

Abstract
Two major C-terminal variants ending at Val40 and Ala42 constitute the majority of amyloid β-protein (Aβ), which undergoes postsecretory aggregation and deposition in the Alzheimer disease (AD) brain. To probe the differential pathobiology of the two Aβ variants, we used an in vivo paradigm in which freshly solubilized Aβ1–40 or Aβ1–42 was injected into rat brains, followed by examination using Congo red birefringence, Aβ immunohistochemistry, and electron microscopy. In the rat brain, soluble Aβ 1–40 and Aβ1–42 formed aggregates, and the Aβ1–40 but not the Aβ1–42 aggregates showed Congo red birefringence. Electron microscopy revealed that the Aβ1–40 aggregates contained fibrillar structures similar to the amyloid fibrils of AD, whereas the Aβ1–42 aggregates contained nonfibrillar amorphous material. Preincubation of Aβ1–42 solutionin vitro led to the formation of birefringent aggregates, and after injection of the preincubated Aβ1–42, the aggregates remained birefringent in the rat brain. Thus, a factor or factors might exist in the rat brain that inhibit the fibrillar assembly of soluble Aβ1–42. To analyze the postsecretory processing of Aβ, we used the same in vivo paradigm and showed that Aβ1–40 and Aβ1–42 were processed at their N termini to yield variants starting at pyroglutamate, and at their C termini to yield variants ending at Val40 and at Val39. Thus the normal rat brain could produce enzymes that mediate the conversion of Aβ 1–40/1–42 into processed variants similar to those in AD. This experimental paradigm may facilitate efforts to elucidate mechanisms of Aβ deposition evolving into amyloid plaques in AD.
Keywords: Alzheimer disease, senile plaques, amyloid β-protein, amyloid fibrils, in vivo, in vitro
Deposition of amyloid β-protein (Aβ) as amyloid fibrils or nonfibrillar amorphous aggregates in senile plaques characterizes the Alzheimer disease (AD) brain. There are two types of senile plaques, i.e., diffuse and dense-cored plaques. The diffuse plaques are composed of nonfibrillar amorphous Aβ aggregates that are not associated with degenerative changes, whereas the cored plaques contain abundant Aβ fibrils that are associated with pathological changes in the surrounding brain parenchyma (for review, see Selkoe, 1994).
Aβ peptides that are composed of 39–43 amino acids derived from the amyloid β-protein precursor (APP) (Kang et al., 1987) are produced as soluble metabolic products of the APP, and they are constitutively secreted into culture medium and cerebrospinal fluids (Haas et al., 1992; Shoji et al., 1992). The majority of the secreted soluble Aβ species includes Aβ1–40 and Aβ1–42, which start at Asp1 and end at either Val40 or Ala42, respectively (Vigo-Pelfrey et al., 1993;Suzuki et al., 1994).
In the AD brain, these soluble Aβ peptides undergo aggregation and are deposited as several variants. The major N-terminal Aβ variants include AβN1, which bears the standard N terminus, and AβN3(pE), which bears an N-terminal pyroglutamate (Mori et al., 1992; Saido et al., 1995, 1996). Deposition of the AβN3(pE) prevails at higher density than the AβN1 and precedes that of the AβN1 in plaques (Saido et al., 1995). The major C-terminal variants include AβC40 and AβC42, which retain C-terminal amino acids identical to those of the secreted Aβ1–40 and Aβ1–42, respectively (Mori et al., 1992;Miller et al., 1993; Roher et al., 1993a,b). In the parenchymal amyloid deposits, AβC42 is deposited in greater density than AβC40 (Iwatsubo et al., 1994, 1995), although soluble Aβ1–40 is more abundantly secreted than soluble Aβ1–42 (Seubert et al., 1992; Dovey et al., 1993; Vigo-Pelfrey et al., 1993; Suzuki et al., 1994). Thus, these distinct Aβ species seem to be metabolized differently and may play different roles in the deposition of Aβ.
Previous in vitro studies have provided important information on the pathobiology of Aβ1–40 and Aβ1–42 and their relevance to the pathological processes in AD, but there has been little effort to extend these studies to the in vivo brain environment of experimental animals. In view of the fact that conditions in vitro and in vivo are quite different, and given the remarkable discrepancies between the apparent toxicity of Aβ peptides in vitro versus in vivo, it is important to undertake in vivo studies of the fate of Aβ peptides. Therefore, we used an in vivoparadigm in which Aβ1–40 or Aβ1–42 was injected into rat brain to dissect their differential pathobiology in AD. Herein we demonstrate that Aβ1–40 and Aβ1–42 differ in their ability to form amyloid fibrils in vivo as well as in the postinjection processing at their C termini. Notably Aβ1–40 but not Aβ1–42 was competent to form amyloid fibrils in vivo. In addition, Aβ1–42 was subjected to rapid C-terminal proteolysis, whereas the C-terminal proteolysis of Aβ1–40 was delayed. Thus, this animal model could be used to understand the fibrillogenesis of and postsecretory processing of Aβ in AD.
