

Abstract
Drugs that stimulate dopamine and glutamate receptors have been shown to induce the expression of AP-1 proteins (such as c-Fos and c-Jun) in the striatum and to induce binding of these proteins to AP-1 sites on DNA, leading to the hypothesis that AP-1-mediated transcription contributes to the long-term effects of these drugs. To examine this hypothesis, we compared the regulation of AP-1-mediated transcription to the inductions of AP-1-binding activity and genes encoding AP-1 proteins in primary cultures of striatal neurons. Although glutamate, dopamine, and forskolin (an activator of adenylate cyclase) all induce c-fos mRNA and AP-1 binding, we found, surprisingly, that only glutamate induces transcription of a transfected AP-1-driven fusion gene. To explore the basis for this discrepancy, we investigated the possibility that the phosphorylation of c-Jun may also be required for AP-1-mediated transcription in striatal neurons. Glutamate, but neither dopamine nor forskolin, raises the levels of phosphorylated c-Jun as well as the activity of a Jun kinase (SAPK/JNK) in striatal cultures. Both the glutamatergic induction of AP-1-mediated transcription and activation of SAPK/JNK appear to be mediated, at least in part, via NMDA receptors. In striatal neurons, the phosphorylation of AP-1 proteins produced by glutamate may be required to convert AP-1 protein expression and binding to transcriptional activation.
Keywords: glutamate, NMDA, SAPK, JNK, c-fos, c-jun, AP-1, transcription, striatum, cell culture
Corticostriatal glutamate projections and mesostriatal dopamine projections are the predominant afferents innervating the medium spiny neurons of the striatum. Medium spiny neurons, which make up >90% of all striatal neurons, project out of the striatum via two major pathways, the striatonigral and striatopallidal pathways. Medium spiny neurons giving rise to the striatonigral pathway express largely D1 dopamine receptors (Gerfen et al., 1990; Le Moine et al., 1991), which are positively coupled to adenylate cyclase; those giving rise to the striatopallidal pathway express largely D2 dopamine receptors (Gerfen et al., 1990; Le Moine et al., 1990), which are negatively coupled to adenylate cyclase. Medium spiny neurons in both output pathways also express a variety of NMDA (Standaert et al., 1994; Landwehrmeyer et al., 1995) and non-NMDA (Martin et al., 1993; Tallaksen-Greene and Albin, 1994) ionotropic glutamate receptors as well as metabotropic glutamate receptors (Testa et al., 1994, 1995).
Stimulation of dopaminergic or glutamatergic receptors on striatal neurons elicits not only short-term physiological responses, but also long-term responses that result from second messenger regulation with subsequent alterations in gene expression. Regulation of gene expression in striatum by dopaminergic drugs has been widely reported, with many groups describing induction of both immediate early genes and neuropeptide genes. For example, the indirect dopamine agonists amphetamine and cocaine have been shown to induce expression of c-fos mRNA and its protein product in striatum in a D1 dopamine receptor-dependent manner (Graybiel et al., 1990; Young et al., 1991; Nguyen et al., 1992; Konradi et al., 1994). Both amphetamine (Nguyen et al., 1992) and cocaine (Hope et al., 1992, 1994) also induce AP-1-binding activity, of which c-Fos is a component, in striatal cell extracts.
Glutamatergic stimulation has been shown to induce mRNAs encoding AP-1 proteins in a variety of neuronal systems, including cultured hippocampal neurons (Lerea et al., 1992, 1993; Bading et al., 1993,1995) and cultured cerebellar granule cells (Szekely, 1989). In primary cultures of cortical and striatal neurons, activation of NMDA receptors results in induction of c-fos, fosB, c-jun, andjunB mRNA (Vaccarino et al., 1992; Condorelli et al., 1994). Glutamate stimulation also results in an NMDA receptor-dependent increase in AP-1-binding activity in cultured neurons (Condorelli et al., 1994). In vivo stimulation of NMDA receptors by intrastriatal injection of quinolinic acid results in the induction of Fos protein in the majority of striatal projection neurons (Berretta et al., 1992).
Overall, there now exists a body of data on the induction of c-Fos and other AP-1 proteins, as well as on AP-1-binding activity by dopaminergic and glutamatergic stimuli in striatum. It has been hypothesized that induction of c-Fos and AP-1 binding in striatal neurons would lead to significant alterations in transcription of AP-1-regulated target genes. However, this hypothesis has not been tested directly. We have undertaken the present study to investigate the mechanisms and transcriptional consequences of those inductions in a primary neuronal culture model system that permits transfection analysis. We report that although glutamate, dopamine, and forskolin all induce c-fos expression and AP-1 binding, only glutamate activates AP-1-mediated transcription. We identify the activation of a Jun kinase (SAPK/JNK) and the phosphorylation of c-Jun as a potential mechanism for the glutamatergic induction of AP-1-mediated transcription. The specificity of the SAPK/JNK signaling cascade for glutamate versus dopamine may have implications for drug action in the striatum in vivo. Moreover, our data raise new questions regarding the involvement of SAPK/JNK pathways in glutamate-regulated developmental, neurodegenerative, and neurotoxic processes in the CNS.
MATERIALS AND METHODS
Primary striatal culture. Primary striatal cell cultures were prepared as described (Konradi et al., 1994), with the modification that cells were placed in defined media just 4 hr after plating to further curb proliferation of glial cells. The defined medium (DMEM/F12 with B-27 supplementation, Life Technologies, Gaithersburg, MD) includes 1.0 mm calcium, 0.7 mm magnesium, 4.2 mm potassium, and 40 μm glutamate. However, as determined by HPLC analysis, glutamate levels in the medium just before drug treatment ranged from 1 to 5 μm (Konradi et al., 1996). Experiments were performed with cells after 5 d in culture. Drugs for treatment of cultures included dopamine (RBI, Natick, MA), glutamate (Sigma, St. Louis, MO; all other glutamatergic agents from RBI), forskolin (Sigma), and 12-O-tetradecanoylphorbol-13-acetate (TPA, Sigma). Glial cell cultures were prepared as with the standard neuronal cultures, except that striatal cell suspensions were seeded onto uncoated six-well tissue culture plates (Costar, Cambridge MA), and cultures were maintained in 10% Nu-serum (Collaborative Biomedical Products, Bedford, MA) media to allow glial growth from sparse (∼1% surface coverage) on day 1 in culture to near confluence on day 9 in culture.
Calcium phosphate transfection. A construct containing four consecutive AP-1 elements fused to the luciferase reporter gene was transfected into primary striatal cultures using a serum-free calcium phosphate transfection protocol. The AP-1 core consensus sequence is identical to that described below for the substance P (SP) AP-1 oligonucleotide. Briefly, on day 5 in culture cells were fed with DMEM (Life Technologies) and the conditioned medium was saved and stored at 4°C. To transfect, 5 μg of DNA in 80 μl of total calcium phosphate precipitate was added to each 35 mm well for 1 hr, at which time cells were washed twice with DMEM, and 1.5 ml of conditioned medium was added back and the cells were returned to the incubator. Drugs were added ∼36 hr after transfection, and cells were harvested 6 hr after the drug treatments and assayed for luciferase activity. All experiments were performed in triplicate. Overall transfection efficiency ranged between 1 and 5%, determined by cytochemistry after transfection of an RSV-βgal expression plasmid (data not shown).
Electrophoretic mobility shift assay. Electrophoretic mobility shift assays (EMSA) were performed as described previously (Korner et al., 1989). Cells were lysed in EMSA buffer containing NP-40 (1%) and the phosphatase inhibitors sodium fluoride (1 μm, Sigma) and microcystin (5 μm, Life Technologies). Approximately 5 μg of protein was loaded per lane. Oligonucleotides were synthesized with an overhang and annealed in the presence of 20 mm NaPO4, 1 mm EDTA, and 100 mm KCl. The overhangs of the double-stranded oligonucleotides were then filled in, using Superscript Reverse Transcriptase (Life Technologies), with 32P-labeled dCTP. All experiments were performed in an excess of probe. The sequence of oligonucleotide used in electrophoretic mobility shift assays was AP-1-human SP promoter or human metallothionein (hMT) promotor, 5′-GATCAGCATGAGTCACTTC-3′ or 5′-GATCCGCGTGACTCAGCGC-3′, respectively (Konradi et al., 1994); CRE, 5′-GATCGCTGACGTCAGGG-3′ (Hoeffler et al., 1988; Hai et al., 1989); AP-4 (human proenkephalin gene), 5′-GATCGTCAGCTGCGGG-3′ (Comb et al., 1988); jun2TRE (rat c-jun promoter), 5′-AGCTAGCATTACCTCATCCC-3′ (Morooka et al., 1995). The overhang is shown in italics, core consensus sequences in bold. Note that the core AP-1 motifs within the SP and hMT promoters are identical on their opposite DNA strands and, thus, the two AP-1 oligonucleotides differ only in flanking sequence. Although the binding patterns of the two AP-1 probes are virtually identical for the experiments described here and previously (Konradi et al., 1993), the difference in flanking sequence does promote the binding of an additional protein complex to the hMT AP-1 element. This binding complex is nonspecific, because it is not altered by drug treatment or competition with at least 10-fold excess unlabeled probe (Konradi et al., 1995).
For competition assays, the unlabeled and labeled probes were combined before the addition of lysates, after which incubation was performed at 4°C for 10 min and then at room temperature for 10 min before loading the reaction mixture on a gel. For supershift analysis, antibodies (1 μg) together with whole-cell extract proteins (5 μg) were incubated in a 30 μl volume at 4°C for 10 min, followed by incubation with labeled probe (1 ng) at 4°C for 10 min and then at room temperature for 10 min before loading (25 μl). Antibodies used in supershift analysis include those directed against c-Jun (sc-45 X, Santa Cruz Biotechnology, Santa Cruz, CA), ATF-2 (sc-187 X, Santa Cruz Biotechnology), and c-Fos (Ab-5, Oncogene Science, Uniondale, NY).
