

Abstract
Synthesis of GAP-43 (also known as neuromodulin) in neurons is induced during axon growth, and high concentrations (estimated between 50 and 100 μm) accumulate in the growth cone. GAP-43 is tightly associated with the growth cone membrane skeleton, the structure that transduces extracellular guidance cues into alterations in morphology by spatially regulating polymerization of actin filaments, thereby causing directional changes in axon growth. GAP-43 cosediments with actin filaments, and its phosphorylation on serine 41 by PKC, too, is spatially regulated so that phosphorylated GAP-43 is found in areas where growth cones make productive, stable contacts with other cells. In contrast, unphosphorylated GAP-43, which binds calmodulin, is always found in parts of the growth cone that are retracting. Here we have used a cell-free assay to investigate how the phosphorylation status of GAP-43 affects its interactions with actin and show that both phosphorylated and unphosphorylated GAP-43 have different, independent effects on actin filament structure. Phosphorylated GAP-43 stabilizes long actin filaments (Kd = 161 nm), and antibodies to phosphorylated GAP-43 inhibit binding of actin to phalloidin, implying a lateral interaction with filaments. In contrast, unphosphorylated GAP-43 reduces filament length distribution (Kd= 1.2 μm) and increases the critical concentration for polymerization. Prebinding calmodulin potentiates this effect. The results show that spatially regulated post-translational modifications of GAP-43 within the growth cone, which can be regulated in response to extracellular signals, have the ability to directly influence the structure of the actin cytoskeleton.
Keywords: GAP-43, neuromodulin, actin filaments, growth cones, capping proteins, PKC phosphorylation
Growth cone advance in response to extracellular guidance cues is directed by the elaboration and retraction of filopodia and lamellae under the control of regulated actin polymerization (Forscher et al., 1992; reviewed by Tanaka and Sabry, 1995; Mitchison and Cramer, 1996). Very little is known about how guidance cues control actin behavior, but it is clear that important components of the response will be proteins that are able to regulate interactions between the plasma membrane and the actin cytoskeleton under the direction of second messengers, especially Ca2+. One growth cone component that is a candidate to act in this manner is the nervous system-specific protein GAP-43 (also known as neuromodulin, B-50, and F1), the synthesis of which is highly induced in vivo in all neurons that are extending axons (Skene, 1989).
During axonogenesis, extremely high levels of GAP-43 (estimated between 50 and 100 μm; Apel and Storm, 1992) are targeted specifically to the growth cone where it associates tightly with the cortical membrane skeleton, the structure responsible for modulating interactions between plasma membrane and actin cytoskeleton (Meiri and Gordon-Weeks, 1990). GAP-43 is the major growth cone substrate of protein kinase C (PKC), which phosphorylates it on a single site, serine 41 (Coggins and Zwiers, 1989; Apel et al. 1991). In vivo, PKC phosphorylation of GAP-43 is restricted to the distal axon and growth cone where it can be stimulated by target-derived chemotropic factors as well as membrane-associated molecules such as neuronal cell adhesion molecule (NCAM) and L1 (Meiri et al., 1991; K. Meiri, J. Saffell, P. Doherty, and F. Walsh, unpublished results). Within growth cones, phosphorylated GAP-43 is found in stable areas having increased attachment to the substrate (Dent and Meiri, 1992). In contrast, unphosphorylated GAP-43 binds calmodulin via an IQ motif (CaM, Alexander et al. 1987; Chapman et al. 1991; Cheney and Mooseker, 1992) and is always found in areas of growth cone retraction (Dent and Meiri, 1992). Moreover, phosphorylation of serine 41 completely inhibits CaM binding. The consequences of this molecular switch are not understood. On the one hand, it has been suggested that GAP-43 phosphorylation primarily modulates CaM availability at the growth cone plasma membrane (Apel and Storm, 1992). On the other, our study and other studies suggest that the phosphorylation status of GAP-43 may modulate its interactions with structural components of the growth cone directly (Aigner et al., 1995; Meiri et al., 1996).
The accumulation of f-actin at sites of productive contact between growth cones and other cells in culture is an important prerequisite for changes in growth cone direction (Lin and Forscher, 1993; reviewed by Bentley and O’Connor, 1994; Tanaka and Sabry, 1995), and we have shown increased GAP-43 phosphorylation at such sites too (Meiri et al., 1991; Dent and Meiri, 1992). GAP-43 cosediments with actin filamentsin vitro (Strittmatter et al., 1992; Hens et al., 1993), and f-actin levels in growth cones were decreased when GAP-43 was depleted with antisense oligonucleotides, suggesting that the interaction is functionally significant (Aigner and Caroni, 1994). Together, these results led us to hypothesize that PKC phosphorylation may regulate the interaction between GAP-43 and actin, and to test this hypothesis, we have used a cell-free system to show that both phosphorylated and unphosphorylated GAP-43 interact with actin filaments independently. We show here that the interactions have different but biologically relevant affinities and result in distinct effects on filament structure, providing a means whereby extracellular guidance cues may regulate the growth cone cytoskeleton.
MATERIALS AND METHODS
Materials. Protein kinase C was obtained from Upstate Biotechnology (Lake Placid, NY); calmodulin was from Calbiochem (San Diego, CA); and calf intestinal alkaline phosphatase was from Boehringer Mannheim (Indianapolis, IN). Sephadex G-150 was from Pharmacia (Piscataway NJ). 125I-Anti-IgG antibodies were purchased from Amersham (Arlington Heights, IL). Rhodamine phalloidin and N-(1-pyrenyl)-iodoacetamide were from Molecular Probes (Eugene, OR). The monoclonal antibodies (mAbs) 2G12/C7 and 7B10/C4, used to detect GAP-43 immunoreactivity, have been described previously (Meiri et al., 1991). All other reagents were of the highest quality and were obtained from Sigma (St. Louis, MO).
Preparation of GAP-43 and use of antibodies. GAP-43 was freshly purified from fresh or frozen neonatal rat brain using reverse-phase HPLC, as described previously (Meiri et al., 1991), and was stored at 4°C in 10 mm Tris, pH 7.6. Dephosphorylation using 0.3 U/μl alkaline phosphatase was performed for 1 hr at 37°C and was confirmed by loss of immunoreactivity with the 2G12 mAb. Stoichiometric rephosphorylation by PKC was performed in 50 mm Tris, pH 7.5, containing 100 μmCaCl2, 2 mm DTT, and 20 μg/ml phosphatidylserine. Phosphorylation was started by the addition of 200 μm [γ-32P]ATP (specific activity, 5 Ci/mmol) and allowed to proceed for 30 min at 30°C. Using these conditions, the stoichiometry of phosphorylation was usually 0.7–0.9. (Meiri et al., 1991). In experiments in which enzymatically phosphorylated or dephosphorylated GAP-43 were used, appropriate controls were included to show that neither of the enzymes themselves affected actin polymerization. In some experiments, unphosphorylated GAP-43 was prebound to CaM before use. In these instances, unphosphorylated GAP-43 and CaM were incubated for 2 hr at 4°C at a molar ratio of 1:2 with agitation. Before use any unbound CaM was removed by centrifugation through a Centricon filter (Amicon) according to the manufacturer’s directions; the GAP-43 content in the complex retained by the filter was determined by SDS-PAGE by comparison with standard curves. In these experiments, controls included CaM alone.
Actin purification and derivatization withN-(1-pyrenyl)-iodoacetamide. Skeletal muscle actin was freshly purified from rabbit leg muscles by extraction from an acetone powder essentially using the method of Spudich and Watt (1971), with an additional Sephadex G-150 gel filtration step (McLean-Fletcher and Pollard, 1980). The purified actin was >96% polymerization competent, as judged by its ability to sediment when centrifuged at 132,000 × g for 15 min and its appearance under negative staining electron microscopy (see below). G-actin purified in this way showed a single band when 10 μg of the final preparation was run on a single lane on an SDS-PAGE gel and stained with Coomassie blue. G-Actin was stored at 4°C in 10 mm Tris, pH 7.6, and its polymerization competence was quantitated before each experiment. Actin was used within 1 month of preparation.