MATERIALS AND METHODS
Human analog peptides corresponding to Aβ1–40 and Aβ1–42 were synthesized by solid phase methods on an Applied Biosystems (Foster City, CA) synthesizer Model 430A according to the manufacturer’s procedures. The synthesized peptides were purified by C18 reverse-phase HPLC with the corresponding standards obtained from Bachem (Torrance, CA) for comparison. The synthesized peptide samples co-eluted with the corresponding standards, indicating identity with each other. The Aβ samples were verified further by amino acid sequencing. The Aβ peptides were lyophilized as stock sample and used throughout the following experiments.
The lyophilized samples were freshly solubilized in 0.15 mTris buffer, pH 8.8, filtered (0.22 μm), and adjusted to 10 μg/μl (referred to hereafter as “freshly solubilized” Aβ1–40 and Aβ1–42) immediately before each injection. Five microliters of these Aβ1–40 (10 μg/μl) or Aβ1–42 (10 μg/μl) were injected into the right side of the hippocampus or deep cerebral cortex of female Sprague Dawley rats (200–300 gm; n = 50). After different postinjection survival times, the brains removed from the rats were fixed in 70% ethanol/0.15 m NaCl and embedded in paraffin as reported previously (Shin et al., 1993, 1994). The postinjection survival times included 1 d (n = 5), 1 week (n = 5), 3 weeks (n = 5), 5 weeks (n = 5), and 7 weeks (n = 5) for Aβ1–40 as well as for Aβ1–42. Paraffin sections were examined by Congo red staining and immunohistochemistry using several antibodies that specifically recognize different epitopes in Aβ. The antibodies used here are summarized in Table 1. The anti-C39 was newly produced in a rabbit against a synthetic peptide CMVGGV to which keyhole limpet hemocyanin was conjugated, and was affinity-purified as described previously (Saido et al., 1995). Its specificity was confirmed by dot and Western analyses using Aβ1–39, Aβ1–40, Aβ1–41, Aβ1–42, and Aβ1–43 peptides. Immunostaining of AD sections using the anti-C39 antiserum revealed that AβC39 is present in diffuse and cored plaques of the AD brain. 4G8 was used to recognize authentic Aβ, regardless of its processing at the N or C terminus. Anti-N1D and anti-N3(pE) were used to analyze N-terminal processing of Aβ, and BC05, anti-C42, BA27, anti-C40, and anti-C39 were used to analyze C-terminal processing of Aβ.
Table 1.
Summary of antibodies to Aβ
| Antibody | Specificity | Type | Reference |
|---|---|---|---|
| 4G8 | Aβ17–24 | M | Kim et al., 1990 |
| Anti-N1D | Standard Aβ bearing the first N-terminal residue, AβN1D | P | Saido et al., 1995 |
| Anti-N3 (pE) | Modified Aβ in which the first and second N-terminal residues are deleted and the third Glu is converted to pyroGlu, AβN3 (pE) | P | Saido et al., 1995 |
| BC05 | Aβ ending at the forty-second C-terminal residue, AβC42 | M | Suzuki et al., 1994 |
| Anti-C42 | Aβ ending at the forty-second C-terminal residue, AβC42 | P | Saido et al., 1995 |
| BA27 | Aβ ending at the fortieth C-terminal residue, AβC40 | M | Suzuki et al., 1994 |
| Anti-C40 | Aβ ending at the fortieth C-terminal residue, AβC40 | P | Saido et al., 1995 |
| Anti-C39 | Aβ ending at the thirty-ninth C-terminal residue, AβC39 | P | This study |
M, Monoclonal antibody; P, polyclonal antibody.
For electron microscopic study, the same amounts of the freshly solubilized Aβ1–40 (10 μg/μl) and Aβ1–42 (10 μg/μl) were injected into the right and left sides of the same rat brain (n = 18), respectively. After different postinjection survival times, the rats were perfused with PBS, followed by perfusion with 4% glutaraldehyde/0.1 m cachodylate buffer, pH 7.4, as described (Shin et al., 1993). The postinjection survival times included 1 week (n = 8), 2 weeks (n = 5), and 3 weeks (n = 5). The brains were removed, cut coronally, and dissected into small blocks that contained the injection sites. The blocks were further fixed in 1% osmium tetroxide and then dehydrated through a graded series of ethanol, immersed in propylene oxide, and embedded in Epoxy resin. One micrometer semithin sections were cut and stained with toluidine blue. Selected areas were thin-sectioned with a diamond knife, stained with uranyl acetate and lead citrate, and examined with a Hitachi H-7000 electron microscope at 75 kV as reported previously (Kondo et al., 1996).