Northern blot analysis. Northern blot analysis was performed as described (Cole et al., 1995). Briefly, cells were lysed in 400 μl of NP-40 lysis buffer, and phenol-chloroform extraction was used to isolate the RNA. Equal amounts of RNA (2 μg) were loaded per lane. Blots were hybridized with a c-fos or c-junriboprobe (Gemini system, Promega, Madison, WI). Hybridization of a cyclophilin cDNA probe (Danielson et al., 1988) was used as an internal control.
Western blot analysis. Cells were lysed in their wells as they were thawed on ice in 100 μl of a 1× Laemmli buffer. Lysates were sonicated, boiled, separated on polyacrylamide gels, and then electroblotted onto Immobilon-P membranes (Millipore, Bedford, MA). After blocking in 5% milk/0.05% Tween 20 in PBS, the membranes were probed with primary antibody in otherwise identical buffer except with 0.5% milk and 1:10,000 polyclonal anti-SAPKβ (which cross-reacts with α and γ forms) (Kyriakis et al., 1994) and/or anti-JNK1 (C-17, Santa Cruz Biotechnology), 1:1000 anti-phospho(Ser73)-c-Jun (New England Biolabs, Beverly, MA), 1:2000 anti-c-Jun [Santa Cruz’s H-79 was used as NEB’s anti-c-Jun antibody (#9162)] did not detect any inducible immunoreactivity), 1:500 anti-phospho-CREB (UBI, Lake Placid, NY), and 1:1000 anti-CREB (New England Biolabs) for 1 hr. After multiple washes membranes, were treated with 1:5000 donkey anti-rabbit IgG (Amersham, Arlington Heights, IL) in the same buffer for 1 hr. After multiple additional washes in the same buffer, except without milk, membranes were processed with ECL reagents per the manufacturer’s protocol (Amersham).
For the Western blots with phosphatase pretreatment, each striatal lysate was prepared using 40 μl of the immune-complex kinase assay’s lysis buffer (see below), except that 400 mm NaCl and 5 μm microcystin were included, and that the other phosphatase inhibitors (sodium vanadate and β-glycerophosphate) were present at 1/100th their standard concentrations. Calf intestinal alkaline phosphatase (Sigma P-0405, 20 U in 1 μl with or without heat inactivation by boiling for 15 min) was added to 20 μl of the lysate supernatant, and the mixture was incubated at 37°C for 30 min before stopping the reaction with 6× Laemmli buffer. The Western blot analysis was completed using anti-phospho(Ser73)-c-Jun, as above.
Immune-complex kinase assays. Treatment of striatal cultures was terminated by aspirating off medium, washing once with chilled PBS, aspirating off the wash, and freezing plates in liquid nitrogen followed by storage at −80°C. The kinase assay was modified fromKyriakis et al. (1994). Lysis buffer (1 ml/well; 20 mm HEPES, pH 7.4, 2 mm EGTA, 1 mmsodium vanadate, 50 mm β-glycerophosphate, 10% glycerol, 1% Triton X-100, 2 μm leupeptin, 400 μmphenylmethylsulfonyl fluoride, 10 KIU/ml aprotinin, 400 μm diisopropyl fluorophosphate, and 1 mmdithiothreitol) was added to the frozen cells, which were thawed and lysed on ice for 15 min. The lysate was spun in a microfuge (15,000g) for 5 min at 4°C, and the supernatant was transferred to 40 μl of 1:1 lysis buffer/swollen protein A–Sepharose CL-4B beads (Sigma) to which an anti-kinase antibody had been added. Polyclonal anti-SAPKβ, anti-JNK1(C-17), or anti-p38 (anti-XMpk2) (Rouse et al., 1994) antibodies at 1:1000 final concentration were used. Immunoprecipitation was performed at 4°C, with rotation of samples for 2–12 hr. Beads were washed once in lysis buffer, twice in high ionic strength buffer (500 mm lithium chloride, 100 mm Tris, pH 7.8, 0.1% Triton X-100, and 1 mmdithiothreitol), and three times in kinase buffer (20 mmMOPS, pH 7.2, 10 mm magnesium chloride, 2 mmEGTA, 0.1% Triton X-100, and 1 mm dithiothreitol). To the 20 μl of beads with 20 μl of overlying kinase buffer was added 20 μl of substrate [0.3 mg/ml c-Jun(1–135)-GST or 0.2 mg/ml myelin basic protein (Sigma)] in kinase buffer. The kinase reaction was initiated by adding 15 μl of 50 mmmagnesium chloride and 125 μm total ATP including 4–10 μCi γ32P-ATP per sample. After 20 min at 30°C with frequent mixings, the reactions were stopped by the addition of 20 μl 6× Laemmli buffer. Samples were boiled 5 min before running on a 12% polyacrylamide gel. Radiolabeled (phosphorylated) Jun substrate was detected in a band corresponding to the predicted 40 kDa using a Phosphorimager detection system (Molecular Dynamics, Sunnyvale, CA; used for all radioassays).
In-gel kinase assay. Striatal Jun kinase activity was characterized by immunoprecipitating SAPK/JNK (with anti-JNK1[C-17] at 1:1000 as in the immune complex kinase assay above). The immunoprecipitated kinase was then resolved electrophoretically under denaturing conditions in a polyacrylamide gel polymerized with 40 μg/ml c-Jun(1–135)-GST, renatured, and then incubated in the gel with 100 μCi/ml γ32P-ATP, as described previously (Cano et al., 1994).
RESULTS
Glutamate, but not dopamine or forskolin, induces AP-1-mediated transcription
We compared the effects of known inducers of AP-1 binding on transcription of a transfected 4× AP-1 luciferase construct in neuron-enriched striatal cultures derived from embryonic day 18 (E18) rats. The AP-1 sequence used is a consensus sequence found within the promoter of the preprotachykinin gene, which is expressed in D1 dopamine receptor-expressing striatal neurons. Like the phorbol ester TPA (the classic activator of AP-1-mediated transcription), glutamate (100 μm) stimulates the activity of the luciferase reporter gene 7- to 10-fold (Fig.1A,B). This glutamate-induced transcriptional activation is blocked by pretreatment with the NMDA receptor antagonist MK-801 (Fig. 1B). In contrast, dopamine (100 μm) and forskolin (a direct activator of adenylate cyclase, 10 μm) do not induce transcriptional activation of this AP-1-driven construct. In fact, dopamine and forskolin result in a slight, but reproducible, decrease of luciferase activity to levels below basal (Fig.1A) (data not shown).
Fig. 1.

Glutamate, but not dopamine or forskolin, induces expression of a transfected 4× AP-1-luciferase fusion construct.A, Glutamate (Glu, 100 μm) and TPA (100 nm), but not dopamine (DA, 100 μm) or forskolin (F, 10 μm), activate transcription of a 4× AP-1 luciferase construct. Values for luciferase activity are given as fold induction, and data are represented as mean ± SEM percentage of transfected control values (n = 3). Background luciferase activity determined from untransfected controls was subtracted; *p < 0.05 compared with control group.B, The glutamate activation of AP-1 transcription is blocked by MK-801 (1 μm, added 10 min before glutamate); *p < 0.05 compared with control or glutamate plus MK-801 groups.
Glutamate, dopamine, and forskolin all increase AP-1-binding activity in extracts of the striatal cultures
To investigate whether the inability of dopamine and forskolin to induce Ap-1-mediated transcription results from an inability to induce AP-1 binding in the striatal cultures used here, we measured AP-1-binding activity using EMSA. Glutamate (100 μm) induces binding to the consensus AP-1 sequence with peak binding activity occurring after 2 hr of treatment (Fig.2A–C). The specificity of binding and the relative affinities of the bound proteins for the AP-1 element compared with other DNA binding sites were examined using competition with unlabeled oligonucleotides (Fig.2B). After a 2 hr glutamate stimulation, unlabeled AP-1 (self) competes for the inducible binding at 5- and 10-fold molar excess, whereas the related jun2TRE element and consensus ATF element only compete for binding at 100-fold molar excess. The unrelated AP-4 element does not compete for AP-1 binding, even at 100-fold molar excess (Fig. 2B). Formation of the glutamate-induced AP-1 complex is blocked by pretreatment with the NMDA receptor antagonist MK-801 (Fig. 2D).
Fig. 2.

AP-1 binding after glutamate, dopamine, and forskolin stimulation in primary striatal cultures. A, AP-1 binding in striatal extracts after stimulation with dopamine (DA, 100 μm), glutamate (Glu, 100 μm), forskolin (Forsk, 10 μm), and TPA (100 nm) for 2 hr. Controls (dash) are from untreated cells. B, The specificity of AP-1-binding activity was determined by gel-shift competition experiments with striatal extracts 2 hr after glutamate stimulation. Unlabeled AP-1 (self), the related oligonucleotides for jun2TRE and CRE, and the unrelated AP-4 element display decreasing potencies, respectively, in their abilities to compete for the upper binding complex (specific band). In addition to the specific band, two less-specific, faster-migrating complexes can be observed (arrowheads). These binding complexes are present in untreated cell extracts and are not glutamate-inducible (see Fig. 3). These complexes are not competed effectively by addition of unlabeled self but can be competed by all the oligonucleotides, including the unrelated AP-4 element, at 100-fold molar excess. C, Time courses of glutamate- and forskolin-induced AP-1 protein binding. After glutamate stimulation, AP-1 binding is induced with a peak at 2–4 hr. Forskolin induction of AP-1 binding is more robust and peaks at 2 hr. D, The induction of AP-1 binding by glutamate stimulation can be blocked by the NMDA receptor antagonist MK-801 (1 μm, added 10 min before glutamate). All data are representative of at least three independent experiments. All experiments were performed with the SP AP-1 oligonucleotide probe.