Pyrene actin was prepared using the procedure of Cooper et al., (1983) as follows. Briefly, polymerized actin was dialyzed against buffer P (1 mm NaHCO3, pH 7.6, 0.1 mmCaCl2, and 0.2 mm ATP) for 48 hr and clarified by centrifugation. The supernatant was diluted to 1 mg/ml with buffer P made 0.1 m in KCl and 1 mm in MgCl2 and incubated at room temperature for 30 min, and then N-(1-pyrenyl)-iodoacetamide dissolved in dimethyl fluoride was added at a molar ratio of 7.5 mol/mol of actin. The mixture was rotated in the dark at room temperature overnight and then dialyzed against buffer A for 48 hr to depolymerize actin filaments. Monomeric pyrene-labeled actin was obtained by chromatographing the supernatant on Sephadex G-150. The concentration of pyrene actin and the labeling ratio was calculated as described by Cooper et al. (1983).
Quantitation of the cosedimentation of GAP-43 and actin filaments. Cosedimentation of GAP-43 and actin filaments used modifications of procedures originally described by Hartwig et al. (1992) and Strittmatter et al. (1992), as follows. The stock solution of g-actin (2–3 mg/ml) was diluted to the appropriate concentration in a polymerization buffer containing (final concentration) 2 mm MgCl2, 100 mmKCl, 0.2 mm DTT, 0.2 mm ATP, and 2 mm Tris, pH 7.6, at 4°C. In some experiments, this buffer also contained 0.2 mm CaCl2, and in some cases, GAP-43 and/or calmodulin was also added. Polymerization was allowed to proceed for 30 min at 24°C, and then the mixture was centrifuged for 15 min in a Beckman airfuge at 30 psi (132,000 ×g).
Aliquots of either pellet or supernatant were run on SDS gels and Western blotted exactly as described previously (Meiri and Beverly, 1994). Blots were incubated with a 125I-conjugated anti-mouse IgG (15 μCi/μg) for 2 hr at room temperature and were then exposed to X-Omat AR film. For each experiment, we first established standard curves that determined the range over which binding of the 125I secondary antibody to pure GAP-43 was linear. This range was similar for both 7B10 and 2G12 (results not shown). Thereafter, exposure of the autoradiographs was always adjusted so that the density of the bands fell within this range, and all blots from any particular experiment were exposed on the same film.
Critical concentration and kinetics of actin polymerization. The critical concentration for actin polymerization was measured from the steady state fluorescence of serially diluted actin as follows. Pyrene actin (5 μm, 5% labeled) was polymerized with or without GAP-43 for 30 min at room temperature. Samples were then diluted in polymerizing buffer (as above) to final concentrations of 0.5, 0.9, and 1 μmwhile keeping the concentration of GAP-43 constant at 300 nm. The samples were allowed to equilibrate overnight at 4°C, and the fluorescence was measured on a Perkin-Elmer MPF-66 fluorescence spectrophotometer, at excitation and emission wavelengths of 365 and 407 nm, respectively. The excitation and emission slits were set at 2.5 and 10 nm, respectively. The polymerization kinetics of f-actin were monitored as described previously (Cooper et al., 1983). Either phosphorylated or unphosphorylated GAP-43 was added to samples containing 1 μm actin, 5% labeled, immediately before polymerization. Fluorescence data were plotted as arbitrary units with respect to the basal fluorescence before polymerization. In these experiments, the data were fit to the equation: [f-actin] =Xo × [1 − cosh(pt)]−2/n to model the kinetics of polymerization using Sigmaplot (see Fesce et al., 1992).
Negative staining electron microscopy. Actin (with or without GAP-43) was polymerized for 40 min at 24°C in polymerization buffer, as above. Aliquots were spotted onto glow-discharged, Formvar-coated 400 mesh grids, fixed for 30 sec in 0.5% glutaraldehyde in polymerization buffer, and then counterstained with 1% uranyl acetate. After drying, grids were viewed with a JEOL 100cx electron microscope at 80 kV.
Immunocytochemistry of dorsal root ganglia cultures.Dissociated cultures of embryonic day 15–18 rat dorsal root ganglion cells were prepared exactly as described previously (Dent and Meiri, 1992). Cultures were fixed for 15 min in 50% Bouins fixative in a solution containing (final concentrations) 145 mm NaCl, 5 mm KCl, 1.2 mm CaCl2, 1.3 mm MgCl2, 1.2 mmNaH2P04, 10 mm glucose, and 20 mm HEPES, pH 7.4. Fixed cells were permeabilized for 20 min in 0.01% digitonin in goat block (10% normal goat serum/4% BSA in PBS) before incubation in primary antibodies. Visualization of specifically bound antibody used fluorescein-conjugated secondary antibodies. In some cases, rhodamine phalloidin (0.33 μm) was added either alone or after incubation with secondary antibody. All cultures were photographed on a Nikon Microphot microscope fitted with B2E and G1A filter sets.
Viscometry. The effect of addition of GAP-43 on the viscosity of actin filaments was measured using falling ball viscometry, essentially as described previously (Pollard, 1982). Briefly, a falling ball viscometer was constructed from a 100 μl glass micropipette, supported at fixed angles between 10 and 50° in a closed Plexiglas chamber equipped with a thermocouple and heater to regulate temperature at 24°C and a stainless steel ball (0.64 mm diameter; density, 7.2 gm/cm2; grade, 10; gauge deviation, 0.000064 mm; a generous gift of Microball Company, Peterborough, NH). The viscometer was calibrated with glycerol/water mixtures of known viscosity. Then, actin (0.5 mg/ml) in polymerization buffer, with or without either phosphorylated or dephosphorylated GAP-43, was allowed to polymerize in the capillary tubes for 30 min at 24°C. In some experiments, calmodulin was preincubated with dephosphorylated GAP-43 before addition to the actin solution. The time taken for the ball to fall between two fixed points was measured, and the specific viscosity was calculated as described previously.
Other procedures. Protein concentrations were determined by the method of Bradford (1976). PAGE was performed according to the method of Laemmli (1970). Scanning absorbance densitometry at 415 nm used a Shimadzu CS-9000 densitometer (Meiri and Burdick, 1991).
RESULTS
Saturation binding kinetics of phosphorylated and unphosphorylated GAP-43 to actin filaments
We first used two specific mAbs to establish the affinity of phosphorylated and unphosphorylated GAP-43 for f-actin, because saturation binding kinetics of GAP-43 for actin have not yet been reported (Strittmatter et al., 1992; Hens et al., 1993). The mAb 7B10 recognizes all post-translationally modified forms of GAP-43, whereas 2G12 specifically recognizes an epitope that includes phosphoserine 41 and, therefore, serves as a reporter for PKC phosphorylation (Meiri et al., 1991). In this study, phosphorylated (phospho)-GAP-43 refers to phosphorylation on serine 41. The selection was made on the basis of two criteria. First, the antibodies bind GAP-43 with an affinity higher than its affinity for actin, and second, they have similar affinities for GAP-43 (Meiri and Beverly, 1994). A further advantage was that 7B10 and 2G12 recognize different epitopes, so that their immunoreactivity in combination is additive, enabling the behavior of unphosphorylated GAP-43 to be calculated from the difference between 7B10 and 2G12 immunoreactivity (Meiri and Burdick, 1991). This was necessary because the purified GAP-43 used in this experiment contained both phosphorylated and unphosphorylated forms. Our previous studies have shown that at this age, approximately 50% of GAP-43 from neonatal rat brain is phosphorylated (Meiri and Burdick, 1991). The advantage of performing the analysis in this way, rather than by enzymatically dephosphorylating GAP-43 to isolate the unphosphorylated form, is that other phosphorylation sites were not affected, thereby allowing us to restrict our attention specifically to the behavior of serine 41, the PKC site. Both GAP-43 and actin have acidic pI values (4.3 and 4.6, respectively), and previous studies have confirmed that their cosedimentation is unlikely attributable to nonspecific electrostatic interactions. Cosedimentation of GAP-43 with f-actin was detected after polymerization of actin in the presence of GAP-43 by specific immunoreactivity in the pellet after centrifugation (Fig.1A,B). The amount of GAP-43 cosedimenting was quantified by comparison with the immunoreactivity obtained when standard curves of known GAP-43 concentrations were run in parallel. The results showed that GAP-43 binding to f-actin was specific and saturable (Fig.1C). The amount of unphosphorylated GAP-43 binding was determined by subtracting the specific binding of phosphorylated GAP-43 (detected with the 2G12 mAb) from the total amount (detected with the 7B10 mAb, which recognizes both forms). Scatchard analysis determined that the Kd of phospho-GAP-43 for f-actin was 160.8 nm, whereas that of the unphosphorylated form was higher, Kd = 1.2 μm (Fig.1D), while the appearance of the Scatchard plot implied that phosphorylated and unphosphorylated GAP-43 interact with different sites. From equilibrium binding, the stoichiometry of the interaction between phospho-GAP-43 and actin filaments was calculated as 1:27, whereas that of dephosphorylated (dephospho)-GAP-43/actin was 1:77. In all of these experiments, GAP-43 was added to g-actin immediately before polymerization was induced; however, the results were similar if it was added after polymerization of f-actin had occurred. Likewise, the presence of Ca2+ had no apparent effects on the binding affinities (not shown).