The Aβ peptides were tested to determine whether they were competent to assemble into fibrils in vitro under conditions that are known to favor Aβ fibrillogenesis as described previously (Lorenzo and Yankner, 1994), with minor modification. Briefly, 10 mg of the Aβ stock samples was dissolved in double-distilled water, filtered (0.22 μm), and diluted with the same volume of PBS to 350 μm. The same amount of Aβ1–42 was dissolved in double-distilled water, filtered (0.22 μm), and adjusted to 350 μm. The Aβ solutions were incubated at 37°C for 5 d, followed by centrifugation at 30,000 × g for 20 min. The resultant pellet was resuspended in PBS to 10 μg/μl (referred to hereafter as “in vitro preincubated” Aβ1–40 and Aβ1–42). Aliquots of these samples were plated onto poly-l-lysine-coated glass plates, dried, stained with Congo red, and viewed under polarized microscopy. Other aliquots of thein vitro preincubated Aβ1–40 and Aβ1–42 peptides were also used for injection into rat brains (n = 40) and examined as described above, with postinjection survival times of 1 d (n = 5), 1 week (n = 5), 3 weeks (n = 5), and 5 weeks (n = 5) for Aβ1–40 as well as for Aβ1–42. In addition, the Aβ1–40 and Aβ1–42 peptides were freshly solubilized in the same buffer and in the same concentration as those that were used for injection into rat brain to test further the ability of these peptides to undergo fibrillogenesis in vitro. After incubation at 37°C for 5 d, the Aβ samples were plated onto poly-l-lysine-coated glass plates and examined as described above.
RESULTS
The near-serial sections through the injection sites in the rat brains were immunostained with 4G8 (Figs.1A,D, 3A,D,4A,E) to document the location of the injected material. The Aβ1–40 and Aβ1–42 samples that were soluble before injection formed 4G8-positive aggregates in the rat brain at the earliest postinjection survival time (1 d) (Figs. 1D,3A,D, 4A,E). Although these aggregates persisted for a prolonged period, they were cleared from the rat brains by ∼5–7 weeks postinjection. Thus, in the experimental system used here, the temporal profiles of aggregation, persistence, and clearance of the injected Aβ peptides were the same for Aβ1–40 and Aβ1–42, except that the abundance of the Aβ1–40 aggregates was greater than the Aβ1–42 aggregates at the temporal stage between injection and final clearance.
Fig. 1.

Photomicrographs of rat brain sections showing aggregates formed after intracerebral injection of freshly solubilized Aβ1–40 (A, B) and Aβ1–42 (D, E) at 3 weeks (A, B) and 1 d (D, E) postinjection survival times. The sections were probed by immunohistochemistry with 4G8 (A, D) and by Congo red staining (CR) (B, E). The preparations of Aβ1–40 (C) and Aβ1–42 (F) were preincubated in vitro to form fibrils, followed by staining with Congo red. Photomicrographs (B, C, E, F) were taken under illumination with polarized light.
Fig. 3.
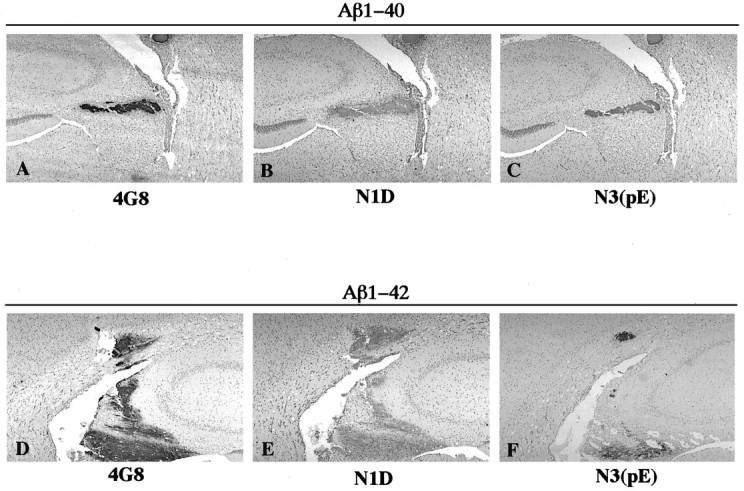
Photomicrographs of rat brain sections showing aggregates formed after injection of freshly solubilized Aβ1–40 (A–C) and Aβ1–42 (D–F) at 1 d postinjection survival time. The sections were immunostained with 4G8 (A, D), anti-N1D (B, E), and anti-N3(pE) (C, F).