Stimulation by dopamine results in a small but reproducible induction of AP-1 binding, with an increase that also peaks at 2 hr (Fig.2A). In other experiments, it was observed that dopamine can also induce a modest increase in a slightly more slowly migrating AP-1 complex (data not shown). Many D1 dopamine receptor actions are mediated by the cAMP pathway. In striatum in vivo, ∼50% of medium spiny neurons express D1 receptors (Gerfen et al., 1990). Thus, dopamine would only stimulate adenylate cyclase in a subset of the cells in the cultures and perhaps not fully stimulate adenylate cyclase, even in those cells. Forskolin, which directly stimulates adenylate cyclase in a receptor-independent manner and presumably in all cells, thus would be expected to produce a greater activation of the cAMP pathway than would dopamine. Indeed, forskolin (10 μm), like glutamate and TPA, results in a robust increase in AP-1 binding that also peaks at 2 hr (Fig.2A,C).
Fos and Jun, but not ATF-2, are present in induced AP-1 complexes
Antisera directed against AP-1 proteins were used to investigate the composition of glutamate- and forskolin-induced AP-1 complexes bound to the consensus AP-1 element (Fig.3A). Two hours after glutamate or forskolin stimulation, the specific AP-1 complex is recognized and supershifted by c-Fos antibodies, indicating that c-Fos is present in both complexes. With a specific antiserum directed against c-Jun, evidence for inclusion of c-Jun in the AP-1 complex was detected reproducibly in extracts of striatal neurons that had been treated with glutamate. A small supershifted complex associated with a diminution in the specific band is observable after glutamate and, to a lesser extent, after forskolin stimulation. The ability of this antibody to recognize adequately the c-Jun component of AP-1 complexes was verified by nearly complete supershifts of the labeled AP-1 probe incubated in the presence of synthetic c-Jun and c-Fos prepared in reticulocyte lysates (data not shown).
Fig. 3.
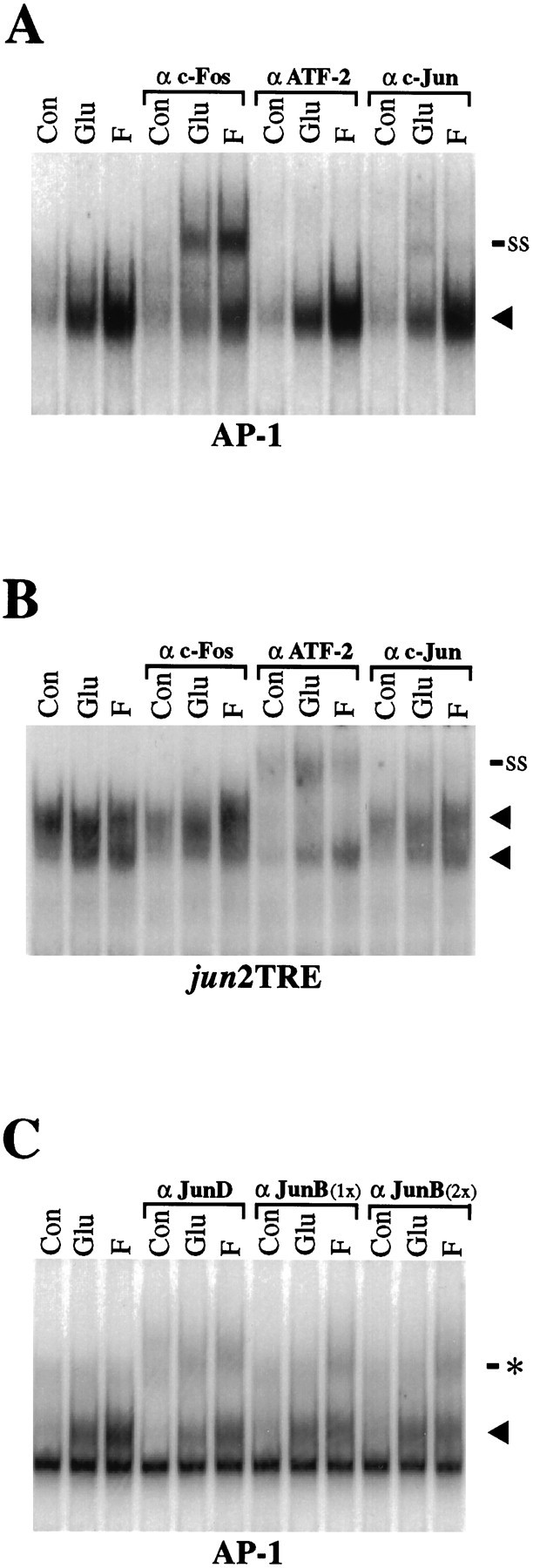
Supershift analysis of AP-1 and jun2TRE binding complexes after glutamate and forskolin stimulation.A, After 2 hr of glutamate (Glu) and forskolin (F) stimulation, AP-1-binding activity is increased over control (Con, no drug treatment), as indicated by the increased intensity of the specific complex (arrowhead). Addition of anti-Fos antibodies (α c-Fos) results in the appearance of a supershifted band (ss), indicating that Fos protein is present in the glutamate- and forskolin-induced AP-1-binding complex. The ATF-2 antibody (α ATF) has no effect on AP-1 binding. The c-Jun antibody (α c-Jun) results in a reduction in the overall levels of AP-1 binding, suggesting that addition of the antibody is interfering with formation of the DNA/protein complex. In addition, a minor supershifted band is observable after glutamate and, to a lesser extent, after forskolin stimulation. B, In contrast to AP-1 binding (A), protein binding to the jun2TRE after 2 hr of glutamate and forskolin stimulation can be supershifted by addition of ATF-2 antibodies, but not by addition of c-Fos antibodies.Arrowheads indicate two specific protein–oligonucleotide complexes. A modest reduction injun2TRE binding and a minor supershifted band are also seen after addition of c-Jun antibodies. C, Binding to the AP-1 element can be disrupted by addition of either a JunD (αJunD) or a JunB (α JunB) antibody. Addition of the JunD antibody reduces basal binding as well as glutamate- and forskolin-induced binding. The antibody directed against JunB selectively reduces the forskolin-induced binding complex. No difference is apparent if 1 μg (1×) or 2 μg (2×) of JunB antibody is included per lane. These reductions in the induced specific complexes (arrowhead) are associated with the appearance of modest probable supershift bands (asterisk). The SP AP-1 and hMT AP-1 oligonucleotides were used in A andC, respectively. A nonspecific binding complex is apparent below the specific band in C (not present inA) and is attributable to flanking sequences of the hMT probe, which are distinct from those of the SP probe (see Materials and Methods). Each experiment is representative and was performed at least four times (A, B) or twice (C).
Because AP-1 complexes may differ in their abilities to activate transcription depending on which Jun family members they contain, we also tested antibodies to other members of the Jun family. Although anti-JunD antibodies disrupt formation of glutamate- and forskolin-induced complexes proportionally, anti-JunB antibodies (at two concentrations) reduce levels of AP-1 complexes induced by forskolin to a greater extent than those induced by glutamate (Fig.3C). The diminutions in these specific bands correspond to the appearance of probable supershifted complexes. Although faint, these more slowly migrating complexes appear of increased intensity after glutamate treatment using anti-JunD antibodies and after forskolin treatment using either anti-JunD or anti-JunB antibodies. These data suggest that although glutamate and forskolin similarly enhance the formation of AP-1 complexes that recruit JunD (which is constitutively present in striatal neurons) (data not shown), forskolin, to a greater extent than glutamate, induces JunB as a component of these complexes.
Antibodies specifically directed against another protein, ATF-2, do not recognize the complexes binding the consensus AP-1 element (Fig.3A), although they supershift completely a complex binding to a jun2TRE oligonucleotide probe (Fig. 3B). Thejun2TRE is a variant AP-1 promoter element, which binds preferentially c-Jun/ATF-2 heterodimers instead of c-Fos/c-Jun heterodimers (van Dam et al., 1993).
Glutamate, dopamine, and forskolin induce c-fos mRNA
The inductions of Fos-containing AP-1 complexes may be attributable to inductions of c-fos gene expression. We have shown previously that dopamine induces c-fos mRNA in primary cultures of striatal neurons in a D1 dopamine receptor-dependent fashion (Konradi et al., 1994; Cole et al., 1995) (Fig.4C). Forskolin (10 μm) produces an even stronger induction of c-fos mRNA. Glutamate (100 μm) also induces c-fos mRNA in the cultures (Fig. 4A,B). The increase in c-fos mRNA stimulated by glutamate peaks after 30–90 min and returns to basal levels by ∼6 hr. A similar time course of c-fos mRNA induction was observed after dopamine or forskolin stimulation. These inductions of c-fos mRNA, which precede those of AP-1 binding, support further the possibility that increased c-Fos levels account, at least in part, for the increased AP-1 binding observed in striatal cultures after stimulation of both glutamatergic and dopaminergic pathways.
Fig. 4.

Glutamate, dopamine, and forskolin all induce c-fos mRNA, whereas only glutamate induces c-jun mRNA. A, Northern blot analysis for c-jun mRNA and c-fos mRNA induced by glutamate (100 μm). c-jun mRNA levels peak at an 8.5-fold induction 1.5 hr after glutamate stimulation. The more rapidly and more highly inducible c-fos mRNA peaks at 65-fold induction. Mean values ± SEM represent percentages of control levels (n = 3). B,C, Time courses for the effects of glutamate (100 μm), forskolin (10 μm), and dopamine (100 μm) on c-jun, c-fos, andcyclophilin mRNA. Northern blots were first hybridized with a c-jun riboprobe (3.2 and 2.5 kb transcripts), stripped, rehybridized with a c-fos riboprobe (2.2 kb transcript), and then hybridized for the third time with a cyclophilin cDNA probe. Cyclophilin (cyclo) mRNA was used as an internal loading control. All data are representative of at least three independent experiments.
