

Abstract
Newborn mice carrying targeted mutations in genes encoding neurotrophins or their signaling Trk receptors display severe neuronal deficits in the peripheral nervous system but not in the CNS. In this study, we show that trkB (−/−) mice have a significant increase in apoptotic cell death in different regions of the brain during early postnatal life. The most affected region in the brain is the dentate gyrus of the hippocampus, although elevated levels of pyknotic nuclei were also detected in cortical layers II and III and V and VI, the striatum, and the thalamus. Furthermore, axotomized hippocampal and motor neurons of trkB (−/−) mice have significantly lower survival rates than those of wild-type littermates. These results suggest that neurotrophin signaling through TrkB receptors plays a role in the survival of CNS neurons during postnatal development. Moreover, they indicate that TrkB receptor signaling protects subpopulations of CNS neurons from injury- and axotomy-induced cell death.
Keywords: TrkB, CNS, cell death, axotomy, hippocampus, motor neuron
Neurotrophins, including nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), NT-4/5, and NT-6, have been shown to promote neuronal survival of a variety of neuronal populations (Fariñas and Reichardt, 1996). Mutant mice lacking the genes encoding each of these neurotrophins or their receptors have illustrated the exquisite requirement of neurotrophin signaling for the survival of distinct neuronal populations in the peripheral nervous system during embryonic development (Snider, 1994; Barbacid, 1995; Fariñas and Reichardt, 1996). However, very few defects have been detected in the CNS of these mutant mice.
It has been suggested (Snider, 1994; Fariñas and Reichardt, 1996) that the absence of CNS defects in these animals might be attributable to the significant overlap in the pattern of TrkB and TrkC receptors in CNS neurons (Ernfors et al., 1992; Merlio et al., 1992). However, this hypothesis seems unlikely, because double mutant mice lacking both of these Trk receptors do not show any obvious defects in the CNS, at least during embryonic development (Silos-Santiago et al., 1997). These observations suggest that the growth factor requirements of CNS neurons are more complex than those of the periphery. Another possibility is that CNS neurons require only neurotrophin support during postnatal development or even in adult animals. Alternatively, the role of neurotrophins in the CNS might involve aspects of neuronal function other than survival. Indeed, neurotrophin signaling has been implicated in physiological events such as synthesis of neuroactive substances (Lindsay and Harmar, 1989; Ip et al., 1993; Nawa et al., 1993, 1994;Jones et al., 1994; Marty et al., 1996), synaptic efficacy and rearrangement (Lohof et al., 1993; Cabelli et al., 1995; Kang and Schuman, 1995; Korte et al., 1995; Lesser and Lo, 1995; Levine et al., 1995; Lo, 1995; Thoenen, 1995; Patterson et al., 1996) and modulation of dendritic and axonal growth (Diamond et al., 1992; Schnell et al., 1994; Cohen-Cory and Fraser, 1995; McAllister et al., 1995).
Neurotrophin signaling may also play a primary role in protecting CNS neurons from insults during postnatal life. Indeed, there is abundant evidence supporting the concept that neurotrophins and their receptors have a protective effect on many neuronal populations after injury (Sendtner et al., 1992; Yan et al., 1992, 1993; Koliatsos et al., 1993; Arenas and Persson, 1994; Li et al., 1994). For instance, it has been shown that axotomy results in changes in the expression of neurotrophins and p75 and Trk receptors in neurons, glial cells, and target tissues (Heumann et al., 1987; Ernfors et al., 1989;Frisén et al., 1992, 1993; Meyer et al., 1992; Beck et al., 1993;Funakoshi et al., 1993; Koliatsos et al., 1993; Mearow et al., 1993;Merlio et al., 1993; Piehl et al., 1994; Kobayashi et al., 1996). Furthermore, exogenously administered neurotrophins can rescue neurons from injury-induced cell death in axotomy paradigms (Sendtner et al., 1992; Yan et al., 1992; Koliatsos et al., 1993, 1994; Yan et al., 1993;Arenas and Persson, 1994).
In this study, we have analyzed CNS neurons of mice defective for TrkB tyrosine kinase receptors. Most of these mice die during their first postnatal week (Klein et al., 1993). A few animals, however, survive for 2–3 weeks. We report here that these “older” trkB (−/−) mutant mice display increased levels of pyknotic nuclei in the neocortex and hippocampus well after the period of naturally occurring cell death. Moreover, we show that in the absence of TrkB receptors axotomy results in decreased survival of hippocampal and facial motor neurons. These data demonstrate, for the first time, a role of TrkB receptors in the survival of subpopulations of postnatal CNS neurons and provide genetic support for the concept that TrkB signaling protects CNS neurons from injury- and axotomy-induced cell death.
MATERIALS AND METHODS
Mice
trkB (−/−) mice carrying a targeted mutation in those sequences encoding the tyrosine kinase domain of the TrkB signaling receptor have been described (Klein et al., 1993). Postnatal mice were fixed in 4% paraformaldehyde in PBS, pH 7.4, or 1% paraformaldehyde and 1% glutaraldehyde in 0.12 m phosphate buffer, pH 7.4. All mice were perfused transcardially, post-fixed in fresh fixative overnight, and then processed separately for paraffin embedding, immunohistochemistry, terminal deoxytransferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL), or electron microscopy. Mice were analyzed at different developmental stages: postnatal day 5 (P5)–P8 (n = 8), P10 and P11 (n = 13), and P13–P18 (n = 21).
Immunohistochemistry
Brains of mice fixed in 4% paraformaldehyde were post-fixed in fresh fixative and cryoprotected in 30% sucrose. Cryostat or vibratome brain coronal sections were cut at a 50 μm thickness. Free-floating sections were treated with methanol and hydrogen peroxide to inhibit endogenous peroxidases and blocked in 10% normal goat serum. Sections were incubated overnight with antibodies elicited against calbindin (1:4000 dilution), parvalbumin (1:4000 dilution), or calretinin (1:2000 dilution; Swant Antibodies, Bellinzona, Switzerland) and calcitonin gene-related peptide (1:750 dilution; Peninsula, Belmont, CA), substance P (1:500 dilution; Zymed, San Francisco, CA), TrkA and p75 receptors (1:5000 dilution; a gift from L. Reichardt), GAP-43 (1:2000 dilution; Boehringer Mannheim, Indianapolis, IN), and c-Jun (1:1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA). Primary antibodies were detected using the avidin–biotin complex method as indicated by the manufacturer (Vector Laboratories, Burlingame, CA). After development with 3,3′-diaminobenzidine (DAB; 0.25 mg/ml; Sigma, St. Louis, MO) and hydrogen peroxide, sections were stained with cresyl violet before viewing with a Zeiss Axiophot microscope.
TUNEL
Cryostat or vibratome sections were processed for DNA nick end-labeling as described (Gavrielli et al., 1992). Briefly, sections were mounted onto slides, dried overnight at 30°C, digested with proteinase K (20 μm/ml), and incubated with terminal transferase (TdT; 0.3 enzymatic unit/μl) and biotinylated deoxyuridine triphosphate at 37°C for 90 min in TdT (Boehringer Mannheim). Sections were incubated for 1 hr with 2% bovine serum albumin (BSA) and the avidin–biotin complex (1:100 dilution) for 2 hr. Peroxidase activity was developed with 3,3′-DAB, hydrogen peroxide, and 0.2% nickel ammonium sulfate.
Electron microscopy
Vibratome sections from mice perfused with 1% paraformaldehyde and 1% glutaraldehyde were post-fixed with 2% osmium tetroxide, stained en bloc with 2% aqueous uranyl acetate, dehydrated in ethanol, and flat embedded in Araldite. Thin sections were mounted onto Formvar-coated slot grids poststained with uranyl acetate and lead citrate before viewing with an electron microscope.
Morphometric analysis
Neuronal counts. After post-fixation, brains were embedded in agar and sectioned coronally (50 μm thick) in a vibratome. Nissl-stained sections were analyzed using a 40× objective and a millimetric eyepiece. The number of pyknotic nuclei was counted in the dentate gyrus, CA1 and CA3 hippocampal fields, supragranular (II and III) and infragranular (V and VI) neocortical layers of somatosensory cortex (first parietal), striatum (caudate-putamen), and thalamus (the ventroposteror medialis and the reticular thalamic nuclei). Counts were performed in six to eight sections per animal (2 brain 100,000 μm2 fields per section for each region or layer). Brainstems from axotomized mice were embedded in paraffin, sectioned coronally at 8 μm, and stained with cresyl violet. Facial motor neurons were counted using a 40× objective. The total neuronal number was determined by counting only cells exhibiting an obvious nucleus and nucleolus in every fifth section. Raw neuronal counts were corrected for split nucleoli with a multiplication factor using section thickness and the average nuclear diameter. Motor neuron survival was estimated by comparing the percentage of remaining neurons in the injured side with that in the uninjured side. Values derived from mutant mice were compared with those in their control littermates using an unpaired Student’s t test.