Fig. 4.

Photomicrographs of rat brain sections showing aggregates formed after injection of freshly solubilized Aβ1–40 (A–D) and Aβ1–42 (E–H) at 1 d (A–C, E–H) and 3 weeks (D) postinjection survival times. The sections were immunostained with 4G8 (A, E), BA27 (B), anti-C39 (C, D, H), BC05 (F), and anti-C40 (G).
Although it is plausible that endogenous rodent Aβ species contribute to the Aβ aggregates detected here, there were significant differences in the immunohistochemical and histochemical profile of the aggregates induced by injections of Aβ1–40 versus Aβ1–42. Thus, for simplicity, we consider the immunohistochemical and other data described here to be a consequence of the injected human Aβ.
Freshly solubilized Aβ1–40 but not Aβ1–42 is assembled into amyloid fibrils in rat brain
The in vivo paradigm of extracellular injection of freshly solubilized Aβs in rat brain was analyzed for the ability of these peptides to assemble into amyloid fibrils. The rat brain sections including the Aβ aggregates were stained with Congo red. The Aβ1–40 aggregates demonstrated intense birefringence under polarized microscopy (Fig. 1B), indicating the formation of amyloid fibrils by the Aβ1–40 peptides. The birefringence in the Aβ1–40 aggregates appeared at 1 d postinjection survival time, and this birefringence persisted until the aggregates were cleared away by 5–7 weeks postinjection survival times (not shown). In contrast, the aggregates formed by the Aβ1–42 peptides were not birefringent after Congo red staining at any of the postinjection survival times (Fig. 1E), indicating that these peptides did not form amyloid fibrils.
To confirm that the birefringent Aβ aggregates contained amyloid fibrils, we performed electron microscopy on sections of rat brains injected with freshly solubilized Aβ peptides. In the right side of the rat brains injected with Aβ1–40 peptides, 5–10 nm fibrillar structures were observed (Fig.2A,B) that appeared similar to the fibrils seen in amyloid plaques of the AD brain. However, in the left side of the rat brains injected with Aβ1–42 peptides, only amorphous material was found (Fig. 2C,D). Thus, these electron microscopic studies demonstrate that freshly solubilized Aβ1–40, but not Aβ1–42, is assembled into amyloid fibrils after injection into the rat brain, consistent with the results obtained by the examination using Congo red birefringence.
Fig. 2.
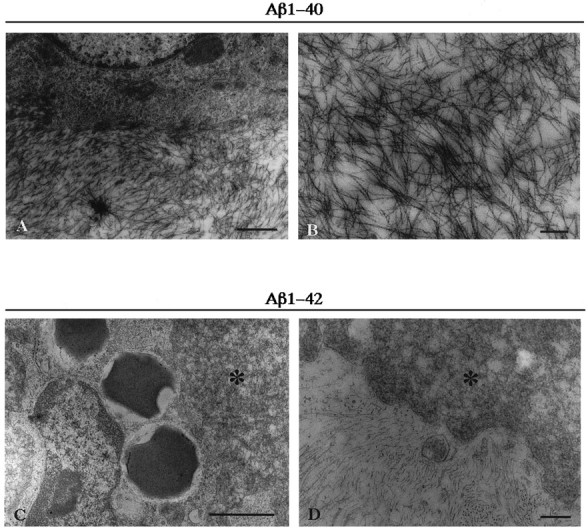
Electron micrographs of rat brain injected with freshly solubilized Aβ1–40 (A, B) and Aβ1–42 (C, D) at 1 week postinjection survival time. Injected Aβ1–40 consists of 5–10 nm fibrils, and injected Aβ1–42 consists of nonfibrillar amorphous material (indicated as *). Note that Aβ1–42 amorphous aggregates are intimately intermingled with macrophages (C) and astrocytes (C, D). Scale bars: A, 1 μm; B, 250 nm; C, 1 μm; D, 250 nm.
Because previous in vitro experiments showed that both Aβ1–40 and Aβ1–42 peptides self-assemble into amyloid fibrils (Fraser et al., 1992; Bush et al., 1994; Lorenzo and Yankner, 1994; Ma et al., 1994), and biophysical experiments indicated that Aβ1–42 forms amyloid fibrils even more readily than Aβ1–40 (Jarret and Lansbury, 1993; Jarret et al., 1993), we examined the ability of each of the species of Aβ peptides to form amyloid fibrils in vitro under two different conditions, as described in Materials and Methods. After incubation under each condition, we observed that both Aβ1–40 and Aβ1–42 peptides formed Congo red-stained birefringent aggregates (Fig. 1C,F), indicating that they assembled into amyloid fibrils. Thus, Aβ1–40 is competent to form amyloid fibrils in vitro and in vivo, whereas Aβ1–42 is competent to form amyloid fibrils in vitro but not in vivo.