Glutamate, but not dopamine or forskolin, increases c-jun mRNA levels
Although the c-fos promoter contains multiple cAMP response elements (CREs) (Berkowitz et al., 1989; Fisch et al., 1989) and a serum response element (SRE), which may account for the inductions of c-fos mRNA by dopamine, forskolin, and glutamate (see Discussion), the c-jun promoter contains neither a CRE nor an SRE. Rather, major regulatory targets for extracellular signals within the c-jun promoter are two TPA response elements, the jun1TRE and jun2TRE (Angel et al., 1988; van Dam et al., 1995). In contrast to c-fosmRNA, c-jun mRNA is not increased in response to either dopamine or forskolin in the striatal cultures (Fig.4B,C), although we have shown previously that junB mRNA is induced by dopamine in these cultures (Konradi et al., 1996). Unlike dopamine and forskolin, glutamate markedly increased levels of c-jun mRNA with a nearly ninefold induction observed at 60–90 min (Fig.4A). The specificity for glutamate of the induction of c-jun mRNA may account for the greater induction of a c-Jun component in the AP-1-binding complex by glutamate compared with forskolin (Fig. 3A).
Glutamate, but not dopamine or forskolin, increases phosphorylated c-Jun
We have demonstrated that in striatal cultures, dopamine, forskolin, and glutamate all induce c-fos expression followed by increased binding of a Fos-containing AP-1 protein complex to consensus AP-1 sites. Surprisingly, however, only glutamate increases transcription through this AP-1 element, indicating that the induction of AP-1 binding does not suffice to induce AP-1 transcription. In several non-neuronal cell types, AP-1-mediated transcription has been shown to require the phosphorylation of proteins within AP-1 complexes (Binétruy et al., 1991; Deng and Karin, 1994). Phosphorylation of c-Jun on Ser-63 and Ser-73 within its N-terminal activation domain has been shown to enhance markedly its ability to activate transcription without affecting its ability to form dimers or bind DNA (Dérijard et al., 1994; Karin, 1995).
We investigated whether the levels of phosphorylated c-Jun correlate with the inductions of AP-1-mediated transcription in striatal cultures. Using an antiserum that recognizes phospho(Ser73)-c-Jun, but not the dephosphorylated form, Western blot analysis demonstrates that glutamate increases levels of phospho(Ser73)-c-Jun-like immunoreactivity in three bands with approximate molecular weights of 40, 43, and 45 kDa, which correspond to the range of reported molecular weights of c-Jun (Lamph et al., 1988) (Fig. 5A). The uppermost induced band (∼45 kDa) likely represents phospho-c-Jun, because it coincides in molecular weight with the major immunoreactive band for total (unphosphorylated and phosphorylated) c-Jun (Fig.5A,B, bottom blots) and with a similarly induced band of phospho(Ser63)-c-Jun immunoreactivity (data not shown). Furthermore, the induced band at ∼45 kDa (but not at ∼43 kDa) was eliminated by phosphatase treatment of lysates (see Materials and Methods), indicating that it represents a phospho-antigen. This putative 45 kDa phospho-c-Jun band increases within 30 min and peaks after 60–90 min of glutamate treatment (Fig. 5B, top blot). Increases in total c-Jun protein were also induced by glutamate, but did not appear before 1 hr and did not peak until 3 hr of glutamate treatment (Fig.5B, bottom blot). Thus, although a rise in c-Jun protein may be contributing to increased levels of phospho-c-Jun measured at later time points, it cannot account for the initial increase in phospho-c-Jun levels.
Fig. 5.
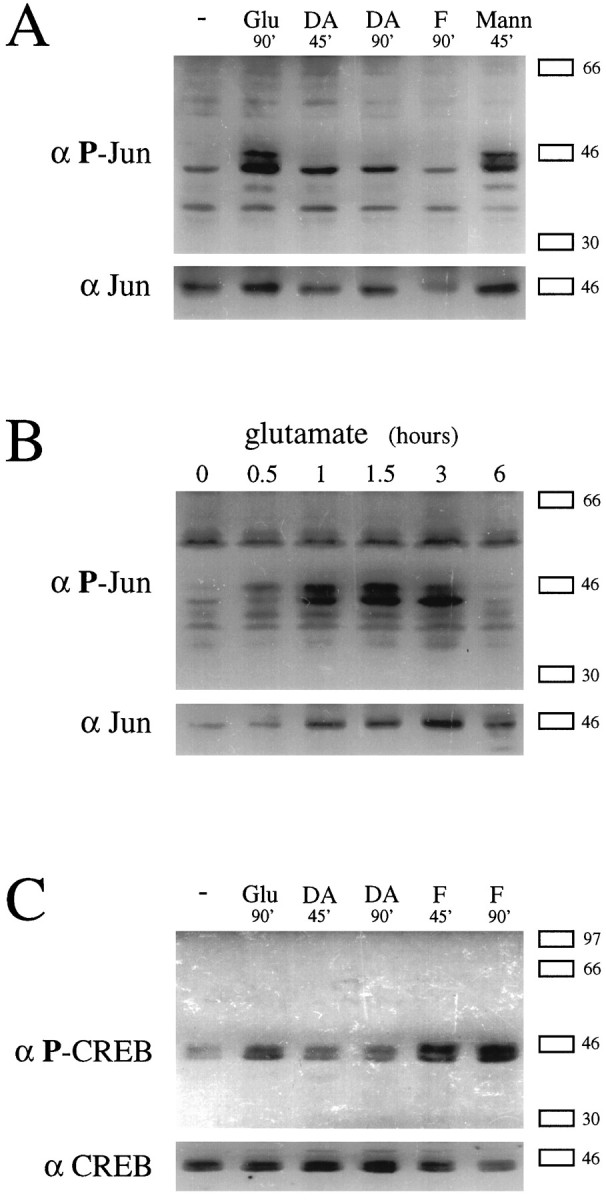
Elevation of phosphorylated c-Jun levels by glutamate, but not by dopamine or forskolin. A, Western blots probed with anti-phospho(Ser73)-c-Jun (top) and anti-c-Jun (bottom) antibodies performed on the same lysates of striatal cultures treated with or without glutamate (100 μm), dopamine (100 μm), or mannitol (300 mm) for the times indicated. B, Western blots probed as in A on lysates of striatal cultures treated with glutamate (100 μm) for the times indicated.C, Western blots probed with anti-phospho-CREB (top) and anti-CREB (bottom) antibodies performed on lysates of striatal cultures treated as inA. In A–C, the same blot was probed with antibodies directed toward the phosphorylated transcription factor and then reprobed with the antibodies directed toward the total form. The positions of flanking molecular weight markers are indicated on the right. All results were consistently reproducible in independent experiments.
Dopamine and forskolin, which fail to increase AP-1-mediated transcription (see Fig. 1), also fail to elevate levels of phospho-c-Jun at 45 or 90 min (Fig. 5A). In these cultures, however, forskolin is capable of raising levels of Ser133phosphorylated CREB (CRE binding protein), which, like c-Jun, is a member of the superfamily of basic leucine zipper transcription factors (Fig. 5C). Interestingly, glutamate increases levels of phospho-CREB in striatal cultures, although to a lesser extent than forskolin. Glutamate-induced phosphorylation of CREB may reflect the known effects on this transcription factor of calcium/calmodulin-dependent protein kinases (Sheng et al., 1991) or the Ras-MAPK pathway (Xing et al., 1996).
Glutamate, but not dopamine or forskolin, activates SAPK/JNK
Phosphorylation and activation of c-Jun results from the action of JNK, also called SAPK, because it was shown initially to be activated by cellular stressors such as ultraviolet radiation, heat shock, and hyperosmolar conditions (Dérijard et al., 1994,Kyriakis et al., 1994). SAPK/JNK is a member of the MAPK/extracellular signal regulated kinase (ERK) family of protein kinases, the mammalian members of which include the ERKs, p38, and SAPK/JNK. SAPK/JNK comprises several gene products called SAPKα, β, and γ in the rat and JNK1 and JNK2 in humans (Kyriakis and Avruch, 1996). In addition to cellular stressors, the inflammatory cytokines IL-1β and TNFα have been shown to activate both SAPK/JNK and p38 (Freshney et al., 1994;Han et al., 1994; Rouse et al., 1994). SAPK/JNK also can be activated by stimulation of muscarinic acetylcholine receptors expressed by stable transfection of a fibroblast cell line (Mitchell et al., 1995).
We investigated whether the activation of SAPK/JNK might account for the phosphorylation of c-Jun and the activation of AP-1-mediated transcription in cultured striatal neurons. Glutamate rapidly increases SAPK/JNK activity, as measured by in vitro phosphorylation of a c-Jun N-terminal substrate by anti-SAPK/JNK immunoprecipitates from treated cells (Fig. 6A). The antibodies used to immunoprecipitate SAPK/JNK activity recognize two major bands on Western blots prepared from lysates of striatal cultures (Fig. 6B) corresponding to the 54 and 46 kDa isoforms of SAPK/JNK (Hibi et al., 1993; Dérijard et al., 1994; Kyriakis et al., 1994). The lack of alteration of SAPK/JNK protein levels during the period when its activity is maximally increased suggests that increased activity is attributable to a post-translational modification of the kinase rather than increased protein levels. In-gel kinase assays (Fig. 6B) demonstrate that the glutamate-inducible c-Jun kinase activity in lysates of primary striatal culture corresponds to proteins of the predicted sizes, with the 46 kDa form predominating.
Fig. 6.