Neuronal size. For determination of neuronal cross-sectional areas, profiles of neurons in layer V (somatosensory neocortex), the hilus (hippocampus) of trkB (−/−) and wild-type mice (P13–P18, 4 animals each, 40–50 neurons per animal), were traced on a digitizing tablet interfaced with computer software designed to calculate areas (SigmaPlot, Jandel Scientific, San Rafael, CA). Areas from axotomized facial motor neurons were also measured (three animals per genotype, 100 cells per animal). Cross-sectional areas were compared using an unpaired Student’s t test.
Hippocampal slice cultures
Hippocampal slice cultures were prepared from newborn mice as described elsewhere (Stoppini et al., 1991; Del Río et al., 1996). Briefly, animals were anesthetized by hypothermia, brains were aseptically removed, and the hippocampus was dissected out under a dissecting microscope. Tissue pieces were cut into transverse slices (400 μm thick) using a tissue chopper and cultured using the membrane interphase technique. Tissue slices were placed on 30 mm sterile membranes (Millicell-CM, Millipore, Bedford, MA) and transferred into six-well tissue culture trays. Cultures were maintained in 0.8 ml of culture medium (50% minimum essential medium and 25% horse serum) containing 2 mm glutamine and 0.044% NaHCO3adjusted to pH 7.3. In another set of experiments, cultures from wild-type newborn mice were treated for 3 d with BDNF (Promega, Madison, WI), NT-4 (Genzyme, Cambridge, MA), NGF (Boerhinger Mannheim), recombinant NT-3 (Bristol-Myers Squibb, Princeton, NJ), or vehicle solution (9–10 cultures per group). Neurotrophins (25–50 ng/μl) were diluted in PBS (0.1 m, pH 7.4) containing 0.2% BSA (Sigma) and added directly on each slice culture daily (100 ng/culture) from the explantation day (Marty et al., 1996). Cultures were fixed after 3 or 5 d with 4% paraformaldehyde in 0.1 m PBS. Vibratome sections (50 μm thick) were stained with cresyl violet. Pyknotic nuclei in the dentate granule cell layer and pyramidal layer of CA3, CA1, and subicular regions were counted as described above (4–6 hippocampal 25,000 μm2 fields per culture for each region).
Facial axotomy
P5 mice were anesthetized by hypothermia, and the left facial nerve was transected just after its exit through the stylomastoid foramen. Mice were allowed to survive until P10. At this time, they were anesthetized with avertin (200 mg/kg) and perfused transcardially with PBS followed by Bouin’s fixative (Polysciences, Warrington, PA). Brainstems were dissected out and embedded in paraffin for neuronal counts. Brainstems from P7 animals were cut in a cryostat and processed for immunohistochemistry (see above) or acetylcholinesterase histochemistry as described previously (Hedreen et al., 1985).
RESULTS
Limited morphological alterations in postnatal mice lacking TrkB receptors
Mice lacking TrkB tyrosine kinase receptors survive up to 3 weeks after birth. Brain sections from late postnatal (P10–P18)trkB (−/−) mice exhibit normal cytoarchitectonics when compared with their wild-type littermates, as revealed by Nissl staining and immunolabeling with several neuronal markers (Fig.1). For instance, the typical layering of the neocortex and hippocampus is well preserved in the mutant animals (Fig.1B,D,G). Likewise, the olfactory bulb, as well as different nuclei of the thalamus, basal ganglia, and other forebrain regions, did not display apparent morphological defects (data not shown). However, most neuronal populations in trkB (−/−) mice displayed significantly smaller somas than their control littermates, probably as a consequence of the overall smaller size of these mutant mice. For instance, the soma areas of hilar neurons in the hippocampus and pyramidal neurons in layer V of the somatosensory cortex are 233 ± 5.4 and 195 ± 4.1 μm2, respectively, in wild-type mice versus 160 ± 4.1 and 146 ± 3.2 μm2 in trkB (−/−) mice (p < 0.001). Moreover, some of the oldest surviving trkB (−/−) mice (P13–P18) revealed small, empty areas in the gray matter that were devoid of neurons. These areas were more prominent in the neocortex, where they seemed to be randomly distributed (Fig. 1E).
Fig. 1.
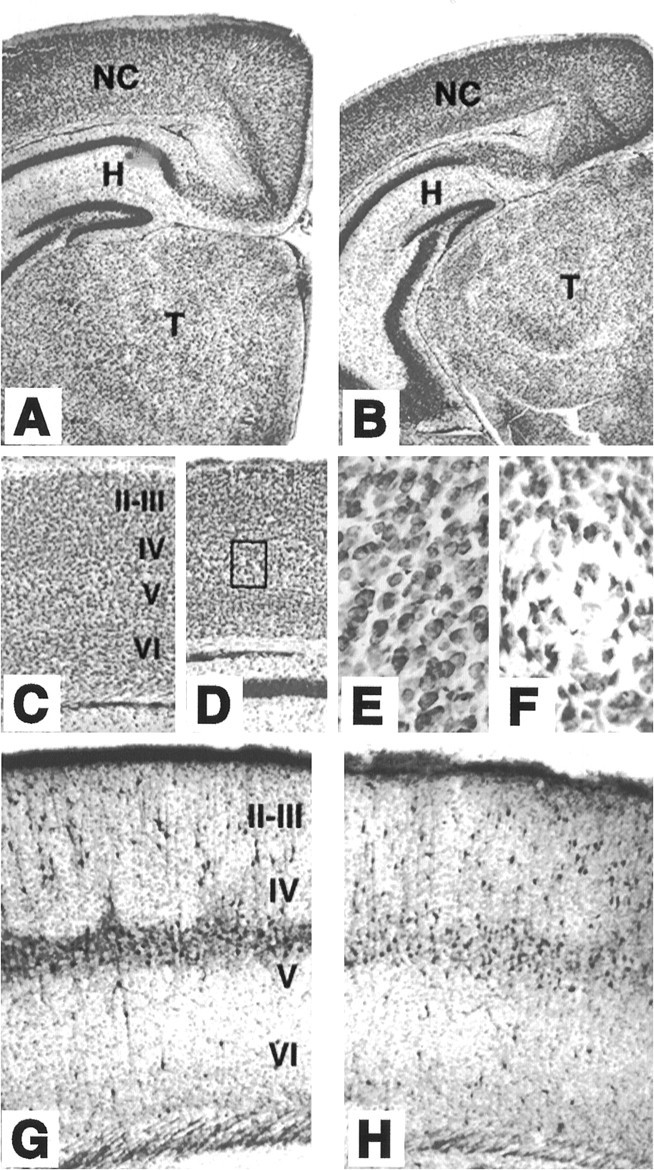
Cytoarchitecture of brain structures of postnataltrkB (−/−) mutant mice. A–D, Low-power micrographs of the brain of wild-type (A, C) andtrkB (−/−) (B, D) mice. Notice the normal appearance of Nissl-stained brain sections from P13 (B) and P16 (D) trkB (−/−) mice compared with their age-mated control littermates (A, C, respectively). E, F, High-power magnification of the neocortex of P16 wild-type (E) andtrkB (−/−) (F) mice. Notice the presence of empty areas devoid of neurons in trkB (−/−) mice (F) (from D, inset).G, H, Calretinin immunoreactivity in the neocortex of P10 wild-type (G) and trkB (−/−) (H) mice. Cortical layers are denoted byroman numerals. NC, Neocortex;H, hippocampus; T, thalamus. Magnification: A–D, 2.5×; E, F, 40×;G, H, 10×.
The distribution of forebrain neurons expressing different calcium-binding proteins, including calretinin (Fig.1F,G), calbindin, and parvalbumin (not shown), did not reveal major differences between wild-type andtrkB (−/−) mice. However, the expression of parvalbumin seems to be delayed in the neocortex and hippocampus of thetrkB (−/−) mutant mice (data not shown). Similar results have been reported previously in mice deficient in BDNF (Jones et al., 1994).