To examine the in vivo properties of these Aβ1–40 and Aβ1–42 fibrils generated in vitro, we injected aliquots of these fibrillar Aβ samples into rat brains, and we observed aggregates of these Aβ fibrils that persisted in abundance much more than aggregates of Aβ that formed after injection of freshly solubilized Aβ samples at all postinjection survival times examined (1 d to 5 weeks). The aggregates of in vitro preincubated Aβ1–40 and Aβ1–42 were intensely birefringent after Congo red staining (not shown), and they appeared similar to those produced with injections of freshly solubilized Aβ1–40 (Fig.1B).
In vivo N- and C-terminal processing of freshly solubilized Aβ1–40 and Aβ1–42
To gain insights into the postsecretory processing of Aβ1–40 and Aβ1–42 in the in vivo brain, we next examined whether freshly solubilized Aβs injected into the rat neocortex and hippocampus were modified as were Aβ deposits in amyloid plaques of the AD brain. For this purpose, we performed immunohistochemistry using antibodies that specifically recognize different N-terminal variants of Aβ, including Aβ1N and AβN3(pE), as well as C-terminal variants of Aβ, including AβC42, AβC40, and AβC39, on rat brain sections containing aggregates of the Aβ peptides (see Table1 for a summary of the specificities of these antibodies). The Aβ aggregates were positively immunostained with the anti-N1 as well as with the anti-N3(pE) antibodies as early as 1 d postinjection (Fig.3B,C,E,F). Because aggregates of injected Aβ1–40 and Aβ1–42 were promptly processed at their N termini to yield the AβN3(pE) variant similar to Aβ deposited in amyloid plaques of the AD brain (Mori et al., 1992; Saido et al., 1995, 1996), both Aβ1–40 and Aβ1–42 appear to be equally susceptible to this modification. Furthermore, the Aβ1–42 aggregates were positively immunostained with the BA27, the anti-C40, and the anti-C39 antibodies at the earliest postinjection survival time (1 d) (Fig. 4F–H), suggesting that injected Aβ1–42 peptides underwent rapid C-terminal proteolysis to yield AβC40 and AβC39. In contrast, the aggregates of injected Aβ1–40 were not immunostained with the anti-C39 at 1 d postinjection survival time (Fig. 4C), but were positively immunostained with this antibody at 1–3 weeks postinjection survival times (Fig. 4D). This suggests that injected Aβ1–40 yielded AβC39 after a more attenuated process of C-terminal proteolysis. Thus, Aβ1–40 and Aβ1–42 are processed at their C termini, but this processing occurs more rapidly for Aβ1–42 than for Aβ1–40.
DISCUSSION
In 1987, Kang et al. first reported biochemical evidence that Aβ1–42/43 consists of the neuritic plaques (Kang et al., 1987). In 1993, Jarret and Lansbury suggested the “seeding” hypothesis wherein Aβ1–42 serves as a seed for plaque formation and Aβ1–40 is incorporated later as Aβ progressively deposits in the AD brain (Jarret and Lansbury, 1993; Jarret et al., 1993). Since then, considerable attention has focused on the differential amyloidogenic capabilities of Aβ species with variable C termini, especially the AβC40 and AβC42 variants. Indeed, support for this concept has come from immunohistochemical studies (Iwatsubo et al., 1994, 1995) conducted with antibodies that specifically recognize AβC40 versus AβC42 (Suzuki et al., 1994). For example, in the brains of patients with AD or Down’s syndrome, AβC42 is deposited before AβC40, and AβC42 is the predominant species of Aβ in amyloid plaques at all stages of these diseases. Furthermore, prominent accumulations of AβC42 are seen in the diffuse plaques that are thought to represent an early stage in the formation of amyloid plaques, whereas accumulations of AβC40 are more characteristic of the cored plaques that are believed to form later in the process of amyloidogenesis. Aβ17–42 is a unique proteolytic fragment of Aβ in that it is biochemically extractable from the diffuse plaques but not from the cored plaques (Gowing et al., 1994). Taken together, these observations suggest that AβC40 and AβC42 may play distinct roles in the progressive deposition of Aβ in the AD brain.
