain Res. 1990;536:124–132. doi: 10.1016/0006-8993(90)90016-5. [DOI] [PubMed] [Google Scholar]
- 48.Tatro JB. Melanotropin receptors of the brain. In: Conn PM, editor. Methods in neurosciences, Vol 11. Academic; New York: 1996. pp. 87–104. [Google Scholar]
- 49.Tatro JB (1996) Receptor biology of the melanocortins, a family of neuroimmunomodulatory peptides. Neuroimmunomodulation, in press. [DOI] [PubMed]
- 50.Tatro JB, Entwistle ML. Distribution of melanocortin receptors in the lower brainstem of the rat. Ann NY Acad Sci. 1994a;739:311–314. doi: 10.1111/j.1749-6632.1994.tb19833.x. [DOI] [PubMed] [Google Scholar]
- 51.Tatro JB, Entwistle ML. Heterogeneity of brain melanocortin receptors suggested by differential ligand binding in situ. Brain Res. 1994b;635:148–158. doi: 10.1016/0006-8993(94)91434-6. [DOI] [PubMed] [Google Scholar]
- 52.Tatro JB, Reichlin S. Specific receptors for α-melanocyte-stimulating hormone are widely distributed in tissues of rodents. Endocrinology. 1987;121:1900–1907. doi: 10.1210/endo-121-5-1900. [DOI] [PubMed] [Google Scholar]
- 53.Tatro JB, Entwistle ML, Lester BR, Reichlin S. Melanotropin receptors of murine melanoma characterized in cultured cells and demonstrated in experimental tumors in situ. Cancer Res. 1990;50:1237–1242. [PubMed] [Google Scholar]
- 54.Tatro JB, Entwistle ML, Alvaro JD, Duman RS, Sharma S, Hruby VJ, Huang Q-H. Antipyretic role of endogenous brain melanocortins during fever. Soc Neurosci Abstr. 1996;22:1456. doi: 10.1523/JNEUROSCI.17-09-03343.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thorell JI, Johansson BG. Enzymatic iodination of polypeptides with 125I to high specific activity. Biochim Biophys Acta. 1971;251:363–369. doi: 10.1016/0005-2795(71)90123-1. [DOI] [PubMed] [Google Scholar]
- 56.Villar M, Perassi N, Cellis ME. Central and peripheral actions of alpha-MSH in the thermoregulation of rats. Peptides. 1991;12:1441–1443. doi: 10.1016/0196-9781(91)90231-d. [DOI] [PubMed] [Google Scholar]
- 57.Vriend CA, Janz L, Jansen A, Murray L, Greenberg AH, Nance DM. Alpha-MSH activates the splenic nerve and is depleted from the arcuate nucleus following IL-1β injections. Soc Neurosci Abstr. 1994;20:950. [Google Scholar]
- 58.Wilson JF. Low permeability of the blood-brain barrier to nanomolar concentrations of immunoreactive alpha-melanotropin. Psychopharmacology (Berl) 1988;96:262–266. doi: 10.1007/BF00177572. [DOI] [PubMed] [Google Scholar]