Increased apoptotic cell death in trkB (−/−)-defective mice
Naturally occurring cell death in the brain occurs mainly during the first postnatal week and decreases markedly thereafter (Gould et al., 1991; Ferrer et al., 1992). To determine whether the absence of TrkB tyrosine kinase receptors affects survival of CNS neurons postnatally, we analyzed Nissl-stained sections from P5–P18trkB (−/−) mice for the presence of pyknotic nuclei. As illustrated in Figure 2, trkB (−/−) mice showed a significant increase in the number of pyknotic nuclei in most of the brain regions analyzed, including the neocortex, dentate gyrus, striatum, septum, thalamus, and olfactory bulb. These pyknotic nuclei were extremely shrunken, dark, and surrounded by an almost absent cytoplasm, suggesting that they corresponded to apoptotic cells. In the neocortex, the pyknotic nuclei were distributed throughout all cortical layers (Fig. 2A,B), although some differences in the number of pyknotic nuclei were observed between different cortical areas (see below). In the hippocampus, they were present in the hippocampal CA1 and CA3 subfields and were particularly abundant in the deep tier of the granule cell layer of the dentate gyrus (Fig.2C,D).
Fig. 2.
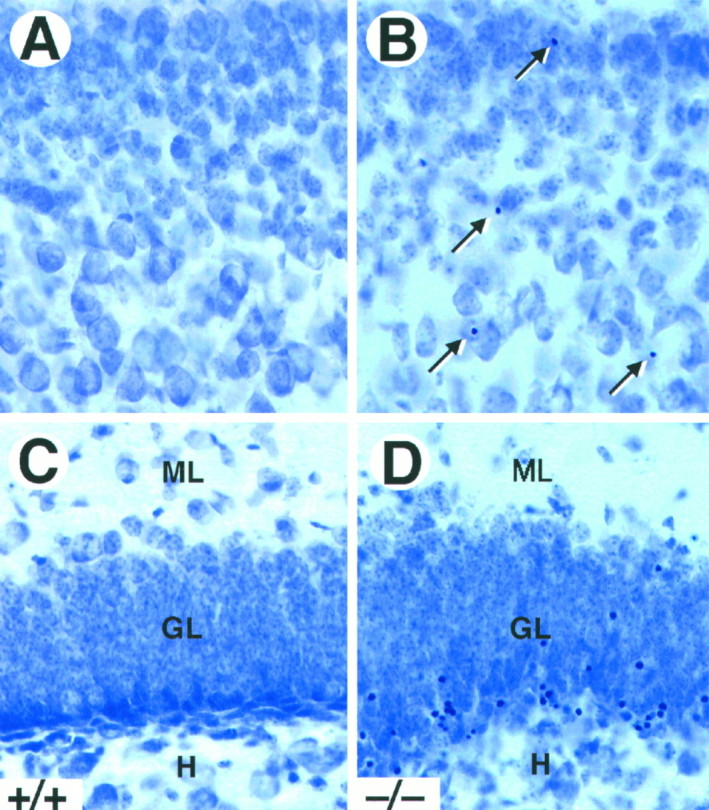
Increased cell death in trkB (−/−) mice. A–D, Cresyl violet-stained sections of the neocortex (A, B) and dentate gyrus (C, D) of P16 control (A, C) andtrkB (−/−) (B, D) mice. Notice the presence of pyknotic nuclei in layers II and III of the neocortex (B, arrows) and the granular layer of the dentate gyrus (D) in the trkB (−/−) mutant mice.GL, Granular layer; H, hilus;ML, molecular layer. Magnification, 40×.
To determine the nature of the observed cell death in the forebrain oftrkB (−/−) mice, sections from these mutant mice as well as from their wild-type littermates were processed for c-Jun immunohistochemistry, TUNEL staining, and electron microscopy. Staining with antibodies against c-Jun, the product of a proto-oncogene upregulated in dying neurons (Ferrer et al., 1996), showed an increased number of immunopositive cells in the trkB (−/−) mice. Most c-Jun-positive cells displayed a morphology characteristic of apoptotic cells (Fig. 3A). However, in other cases c-Jun-immunoreactive neurons exhibited several processes resembling dendrites and soma sizes typical of neurons (Fig.3B), thus suggesting that c-Jun upregulation may precede the onset of apoptosis. Similarly, brain sections derived fromtrkB (−/−) mice and processed for TUNEL staining showed increased numbers of apoptotic cells (Fig. 3D–G). The number of TUNEL-positive neurons correlated well with the number of pyknotic nuclei observed by Nissl staining. Finally, the presence of apoptotic cells was confirmed by electron microscopy. As illustrated in Figure 3C, dying neurons of the neocortex and hippocampus exhibited a high degree of chromatin and cytoplasm condensation and were often engulfed by neighboring cells.
Fig. 3.
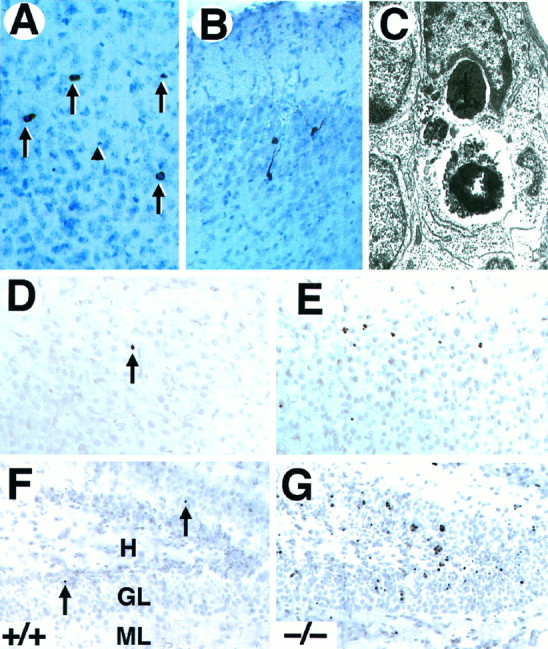
Neurons in trkB (−/−) mice die by apoptosis. A, B, Immunolabeling with c-Jun antibody reveals the presence of pyknotic cells in layers II and III (A) and degenerating neurons in the pyriform cortex (B) in P10 trkB (−/−) mice.C, Electron micrograph showing the characteristic morphology of an apoptotic cell in the dentate gyrus that is being engulfed by a neighboring cell in a trkB (−/−) mouse.D–G, TUNEL staining of neocortex (layers II and III) (D, E) and hippocampus (F, G); sections from P13 control (D, F) and trkB (−/−) (E, G) mice. Notice the increase in TUNEL-positive nuclei in the trkB (−/−) mice.Arrows in A denote pyknotic nuclei. Thearrowhead denotes a pyknotic nucleus negative for c-Jun staining. Sections in A and B are counterstained with cresyl violet. GL, Granular layer;H, hilus; ML, molecular layer. Magnification: A, B, 30×; C, 15,000×;D–G, 40×.
To determine whether the dying cells in the trkB (−/−) mice corresponded to particular neuronal populations, we examined the expression of several calcium-binding proteins, including calbindin, calretinin, and parvalbumin. We observed that some immunoreactive cells had shrunken perikarya and extremely beaded atrophic dendrites, features also associated with dying neurons (Fig.4A,B). Such atrophic immunoreactive cells were present in the neocortex and hippocampus of trkB (−/−) mice, but they were rarely observed in wild-type mice. Furthermore, we also detected pyknotic cells immunoreactive for parvalbumin (data not shown), calbindin, or calretinin (Fig.4C–E). These observations strongly suggest that a majority of the dying cells in the CNS of trkB (−/−) mice are neurons.
Fig. 4.

Expression of calcium-binding proteins in degenerating neurons in trkB (−/−) mutant mice.A–D, Calretinin immunoreactivity in layers II and III of the neocortex (A, B, D) and the stratum oriens of the hippocampus (C) of P10 trkB (−/−) mutant mice. Presumably, degenerating neurons with atrophic somata and beaded dendrites are immunoreactive for calretinin staining (A, B, arrows). E, Calbindin expression in layers II and III of the neocortex of a P13 trkB (−/−) mice. Arrows in C–E denote pyknotic cells immunoreactive for different calcium-binding proteins.Arrowheads denote unlabeled pyknotic cells. Sections are counterstained with cresyl violet. Magnification: A, B,35×; C, D, 40×; E, 45×.