The studies described here were designed to gain insight into the differential pathobiology of Aβ1–40 versus Aβ1–42 in the brains of living mammals. To this end, we studied synthetic Aβ1–40 or Aβ1–42 peptides that were injected into the neocortex and hippocampus of rats, and we showed here that these Aβ variants exhibited striking differences in their ability to form amyloid fibrils. Specifically, fibrils were generated consistently from Aβ1–40 but not from Aβ1–42. Although previous in vivostudies (Rush et al., 1992; Snow et al., 1994) reported the formation of amyloid fibrils in the rat brain after injections of Aβ1–40, and other in vivo studies (Waite et al., 1992) of injections of Aβ1–42 failed to produce amyloid fibrils, our study is novel because it directly assessed the differential amyloidogenic capabilities of these two important species of Aβ in vivo, and this has enabled us to reconcile a critical discrepancy in the literature on amyloidogenesis in the AD brain. Indeed, our data are consistent with studies showing that AβC42 dominates in diffuse plaques with few amyloid fibrils (Yamaguchi et al., 1989; Davies and Mann, 1993;Iwatsubo et al., 1994, 1995), whereas AβC40 is most prominent in cored plaques with abundant amyloid fibrils (Iwatsubo et al., 1994,1995). Thus our observations on the preferential contribution of Aβ1–40 to the formation of Aβ fibrils in the rat brain suggest the these findings may reflect authentic molecular events underlying the fibrillogenesis of Aβ in the AD brain. It should be emphasized, however, that these findings do not contradict the putative role of Aβ1–42 in the pathogenesis of AD; this Aβ variant is shown to be the earliest and most abundant species of Aβ deposited in the amyloid-rich senile plaques in AD brains, and the production of more Aβ1–42 has been linked to the onset of familial AD attributable to mutations in the presenilin and APP genes (Hardy, 1997). Therefore, initial deposition of Aβ1–42 is a necessary early pathological process, but it is not sufficient to develop mature amyloid plaques unless succeeded by further deposition of Aβ1–40.
Although Aβ1–42 failed to form fibrils in vivo as we observed in the present experimental animal system, it spontaneously assembled into fibrils after preincubation in vitro, and this is consistent in turn with previous in vitro studies (Lorenzo and Yankner, 1994; Ma et al., 1994; Buscioglio et al., 1995). The mechanism that accounts for this differential assembly of Aβ1–42 into fibrils in vitro but not in vivo is currently unknown, but it is plausible that a factor or factors exist in the rat brain that inhibit the assembly of Aβ1–42 into amyloid fibrils. However, such factors do not induce Aβ1–42 fibrils formedin vitro to disassemble after injection into the rat brain, because these Aβ1–42 fibrils retained their Congo red birefringence for prolonged intervals in vivo. On the basis of the data reported here, we speculate that our model system could be used to identify factors that prevent the assembly of soluble Aβ1–42 into amyloid fibrils in the mammalian brain. For example, cells of the macrophage/microglia lineage and glial cells in the brain could secrete such factors into the extracellular space because these cells accumulate around plaques in the AD brain and appear to intermingle preferably with the Aβ1–42 aggregates in the rat brain (our unpublished results). Lending support for this hypothesis, a recentin vitro study showed that microglia adhere to fibrillar Aβ1–42 via their scavenger receptors, followed by secretion of reactive oxygen species from the microglia, leading to clearance of the fibrillar Aβ1–42 (Khoury et al., 1996). Because in vitroexperiments (Lorenzo et al., 1994) suggest that the neurotoxicity of Aβ is mediated by the fibrillar rather than the amorphous forms of the peptides, and fibril-rich amyloid plaques induce the most reactive changes in the AD brain, the assembly of Aβ into fibrils rather than the mere accumulation of Aβ as amorphous deposits in the extracellular space may be a critical event that leads to the degeneration of neurons in AD. Thus, the model system described here may enable the elucidation of the mechanisms that regulate the acquisition of Aβ neurotoxicity via fibrillogenesis in the AD brain.
In view of current uncertainties about the biological consequences of Aβ deposition in the AD brain, it is important to elucidate the mechanisms that regulate production, aggregation, and proteolysis of Aβ. The model system described here could be used to unravel the molecular basis of the postsecretory processing of Aβ. In the rat brain, injected Aβ1–40 and Aβ1–42 were similarly processed at their N termini to yield AβN3(pE), whereas they were processed differently at their C termini where Aβ1–42 was rapidly processed to yield AβC40 and AβC39, but the proteolysis of Aβ1–40 to yield AβC39 occurred more slowly. Taken together, these data suggest that the normal rat brain secretes enzymes that differentially process Aβ1–40 and Aβ1–42 to yield AβN3(pE), AβC40, and AβC39. Thus, the N- and C-terminal processing of Aβ occurs in the rat brain, and this processing is not a pathological process unique to the AD brain. Additional studies of the processing and fate of human Aβ peptides injected into the rodent brain may clarify the mechanisms responsible for fibrillogenesis and neurodegenerative processes in the AD brain.