Rapid and specific activation of SAPK/JNK by glutamate. A, Immune-complex kinase assay for SAPK/JNK activity at different times after treatment of primary striatal cultures with glutamate (100 μm). The bands indicate c-Jun(1–135)-GST substrate (∼40 kDa) that was phosphorylated by anti-SAPK/JNK immunoprecipitates before separation in a polyacrylamide gel. The last lane shows the reaction product using cells that were treated with an additional 100 μmglutamate for the last 45 min of a 6 hr treatment. Fold increases are shown above each lane. B, On theleft, a Western blot for SAPK/JNK performed on lysates of untreated striatal cultures and those treated with glutamate (100 μm) for 1 or 2 hr. On the right, an in-gel kinase assay on anti-SAPK/JNK immunoprecipitates from striatal cultures. The phosphorylation of c-Jun(1–135)-GST substrate, which had been incorporated into the polyacrylamide gel, was performed in the gel after separation of immunoprecipitate components. When parallel in-gel kinase assays were performed without c-Jun substrate polymerized into the gel, kinase activity was reduced >100-fold (data not shown). The positions of flanking molecular weight markers are indicated forA and B. C, Comparison of immune-complex kinase assays for SAPK/JNK activity (top graph; using c-Jun(1–135)-GST as substrate) and p38 activity (bottom graph; using myelin basic protein as substrate) in immunoprecipitates of striatal cultures. In B(right) and C, treatments lasted 45 min with glutamate (Glu), dopamine (DA), and carbachol (Carb) at 100 μm; forskolin (Fors) at 10 μm; and mannitol (Mann) at 300 mm. The data inA and B were typical of at least three experiments. The data in C were pooled from two experiments, with n = 6 (except for the forskolin group, in which n = 3); * greater than control, with p < 0.05; ** less than control (by 10%), with p < 0.05.
SAPK/JNK activation is maximal (typically two- to threefold over basal levels) 45 min after initiation of treatment with glutamate (100 μm) or 15–45 min before the maximal glutamatergic induction of phospho-c-jun levels (Figs. 5B,6A). The kinase activation is prolonged, returning to baseline over 6 hr. To address the question of whether kinase activation becomes desensitized under these conditions, the medium was repleted with additional glutamate (100 μm) 45 min before the end of a 6 hr incubation. This second addition led to a modest induction of SAPK/JNK activity, closer to the basal than the maximally activated level (Fig. 6A, last lane). This result suggests that even if the decrease in SAPK/JNK activity over 6 hr is partly attributable to loss of glutamate from the medium, there also appears to be desensitization of the glutamatergic activation of SAPK/JNK.
Among the major neurotransmitters known to act on striatal neurons, only glutamate activated SAPK/JNK. Neither dopamine nor carbachol, an agonist at muscarinic acetylcholine receptors, increased SAPK/JNK activity (Fig. 6C). In fact, dopamine as well as forskolin slightly but consistently decreased activity in primary striatal cultures to levels below basal (Fig.6B,C) (data not shown), as they had for levels of AP-1-mediated transcription (Fig. 1A). To assess the possibility that a neurotransmitter other than dopamine or acetylcholine might be released from cells in the cultures in response to glutamate and then, in turn, activates SAPK/JNK, we also measured the effect of glutamate in cultures treated with tetrodotoxin, at a concentration (1 μm) that prevents action potential propagation. Tetrodotoxin had no effect on the glutamatergic activation of SAPK/JNK (data not shown), suggesting that glutamate acts directly on the cells in which the kinase is activated, rather than by relying on the release of an intermediary neurotransmitter.
Another proline-directed MAPK family member, p38, is closely related to SAPK/JNK and has been shown to be activated by the same cellular stresses, proinflammatory cytokines, and intracellular signaling cascades that activate SAPK/JNK (Freshney et al., 1994; Han et al., 1994; Rouse et al., 1994; Lin et al., 1995; Minden et al., 1995). Indeed, in primary cultures of striatal neurons, both p38 and SAPK/JNK activities increase three- to fourfold after exposure to hyperosmolar conditions with 300 mm mannitol (Fig.6C), which also raised phosphorylated c-Jun levels (Fig.5A). p38 Activity was measured by in vitrophosphorylation of a myelin basic protein substrate by anti-p38 immunoprecipitates from treated cells. Surprisingly, p38 activity was not increased by glutamate in striatal neurons, even when measured in the same lysates that contained elevated SAPK/JNK activity.
Glutamate activates SAPK/JNK with a high-potency (EC50 ∼ 20 μm) (Fig. 7A) (data not shown) and a pharmacological profile typical of the NMDA subtype of ionotropic glutamate receptors (Fig. 7B). Although NMDA and the non-NMDA ionotropic glutamate receptor agonist kainate both strongly induce SAPK/JNK activity in the cultures, NMDA antagonists are much more effective than a non-NMDA antagonist in blocking activation by glutamate, the presumed neurotransmitter. Specifically, the noncompetitive NMDA receptor blocker MK-801 and the competitive blocker APV almost completely block the glutamate- (and NMDA-) induced activation of SAPK/JNK. DNQX, a competitive non-NMDA ionotropic glutamate receptor antagonist, has no effect on the NMDA-induced activation and only slightly (but reproducibly) reduced the glutamate-induced activation. Stimulation of SAPK/JNK activity by kainate, however, is minimally attenuated by the NMDA antagonists and is completely blocked by DNQX. The metabotropic glutamate receptor agonist tACPD has no effect on SAPK/JNK activity.
Fig. 7.

Pharmacology of glutamatergic activation of SAPK/JNK and comparison of neuronal and glial cultures.A, Concentration–response curve for glutamatergic activation of SAPK/JNK in striatal cultures using immune-complex kinase assay. Striatal cultures were treated for 45 min at the concentrations indicated. B, Effects of glutamate agonists and antagonists on SAPK/JNK activity in striatal cultures. Immune-complex kinase assays were performed on cultures treated with glutamate (20 μm), NMDA (500 μm), kainate (50 μm), or tACPD (500 μm). The antagonists MK-801 (1 μm), APV (100 μm), and DNQX (100 μm) were added as indicated 10 min before the 45 min incubation with agonist. Data in A and Bwere typical of multiple experiments. NMDA,N-methyl-d-aspartate;tACPD,trans-(1S,3R)−1-aminocyclopentane-1.3-dicarboxylate;APV, 2-amino-5-phosphonovalerate; DNQX, 6,7-dinitroquinoxaline-2,3-dione. C, The effects of glutamate (100 μm) and mannitol (300 mm) for 45 min on SAPK/JNK activity measured by immune-complex kinase assay in standard (predominantly neuronal) striatal cultures (left) and glial striatal cultures (right). E18 striatal cell suspensions were seeded onto cell culture plates with or without a precoating of polyethylenimine for neuronal or glial cultures, respectively. Glial cells were cultured for 9 d in the presence of fetal calf serum until near confluent. Standard neuronal cultures (normally incubated in the presence of serum-containing medium for <1 d before it is replaced with defined medium) that were incubated instead in serum-containing medium still displayed a two- to threefold induction of SAPK/JNK activity with glutamate (data not shown).
We considered the possibility that NMDA receptors might be expressed by some glia as well as by neurons under conditions of culture. Although our cultures are highly neuron-enriched, containing ∼95% neurons based on staining with neuron-specific enolase and GFAP antisera (Konradi et al., 1996), we investigated the possibility that glia might contribute to the SAPK/JNK activation by comparing our standard cultures with pure cultures of striatal glia. These glial cultures are grown from the same striatal cell suspensions used to generate the predominantly neuronal glutamate-responsive cultures. Although SAPK/JNK activity was robustly induced in both neuron-enriched and glial cultures by mannitol (Fig. 7C), only the neuronal cultures responded to glutamate.
DISCUSSION
We provide evidence that in primary cultures of striatal neurons, glutamate increases SAPK/JNK activity, phospho-c-Jun levels, and transcription of both a transfected AP-1-driven reporter construct and an endogenous AP-1 (junTRE)-driven immediate early gene, c-jun. Moreover, the sequential time courses of these events and their specificity for glutamate over dopamine or forskolin support a role for the SAPK/JNK pathway in the glutamatergic activation of AP-1-regulated genes. We propose that SAPK/JNK activation and the associated rise in phosphorylated c-Jun levels may mediate the glutamatergic induction of AP-1-mediated transcription in these neurons and, in addition, that glutamatergic stimulation may convert AP-1 binding induced by other neurotransmitters into a transcriptionally active form.
Glutamatergic activation of SAPK/JNK
SAPK/JNK, a critical mediator of responses to a variety of cellular stressors and inflammatory cytokines (Kyriakis and Avruch, 1996), was first cloned from fetal brain (Dérijard et al., 1994), and at least one isoform appears to be expressed exclusively in the CNS (Mohit et al., 1995). However, SAPK/JNK regulation has not been described previously in the nervous system. Here, we demonstrate that glutamate, a major excitatory amino acid neurotransmitter, activates SAPK/JNK and several of its transcriptional targets in neuron-enriched cultures from E18 striatum.
The pharmacological profile of the glutamatergic activation of SAPK/JNK demonstrates prominent involvement of an NMDA receptor. However, this action of glutamate is also mimicked by a non-NMDA ionotropic receptor agonist and is partially blocked by a non-NMDA receptor antagonist, suggesting the involvement of a non-NMDA receptor component as well. Non-NMDA receptor activation can lead to membrane depolarization that may relieve the voltage-dependent Mg2+ block of NMDA receptors and, in doing so, may enhance NMDA receptor activation attributable to low levels of ambient glutamate in the culture medium. Alternatively, there may be a component of NMDA receptor-independent glutamate activation of this pathway. In addition, a single hybrid receptor comprising NMDA and non-NMDA receptor subunits (Henley et al., 1992) could account for the observed pharmacology (Marin et al., 1993).
The neurotransmitter activation of SAPK/JNK in striatal neurons appears specific for glutamate, with neither dopamine nor a muscarinic agonist displaying any stimulatory effect. Indeed, SAPK/JNK regulation provides an opportunity for an antagonistic interaction between dopamine and glutamate, because dopamine slightly reduces basal SAPK/JNK activity. Activation of the cAMP pathway may mediate this dopaminergic inhibition, because forskolin also decreases basal SAPK/JNK activity in striatal cultures and has been shown to inhibit stimulated SAPK/JNK activity in T lymphocytes (Hsueh and Lai, 1995). However, depending on the sequence of events, the possible interactions are complex.