Developmental pattern of cell death in the CNS of trkB (−/−) mice
We next analyzed the developmental course of cell death in the hippocampus, striatum, thalamus, and parietal neocortex oftrkB (−/−) mice by quantifying the number of pyknotic nuclei at different stages of postnatal development. No significant differences were observed in the number of pyknotic nuclei between P5 and P8 trkB (−/−) and their control littermates (Fig.5A) for most of the regions analyzed in the brain. However, the striatum and the reticular thalamic nucleus display significant increases in the number of pyknotic nuclei in this first postnatal week. trkB (−/−) animals that survived their first postnatal week (P10–P12) revealed a significant increase in pyknotic nuclei in the majority of the brain regions we analyzed. This increase was more dramatic in the dentate gyrus. A more moderate increase was also observed in the neocortex, the striatum, and the reticular thalamic nucleus of these mutant mice (Fig. 5B). The difference between the number of pyknotic nuclei between control and trkB (−/−) animals became more evident in animals that survive their second week of life (P13–P18). For instance, these animals had two to four times more pyknotic nuclei in their hippocampal CA3 region, neocortex, and the ventroposterior medialis thalamic nucleus than their control littermates. This difference was much more dramatic, a ninefold increase, in the dentate gyrus (Fig.5C). Interestingly, after the second postnatal week no significant differences in cell death could be observed in striatum and reticular thalamic nucleus (Fig. 5C). These observations indicate that the number of pyknotic nuclei increases progressively from P10 to P18 within all cortical layers and hippocampal subfields oftrkB (−/−) mice, a time subsequent to the period of naturally occurring cell death in these forebrain regions. Furthermore, our results indicate that the increased rate of cell death intrkB (−/−) mice follows a specific temporal pattern in different regions of the CNS.
Fig. 5.

Number of pyknotic nuclei in different regions of the brain at several developmental stages (expressed as number per 100,000 μm2). A, P5–P8 mice (control mice, n = 4; trkB (−/−) mice,n = 4). B, P10–P12 mice (control mice, n = 6; trkB (−/−) mice,n = 7). C, P13–P18 mice (control mice, n = 10; trkB (−/−) mice,n = 11). Black bars, Control mice;white bars, trkB (−/−) mutant mice. DG,Dentate gyrus; CA3, CA3 hippocampal field;II–III and V–VI, cortical layers II and III and V and VI, respectively; VPM, ventroposterior medialis thalamic nucleus; RET, reticular thalamic nucleus; STR, striatum. Error bars indicate SEM;asterisks indicate that there are significant differences between control and trkB (−/−) mutant mice (*p < 0.01; **p < 0.001).
Increased neuronal cell death in organotypic slice cultures oftrkB (−/−) mice
To investigate whether TrkB receptors play a role in regulating the survival of CNS neurons after injury, we measured the rate of neuronal cell death in hippocampal slice preparations. Organotypic slice cultures were prepared from newborn mice and examined after 3 or 5 d in culture. In addition, to induce injury, coronal transection of the neonatal hippocampus also results in axotomy of those projection neurons (e.g., CA3 region and subiculum) that have already reached their target fields (Bayer, 1980; Supér and Soriano, 1994). After 3 d, slices from wild-type, trkB (+/−), ortrkB (−/−) newborn mice display a well preserved cytoarchitectonic pattern in which all major hippocampal subdivisions and layers were recognizable (Fig. 6). However, substantial numbers of pyknotic nuclei were observed in all hippocampal subfields, suggesting that neuronal cell death occurs as a consequence of the injury caused during preparation of these organotypic slices. Quantitative analysis of the number of pyknotic nuclei revealed a marked increase in the hippocampal CA3 and subicular regions oftrkB (−/−) mice when compared with those present in cultures derived from either heterozygous or wild-type animals (Fig.7).
Fig. 6.
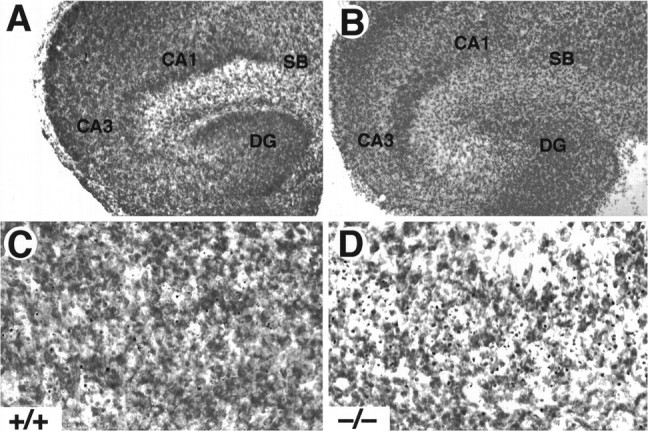
Increased cell death in hippocampal slice cultures from trkB (−/−) mice. A, B, Cresyl violet-stained sections show the normal structure of hippocampal slices in both control (A, C) and trkB (−/−) mice (B, D) after 3 d in culture. C, D, Higher-magnification micrographs show an increase in pyknotic nuclei in the CA3 region of trkB (−/−) mutant mice (D) when compared with control mice (C). CA1, Pyramidal layer CA1;CA3, pyramidal layer CA3; DG, dentate gyrus; SB, subiculum. Magnification: A, B, 10×; C, D, 40×.
Fig. 7.
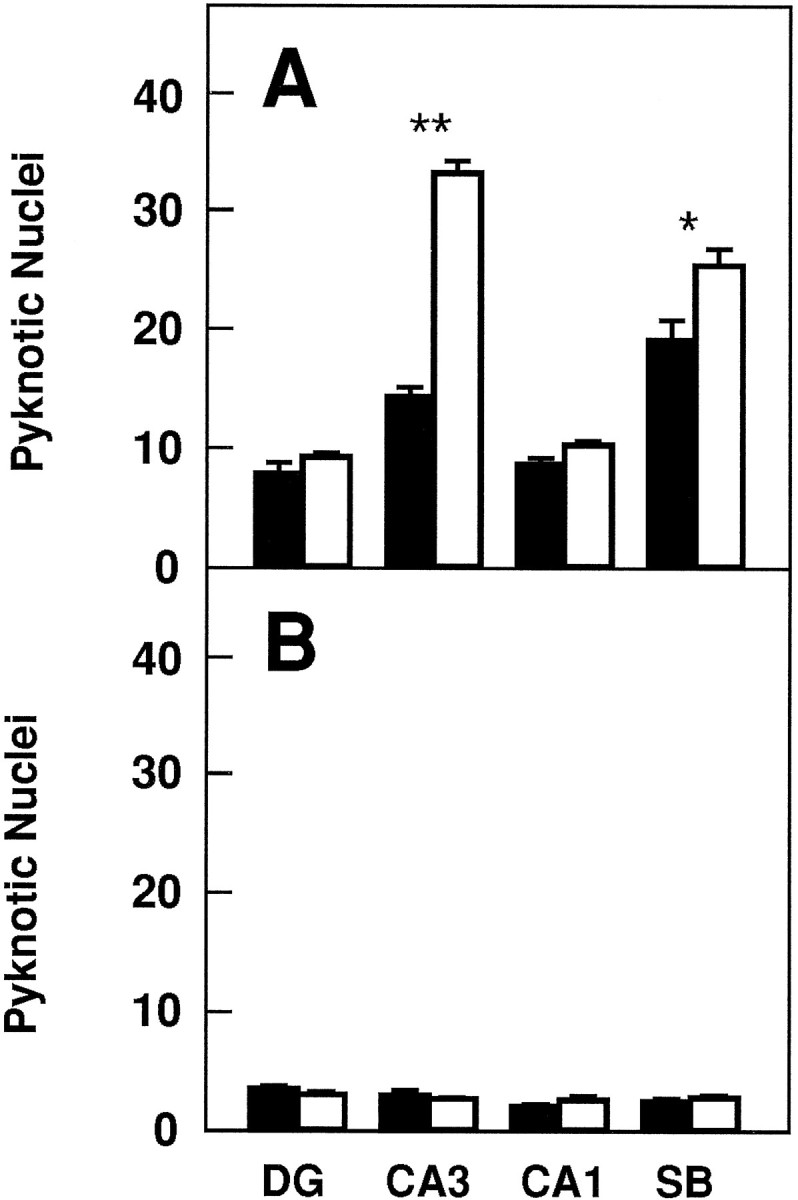
Number of pyknotic nuclei in different regions of the hippocampus after 3 (A) and 5 (B) d in culture (expressed as number per 25,000 μm2).Black bars, Control mice (n = 8 in each group of cultures); white bars, trkB (−/−) mutant mice (n = 8 in each group of cultures).DG, Dentate gyrus; CA1 andCA3, CA1 and CA3 hippocampal fields, respectively;SB, subiculum. Error bars indicate SEM;asterisks indicate that there are significant differences between control and trkB (−/−) mutant mice (*p < 0.001; **p < 0.0001).