Footnotes
This research was supported by Grants-in-Aid from the Japanese Ministry of Education (R.-W.S, T.K.) and by a Grant from the Japan Brain Foundation (R.-W.S, T.K.). This study was conducted in accordance with the Guide for Animal Experimentation, Tohoku University and Tohoku University School of Medicine. We thank Drs. K. S. Kim and H. M. Wisniewski, New York State Institute for Basic Research in Developmental Disabilities, for providing 4G8, and Drs. M. Umemiya, S. Shibuya, and J. Higuchi, and Ms. H. Kudo, Tohoku University, for comments and technical assistance.
Correspondence should be addressed to Ryong-Woon Shin, Department of Neurological Science, Tohoku University School of Medicine, Seiryo-machi 2–1, Sendai 980, Japan.
REFERENCES
- 1.Buscioglio J, Lorenzo A, Yeh J, Yankner BA. β-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995;14:879–888. doi: 10.1016/0896-6273(95)90232-5. [DOI] [PubMed] [Google Scholar]
- 2.Bush AI, Pettingell WH, Multhaup G, Paradis MD, Vonsattel JP, Gusella JF, Beyreuther K, Masters CL, Tanzi RE. Rapid induction of Alzheimer Aβ amyloid formation by zinc. Science. 1994;265:1464–1467. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- 3.Davies CA, Mann DM. Is the “preamyloid” of diffuse plaques in Alzheimer’s disease really nonfibrillar? Am J Pathol. 1993;143:1594–1605. [PMC free article] [PubMed] [Google Scholar]
- 4.Dovey HF, Suomesaari-Chrisler S, Lieberburg I, Sinha S, Kiem PS. Cells with a familial Alzheimer’s disease mutation produce authentic β-peptide. NeuroReport. 1993;4:1039–1042. doi: 10.1097/00001756-199308000-00011. [DOI] [PubMed] [Google Scholar]
- 5.Fraser PE, Nguyen JT, Inouey H, Surewicz WK, Selkoe DJ, Podlisny MB, Kirschner DA. Fibril formation by primate, rodent, and Dutch-hemorrhagic analogues of Alzheimer amyloid β-protein. Biochemistry. 1992;31:10716–10723. doi: 10.1021/bi00159a011. [DOI] [PubMed] [Google Scholar]
- 6.Gowing E, Roher AE, Woods AS, Cotter RJ, Chaney M, Little SP, Ball MJ. Chemical characterization of Ab17–42 peptide, a component of diffuse amyloid deposits of Alzheimer disease. J Biol Chem. 1994;269:10987–10990. [PubMed] [Google Scholar]
- 7.Haas C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, Selkoe DJ. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 8.Hardy J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 9.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43). Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 10.Iwatsubo T, Mann DMA, Odaka A, Suzuki N, Ihara Y. Amyloid β protein (Aβ) deposition: Aβ42 (43) precedes Aβ40 in Down syndrome. Ann Neurol. 1995;37:294–299. doi: 10.1002/ana.410370305. [DOI] [PubMed] [Google Scholar]
- 11.Jarret JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenetic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 12.Jarret JT, Berger EP, Lansbury PT., Jr The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 13.Kang J, Lemaire H-G, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 14.Khoury JEI, Hickman SE, Christian AT, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature. 1996;382:716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- 15.Kim KS, Wen GY, Bancher C, Chen CMJ, Sapienza VJ, Hong H, Wisniewski HM. Detection and quantitation of amyloid β-peptide with two monoclonal antibodies. Neurosci Res Commun. 1990;7:113–122. [Google Scholar]
- 16.Kondo A, Baba S, Iwaki T, Harai H, Koga H, Kimura T, Takamatsu J. Hyperbaric oxygenation prevents delayed neuronal death following transient ischemia in the gerbil hippocampus. Neuropathol Appl Neurobiol. 1996;22:350–360. doi: 10.1111/j.1365-2990.1996.tb01114.x. [DOI] [PubMed] [Google Scholar]
- 17.Lorenzo A, Yankner BA. β-Amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc Natl Acad Sci USA. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma J, Yee A, Brewer HB, Jr, Das S, Potter H. Amyloid-associated proteins α1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature. 1994;372:92–94. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- 19.Miller DL, Papayannoopoulos IA, Styles J, Bobin SA, Lin YY, Biemann K, Iqbal K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch Biochem Biophys. 1993;301:41–52. doi: 10.1006/abbi.1993.1112. [DOI] [PubMed] [Google Scholar]