The selectivity of the glutamate stimulation for SAPK/JNK versus p38 in the striatal cultures differs from the parallel activation of these kinases by cell stress or cytokines. The basis for the previously reported coordinate regulation of SAPK/JNK and p38 may involve upstream kinases that utilize p38 as well as SAPK/JNK as their substrates. Indeed, the best-characterized SAPK/JNK kinase (known as SEK1) phosphorylates p38 as well (Lin et al., 1995). However, Moriguchi et al. (1995) presented biochemical evidence that SEK1 may make up only a minor component of the total SAPK/JNK kinase activity induced by cellular stress. Moreover, they identified several distinct kinase activities that show substrate specificity for SAPK/JNK over p38. Thus, glutamate may activate a unique SAPK/JNK kinase, which, in contrast to SEK1, is selective for SAPK/JNK versus p38.
The known neurotoxic effects of glutamate and NMDA raise the possibility that the SAPK/JNK activation is secondary to membrane damage or metabolic insult. For example, glutamate and NMDA induce marked swelling of striatal neurons (Colwell et al., 1996), and membrane stretching has in fact been shown to activate SAPK/JNK in cardiac myocytes (Komuro et al., 1996). However, glutamate at a concentration of 100 μm, which maximally activates SAPK/JNK (Fig. 7A), does not produce signs of cellular toxicity or death in our striatal cultures (M. Schwarzschild and S. Hyman, unpublished observations). Others have also reported minimal or no toxicity of glutamate in similar short-term cultures of striatal neurons (Galarraga et al., 1990). In addition, the selectivity for glutamate at this concentration for SAPK/JNK over p38 argues further for a more discrete mechanism of SAPK/JNK activation by glutamate. Whether this regulation is mediated by cation influx, traditionally associated with NMDA receptor signaling, or by more recently identified direct coupling of NMDA receptors to potential signaling proteins (Niethammer et al., 1996) remains to be explored.
Regulation of AP-1 proteins and their DNA binding activity
Glutamate and dopamine differentially regulate c-fosand c-jun mRNA in rat striatal neurons in culture. Although both neurotransmitters induce c-fos mRNA, only glutamate induces c-jun mRNA. This is likely attributable to differences in the cis-regulatory elements located in the promoter regions of these genes. The c-fos promoter contains an SRE that can mediate glutamatergic induction of c-fosmRNA (Xia et al., 1996). It also contains several CREs, which likely mediate the effects of dopamine and forskolin (Konradi et al., 1994) and may, in addition, contribute to the calcium-dependent effects of glutamate (Sheng et al., 1990; Xing et al., 1996).
In contrast, c-jun expression appears to be regulated through TREs rather than through a CRE or SRE. The c-junpromoter elements jun1TRE and jun2TRE share homology with the consensus AP-1 element, bind AP-1 proteins (Angel et al., 1988; Stein et al., 1992; Rozek and Pfeifer, 1993), and are differentially involved in the induction of c-jun by phorbol esters, E1A protein, and UV irradiation (Angel et al., 1988; Devary et al., 1991; Stein et al., 1992; van Dam et al., 1993). Although the proteins binding to jun1TRE have not been characterized fully, the jun2TRE has been shown to preferentially bind a heterodimer composed of c-Jun and ATF-2 proteins (Morooka et al., 1995; van Dam, 1995). Because c-Jun and ATF-2 both can be phosphorylated and activated by SAPK/JNK (Hibi et al., 1993; Dérijard et al., 1994;Gupta et al., 1995), the glutamate-specific induction of c-jun mRNA in our cultures may be mediated by the activation of SAPK/JNK.
Differential regulation of AP-1-mediated transcription
In contrast to glutamate, dopamine and forskolin do not stimulate AP-1-mediated transcription, despite their ability to induce AP-1 binding. These results indicate that in striatal neurons, an induction of AP-1 binding does not necessarily produce transcriptional activation. A simple interpretation of this dissociation might suggest that the AP-1 binding complexes induced by dopamine or forskolin stimulation are transcriptionally inactive. Increased transcription through the AP-1 site may require the phosphorylation of AP-1 complex components, one example of which is provided by glutamate activation of SAPK/JNK and phosphorylation of c-Jun. Alternatively, because supershift analysis suggests that c-Jun is not a major component of the AP-1 complex induced by forskolin, acute activation of cAMP pathways may promote pairing of c-Fos with inhibitory partners. For example, dopaminergic stimulation of these cultures has been shown to inducejunB mRNA (Konradi et al., 1996), which is not activated by SAPK/JNK (Karin, 1995; Kallunki et al., 1996). Indeed, our data suggest that forskolin preferentially induces JunB rather than c-Jun in AP-1 complexes, whereas glutamate may induce c-Jun to a greater extent than JunB in these complexes.
Other inhibitory effects of cAMP in striatal cell culture may contribute to their lack of efficacy for stimulating AP-1-mediated transcription. Inhibition could occur upstream of AP-1 protein phosphorylation at the level of SAPK/JNK, where we and others have identified an inhibitory effect of cAMP signaling (as described above). Conversely, cAMP-mediated inhibition of AP-1 transcription may occur downstream of c-Jun phosphorylation at the level of CREB binding protein, which mediates transcriptional activation through AP-1 as well as through CRE sites (Kamei et al., 1996).
The inability of dopamine or forskolin alone to induce AP-1-mediated transcription was observed in primary striatal cultures, which are necessarily devoid of the dopaminergic and glutamatergic afferents that exist in the intact striatum. However, indirect dopamine agonists such as cocaine and amphetamine and dopamine receptor antagonists such as haloperidol may act in vivo to modulate glutamate-induced AP-1 transcription. Interactions between dopamine and glutamate are suggested by the close anatomic arrangement of corticostriatal and nigrostriatal nerve terminals synapsing on the dendritic spines of striatal neurons (Freund et al., 1984; Smith and Bolam, 1990). In addition, pharmacological evidence implicates glutamate receptors in cocaine- and amphetamine-induced gene expression in the striatum (Snyder-Keller, 1991; Ohno et al., 1994; Wang et al., 1994; Konradi et al., 1996). Thus, our findings in culture raise additional questions about the in vivo interactions of excitatory amino acids with endogenous and exogenous dopaminergic signals. For example, dopaminergic psychostimulant and antipsychotic drugs might raise the levels of AP-1 transcription factors in the intact striatum in a manner that primes or amplifies a subsequent glutamatergic induction of AP-1 transcriptional effects.
Our findings also raise the possibility that SAPK/JNK, as well as its AP-1 targets, plays a role in physiological and pathological effects of glutamate. Recently, SAPK/JNK activation has been shown under certain circumstances to be essential for apoptotic cell death (Xia et al., 1995; Verheij et al., 1996). For example, in a cell line bearing similarities to sympathetic neurons, SAPK/JNK-dependent apoptosis is induced by withdrawing neurotrophic factor support (Xia et al., 1995). These results, together with our own, lead to the hypothesis that SAPK/JNK may mediate the glutamate-induced cell death implicated in both normal neural development and neurodegenerative disease. Interestingly, riluzole, an antagonist of postsynaptic glutamate function used clinically to slow progression of amyotrophic lateral sclerosis (Bensimon et al., 1994), completely blocks the glutamatergic induction of SAPK/JNK and phospho-c-jun in our system (M. Schwarzschild and S. Hyman, unpublished results). Because cytoskeletal proteins, in addition to transcription factors, can also serve as substrates for this kinase (Kyriakis and Avruch, 1990), the NMDA-dependent glutamatergic activation of SAPK/JNK observed here in embryonic striatal neuron cultures may also be relevant to NMDA-dependent changes in neuronal connectivity observed during development (Scheetz and Constantine-Paton, 1994). Finally, more acute NMDA-dependent neuronal damage, such as that seen in ischemic stroke, may also involve SAPK/JNK, because renal and cardiac ischemia/reperfusion has been shown to activate SAPK/JNK (Pombo et al., 1994; Knight and Buxton, 1996).
M.A.S. and R.L.C. contributed equally to this manuscript.
This work was supported by Public Health Service Grants DA07134, DA00257, and NS01729. We thank John Kyriakis for anti-SAPKβ antibody, c-jun(1–135)-GST construct, and helpful suggestions; Angel Nebreda for anti-XMpk2 antibody; Steve Fink for the AP-1-luciferase construct; and Zhengui Xia and Michael Greenberg for discussion of transfection methods. We also thank Melissa Meyers and Allison Wong for technical assistance and Bruce Hope, Linda Kobierski, Christine Konradi, and Susan Lewis for valuable discussions.
Correspondence should be addressed to Dr. Steven E. Hyman at his current address: National Institute of Mental Health, Room 17-99, Parklawn Building, 5600 Fishers Lane, Rockville, MD 20857.