Fig. 1.
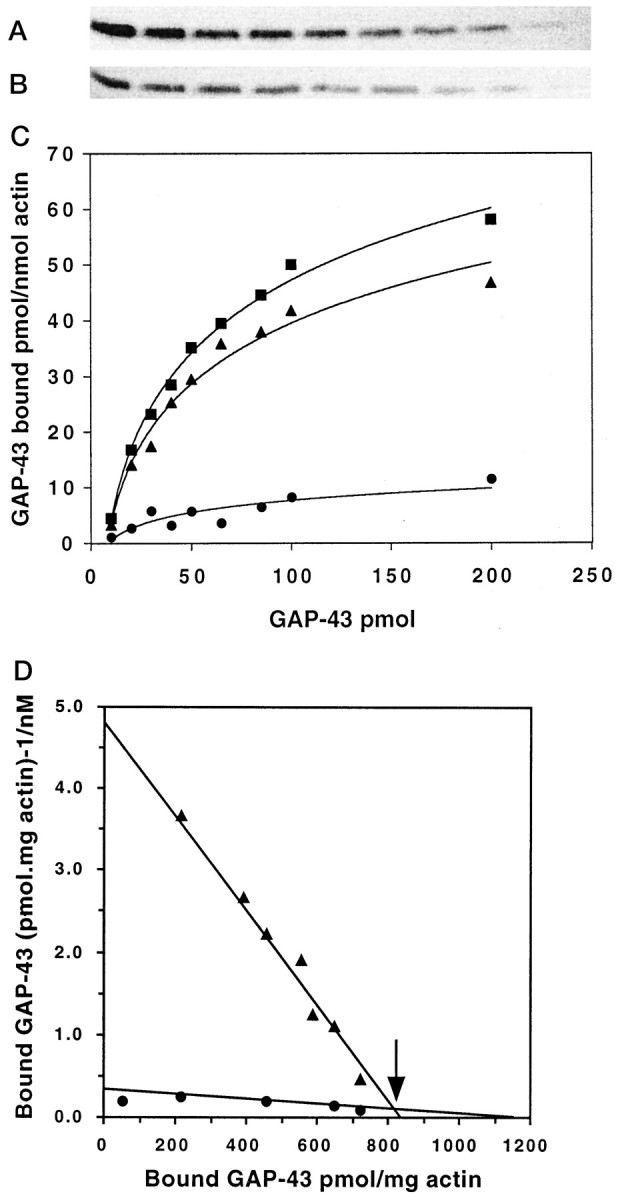
Saturation binding kinetics of GAP-43 to actin filaments. Actin (15 μm) was incubated in polymerizing buffer for 20 min with (left toright) 1000, 850, 650, 500, 400, 300, 200, and 100 nm GAP-43 before centrifugation. The pellet containing f-actin and bound GAP-43 was quantitated by immunoblotting with either 7B10 (A) or 2G12 (B) mAb followed by 125I secondary antibody, autoradiography, and densitometry. C, Equilibrium binding of total (phosphorylated and unphosphorylated) GAP-43 (▪) and phosphorylated GAP-43 (▴) to actin, calculated from data including that presented inA and B (n = 4 independent experiments). The binding of unphosphorylated GAP-43 to actin (•) was calculated from the difference between total GAP-43 and the phosphorylated form (see Results). D, Scatchard analysis of the binding of phosphorylated (▴) and unphosphorylated (•) GAP-43 to actin. The arrow indicates the break point in the Scatchard plot, indicating two independent binding sites of GAP-43 for actin.
Phosphorylated and unphosphorylated GAP-43 have different effects on the critical concentration and kinetics of actin polymerization
The kinetics of actin filament assembly in the presence of GAP-43 were determined from increases in fluorescence during polymerization of pyrene-conjugated actin (Cooper et al., 1983). In these experiments, GAP-43 was enzymatically dephosphorylated with alkaline phosphatase and, in some cases, then rephosphorylated with PKC before use as we have done previously (Meiri et al., 1991) (see Materials and Methods). Enzymatic dephosphorylation did not alter the binding of GAP-43 to actin, determined in the previous experiments, suggesting that other phosphorylation sites do not affect the interaction of the unphosphorylated GAP-43 for actin, which could not have been predicted. To determine the effects of either form of GAP-43 on actin polymerization kinetics, first the critical concentration for actin polymerization itself was calculated by serially diluting pyrene actin in polymerization buffer, using the method described by Benfenati et al. (1992) (Fig. 2A). The critical concentration for actin polymerized alone, 231 ± 10 (SD) nm (n = 5), was not significantly altered by 300 nm phospho-GAP-43 [212 ± 20 (SD) nm (n = 3)]. In contrast, when 300 nm dephospho-GAP-43 was present, it increased to 435 ± 6 (SD) nm (n = 3; p < 0.002 compared with either). The total amount of fluorescence, which indicates the amount of f-actin present, was significantly increased in the presence of phospho-GAP-43 compared with actin, in agreement with subsequent results (see below). These results suggest that the presence of phospho-GAP-43 sequesters a population of actin so that it can no longer participate in the polymerization–depolymerization cycle. In contrast, there was less f-actin present in the presence of dephosphorylated GAP-43, suggesting that this form of GAP-43 may also affect nucleation of filaments.
Fig. 2.