In contrast, we did not observe increased numbers of dying cells in other hippocampal areas of the trkB (−/−) mice such as the CA1 region and the dentate gyrus in this culture model. These observations might be attributable to the fact that neurons in the dentate gyrus are not axotomized in this paradigm. Likewise, most CA1 neurons are still migrating in newborn mice and may have not reached their targets (Supér and Soriano, 1994). These results suggest a protective role of TrkB receptors in those hippocampal neurons that undergo cell death after axotomy. After 5 d in culture, the number of pyknotic nuclei becomes significantly reduced in the control cultures. At this time, no significant increase in cell death could be observed in those slices derived from the mutant trkB (−/−) mice (Fig. 7), suggesting that those neurons that are resistant to cell death in this paradigm are not affected by the absence of TrkB receptors.
To determine whether the cell death induced in this paradigm can be rescued with different neurotrophins, we treated control hippocampal slice cultures with NGF, BDNF, NT-3, NT-4, or vehicle solution. Both BDNF and NT-4 rescued the cell death observed in the subiculum and CA3 in control cultures (p < 0.01; Fig.8). Interestingly, NT-3 and NGF were able to rescue the cell death observed in CA3 (p < 0.01; Fig. 8). None of the neurotrophins, however, were able of rescuing the cell death in the dentate gyrus or in CA1 hippocampal fields.
Fig. 8.

Number of pyknotic nuclei in different regions of the hippocampus after 3 d of treatment with neurotrophins in culture (expressed as number per 25,000 μm2).Black bars, Vehicle; dotted bars, BDNF;gray bars, NT-3; white bars, NT-4;striped bars, NGF (n = 8 or 9 cultures per group). DG, Dentate gyrus;CA1 and CA3, CA1 and CA3 hippocampal fields, respectively; SB, subiculum. Error bars indicate SEM; asterisks indicate that there are significant differences between the different treatments (*p < 0.01, ANOVA with Fisher’s protected least significant difference forpost hoc comparisons).
TrkB signaling supports motor neuron survival after axotomyin vivo
Neither TrkB nor TrkC receptors are required for the survival of facial or spinal cord motor neurons during embryonic development (Silos-Santiago et al., 1997). After motor neuron axotomy, the levels of neurotrophin expression are elevated in peripheral nerves and in denervated muscle (Heumann et al., 1987; Meyer et al., 1992; Funakoshi et al., 1993; Koliatsos et al., 1993). Furthermore, the expression of p75 and TrkB receptors is increased in axotomized motor neurons (Ernfors et al., 1989; Piehl et al., 1994; Kobayashi et al., 1996). These observation suggest that neurotrophin receptor signaling may mediate survival of motor neurons or may promote axonal regeneration after injury.
To elucidate the role of TrkB signaling in promoting the survival of postnatal motor neurons after a lesion, we quantified the number of facial motor neurons after axotomy in wild-type, trkB (+/−), and trkB (−/−) mice. Because most trkB (−/−) mice do not survive to more than 2 weeks of age, the left facial nerve was transected at P5, and the animals were allowed to survive until P10. Surviving motor neurons in the trkB (−/−) mice looked morphologically indistinguishable from those of wild-type mice (Fig. 9A–F). Moreover, the absence of TrkB receptors in these axotomized motor neurons had no effect on the expression of a variety of markers, including p75, calcitonin gene-related peptide, and GAP-43 immunoreactivity (not shown), as well as acetylcholinesterase activity (Fig.9G,H). Although the soma area of these motor neurons is significantly (p < 0.02) smaller (250 ± 3.4 μm2) in trkB (−/−) mice when compared with their wild-type littermates (261 ± 3.9 μm2), size–frequency histograms in trkB (−/−) animals seem similar to those in wild-type mice (data not shown). Furthermore, the percentage of facial motor neurons that survived in the operated side was significantly decreased in trkB (−/−) mice (27%) when compared with their wild-type littermates (53%; Table1). In agreement with our recent studies (Silos-Santiago et al., 1997), no differences in the number of facial motor neurons were observed in the unoperated sides of wild-type andtrkB (−/−) animals (Table 1). These results provide further support to the concept that TrkB receptor signaling protects postnatal CNS neurons from axotomy-induced death.
Fig. 9.
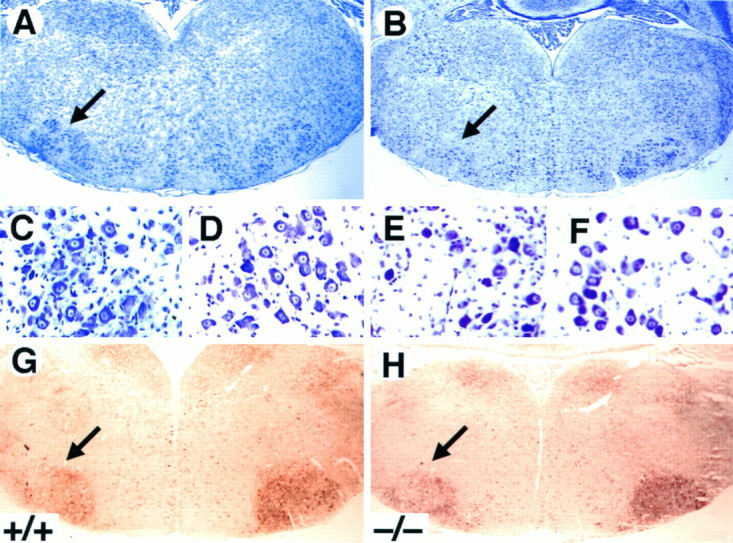
Decrease in the number of facial motor neurons after axotomy in trkB (−/−) mice. A–F,Cresyl violet-stained sections show the reduction in the size of the axotomized facial motor nucleus (B) but the normal appearance of the surviving motor neurons (E) in P10trkB (−/−) mice. A, C, D, Wild-type mice. B, E, F, trkB (−/−) mice. G, H,Acetylcholinesterase activity in P7 wild-type (G) andtrkB (−/−) (H) mice. Similar activity was observed in the trkB (−/−) mutant mice when compared with wild-type mice. Magnification: A, B, G, H, 2.5×; C–F, 100×.
Table 1.
Motor neuron number in facial nucleus after axotomy
| Wild type | trkB (+/−) | trkB (−/−) | |
|---|---|---|---|
| Contralateral | 2260 ± 142 | 2518 ± 72 | 2484 ± 128 |
| Ipsilateral | 1209 ± 134 | 1282 ± 47 | 680 ± 90 |
| Survival (%) | 53 ± 4 | 52 ± 2 | 27 ± 3 |
| n | 7 | 4 | 7 |
DISCUSSION
Analysis of mutant mice lacking TrkB tyrosine kinase receptors has illustrated the critical role that TrkB signaling plays in the generation and/or survival of a variety of sensory neurons (Klein et al., 1993; Minichiello et al., 1995; Schimmang et al., 1995) (Silos-Santiago et al., 1997). Similar results have been obtained with mice lacking BDNF and, to a much lesser extent, NT-4, the two primary ligands of TrkB receptors (Ernfors et al., 1994, 1995; Jones et al., 1994; Conover et al., 1995; Liu et al., 1995). These neuronal deficits, however, have not been observed in CNS neurons, despite the widespread expression of TrkB receptors in these neurons during development. An earlier report from our laboratory describing partial loss of facial and spinal cord motor neurons in neonatal trkB (−/−) mice (Klein et al., 1993) has not been confirmed in our subsequent studies (Silos-Santiago et al., 1997). Double mutant mice lacking BDNF and NT-4 ligands also failed to reveal motor neuron deficits (Conover et al., 1995; Liu et al., 1995). It has been argued that these results might be a consequence of compensatory effects provided by coexpression of the related TrkC tyrosine kinase receptors in thesetrkB-targeted mice. However, this hypothesis is unlikely, considering the absence of detectable neuronal deficits in the CNS of newborn mice lacking both TrkB and TrkC signaling receptors (Silos-Santiago et al., 1997).
To date, the most significant CNS defect observed in mice lacking either neurotrophins or their receptors corresponds to those animals devoid of TrkA receptors. By 4 weeks of age, these mutant mice display a significant reduction in their septal cholinergic projection, a defect not observed in younger trkA (−/−) animals (Smeyne et al., 1994). BDNF-deficient mice have reduced numbers of CNS neurons expressing calbindin and parvalbumin (Jones et al., 1994). However, no increase in cell death has been described in the CNS of these mice or in those lacking both BDNF and NT-4 (Ernfors et al., 1994; Jones et al., 1994; Conover et al., 1995; Liu et al., 1995). Gross examination of brains from early postnatal trkB (−/−) mice revealed a normal cytoarchitecture in all the CNS regions analyzed. Furthermore, immunostaining with several neuronal markers showed a rather normal distribution of immunopositive neurons. However, trkB (−/−) mice undergo a period of increased cell death during early postnatal life. In certain regions, this phenomenon becomes more prominent as these mutant animals become older. For instance, 2- to 3-week-old mice display an increase of two- to ninefold in the number of pyknotic nuclei in different regions of the forebrain, particularly in the hippocampus and the neocortex. These pyknotic nuclei are likely to correspond to cells undergoing apoptosis, because these mutant mice display a similar number of TUNEL-immunoreactive cells in the same CNS structures. Unfortunately, trkB (−/−) mice do not survive beyond 3 weeks, thus preventing us from analyzing these deficits in older animals.