- 20.Mori H, Takio K, Ogawara M, Selkoe DJ. Mass spectrometry of purified amyloid β protein in Alzheimer’s disease. J Biol Chem. 1992;267:17082–17086. [PubMed] [Google Scholar]
- 21.Roher AE, Lowenson JD, Clarke S, Wolkow C, Wang R, Cotter RJ, Reardon IM, Zurcher-Neely HA, Heinrikson RL, Ball MJ, Greenberg BD. Structural alterations in the peptide backbone of β-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J Biol Chem. 1993a;268:3072–3083. [PubMed] [Google Scholar]
- 22.Roher AE, Lowenson JD, Clarke S, Wood AS, Cotter RJ, Gowing E, Ball MJ. β-amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci USA. 1993b;90:10836–10840. doi: 10.1073/pnas.90.22.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rush DK, Aschmies S, Merriman MC. Intracerebral β-amyloid (25–35) produces tissue damage. Is it neurotoxic? Neurobiol Aging. 1992;13:591–594. doi: 10.1016/0197-4580(92)90061-2. [DOI] [PubMed] [Google Scholar]
- 24.Saido TC, Iwatsubo T, Mann DMA, Shimada H, Ihara Y, Kawashima S. Dominant and differential deposition of distinct β-amyloid peptide species, AβN3(pE), in senile plaques. Neuron. 1995;14:457–466. doi: 10.1016/0896-6273(95)90301-1. [DOI] [PubMed] [Google Scholar]
- 25.Saido TC, Yamao-Harigaya W, Iwatsubo T, Kawashima S. Amino- and carboxyl-terminal heterogeneity of β-amyloid peptides deposited in human brain. Neurosci Lett. 1996;215:173–176. doi: 10.1016/0304-3940(96)12970-0. [DOI] [PubMed] [Google Scholar]
- 26.Selkoe DJ. Normal and abnormal biology of the β-amyloid precursor protein. Annu Rev Neurosci. 1994;17:489–517. doi: 10.1146/annurev.ne.17.030194.002421. [DOI] [PubMed] [Google Scholar]
- 27.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, McCormack R, Wolfert R, Selkoe D, Lieberburg I, Schenk D. Isolation and quantification of soluble Alzheimer’s β-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 28.Shin R-W, Bramblett GT, Lee VM-Y, Trojanowski JQ. Alzheimer disease A68 proteins injected into rat brain induce codeposits of β-amyloid, ubiquitin, and α1-antichymotrypsin. Proc Natl Acad Sci USA. 1993;90:6825–6828. doi: 10.1073/pnas.90.14.6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shin R-W, Lee VM-Y, Trojanowski JQ. Aluminum modifies the properties of Alzheimer’s disease PHFτ proteins in vivo and in vitro. J Neurosci. 1994;14:7221–7233. doi: 10.1523/JNEUROSCI.14-11-07221.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Schaffer LM, Cai X-D, McKay D, Tintner R, Frangione B, Younkin SG. Production of the Alzheimer amyloid β protein by normal proteolytic processing. Science. 1992;258:126–129. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- 31.Snow AD, Sekiguchi R, Nochlin D, Fraser P, Kimata K, Mizutani A, Arai M, Scherier WA, Morgan DG. An important role of heparan sulfate proteoglycan (Perlecan) in a model system for the deposition and persistence of fibrillar Aβ-amyloid in rat brain. Neuron. 1994;12:219–234. doi: 10.1016/0896-6273(94)90165-1. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki N, Cheung TT, Cai X-D, Odaka A, Otvos L, Jr, Eckman C, Golde TE, Younkin SG. An increased percentage of long amyloid β protein secreted by familial amyloid β protein precursor (βAPP717) mutants. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 33.Vigo-Pelfrey C, Lee D, Keim P, Lieberburg I, Schenk DB. Characterization of β-amyloid peptide from human cerebrospinal fluid. J Neurochem. 1993;61:1965–1968. doi: 10.1111/j.1471-4159.1993.tb09841.x. [DOI] [PubMed] [Google Scholar]
- 34.Waite J, Cole GM, Frautschy SA, Conner DJ, Thal LJ. Solvent effects of beta protein toxicity in vivo. Neurobiol Aging. 1992;13:595–600. doi: 10.1016/0197-4580(92)90062-3. [DOI] [PubMed] [Google Scholar]
- 35.Yamaguchi H, Nakazato Y, Hirai S, Shoji M, Harigaya Y. Electron micrograph of diffuse plaques: initial stage of senile plaque formation in the Alzheimer brain. Am J Pathol. 1989;135:593–597. [PMC free article] [PubMed] [Google Scholar]