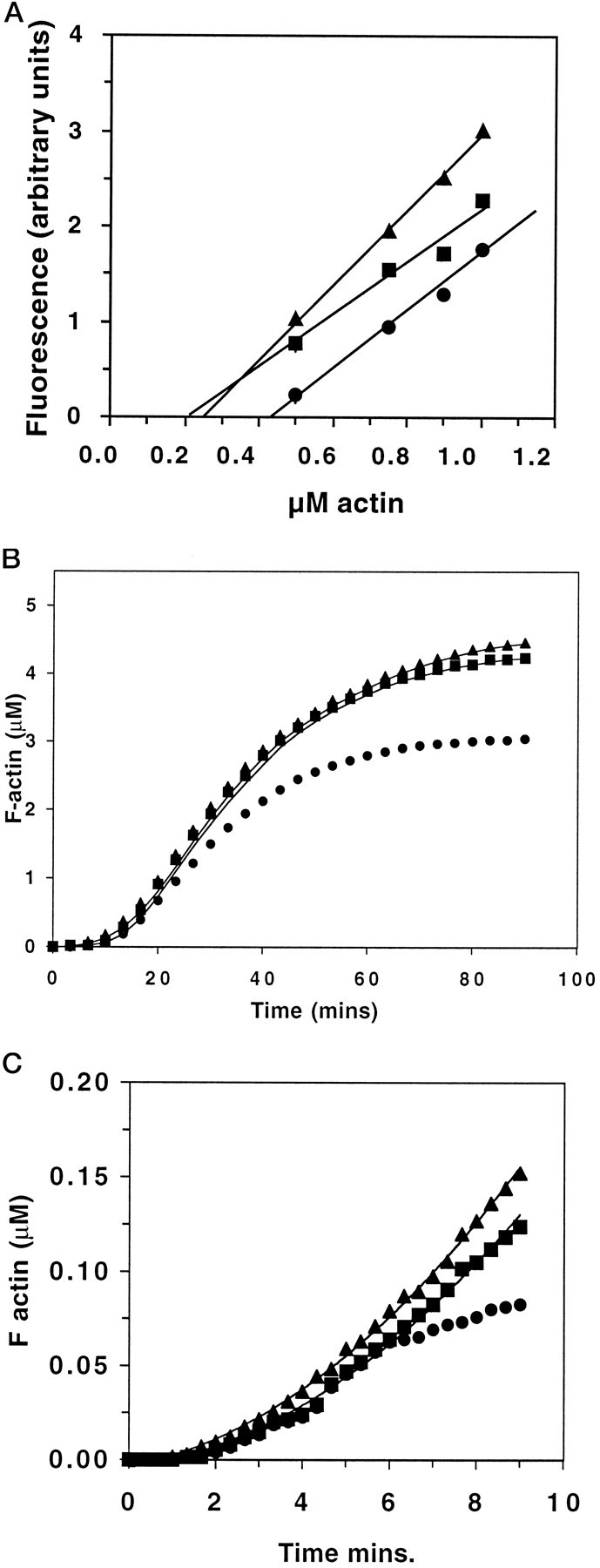
A, Effect of GAP-43 on the critical concentration for actin polymerization. The actin critical concentration was evaluated from the decrease in fluorescence of polymerized pyrenyl-actin (5 μm, 5% labeled) induced by serial dilutions of the samples in polymerization buffer containing 2 mm MgCl2, 100 mm KCl, 15 mm NaCl, and the appropriate concentrations of GAP-43. The decay of f-actin fluorescence as a function of the monomer concentration was fit by least squares linear regression analysis. The intersections between the regression lines and the fluorescence baseline (critical concentration) were: 5 μm actin alone (▪), 231 ± 10 (SD) nm (n = 5); 5 μm actin plus 300 nm phospho-GAP-43 (▴), 212 ± 20 (SD) nm (n = 3); and 5 μm actin plus 300 nm dephospho-GAP-43 (•), 435 ± 6 (SD) nm (n = 3).B, Actin polymer self-assembly in the presence of GAP-43. The self-assembly of pyrenyl-g-actin (5 μm, 5% labeled) was analyzed by measuring the increase in fluorescence related to the g-actin to f-actin transition. Polymerization was triggered at time 0 by the addition of KCl and MgCl2 in the absence (▪) or presence of 0.5 μm phosphorylated GAP-43 (▴) or 0.5 μm dephosphorylated GAP-43 (•).C, Polymerization of actin from f-actin seeds in the presence of GAP-43. The polymerization of pyrenyl-g-actin (1 μm, 5% labeled) in the presence of 0.1 μmf-actin seeds was measured by an increase in fluorescence as before. Polymerization occurred in the absence (▪) or presence of 0.1 μm phosphorylated GAP-43 (▴) or 0.1 μmdephosphorylated GAP-43 (•).
The characteristics of actin polymer self-assembly in the presence of either form of GAP-43 were examined using 5 μm actin (5% pyrene label), and the ratio of actin/GAP-43 was 10:1. The kinetics of polymerization were not significantly affected by the presence of phospho-GAP-43. Furthermore, when the data were fit to an equation that describes both the rate and extent of polymerization (Fesce et al., 1992) (see Materials and Methods), the fit was good (the theoretical curve is given by theline through the points in Fig. 2B). In contrast, the Bmax in the presence of dephospho-GAP-43 was reduced by 30–36% (p < 0.001). Moreover, the theoretical curve could not be fit to the experimental data. To determine whether the poor fit was because of an effect of dephosphorylated GAP-43 on filament nucleation, we substituted numbers other than 4, the experimentally determined nucleation value, into the equation (e.g., see Fesce et al., 1992, and Materials and Methods). However, the fit did not improve, indicating that disruption of filament nucleation does not contribute to the effect of dephospho-GAP-43 on actin filament polymerization.
Finally, the relative initial rates of actin polymerization were determined using a mixture of 1 μm g-actin (5% pyrene labeled) and 0.1 μm f-actin seeds together with similar ratios of GAP-43 as above (Fig. 2C). Under these conditions, neither form of GAP-43 altered the lag time for initiation of polymerization, confirming experimentally that nucleation was not affected. Likewise, the initial rate of actin polymerization (0.339 ± 0.049 (SD) U/min) was only slightly increased in the presence of phospho-GAP-43 to 0.375 ± 0.061 (SD) U/min, whereas with dephospho-GAP-43 present, it decreased slightly to 0.279 ± 0.015 (SD) U/min. Neither difference was significant when compared with the kinetics of actin polymerized alone. At these low concentrations of g-actin and f-actin seeds, the polymerization profile was not yet hyperbolic (Pollard and Cooper, 1986) whether phospho-GAP-43 was present. In contrast, polymerization reached a steady state within 10 min in the presence of dephospho-GAP-43, showing that filament elongation was inhibited, a behavior characteristic of a barbed end-capping protein. Dephospho-GAP-43 had no effect if it was added after polymerization had occurred, demonstrating that it does not sever preformed filaments (not shown).
Direct observation of the effect of GAP-43 on the length of actin filaments
We next used negative stain electron microscopy to examine the appearance of filaments in the presence of either form of GAP-43. Compared with actin polymerized alone (Fig.3A,B), much longer filaments were commonly seen in the presence of phospho-GAP-43 (Fig.3C,D). On the other hand, when actin was copolymerized with dephospho-GAP-43, the most striking feature was the presence of large aggregates (Fig.3E,F) that were not present in the other conditions or when unphosphorylated GAP-43 alone was negatively stained (not shown).
Fig. 3.

Electron microscopy of negatively stained specimens. Actin filaments were polymerized and then negatively stained on electron microscope grids. A, Actin (10 μm); note the smooth contours of the filament bundles. B, Actin (10 μm) at higher power. C, D, Actin (10 μm) polymerized in the presence of 30 μmphosphorylated GAP-43. Note the long filaments commonly seen in this condition but never when unphosphorylated GAP-43 was present. E, F, Actin (10 μm) polymerized in the presence of 30 μmunphosphorylated GAP-43. Note the short filaments andlarge aggregates seen commonly under this condition but only very rarely when actin is polymerized alone or with phosphorylated GAP-43. A, C, E, Actins were from the same experiment. B, D,F, Actins were from independent experiments. Scale bars, 200 nm (A), 100 nm (C), and 500 nm (D–F).
The length distribution of actin filaments in the presence of two different molar ratios of GAP-43 was calculated directly from the EM data. Both forms of GAP-43 significantly affected the mean length of filaments (Fig. 4A). At a stoichiometry of actin/phospho-GAP-43 of 10:1, 79.1 ± 7%, filaments were >1 μm in length (∼3:1 ratio) compared with 70.5 ± 8.5% of actin alone (2.4:1). However, at actin/phospho-GAP-43 of 1:3, the percentage of long filaments significantly increased to 87.2 ± 6.8%, a ratio of 6.8:1 (p < 0.001 compared with either actin or actin/phospho-GAP-43 at the 10:1 ratio). Moreover, under these conditions, there were twice as many filaments longer than 5 μm than seen when actin was polymerized alone (12 ± 3.5% vs 5 ± 3.4%; p < 0.001). For comparison, there were no filaments >4 μm in length at this ratio of dephospho-GAP-43. In the presence of both ratios of dephospho-GAP-43/actin, filaments were significantly shorter than seen with actin alone. At an actin/dephospho-GAP-43 ratio of 10:1, only 53.8 ± 6.9% of filaments were longer than 1 μm (1.1:1), and at an actin/dephospho-GAP-43 ratio of 1:3, 52.7 ± 4.5% were >1 μm in length (1.2:1; p < 0.001 compared with actin alone under both conditions).