Available evidence suggests that most dying cells in thetrkB (−/−) mice are likely to be neurons. For instance, a majority of the pyknotic nuclei are located in the neuron-rich layers of the neocortex and hippocampus. Moreover, they are observed within cells immunoreactive with antibodies elicited against calcium-binding proteins such as calretinin, calbindin, and parvalbumin (Celio, 1990;Gulyás et al., 1992; Résibois and Rogers, 1992). It is possible, however, that some of these pyknotic nuclei may correspond to dying glial cells. In addition to the pyknotic nuclei, trkB (−/−) mice have a significant number of morphologically abnormal cells that are also likely to be dying neurons. These cells display several beaded or varicose processes resembling dendrites and are immunoreactive for c-Jun, a protein known to be upregulated in dying neurons (Ferrer et al., 1996). Based on the distribution of dying cells, it is likely that they include pyramidal and granular projection neurons as well as interneurons.
The increased neuronal death in the trkB (−/−) mice is particularly conspicuous in the dentate gyrus. Consistent with our results is the fact that 2-week-old BDNF-deficient mice have an increase in pyknotic nuclei in this region (I. Fariñas, personal communication). Within the dentate gyrus, pyknotic nuclei could be observed within all of the layers. However, these dying cells predominate in the deep tier of the granule cell layer, which is primarily composed of neurons generated during the postnatal period (Altman and Bayer, 1990a,b). Therefore, it is possible that these young granule neurons are most susceptible to lack of the trophic effects in the trkB (−/−) mice.
Several explanations may account for the role of TrkB receptors on the survival of postnatal but not embryonic CNS neurons. AlthoughtrkB mRNA has been observed during mid and late stages of embryonic development (Klein et al., 1989, 1990; Ernfors et al., 1992;Escandón et al., 1994), expression of the catalytic TrkB tyrosine kinase receptors reaches its peak during the second postnatal week (Dugich-Djordjevic et al., 1993; Knüsel et al., 1994), and their autophosphorylation in response to BDNF and NT-4 is maximal at P7 in the hippocampus (Knüsel et al., 1994). Moreover, it is possible that the TrkB receptors may be primarily activated in postnatal animals, because BDNF transcripts are very low in the embryonic brain, and although NT-4 levels are maximal at embryonic day 13.5, they decrease rapidly, reaching the lowest level around birth (Timmusk et al., 1993). BDNF expression, however, increases postnatally, particularly in the neocortex and dentate gyrus (Dugich-Djordjevic et al., 1992; Friedman et al., 1991; Huntley et al., 1992; Förster et al., 1993; Timmusk et al., 1993, 1994), two of the regions where cell death is most pronounced in the absence of TrkB receptors.
BDNF and NT-4 have been shown previously to prevent death of motor neurons after axotomy (Sendtner et al., 1992; Yan et al., 1992;Koliatsos et al., 1993). Moreover, neurotrophin expression increases as a response to nerve injury. For instance, transection of a peripheral nerve results in increased expression of NGF, BDNF, and NT-4 mRNA in Schwann cells and fibroblasts in the distal nerve stump (Heumann et al., 1987; Meyer et al., 1992; Funakoshi et al., 1993). Similarly, denervated muscle expresses increased levels of BDNF mRNA (Funakoshi et al., 1993; Koliatsos et al., 1993). This increased synthesis of neurotrophins is accompanied by elevated expression of some of their receptors, such as p75 and TrkB, in motor neurons (Ernfors et al., 1989; Piehl et al., 1994; Kobayashi et al., 1996). Indeed, it has been suggested that the increased levels of p75 and TrkB could make these injured motor neurons more sensitive to local and target-derived neurotrophins, optimizing the conditions for their survival (Ernfors et al., 1989; Piehl et al., 1994). Our results provide, for the first time, genetic evidence of a protective role of TrkB receptors after axotomy of facial motor neurons in young (P5) mice. Whereas half of the axotomized motor neurons survived in the trkB (+/−) heterozygous and wild-type mice, only 27% survived in thetrkB (−/−) mutant animals.
In support of these observations, we have also found that TrkB receptors play a role in preventing death of neurons in slice cultures. Organotypic cultures of hippocampal slices of neonatal trkB (−/−) mice display increased cell death in the CA3 region and the subiculum but not in the CA1 region or the dentate gyrus. CA3 and subiculum neurons project to the contralateral hippocampus and entorhinal cortex, respectively, and their axons reach their targets before birth (are among the first to be generated in the hippocampus) (Bayer, 1980; Supèr and Soriano, 1994). Therefore, these neurons are axotomized during the preparation of organotypic hippocampal cultures. In contrast, most CA1 neurons are still migrating at this time, and few of their axons have entered the target fields (Bayer, 1980; Supèr and Soriano, 1994). Likewise, the granular neurons of the dentate gyrus project their axons (the mossy fibers) along a course parallel to the plane of sectioning of these organotypic slices; thus, they have not been axotomized. These results suggest that TrkB receptors prevent death of those hippocampal cells (CA3 and subicular neurons) that become axotomized in these slices. In addition, some of the cell death observed in control cultures is likely to be caused by the general damage or injury caused by the culture procedure (e.g., ischemia, hypoglycemia, or mechanical stress). These observations, taken together, suggest that TrkB receptors play a role in protecting subpopulations of young CNS neurons from injury-induced cell death. This hypothesis is corroborated by the fact that BDNF and NT-4 are able to rescue the cell death induced in this in vitro paradigm. In addition, our data with NT-3 and NGF suggest that these neurotrophins and their receptors may be involved in the regulation of some neuronal cell death in hippocampal slice cultures.
A more complete understanding of the role that TrkB receptor signaling plays in the CNS would require the analysis of trkB (−/−) mice considerably older than those examined here. Unfortunately, the limited survival of these mice prevents such studies. Ectopic expression of TrkB receptors in those peripheral neurons (e.g., nodose–petrosal neurons) known to die in the trkB (−/−) animals is likely to increase their life span. If so, these transgenic mice should prove extremely valuable in ascertaining the role of TrkB tyrosine kinase receptors in the survival and function of the adult CNS.
Footnotes
This work was supported by Spanish Ministries for Education and Health Grants SAF94-743 and PM95-102. J.F. was supported by the Wenner-Gren foundations. We thank M. Garber, J. Gullo-Brown, L. Long, and J. Wolf for excellent technical assistance.
Correspondence should be addressed to Inmaculada Silos-Santiago, Bristol-Myers Squibb Pharmaceutical Research Institute, H24-09, P.O. Box 4000, Princeton, NJ 08543-4000.
Dr. Frisen’s present address: Department of Neuroscience, Karolinska Institute, S-17177 Stockholm, Sweden.