REFERENCES
- 1.Angel P, Hattori K, Smeal T, Karin M. The jun proto-oncogene is positively autoregulated by its product Jun/AP-1. Cell. 1988;55:875–885. doi: 10.1016/0092-8674(88)90143-2. [DOI] [PubMed] [Google Scholar]
- 2.Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 3.Bading H, Segal MM, Sucher NJ, Dudek H, Lipton SA, Greenberg ME. N-methyl-d-aspartate receptors are critical for mediating the effects of glutamate on intracellular calcium concentration and immediate early gene expression in cultured hippocampal neurons. Neuroscience. 1995;64:653–664. doi: 10.1016/0306-4522(94)00462-e. [DOI] [PubMed] [Google Scholar]
- 4.Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med. 1994;330:585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- 5.Berkowitz LA, Riabowol KT, Gilman MZ. Multiple sequence elements of a single functional class are required for cAMP responsiveness of the mouse c-fos promoter. Mol Cell Biol. 1989;9:4272–4281. doi: 10.1128/mcb.9.10.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berretta S, Robertson HA, Graybiel AM. Dopamine and glutamate agonists stimulate neuron-specific expression of Fos-like protein in the striatum. J Neurophysiol. 1992;68:767–777. doi: 10.1152/jn.1992.68.3.767. [DOI] [PubMed] [Google Scholar]
- 7.Binétruy B, Smeal T, Karin M. Ha-Ras augments c-Jun activity and stimulates phosphorylation of its activated domain. Nature. 1991;351:122–127. doi: 10.1038/351122a0. [DOI] [PubMed] [Google Scholar]
- 8.Cano E, Hazzalin CA, Mahadevan LC. Anisomycin-activated protein kinases p45 and p55 but not mitogen-activated protein kinases ERK-1 and -2 are implicated in the induction of c-fos and c-jun. Mol Cell Biol. 1994;14:7352–7362. doi: 10.1128/mcb.14.11.7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cole RL, Konradi C, Douglass J, Hyman SE. Neuronal adaptation to dopamine and amphetamine: molecular mechanisms of prodynorphin gene regulation in rat striatum. Neuron. 1995;14:813–823. doi: 10.1016/0896-6273(95)90225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colwell CS, Altemus KL, Cepeda C, Levine MS. Regulation of N-methyl-d-aspartate-induced toxicity in the neostriatum: a role for metabotropic glutamate receptors? Proc Natl Acad Sci USA. 1996;93:1200–1204. doi: 10.1073/pnas.93.3.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Comb M, Mermod N, Hyman SE, Pearlberg J, Ross M, Goodman HM. Proteins bound at adjacent DNA elements act synergistically to regulate human proenkephalin cAMP inducible transcription. EMBO J. 1988;7:3793–3805. doi: 10.1002/j.1460-2075.1988.tb03264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Condorelli DF, Albani PD, Amico C, Lukasaiuk K, Kaczmarek L, Giuffrida-Stella AM. Glutamate receptor-driven activation of transcription factors in primary neuronal cultures. Neurochem Res. 1994;19:489–499. doi: 10.1007/BF00967329. [DOI] [PubMed] [Google Scholar]
- 13.Danielson PE, Forss-Petter S, Brow MA, Calavetta L, Douglass J, Milner RJ, Sutcliffe JG. p1B15: a cDNA clone of the rat mRNA encoding cyclophilin. DNA. 1988;7:261–267. doi: 10.1089/dna.1988.7.261. [DOI] [PubMed] [Google Scholar]
- 14.Deng T, Karin M. c-Fos transcriptional activity stimulated by H-Ras-activated protein kinase distinct from JNK and ERK. Nature. 1994;371:171–175. doi: 10.1038/371171a0. [DOI] [PubMed] [Google Scholar]
- 15.Dérijard B, Hibi M, Wu I-H, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 16.Devary Y, Gottlieb RA, Lau LF, Karin M. Rapid and preferential activation of the c-jun gene during the mammalian UV response. Mol Cell Biol. 1991;11:2804–2811. doi: 10.1128/mcb.11.5.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fisch TM, Prywes R, Simon MC, Roeder RG. Multiple sequence elements in the c-fos promoter mediate induction by cAMP. Genes Dev. 1989;3:198–211. doi: 10.1101/gad.3.2.198. [DOI] [PubMed] [Google Scholar]
- 18.Freshney NW, Rawlinson L, Guesdon F, Jones E, Cowley S, Hsuan J, Saklatvala J. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell. 1994;78:1039–1049. doi: 10.1016/0092-8674(94)90278-x. [DOI] [PubMed] [Google Scholar]
- 19.Freund TF, Powell JF, Smith AD. Tyrosine hydroxylase-immunoreactive boutons in synaptic contact with identified striatonigral neurons, with particular reference to dendritic spines. Neuroscience. 1984;13:1189–1215. doi: 10.1016/0306-4522(84)90294-x. [DOI] [PubMed] [Google Scholar]
- 20.Galarraga E, Surmeier DJ, Kitai ST. Quinolate and kainate neurotoxicity in neostriatal cultures is potentiated by co-culturing with neocortical neurons. Brain Res. 1990;512:269–276. doi: 10.1016/0006-8993(90)90636-P. [DOI] [PubMed] [Google Scholar]
- 21.Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- 22.Graybiel AM, Moratalla R, Robertson HA. Amphetamine and cocaine induce drug-specific activation of the c-fos gene in striosome-matrix compartments and limbic subdivisions of the striatum. Proc Natl Acad Sci USA. 1990;87:6912–6916. doi: 10.1073/pnas.87.17.6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta S, Campbell D, Dérijard B, Davis RJ. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 24.Hai T, Liu F, Coukos WJ, Green MR. Transcription factor ATF cDNA clones: an extensive family of leucine zipper proteins able to selectively form DNA-binding heterodimers. Genes Dev. 1989;3:2083–2090. doi: 10.1101/gad.3.12b.2083. [DOI] [PubMed] [Google Scholar]
- 25.Han J, Lee J-D, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 26.Henley JM, Ambrosini A, Rodriguez-Ithurralde D, Sudan H, Brackley P, Kerry C, Mellor I, Abutidze K, Usherwood PNR, Barnard EA. Purified unitary kainate/α-amino-3-hydroxy-5-methylisooxazole-proprionate (AMPA) and kainate/AMPA/N-methyl-d-aspartate receptors with interchangeable subunits. Proc Natl Acad Sci USA. 1992;89:4806–4810. doi: 10.1073/pnas.89.11.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 28.Hoeffler JP, Meyer TE, Yun Y, Jameson JL, Habener JF. Cyclic AMP-responsive DNA-binding protein: structure based on a cloned placental DNA. Science. 1988;242:1430–1433. doi: 10.1126/science.2974179. [DOI] [PubMed] [Google Scholar]
- 29.Hope B, Kosofsky B, Hyman SE, Nestler EJ. Regulation of immediate early gene expression and AP-1 binding in the rat nucleus accumbens by chronic cocaine. Proc Natl Acad Sci USA. 1992;89:5764–5768. doi: 10.1073/pnas.89.13.5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hope BT, Nye HE, Kelz MB, Self DW, Iadarola MJ, Nakabeppu Y, Duman RS, Nestler E. Induction of a long-lasting AP-1 complex composed of altered Fos-like proteins in the brain by chronic cocaine and other chronic treatments. Neuron. 1994;13:1235–1244. doi: 10.1016/0896-6273(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 31.Hsueh YP, Lai M-Z. c-Jun N-terminal kinase but not mitogen-activated protein kinase is sensitive to cAMP inhibition in T lymphocytes. J Biol Chem. 1995;270:18094–18098. doi: 10.1074/jbc.270.30.18094. [DOI] [PubMed] [Google Scholar]
- 32.Kallunki T, Deng T, Hibi M, Karin M. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell. 1996;87:929–939. doi: 10.1016/s0092-8674(00)81999-6. [DOI] [PubMed] [Google Scholar]
- 33.Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin S-C, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 34.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 35.Knight RJ, Buxton DB. Stimulation of c-Jun kinase and mitogen-activated protein kinase by ischemia and reperfusion in the perfused rat heart. Biochem Biophys Res Commun. 1996;218:83–88. doi: 10.1006/bbrc.1996.0016. [DOI] [PubMed] [Google Scholar]
- 36.Komuro I, Kudo S, Yamazaki T, Zou Y, Shiojima I, Yazaki Y. Mechanical stretch activates the stress-activated protein kinases in cardiac myocytes. FASEB J. 1996;10:631–636. doi: 10.1096/fasebj.10.5.8621062. [DOI] [PubMed] [Google Scholar]
- 37.Konradi C, Kobierski LA, Nguyen TV, Heckers S, Hyman SE. The cAMP-response-element-binding protein interacts, but Fos protein does not interact, with the proenkephalin enhancer in the rat striatum. Proc Natl Acad Sci USA. 1993;90:7005–7009. doi: 10.1073/pnas.90.15.7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konradi C, Cole RL, Heckers S, Hyman SE. Amphetamine regulates gene expression in rat striatum via transcription factor CREB. J Neurosci. 1994;14:5623–5634. doi: 10.1523/JNEUROSCI.14-09-05623.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Konradi C, Cole RL, Green D, Senatus P, Leveque J-C, Pollack AE, Grossbard SJ, Hyman SE. Analysis of proenkaphalin second messenger-inducible enhancer in rat striatal cultures. J Neurochem. 1995;65:1007–1015. doi: 10.1046/j.1471-4159.1995.65031007.x. [DOI] [PubMed] [Google Scholar]
- 40.Konradi C, Leveque J-C, Hyman SE. Amphetamine and dopamine-induced immediate early gene expression in striatal neurons depends on postsynaptic NMDA receptors and calcium. J Neurosci. 1996;16:4231–4239. doi: 10.1523/JNEUROSCI.16-13-04231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Korner M, Rattner A, Mauxion F, Sen R, Citri Y. A brain-specific transcription activator. Neuron. 1989;3:563–572. doi: 10.1016/0896-6273(89)90266-3. [DOI] [PubMed] [Google Scholar]
- 42.Kyriakis JM, Avruch J. pp54 Microtubule-associated protein 2 kinase. A novel serine/threonine protein kinase regulated by phosphorylation and stimulated by poly-llysine. J Biol Chem. 1990;265:17355–17363. [PubMed] [Google Scholar]