Fig. 4.
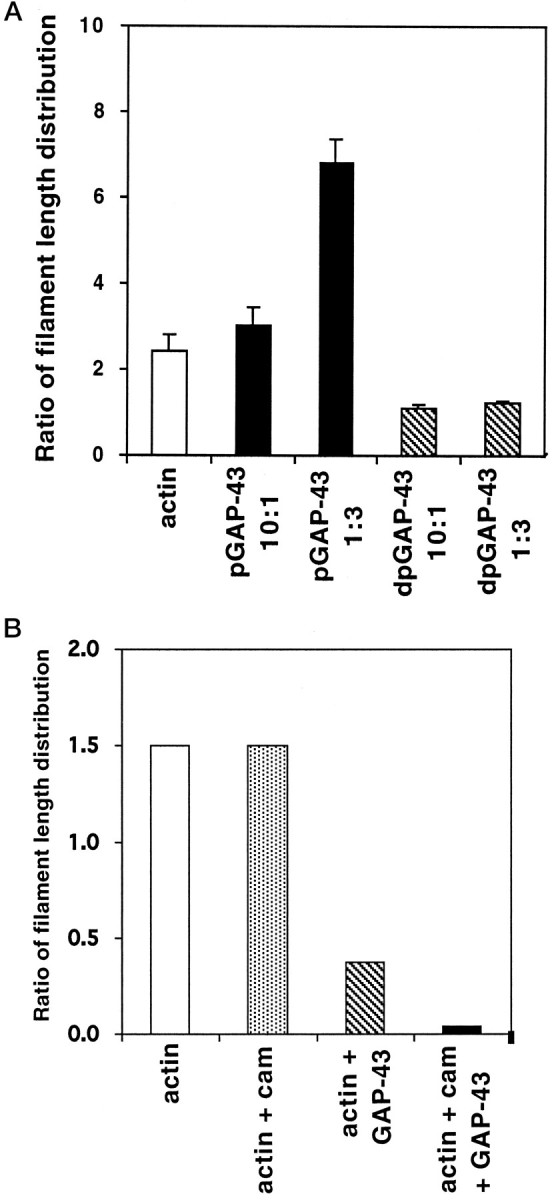
Length distribution of actin filaments in the presence of GAP-43 and CaM. The lengths of actin filaments polymerized in the presence of GAP-43 and negatively stained as above were measured directly from EM grids. A total of at least 400 individual filaments from three independent experiments were measured for each condition. A, The percent ratios of filaments >100 nm were calculated for actin (10 μm; white column), actin polymerized in the presence of phosphorylated GAP-43 at molar ratios of 10:1 and 1:3, respectively (black columns), or dephosphorylated GAP-43 at molar ratios of 10:1 and 1:3, respectively (hatched columns). The mean ± SD is plotted. B, GAP-43 was preincubated with CaM before copolymerization (see Materials and Methods), and the ratios were calculated as before for actin (10 μm; white column), actin polymerized in the presence CaM (gray column), actin polymerized with dephosphorylated GAP-43 at a molar ratio of 1:3 (hatched column), and actin polymerized in the presence of GAP-43 that had been preincubated with CaM at a molar ratio of 1:2 (black column). Two hundred filaments from each condition from a single experiment are depicted.
It was impossible to judge from the electron micrographs whether the addition of GAP-43 potentiated the formation of a filament network, so low shear viscosity was used as an independent means of assessing whether gel formation had occurred (see Materials and Methods). No increase in viscosity was detected in the presence of either form of GAP-43 (negative results not shown), confirming previous results that it does not behave as an actin cross-linking protein (Hens et al., 1993).
Effects of prebinding CaM to GAP-43 on the length of actin filaments
Because GAP-43 unphosphorylated on serine 41 is able to bind CaM, we sought to determine the effect of prebinding CaM on actin filament length. GAP-43 and CaM were first incubated at a molar ratio of 1:2, and unbound CaM was removed by filtration through a Centricon filter (see Materials and Methods) before the complex was allowed to copolymerize with actin, as before. This prebinding of CaM to dephosphorylated GAP-43 before polymerization with actin resulted in further significant decreases in filament lengths; 93% of all actin filaments copolymerized with the CaM/GAP-43 complex were <200 nm, compared with 71% seen with unphosphorylated GAP-43 alone. For comparison, only 40% of all actin filaments were <200 nm when neither CaM/GAP-43 nor GAP-43 was present (Fig. 4B). Again, the presence or absence of Ca2+ did not affect these results, further indicating that CaM is not merely acting as a Ca2+buffer under these circumstances.
Cosedimentation analysis showed that between 94 and 98% of total actin present (4.4–4.8 μm) was polymerization competent in these assays. This was reduced to 84% by the presence of 1 μm unphosphorylated GAP-43 but not by 5 μmCaM alone. However, when both GAP-43 and CaM were present, the total amount of actin pelleting was further reduced to 57% (Fig.5B). Furthermore, under these conditions, unphosphorylated GAP-43 immunoreactivity in the pellet was also reduced by 51% compared with that seen when 1 μm GAP-43 was cosedimented alone (Fig. 5A). These results show that the amount of actin able to polymerize into filaments is also reduced by the presence of GAP-43 that has been prebound to CaM, consistent with the EM data showing an increased incidence of aggregates in the presence of dephospho-GAP-43, and suggest that unphosphorylated GAP-43 may have an effect on g-actin as well.
Fig. 5.

Cosedimentation of f-actin and GAP-43 in the presence of CaM. a, Western blots of pellet formed by cosedimentation of F-actin (5 μm) and 1 μmdephosphorylated GAP-43 (lanes 1–3) or f-actin (5 μm) and 1 μm dephosphorylated GAP-43 that had been preincubated with 2 μm CaM (lanes 4–6). Blots were incubated with 7B10 anti-GAP-43 mAb and visualized with chemiluminescence. The upper band is intact GAP-43, whereas the lower band represents a proteolytic fragment. b, Amount of f-actin appearing in the pellet after cosedimentation in the presence of either GAP-43 or GAP-43 and actin as above. There was significantly less actin and less GAP-43 in the pellet when both GAP-43 and CaM were present.
Phosphorylated GAP-43 colocalizes with actin in primary DRG cultures
We used two methods to examine the localization of GAP-43 and actin in primary neuronal cultures to help us understand the function of GAP-43–actin interactions in growth cones. In initial experiments, DRG cultures were double labeled with the 2G12 mAb, to detect phosphorylated GAP-43, together with rhodamine phalloidin, to label actin filaments. High levels of both anti-phospho-GAP-43 immunoreactivity and rhodamine fluorescence were present (Fig.6A, Hr). The 2G12 immunoreactivity was punctate and was unevenly distributed within growth cones and filopodia, presumably reflecting areas of stable attachment to substrate, as described previously (Dent and Meiri, 1992) (Fig. 6A,D). On the other hand, phalloidin fluorescence, when seen alone, was always most intense at the growth cone margins and in filopodia (Fig.6C,F). Strikingly, in growth cones that were double labeled with both 2G12 and phalloidin, the intensity of phalloidin fluorescence was dramatically attenuated throughout the growth cone (Fig. 6B,E). Comparison with non-neuronal cells in the same cultures that did not react with 2G12 (e.g., Fig. 6E) or with parallel cultures that were incubated with rhodamine phalloidin alone (Fig.6C,F) showed that this was not a nonspecific effect on phalloidin fluorescence because of the presence of the 2G12 antibody.