REFERENCES
- 1.Altman J, Bayer SA. Mosaic organization of the hippocampal neuroepithelium and the multiple germinal sources of dentate granule cells. J Comp Neurol. 1990a;301:325–342. doi: 10.1002/cne.903010302. [DOI] [PubMed] [Google Scholar]
- 2.Altman J, Bayer SA. Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and postnatal periods. J Comp Neurol. 1990b;301:365–381. doi: 10.1002/cne.903010304. [DOI] [PubMed] [Google Scholar]
- 3.Arenas E, Persson H. Neurotrophin-3 prevents the death of adult central noradrenergic neurons in vivo. Nature. 1994;367:368–371. doi: 10.1038/367368a0. [DOI] [PubMed] [Google Scholar]
- 4.Barbacid M. Neurotrophic factors and their receptors. Curr Opin Cell Biol. 1995;7:148–155. doi: 10.1016/0955-0674(95)80022-0. [DOI] [PubMed] [Google Scholar]
- 5.Bayer SA. Development of the hippocampal region of the rat. I. Neurogenesis examined with 3H-thymidine autoradiography. J Comp Neurol. 1980;190:87–114. doi: 10.1002/cne.901900107. [DOI] [PubMed] [Google Scholar]
- 6.Beck KD, Lamballe F, Klein R, Barbacid M, Schauwecker PE, McNeill TH, Finch CE, Hefti F, Day JR. Induction of noncatalytic trkB neurotrophin receptors during axonal sprouting in the adult hippocampus. J Neurosci. 1993;13:4001–4014. doi: 10.1523/JNEUROSCI.13-09-04001.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- 8.Celio MR. Calbindin D-28K and parvalbumin in the rat nervous system. Neuroscience. 1990;35:375–475. doi: 10.1016/0306-4522(90)90091-h. [DOI] [PubMed] [Google Scholar]
- 9.Cohen-Cory S, Frasser SE. Effects of brain-derived neurotrophic factor on optic axon branching and remodeling in vivo. Nature. 1995;378:192–196. doi: 10.1038/378192a0. [DOI] [PubMed] [Google Scholar]
- 10.Conover JC, Erickson JT, Katz DM, Bianchi LM, Poueymirou WT, McClain J, Pan L, Helgren M, Ip NY, Boland P, Friedman B, Wiegand S, Vejsada R, Kato AC, DeChiara TM, Yancopoulos GD. Neuronal deficits, not involving motor neurons, in mice lacking BDNF and/or NT-4. Nature. 1995;375:235–238. doi: 10.1038/375235a0. [DOI] [PubMed] [Google Scholar]
- 11.Del Río JA, Heimrich B, Supèr H, Borrell V, Frotscher M, Soriano E. Differential survival of Cajal-Retzius cells in organotypic cultures of hippocampus and neocortex. J Neurosci. 1996;16:6896–6907. doi: 10.1523/JNEUROSCI.16-21-06896.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diamond J, Holmes M, Coughlin M. Endogenous NGF and nerve impulses regulate the collateral sprouting of sensory axons in the skin of adult rat. J Neurosci. 1992;12:1454–1466. doi: 10.1523/JNEUROSCI.12-04-01454.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dugich-Djordjevic MM, Tocco G, Willoughby DA, Najm I, Pasinetti G, Thompson RF, Baudry M, Lapschak PA, Hefti F. BDNF mRNA expression in the developing rat brain following kainic acid-induced seizure activity. Neuron. 1992;8:1127–1138. doi: 10.1016/0896-6273(92)90133-x. [DOI] [PubMed] [Google Scholar]
- 14.Dugich-Djordjevic MM, Ohsawa F, Hefti F. Transient elevation in catalytic trkB mRNA during postnatal development of the rat brain. NeuroReport. 1993;4:1091–1094. [PubMed] [Google Scholar]
- 15.Ernfors P, Henschen A, Olson L, Persson H. Expression of nerve growth factor receptor mRNA is developmentally regulated and increased after axotomy in rat spinal cord motoneurons. Neuron. 1989;2:1605–1613. doi: 10.1016/0896-6273(89)90049-4. [DOI] [PubMed] [Google Scholar]
- 16.Ernfors P, Merlio J-P, Persson H. Cells expressing mRNA for neurotrophins and their receptors during embryonic rat development. Eur J Neurosci. 1992;4:1140–1158. doi: 10.1111/j.1460-9568.1992.tb00141.x. [DOI] [PubMed] [Google Scholar]
- 17.Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- 18.Ernfors P, Van De Water T, Loring J, Jaenisch R. Complementary roles of BDNF and NT-3 in vestibular and auditory development. Neuron. 1995;14:1153–1164. doi: 10.1016/0896-6273(95)90263-5. [DOI] [PubMed] [Google Scholar]
- 19.Escandón E, Soppet D, Rosenthal A, Mendoza-Ramírez J-L, Szonyi E, Burton LE, Henderson CE, Parada LF, Nikolics K. Regulation of neurotrophin receptor expression during embryonic and postnatal development. J Neurosci. 1994;14:2054–2068. doi: 10.1523/JNEUROSCI.14-04-02054.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fariñas I, Reichardt LF. Neurotrophic factors and their receptors: implications of genetic studies. Semin Neurosci. 1996;8:133–143. [Google Scholar]
- 21.Ferrer I, Soriano E, Del Río JA, Alcántara S, Auladell C. Cell death and removal in the cerebral cortex during development. Prog Neurobiol. 1992;39:1–43. doi: 10.1016/0301-0082(92)90029-e. [DOI] [PubMed] [Google Scholar]
- 22.Ferrer I, Olivé M, Ribera J, Planas AM. Naturally occurring (programmed) and radiation-induced apoptosis are associated with selective c-jun expression in the developing brain. Eur J Neurosci. 1996;8:1286–1298. doi: 10.1111/j.1460-9568.1996.tb01297.x. [DOI] [PubMed] [Google Scholar]
- 23.Förster E, Otten U, Frotscher M. Developmental neurotrophin expression in slice cultures of rat hippocampus. Neurosci Lett. 1993;155:216–219. doi: 10.1016/0304-3940(93)90711-s. [DOI] [PubMed] [Google Scholar]
- 24.Friedman WJ, Olson L, Persson H. Cells that express brain-derived neurotrophic factor mRNA in the developing postnatal rat brain. Eur J Neurosci. 1991;3:688–697. doi: 10.1111/j.1460-9568.1991.tb00854.x. [DOI] [PubMed] [Google Scholar]
- 25.Frisén J, Verge VMK, Cullheim S, Persson H, Fried K, Middlemas DS, Hunter T, Hökfelt T, Risling M. Increased levels of trkB mRNA and trkB protein-like immunoreactivity in the injured rat and cat spinal cord. Proc Natl Acad Sci USA. 1992;89:11282–11286. doi: 10.1073/pnas.89.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frisén J, Verge VMK, Fried K, Risling M, Persson H, Trotter J, Hökfelt T, Lindholm D. Characterization of glial trkB receptors: differential response to injury in the central and peripheral nervous system. Proc Natl Acad Sci USA. 1993;90:4971–4975. doi: 10.1073/pnas.90.11.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Funakoshi H, Frisén J, Barbany G, Timmusk T, Zachrisson O, Verge VMK, Persson H. Differential expression of mRNAs for neurotrophins and their receptors following axotomy of the sciatic nerve. J Cell Biol. 1993;123:455–466. doi: 10.1083/jcb.123.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gavrielli Y, Sherman Y, Ben-Sasson A. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gould E, Woolley CS, McEwen BS. Naturally occurring cell death in developing dentate gyrus of the rat. J Comp Neurol. 1991;304:408–418. doi: 10.1002/cne.903040306. [DOI] [PubMed] [Google Scholar]
- 30.Gulyás AI, Miettinen R, Jacobowitz DM, Freund TF. Calretinin is present in non-pyramidal cells of the rat hippocampus. I. A new type of neuron specially associated with the mossy fiber system. Neuroscience. 1992;48:1–27. doi: 10.1016/0306-4522(92)90334-x. [DOI] [PubMed] [Google Scholar]
- 31.Hedreen JC, Bacon SJ, Price DL. A modified histochemical technique to visualize acetylcholinesterase-containing axons. J Histochem Cytochem. 1985;33:134–140. doi: 10.1177/33.2.2578498. [DOI] [PubMed] [Google Scholar]
- 32.Heumann R, Korsching S, Bandtlow C, Thoenen H. Changes of nerve growth factor synthesis in non-neuronal cells in response to sciatic nerve transection. J Cell Biol. 1987;104:1623–1631. doi: 10.1083/jcb.104.6.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huntley GW, Benson DL, Jones EG, Isackson PJ. Developmental expression of brain-derived neurotrophic factor mRNA by neurons of fetal and adult monkey prefrontal cortex. Dev Brain Res. 1992;70:53–63. doi: 10.1016/0165-3806(92)90103-4. [DOI] [PubMed] [Google Scholar]
- 34.Ip N, Li Y, Yancopoulos GD, Lindsay RM. Cultured hippocampal neurons show responses to BDNF, NT-3, and NT-4, but not NGF. J Neurosci. 1993;13:3394–3405. doi: 10.1523/JNEUROSCI.13-08-03394.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones KR, Fariñas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 37.Klein R, Parada LF, Coulier F, Barbacid M. TrkB, a novel tyrosine protein kinase receptor expressed during mouse neural development. EMBO J. 1989;8:3701–3709. doi: 10.1002/j.1460-2075.1989.tb08545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klein R, Martin-Zanca D, Barbacid M, Parada LF. Expression of the tyrosine kinase receptor gene trkB is confined to the murine embryonic and adult nervous system. Development (Camb) 1990;109:845–850. doi: 10.1242/dev.109.4.845. [DOI] [PubMed] [Google Scholar]