- 43.Kyriakis JM, Avruch J. Sounding the alarm: protein kinase cascades activated by stress and inflammation. J Biol Chem. 1996;271:24313–24316. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- 44.Kyriakis JM, Banerjee P, Nikolakaki E, Dal T, Ruble EA, Ahmad MF, Avruch J, Woodgett JR. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 45.Lamph WW, Wamsley P, Sassone-Corsi P, Verma IM. Induction of proto-oncogene JUN/AP-1 by serum and TPA. Nature. 1988;334:629–631. doi: 10.1038/334629a0. [DOI] [PubMed] [Google Scholar]
- 46.Landwehrmeyer GB, Standaert DG, Testa CM, Penny JB, Young AB. NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J Neurosci. 1995;15:5297–5307. doi: 10.1523/JNEUROSCI.15-07-05297.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Le Moine C, Normand E, Guitteny AF, Fouque B, Teoule R, Bloch B. Dopamine receptor gene expression by enkephalin neurons in rat forebrain. Proc Natl Acad Sci USA. 1990;87:230–234. doi: 10.1073/pnas.87.1.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Le Moine C, Normand E, Bloch B. Phenotypical characterization of the rat striatal neurons expressing the D1 dopamine receptor gene. Proc Natl Acad Sci USA. 1991;88:4205–4209. doi: 10.1073/pnas.88.10.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lerea LS, MacNamara JO. Ionotropic glutamate receptor subtypes activate c-fos transcription by distinct calcium-requiring intracellular signalling pathways. Neuron. 1993;10:31–41. doi: 10.1016/0896-6273(93)90239-n. [DOI] [PubMed] [Google Scholar]
- 50.Lerea LS, Butler LS, MacNamara JO. NMDA and non-NMDA receptor-mediated increase of c-fos mRNA in dentate gyrus neurons involves calcium influx via different routes. J Neurosci. 1992;12:2973–2981. doi: 10.1523/JNEUROSCI.12-08-02973.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin A, Minden A, Martinetto H, Claret F-X, Lange-Carter C, Mercurio F, Johnson GL, Karin M. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. 1995;268:286–290. doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- 52.Marin P, Quignard JF, Lafon-Cazol M, Bockaert J. Non-classical glutamate receptors, blocked by both NMDA and non-NMDA antagonists, stimulate nitric oxide production in neurons. Neuropharmacology. 1993;32:29–36. doi: 10.1016/0028-3908(93)90126-n. [DOI] [PubMed] [Google Scholar]
- 53.Martin LJ, Blackstone CD, Levey AI, Hunigar RL, Price DL. AMPA glutamate receptor subunits are differentially distributed in rat brain. Neuroscience. 1993;53:327–358. doi: 10.1016/0306-4522(93)90199-p. [DOI] [PubMed] [Google Scholar]
- 54.Minden A, Lin A, Claret FX, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 55.Mitchell FM, Russell M, Johnson GL. Differential calcium dependence in the activation of c-Jun kinase and mitogen-activated protein kinase by muscarinic acetylcholine receptors in rat 1a cells. Biochem J. 1995;309:381–384. doi: 10.1042/bj3090381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mohit AA, Martin JH, Miller CA. p493F12 kinase: a novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron. 1995;14:67–78. doi: 10.1016/0896-6273(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 57.Moriguchi T, Kawasaki H, Satoshi M, Gotoh Y, Nishida E. Evidence for multiple activators for stress-activated protein kinases/c-Jun amino-terminal kinases. J Biol Chem. 1995;270:12969–12972. doi: 10.1074/jbc.270.22.12969. [DOI] [PubMed] [Google Scholar]
- 58.Morooka H, Bonventre JV, Pombo CM, Kyriakis JM, Force T. Ischemia and reperfusion enhance ATF-2 and c-Jun binding to cAMP response elements and to an AP-1 binding site from the c-jun promoter. J Biol Chem. 1995;270:30084–30092. doi: 10.1074/jbc.270.50.30084. [DOI] [PubMed] [Google Scholar]
- 59.Niethammer M, Kim E, Sheng M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PDS-95 family of membrane-associated guanylate kinases. J Neurosci. 1996;16:2157–2163. doi: 10.1523/JNEUROSCI.16-07-02157.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nguyen TV, Kosofsky BE, Birnbaum R, Cohen BM, Hyman SE. Differential expression of c-fos and zif268 in rat striatum after haloperidol, clozapine and amphetamine. Proc Natl Acad Sci USA. 1992;89:4270–4274. doi: 10.1073/pnas.89.10.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ohno M, Yoshida H, Watanabe S. NMDA receptor-mediated expression of Fos protein in the rat striatum following methamphetamine administration: relation to behavioral sensitization. Brain Res. 1994;665:135–140. doi: 10.1016/0006-8993(94)91163-0. [DOI] [PubMed] [Google Scholar]
- 62.Pombo CM, Bonventre JV, Avruch J, Woodgett JR, Kyriakis JM, Force T. The stress-activated protein kinases are major c-Jun amino-terminal kinases activated by ischemia and reperfusion. J Biol Chem. 1994;42:26546–26551. [PubMed] [Google Scholar]
- 63.Rouse J, Cohen P, Trigon S, Morange M, Alonso-Liamazares A, Zamanillo D, Hunt T, Nebreda AR. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 64.Rozek D, Pfeiffer GP. In vivo protein-DNA interactions at the c-jun promoter: preformed complexes mediate the UV response. Mol Cell Biol. 1993;13:5490–5499. doi: 10.1128/mcb.13.9.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scheetz AJ, Constantine-Paton M. Modulation of NMDA receptor function: implications for vertebrate neural development. FASEB J. 1994;8:745–752. doi: 10.1096/fasebj.8.10.8050674. [DOI] [PubMed] [Google Scholar]
- 66.Sheng ME, McFadden G, Greenberg ME. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron. 1990;4:571–582. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- 67.Sheng ME, Thompson MA, Greenberg ME. CREB: a Ca2+-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- 68.Smith AD, Bolam JP. The neural network of the basal ganglia as revealed by the study of synaptic connections of identified neurons. Trends Neurosci. 1990;13:259–265. doi: 10.1016/0166-2236(90)90106-k. [DOI] [PubMed] [Google Scholar]
- 69.Snyder-Keller AM. Striatal c-fos induction by drugs and stress in neonatally dopamine-depleted rats given nigral transplants: importance of NMDA activation and relevance to sensitization phenomena. Exp Neurol. 1991;113:155–165. doi: 10.1016/0014-4886(91)90171-8. [DOI] [PubMed] [Google Scholar]
- 70.Standaert DG, Testa CM, Young AB, Penny JB. Organization of N-methyl-d-aspartate glutamate receptor gene expression in the basal ganglia of the rat. J Comp Neurol. 1994;343:1–16. doi: 10.1002/cne.903430102. [DOI] [PubMed] [Google Scholar]
- 71.Stein B, Angel P, van Dam H, Ponta H, Herrlich P, van der Eb A, Rahmsdorf HJ. Ultraviolet-radiation induced c-jun gene transcription: two AP-1 like binding sites mediate the response. Photochem Photobiol. 1992;55:409–415. doi: 10.1111/j.1751-1097.1992.tb04255.x. [DOI] [PubMed] [Google Scholar]
- 72.Szekely AM, Barbaccia ML, Alho H, Costa E. In primary culture of cerebellar granule cells the activation of NMDA-sensitive glutamate receptors induces c-fos mRNA expression. Mol Pharmacol. 1989;35:401–408. [PubMed] [Google Scholar]
- 73.Tallaksen-Greene SJ, Albin RL. Localization of AMPA-selective excitatory amino acid receptor subunits in identified populations of striatal neurons. Neuroscience. 1994;61:509–519. doi: 10.1016/0306-4522(94)90430-8. [DOI] [PubMed] [Google Scholar]
- 74.Testa CM, Standaert DG, Penny JB, Young AB. Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci. 1994;14:3005–3018. doi: 10.1523/JNEUROSCI.14-05-03005.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Testa CM, Standaert DG, Landwehrmeyer GB, Penny JB, Young AB. Differential expression of the mGluR5 metabotropic glutamate receptor by striatal neurons. J Comp Neurol. 1995;354:241–252. doi: 10.1002/cne.903540207. [DOI] [PubMed] [Google Scholar]
- 76.Vaccarino FM, Hayward MD, Nestler EJ, Duman RS, Tallman JF. Differential induction of immediate early genes by excitatory amino acid receptor types in primary cultures of cortical and striatal neurons. Mol Brain Res. 1992;12:233–241. doi: 10.1016/0169-328x(92)90089-t. [DOI] [PubMed] [Google Scholar]
- 77.van Dam H, Duyndam M, Rottier R, Bosch A, de Vries-Smits L, Herrlich P, Zantema A, Angel P, van der Eb AJ. Heterodimer formation of c-jun and ATF-2 is responsible for induction of c-jun by the 243 amino acid adenovirus E1A protein. EMBO J. 1993;12:479–487. doi: 10.1002/j.1460-2075.1993.tb05680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Dam H, Wilhem D, Herr I, Steffen A, Herrlich P, Angel P. ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J. 1995;14:1798–1811. doi: 10.1002/j.1460-2075.1995.tb07168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, Haimovitz-Friedman A, Fuks Z, Kolesnick RN. Requirement for ceramide-initiated SAPK/JNK signaling in stress-induced apoptosis. Nature. 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 80.Wang JQ, Daunais JB, McGinty JF. NMDA receptors mediate amphetamine-induced upregulation of zif/268 and preprodynorphin mRNA expression in rat striatum. Synapse. 1994;18:343–353. doi: 10.1002/syn.890180410. [DOI] [PubMed] [Google Scholar]
- 81.Xia Z, Dickens M, Raingeaud J, Davis R, Greenberg ME. Opposing effects of ERK and JNK-p38 kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 82.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xing J, Ginty DD, Greenberg ME. Coupling of the Ras-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- 84.Young ST, Porrino LJ, Iadarola MJ. Cocaine induces striatal c-fos-immunoreactive proteins via dopaminergic D1 receptors. Proc Natl Acad Sci USA. 1991;88:1291–1295. doi: 10.1073/pnas.88.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]