Fig. 6.

Immunocytochemistry of f-actin and GAP-43 in neuronal cultures. Dorsal root ganglion cultures were plated onto laminin substrates for 24 hr, fixed, and then stained for f-actin with phalloidin and GAP-43 immunoreactivity.A, B,D, E, Double labeling of cultures with the 2G12 antiphosphorylated GAP-43 mAb (A, D) and rhodamine phalloidin (B, E). Growth cones (arrowheads) were heavily reactive with 2G12 and phalloidin. However, the phalloidin staining in the growth cone was attenuated [compare the growth cones with the fibroblast labeled inB (arrow), as well as the parallel cultures in C and F that had been labeled with rhodaminated phalloidin alone]. G,H, High magnification of a growth cone from a DRG culture that had been double labeled with 2G12 (G) and a polyclonal anti-actin antibody (H). Note the colocalization of staining at the lamella (arrowheads). Scale bars, 10 μm (A–F) and 1 μm (G, H).
In other experiments in which actin filaments were identified with an antibody, attenuation of actin immunoreactivity was not seen, presumably because the antibody recognizes a site on f-actin other than that labeled by phalloidin. (This particular actin antibody preferentially labels short actin filaments at growth cone margins and was obtained from Biomedical Technologies, Stoughton MA, but unfortunately it is no longer available.) When it was used to double label growth cones in conjunction with 2G12, a subset of growth cones could be identified in which 2G12 and actin immunoreactivity were clearly colocalized at the growth cone margins. In 86% of all these cases (n = 200), the growth cones had smoothlamellal margins and few filopodia (Fig.6G,H). Given the highly dynamic characteristics of GAP-43 phosphorylation and our previous evidence of the accumulation of phosphorylated GAP-43 in stable areas of growth cones (Dent and Meiri, 1992), the results suggest that the accumulation of these actin filaments at the leading edge may occur subsequent to GAP-43 phosphorylation.
DISCUSSION
GAP-43 levels in growing axons are extremely high (estimated at between 50 and 100 μm), and it is the major substrate of PKC in growth cones, making its phosphorylation in response to extracellular signals a potential mechanism whereby the environment could affect the functional state of the growth cone. Here we have provided evidence that the phosphorylation status of GAP-43 on serine 41, the single PKC phosphorylation site, differentially affects its ability to bind to actin and to modulate the structure and assembly of filaments. Our approach differs in three critical aspects from the study reported by Hens et al. (1993), which also investigated the effects of GAP-43 on actin polymerization. First, by using antibodies that have affinities for GAP-43 that are higher than the affinities of the GAP-43–actin interaction, we were able to measure saturation binding kinetics. Second, by always using actin that was freshly prepared and >90% polymerization competent to perform kinetic experiments, we circumvented variabilities in filament behavior that would have made these data difficult to obtain. Third, by using enzymatic treatment to prepare populations of dephosphorylated and phosphorylated GAP-43, we were able to detect the differences in their effects that would not have been apparent in a mixed population. On the other hand, in experiments using viscosity to measure actin gel formation in the presence of GAP-43, which neither relied on antibodies nor measured polymerization kinetics, both Hens et al. (1993) and ourselves did not detect any cross-linking behavior of GAP-43 to actin filaments.
Effects of GAP-43 on the characteristics of actin filamentsin vitro
The increased fluorescence that occurs during polymerization of pyrene-conjugated actin provided a sensitive assay to measure the effects of GAP-43 on actin polymerization kinetics (Cooper et al., 1983). Because dephospho- and phospho-GAP-43 affected polymerization differently, they will be considered separately.
Three pieces of evidence support the notion that dephospho-GAP-43 may behave as a barbed end-capping protein. First, in the presence of dephospho-GAP-43, the critical concentration for polymerization increased significantly from 200 to 435 nm, closer to that required for polymerization from the pointed end of the filament (600 nm; Pollard and Cooper, 1986; Gaertner et al., 1989). Second, both the self-assembly of actin filaments and the polymerization of low concentrations of monomeric pyrene actin in the presence of f-actin seeds were significantly attenuated when dephospho-GAP-43 was present from the initiation of polymerization, even though nucleation was not affected. Finally, when filaments polymerized in the presence of dephospho-GAP-43 were examined under electron microscopy, >70% were <200 nm long compared with <40% of actin filaments alone. These measurements correlate well with the apparent stoichiometry of 1:77 molecules of actin, calculated from equilibrium binding, which suggests the association of each dephospho-GAP-43 molecule with a filament ∼203 nm long. Dephospho-GAP-43 does not sever preformed actin filaments, thus placing it in the class of f-actin-capping proteins exemplified by capZ, Cap32/34, aginactin, and radixin and not the gelsolin family, which also have filament-severing activity (Carlier, 1991; Weeds and Maciver, 1993; Barkalow and Hartwig, 1995). Dephospho-GAP-43 does not seem to nucleate actin filament polymerization; thus it behaves more like CapZ (Caldwell et al., 1989) than aginactin (Sauterer et al., 1991). Although the pyrene actin studies suggested that dephospho-GAP-43 might be able to sequester g-actin as well as bind to actin filaments, none of these experiments addressed that question directly. A plateau of polymerization such as that which we saw with dephospho-GAP-43 was also detected with the monomer-sequestering protein profilin (Carlier and Pantaloni 1994), and in this case, too, unphosphorylated GAP-43 may be inducing production of ADP–actin. Moreover, copolymerization of actin and dephospho-GAP-43 reduced the amount of actin able to pellet in the cosedimentation assay, suggesting that there may be some interaction with g-actin as well. On the other hand, we failed to detect interaction between GAP-43 and monomeric actin in a yeast two-hybrid system, suggesting that any such interaction is of rather low affinity (Q. He and K. F. Meiri, unpublished results).
The effect of phospho-GAP-43 on actin filament assembly was significantly different from its unphosphorylated counterpart. Phospho-GAP-43 bound to actin with a higher affinity (161 nm compared with 1.2 μm) than the dephosphorylated form but did not affect the critical concentration, self-assembly kinetics, or total extent of actin polymerization in the presence of f-actin seeds. In further contrast with its unphosphorylated counterpart, it bound to filaments with a different stoichiometry (1:27 compared with 1:77), which gave a predicted periodicity of interaction of 63 nm. In this regard, the attenuation of phalloidin labeling of actin filaments seen in the presence of the anti-phospho-GAP-43 antibody 2G12 implies that phospho-GAP-43 may interact with actin around the phalloidin-binding site, which is found at subdomain 1a and which stabilizes interactions between the two strands of the f-actin helix (Vandekerckhove et al., 1985). Finally, EM analysis of filament length distribution in the presence of phospho-GAP-43 showed a significant increase in the average steady state length. Both this EM data and our previous observations of increased levels of phosphorylated GAP-43 in stable filopodia and in areas of the growth cone tightly attached to the substrate (Dent and Meiri, 1992; E. W. Dent and K. F. Meiri, unpublished observations) support the notion that phospho-GAP-43 may be playing a role in the stabilization of actin filament association with the plasma membrane. This mechanism has been proposed as a means to allow forward protrusion of filopodia in a situation in which the net direction of actin polymerization occurs in the cytoplasm (Mitchison and Kirschner, 1988;Lin et al., 1994). Both type I and type II myosins have been suggested as candidates for binding proteins that behave in this way (Mitchison and Kirschner, 1988; Sobue, 1993; Tanaka and Sabry, 1995). Interestingly, both type I and type II myosins also contain the highly conserved IQ motif, which forms the CaM-binding domain and includes the PKC phosphorylation site on GAP-43 (Cheney and Mooseker, 1992).