- 39.Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75:113–122. [PubMed] [Google Scholar]
- 40.Knüsel B, Rabin SJ, Hefti F, Kaplan DR. Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J Neurosci. 1994;14:1542–1554. doi: 10.1523/JNEUROSCI.14-03-01542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kobayashi NR, Bedard AM, Hincke MT, Tetzlaff W. Increased expression of BDNF and trkB mRNA in rat facial motoneurons after axotomy. Eur J Neurosci. 1996;8:1018–1029. doi: 10.1111/j.1460-9568.1996.tb01588.x. [DOI] [PubMed] [Google Scholar]
- 42.Koliatsos VE, Clatterbuck RE, Winslow JW, Cayouette MH, Price D. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron. 1993;10:359–367. doi: 10.1016/0896-6273(93)90326-m. [DOI] [PubMed] [Google Scholar]
- 43.Koliatsos VE, Cayouette MH, Berkemeier LR, Clatterbuck RE, Price DL, Rosenthal A. Neurotrophin 4/5 is a trophic factor for mammalian facial motor neurons. Proc Natl Acad Sci USA. 1994;91:3304–3308. doi: 10.1073/pnas.91.8.3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lesser SS, Lo DC. Regulation of voltage-gated ion channels by NGF and ciliary neurotrophic factor in SK-N-SH neuroblastoma cells. J Neurosci. 1995;15:253–261. doi: 10.1523/JNEUROSCI.15-01-00253.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li L, Oppenheim RW, Lei M, Houenou LJ. Neurotrophic agents prevent motoneuron death following sciatic nerve section in the neonatal mouse. J Neurobiol. 1994;25:759–766. doi: 10.1002/neu.480250702. [DOI] [PubMed] [Google Scholar]
- 48.Lindsay RM, Harmar AJ. Nerve growth factor regulates expression of neuropeptide genes in adult sensory neurons. Nature. 1989;337:362–364. doi: 10.1038/337362a0. [DOI] [PubMed] [Google Scholar]
- 49.Liu X, Ernfors P, Wu H, Jaenisch R. Sensory but not motor neuron deficits in mice lacking NT-4 and BDNF. Nature. 1995;375:238–241. doi: 10.1038/375238a0. [DOI] [PubMed] [Google Scholar]
- 50.Lo DC. Neurotrophic factors and synaptic plasticity. Neuron. 1995;15:979–981. doi: 10.1016/0896-6273(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 51.Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- 52.Marty S, Carroll P, Cellerino A, Castren E, Staiger V, Thoenen H, Lindholm D. Brain-derived neurotrophic factor promotes the differentiation of various hippocampal nonpyramidal neurons, including Cajal-Retzius cells, in organotypic slice cultures. J Neurosci. 1996;15:675–687. doi: 10.1523/JNEUROSCI.16-02-00675.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McAllister AK, Lo DC, Katz L. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 54.Mearow KM, Kril Y, Diamond J. Increased NGF mRNA expression in denervated rat skin. NeuroReport. 1993;4:351–354. doi: 10.1097/00001756-199304000-00002. [DOI] [PubMed] [Google Scholar]
- 55.Merlio J-P, Ernfors P, Jaber M, Persson H. Molecular cloning of rat trkC and distribution of cells expressing messenger RNAs for members of the trk family in the rat central nervous system. Neuroscience. 1992;51:513–532. doi: 10.1016/0306-4522(92)90292-a. [DOI] [PubMed] [Google Scholar]
- 56.Merlio J-P, Ernfors P, Kokaia Z, Middlemas DS, Bengzon J, Kokaia M, Smith M-L, Siesjö BK, Hunter T, Lindvall O, Persson H. Increased production of the trkB protein-tyrosine kinase receptor after brain insults. Neuron. 1993;10:151–164. doi: 10.1016/0896-6273(93)90307-d. [DOI] [PubMed] [Google Scholar]
- 57.Meyer M, Matsuoka I, Wetmore C, Olsson L, Thoenen H. Enhanced synthesis of brain-derived neurotrophic factor in the lesioned peripheral nerve: different mechanisms are responsible for the regulation of BDNF and NGF mRNA. J Cell Biol. 1992;119:45–54. doi: 10.1083/jcb.119.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Minichiello L, Piehl F, Vazquez E, Schimmang T, Hokfelt T, Represa J, Klein R. Differential effects of combined trk receptor mutations on dorsal root ganglion and inner ear sensory neurons. Development (Camb) 1995;121:4067–4075. doi: 10.1242/dev.121.12.4067. [DOI] [PubMed] [Google Scholar]
- 59.Nawa H, Bessho Y, Carnahan J, Nakanishi S, Mizuno K. Regulation of neuropeptide expression in cultured cerebral cortical neurons by brain-derived neurotrophic factor. J Neurochem. 1993;60:772–775. doi: 10.1111/j.1471-4159.1993.tb03216.x. [DOI] [PubMed] [Google Scholar]
- 60.Nawa H, Pelleymounter MA, Carnahan J. Intraventicular administration of BDNF increases neuropeptide expression in newborn rat brain. J Neurosci. 1994;14:3751–3765. doi: 10.1523/JNEUROSCI.14-06-03751.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Patterson SL, Abel T, Deuel TAS, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 62.Piehl F, Frisén J, Risling M, Hökfelt T, Cullheim S. Increased trkB mRNA expression by axotomized motoneurons. NeuroReport. 1994;5:697–700. doi: 10.1097/00001756-199402000-00009. [DOI] [PubMed] [Google Scholar]
- 63.Résibois A, Rogers JH. Calretinin in rat brain: an immunohistochemical study. Neuroscience. 1992;46:101–134. doi: 10.1016/0306-4522(92)90012-q. [DOI] [PubMed] [Google Scholar]
- 64.Schimmang T, Minichiello L, Vazquez E, San Jose I, Giraldez F, Klein R, Represa J. Developing inner ear sensory neurons require TrkB and TrkC receptors for innervation of their peripheral targets. Development (Camb) 1995;121:3381–3391. doi: 10.1242/dev.121.10.3381. [DOI] [PubMed] [Google Scholar]
- 65.Schnell L, Schneider R, Kolbeck R, Barde Y-A, Schwab ME. Neurotrophin-3 enhances sprouting of corticospinal tract during development and after adult spinal cord lesion. Nature. 1994;367:170–173. doi: 10.1038/367170a0. [DOI] [PubMed] [Google Scholar]
- 66.Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin SA, Barbacid M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF. Nature. 1994;368:246–248. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- 67.Sendtner M, Holtmann B, Kolbeck R, Thoenen H, Barde YA. Brain-derived neurotrophic factor prevents the death of motorneurons in newborn rats after nerve section. Nature. 1992;360:757–759. doi: 10.1038/360757a0. [DOI] [PubMed] [Google Scholar]
- 68.Silos-Santiago I, Fagan AM, Garber M, Fritzsch B, Barbacid M (1997) Severe sensory deficits but normal CNS development in newborn mice lacking trkB and trkC tyrosine kinase receptors. Eur J Neurosci, in press. [DOI] [PubMed]
- 69.Snider WD. Function of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 70.Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 71.Supèr H, Soriano E. The organization of the embryonic and early postnatal murine hippocampus. II. Development of the entorhinal, commissural, and septal connections studied with the lipophilic tracer DiI. J Comp Neurol. 1994;344:101–120. doi: 10.1002/cne.903440108. [DOI] [PubMed] [Google Scholar]
- 72.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 73.Timmusk T, Belluardo N, Metsis N, Persson H. Widespread and developmentally regulated expression of neurotrophin-4 mRNA in rat brain and peripheral tissues. Eur J Neurosci. 1993;5:605–613. doi: 10.1111/j.1460-9568.1993.tb00526.x. [DOI] [PubMed] [Google Scholar]
- 74.Timmusk T, Belluardo N, Persson H, Metsis M. Developmental regulation of brain-derived neurotrophic factor messenger RNAs transcribed from different promoters in the rat brain. Neuroscience. 1994;60:287–291. doi: 10.1016/0306-4522(94)90242-9. [DOI] [PubMed] [Google Scholar]
- 75.Yan Q, Elliott J, Snider WD. Brain-derived neurotrophic factor rescues spinal motor neurons from axotomy-induced cell death. Nature. 1992;360:753–755. doi: 10.1038/360753a0. [DOI] [PubMed] [Google Scholar]
- 76.Yan Q, Elliott JL, Matheson C, Sun J, Zhang L, Mu X, Rex KL, Snider WD. Influences of neurotrophins on mammalian motoneurons in vivo. J Neurobiol. 1993;24:1555–1577. doi: 10.1002/neu.480241202. [DOI] [PubMed] [Google Scholar]