Direct control of actin polymerization kinetics is an important means of regulating cell shape and motility (Forscher et al., 1992; Theriot and Mitchison, 1992; Tilney et al., 1992; Mitchison and Cramer, 1996). Recent data using Listeria monocytogenes to model the role of actin-binding proteins in lamellal and filopodial extension (Marchand et al., 1995) supports the hypothesis that barbed ends of actin filaments in vivo may normally be capped, thereby reducing the rate of steady state assembly. According to this model, active elongation of filaments occurs by uncapping the barbed end, thereby shifting the critical concentration from 0.6 μm, found at the pointed ends in the cytoplasm, to 0.1 μm, found at uncapped barbed ends adjacent to the membrane. Barbed-end elongation then occurs from the pool of sequestered monomeric actin (Mitchison and Cramer, 1996). It is clear from these studies that a key regulatory component in the process is the so-called “leaky capper,” the interactions of which with the barbed end of the filament are regulatable in response to extracellular signals. Particularly in the growth cone, extreme fluctuations in the rate and extent of elongation suggest that such capping proteins are often rate limiting. Moreover, discrete zones of highly regulated local actin accumulation and filopodial elaboration (Forscher et al., 1992) also imply that spatially segregated nucleators may mediate filament assembly from barbed ends anchored at the plasma membrane. Our results suggest that phosphorylated GAP-43 may be playing such a role.
We have shown that unphosphorylated GAP-43 also has a direct but different role in the regulation of actin polymerization. First, the kinetic and EM evidence is consistent with the notion that unphosphorylated GAP-43 inhibits actin assembly directly, behaving as a “leaky” barbed end capper, thereby attenuating filopodial extension. Unphosphorylated GAP-43 does not completely shift the critical concentration to the pointed end value, probably reflecting its Kd for actin. However, its significance lies in the extremely high concentration of GAP-43 in growth cones (50–100 μm; Apel and Storm, 1992), of which approximately half is unphosphorylated (Meiri and Burdick, 1991) and which exceeds that of actin (estimated at 15 μm; Marchand et al., 1995). The results are in agreement with our observation that areas of the growth cone that are retracting always contain the unphosphorylated form of GAP-43 (Dent and Meiri, 1992) as well as previous findings that loss of f-actin from anterior parts of the growth cone, an early step in growth cone collapse (Fan et al., 1993), occurs when GAP-43 is depleted (Aigner and Caroni, 1994). The results also show that CaM potentiates the effect of unphosphorylated GAP-43 on actin. Although the mechanisms are not yet clear, the ability of Ca2+ to regulate GAP-43–CaM interactions independently of phosphorylation may be significant (Gerendasy et al., 1995). With regard to the role of phosphorylated GAP-43, the EM evidence is consistent with the notion that it may serve as a lateral stabilizer of actin filaments, thereby favoring filopodial elongation in areas where phosphorylation occurs. This is consistent with our previous findings that stable filopodia are enriched in phosphorylated GAP-43 and that “productive” contacts between growth cones and other cells can stimulate phosphorylation of GAP-43 (Dent and Meiri, 1992), together with other evidence that an important preliminary step in directional outgrowth is the accumulation of actin at areas of the growth cone where directional change will be initiated (reviewed by Tanaka and Sabry, 1995). Moreover, the enrichment of phosphorylated GAP-43 in areas of membrane tightly attached to the substrate implies that additional transmembrane components also contribute to the eventual stabilization of filopodia. To our knowledge, this is the first example of how the modification of a single site on a specific growth cone component, GAP-43, can result in distinct effects on regulation of actin filament behavior. This, together with its potential ability to affect CaM availability, underscores the centrality of this molecule to the molecular integration of diverse growth cone responses.
Footnotes
This study was supported by National Institutes of Health Grant NS26091 (K.F.M.). We thank Drs. Tom Pollard, Roger Morris, and James Schwob for their critical comments on this manuscript and Dr. Barry Knox for his help in fitting theoretical equations to the fluorescence data.
Correspondence should be addressed to Karina Meiri, Department of Pharmacology, SUNY Health Science Center, 750 East Adams Street, Syracuse, NY 13210.
REFERENCES
- 1.Aigner L, Caroni P. Depletion of 43 kDa growth-associated protein in primary sensory neurons leads to diminished formation and spreading of growth cones. J Cell Biol. 1994;123:417–429. doi: 10.1083/jcb.123.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aigner L, Arber S, Kapfhammer J, Laux T, Schneider C, Botteri F, Brenner H-R, Caroni P. Overexpression of the neural growth-associated protein GAP-43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell. 1995;83:269–278. doi: 10.1016/0092-8674(95)90168-x. [DOI] [PubMed] [Google Scholar]
- 3.Alexander K, Cimler B, Meier K, Storm D. Regulation of calmodulin binding to p-57, a neurospecific calmodulin binding protein. J Biol Chem. 1987;262:6108–6113. [PubMed] [Google Scholar]
- 4.Apel ED, Storm D. Functional domains of GAP-43 (GAP-43). Perspect Dev Neurobiol. 1992;1:3–11. [PubMed] [Google Scholar]
- 5.Apel ED, Litchfield DW, Clark RH, Krebs EG, Storm DR. Phosphorylation of GAP-43 (GAP-43) by casein kinase II: identification of phosphorylation sites and regulation by calmodulin. J Biol Chem. 1991;266:10544–10551. [PubMed] [Google Scholar]
- 6.Barkalow K, Hartwig JH. Actin cytoskeleton: setting the pace of cell movement. Curr Biol. 1995;5:1000–1003. doi: 10.1016/s0960-9822(95)00200-4. [DOI] [PubMed] [Google Scholar]
- 7.Benfenati F, Valtorta F, Chieregatti E, Greengard P. Interaction of free and synaptic-vesicle-bound synapsin 1 with f-actin. Neuron. 1992;8:377–386. doi: 10.1016/0896-6273(92)90303-u. [DOI] [PubMed] [Google Scholar]
- 8.Bentley D, O’Connor TP. Cytoskeletal events in growth cone steering. Curr Opin Neurobiol. 1994;4:43–48. doi: 10.1016/0959-4388(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 9.Bradford MM. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 10.Caldwell JE, Heiss SG, Mermall V, Cooper JA. Effects of Capz, an actin capping protein of muscle on the polymerization of actin. Biochemistry. 1989;268:288506–288514. doi: 10.1021/bi00447a036. [DOI] [PubMed] [Google Scholar]
- 11.Carlier M-F. Actin: protein structure and filament dynamics. J Biol Chem. 1991;266:1–4. [PubMed] [Google Scholar]
- 12.Carlier M-F, Pantaloni D. Actin assembly in response to extracellular signals: role of capping proteins, thymosin β4, and profilin. 1994. Semin Cell Biol. 1994;5:183–191. doi: 10.1006/scel.1994.1023. [DOI] [PubMed] [Google Scholar]
- 13.Chapman E, Au D, Alexander K, Nicolson T, Storm D. Characterization of the calmodulin binding domain of GAP-43. J Biol Chem. 1991;266:207–213. [PubMed] [Google Scholar]
- 14.Cheney R, Mooseker M. Unconventional myosins. Curr Opin Cell Biol. 1992;4:27–35. doi: 10.1016/0955-0674(92)90055-h. [DOI] [PubMed] [Google Scholar]
- 15.Coggins P, Zwiers H. Evidence for a single protein kinase C-mediated phosphorylation site in rat brain protein B-50. J Neurochem. 1989;53:1895–1901. doi: 10.1111/j.1471-4159.1989.tb09259.x. [DOI] [PubMed] [Google Scholar]
- 16.Cooper JA, Walker SB, Pollard TD. Pyrene actin: documentation of the validity of a sensitive assay for actin polymerization. J Muscle Res Cell Motil. 1983;4:253–262. doi: 10.1007/BF00712034. [DOI] [PubMed] [Google Scholar]
- 17.Dent EW, Meiri KF. GAP-43 phosphorylation is dynamically regulated in individual growth cones. J Neurobiol. 1992;8:1037–1053. doi: 10.1002/neu.480230809. [DOI] [PubMed] [Google Scholar]
- 18.Fan J, Mansfield SG, Redmond T, Gordon-Weeks P, Raper JA. The organization of F-actin and microtubules in growth cones exposed to a brain-derived collapsing factor. J Cell Biol. 1993;121:867–878. doi: 10.1083/jcb.121.4.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fesce R, Benfenati P, Greengard P, Valtorta F. Effects of the neuronal phosphoprotein synapsin 1 on actin polymerization. J Biol Chem. 1992;267:11289–11299. [PubMed] [Google Scholar]
- 20.Forscher P, Lin C-H, Thompson C. Novel form of growth cone motility involving site-directed actin filament assembly. Nature. 1992;357:515–518. doi: 10.1038/357515a0. [DOI] [PubMed] [Google Scholar]
- 21.Gaertner A, Ruhnau K, Schröer E, Selve N, Wangner M, Wegner A. Probing nucleation, cutting, and capping of actin filaments. J Muscle Res Cell Motil. 1989;10:1–9. doi: 10.1007/BF01739852. [DOI] [PubMed] [Google Scholar]
- 22.Gerendasy DD, Herron S, Jennings PA, Sutcliffe JG. Calmodulin stabilizes an amphiphilic α helix within RC3/neurogranin and GAP-43/GAP-43 only when Ca2+ is absent. J Biol Chem. 1995;270:6741–6750. doi: 10.1074/jbc.270.12.6741. [DOI] [PubMed] [Google Scholar]
- 23.Hartwig JH, Thelen M, Rosen A, Janmey PA, Nairn AC, Aderem A. MARCKS is an actin filament binding protein regulated by protein kinase C and calcium-calmodulin. Nature. 1992;356:618–622. doi: 10.1038/356618a0. [DOI] [PubMed] [Google Scholar]
- 24.Hens JH, Benfenati F, Nielander HB, Valtorta F, Gispen WH, De Graan PNE. B-50/GAP-43 binds to actin filaments without affecting actin polymerization and filament organization. J Neurochem. 1993;61:1530–1533. doi: 10.1111/j.1471-4159.1993.tb13649.x. [DOI] [PubMed] [Google Scholar]
- 25.Laemmli UK. Cleavage of structural proteins during the preassembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 26.Lin C-H, Forscher P. Cytoskeletal remodeling during growth cone-target interactions. J Cell Biol. 1993;121:1369–1383. doi: 10.1083/jcb.121.6.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin C-H, Thompson C, Forscher P. Cytoskeletal organization underlying growth cone motility. Curr Opin Neurobiol. 1994;4:640–647. doi: 10.1016/0959-4388(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 28.Marchand J-B, Moreau P, Paoletti A, Cossart P, Carlier M-F, Pantaloni D. Actin-based movement of Listeria Monocytogenes: actin assembly results from the local maintenance of uncapped filament barbed ends at the bacterium surface. J Cell Biol. 1995;130:331–343. doi: 10.1083/jcb.130.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLean-Fletcher SD, Pollard TD. Identification of a factor in conventional muscle actin preparations which inhibits actin filament self-association. Biochem Biophys Res Commun. 1980;96:18–27. doi: 10.1016/0006-291x(80)91175-4. [DOI] [PubMed] [Google Scholar]
- 30.Meiri KF, Beverly MB. The effects of gangliosides GM1 and GD1a on GAP-43 phosphorylation and dephosphorylation in isolated growth cones. J Neurochem. 1994;67:291–299. doi: 10.1046/j.1471-4159.1994.62010291.x. [DOI] [PubMed] [Google Scholar]
- 31.Meiri KF, Burdick DA. Nerve growth factor stimulation of GAP-43 phosphorylation in intact isolated growth cones. J Neurosci. 1991;11:3155–3164. doi: 10.1523/JNEUROSCI.11-10-03155.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meiri KF, Gordon-Weeks PR. GAP-43 in growth cones is associated with areas of membrane that are tightly attached to the substrate and is a component of a membrane skeleton subcellular fraction. J Neurosci. 1990;10:256–266. doi: 10.1523/JNEUROSCI.10-01-00256.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meiri KF, Bickerstaff LE, Schwob JE. Monoclonal antibodies show that GAP-43 phosphorylation by kinase C during axonogenesis is both spatially and temporally regulated in vivo. J Cell Biol. 1991;112:991–1007. doi: 10.1083/jcb.112.5.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meiri KF, Hammang JP, Dent EW, Baetge EE. Mutagenesis of ser41 to ala inhibits the association of GAP-43 with the membrane skeleton of GAP-43-deficient PC12B cells: effects on cell adhesion and the composition of neurite cytoskeleton and membrane. J Neurobiol. 1996;29:213–232. doi: 10.1002/(SICI)1097-4695(199602)29:2<213::AID-NEU7>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 35.Mitchison T, Cramer LP. Actin-based cell motility and locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- 36.Mitchison T, Kirschner M. Cytoskeletal dynamics and nerve growth. Neuron. 1988;1:761–772. doi: 10.1016/0896-6273(88)90124-9. [DOI] [PubMed] [Google Scholar]
- 37.Pollard TD. A falling ball apparatus to measure filament cross-linking. Methods Cell Biol. 1982;24:301–311. doi: 10.1016/s0091-679x(08)60663-9. [DOI] [PubMed] [Google Scholar]
- 38.Pollard TD, Cooper JA. Actin and actin binding proteins: a critical evaluation of mechanisms and functions. Annu Rev Biochem. 1986;55:987–1035. doi: 10.1146/annurev.bi.55.070186.005011. [DOI] [PubMed] [Google Scholar]
- 39.Sauterer A, Eddy RJ, Hall AL, Condeelis JS. Purification and characterization of aginactin, a newly identified agonist-regulated actin-capping protein from Dictyostelium amoebae. J Biol Chem. 1991;266:24553–24539. [PubMed] [Google Scholar]
- 40.Skene JHP. Axonal growth associated proteins. Annu Rev Neurosci. 1989;12:127–156. doi: 10.1146/annurev.ne.12.030189.001015. [DOI] [PubMed] [Google Scholar]
- 41.Sobue K. Actin-based cytoskeleton in growth cone activity. Neurosci Res. 1993;18:91–102. doi: 10.1016/0168-0102(93)90012-f. [DOI] [PubMed] [Google Scholar]
- 42.Spudich JA, Watt S. The regulation of rabbit skeletal muscle contraction: biochemical studies of the integration of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- 43.Strittmatter SM, Vartanian T, Fishman MC. GAP-43 as a plasticity protein in neuronal form and repair. J Neurobiol. 1992;23:507–520. doi: 10.1002/neu.480230506. [DOI] [PubMed] [Google Scholar]
- 44.Tanaka E, Sabry J. Making the connection: cytoskeletal rearrangements during growth cone guidance. Cell. 1995;83:171–176. doi: 10.1016/0092-8674(95)90158-2. [DOI] [PubMed] [Google Scholar]
- 45.Theriot JA, Mitchison T. The nucleation release model of actin filament dynamics in cell motility. Trends Cell Biol. 1992;2:219–221. doi: 10.1016/0962-8924(92)90298-2. [DOI] [PubMed] [Google Scholar]
- 46.Tilney LG, DeRosier DJ, Weber A, Tilney MS. How Listeria exploits host cell actin to form its own: nucleation, actin filament assembly, and evidence of a pointed end capper. J Cell Biol. 1992;18:83–93. doi: 10.1083/jcb.118.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vandekerckhove J, Deboben A, Nassal M, Wieland T. The phalloidin binding site of f actin. EMBO J. 1985;4:2815–2818. doi: 10.1002/j.1460-2075.1985.tb04008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weeds A, Maciver S. F-actin capping proteins. Curr Opin Cell Biol. 1993;5:63–69. doi: 10.1016/s0955-0674(05)80009-2. [DOI] [PubMed] [Google Scholar]