

Abstract
The enzyme Tpt1 removes the 2′-PO4 at the splice junction generated by fungal tRNA ligase; it does so via a two-step reaction in which (i) the internal RNA 2′-PO4 attacks NAD+ to form an RNA-2′-phospho-ADP-ribosyl intermediate; and (ii) transesterification of the ribose O2″ to the 2′-phosphodiester yields 2′-OH RNA and ADP-ribose-1″,2″-cyclic phosphate products. The role that Tpt1 enzymes play in taxa that have no fungal-type RNA ligase remains obscure. An attractive prospect is that Tpt1 enzymes might catalyze reactions other than internal RNA 2′-PO4 removal, via their unique NAD+-dependent transferase mechanism. This study extends the repertoire of the Tpt1 enzyme family to include the NAD+-dependent conversion of RNA terminal 2′ and 3′ monophosphate ends to 2′-OH and 3′-OH ends, respectively. The salient finding is that different Tpt1 enzymes vary in their capacity and positional specificity for terminal phosphate removal. Clostridium thermocellum and Aeropyrum pernix Tpt1 proteins are active on 2′-PO4 and 3′-PO4 ends, with a 2.4- to 2.6-fold kinetic preference for the 2′-PO4. The accumulation of a terminal 3′-phospho-ADP-ribosylated RNA intermediate during the 3′-phosphotransferase reaction suggests that the geometry of the 3′-p-ADPR adduct is not optimal for the ensuing transesterification step. Chaetomium thermophilum Tpt1 acts specifically on a terminal 2′-PO4 end and not with a 3′-PO4. In contrast, Runella slithyformis Tpt1 and human Tpt1 are ineffective in removing either a 2′-PO4 or 3′-PO4 end.
Keywords: RNA 2'-phosphotransferase, RNA-phospho-ADP-ribose intermediate, nicotinamide adenine dinucleotide, terminal phosphate removal
INTRODUCTION
The enzyme Tpt1 is an essential agent of fungal and plant tRNA splicing that removes the 2′-PO4 at the splice junction generated by fungal and plant tRNA ligases (Culver et al. 1997). Tpt1 catalyzes a unique two-step reaction whereby the internal RNA 2′-PO4 attacks NAD+ to form an RNA-2′-phospho-ADP-ribosyl intermediate that undergoes transesterification to yield 2′-OH RNA and ADP-ribose-1″,2″-cyclic phosphate products (McCraith and Phizicky 1991; Culver et al. 1993; Spinelli et al. 1999; Steiger et al. 2005; Munir et al. 2018a). Tpt1 homologs are distributed widely in archaeal, bacterial, and metazoan taxa (Spinelli et al. 1998). Many of the bacterial species with Tpt1 homologs have no known intron-containing tRNAs and/or no known pathways to generate RNAs with internal 2′-PO4 modifications. In metazoa and archaea, the mechanism of tRNA exon ligation is entirely different from that of fungi and plants and does not result in a junction 2′-PO4 (Popow et al. 2012). Nevertheless, it is clear by the criteria of genetic complementation of yeast tpt1Δ and direct biochemical assay of recombinant Tpt1 proteins that Tpt1 enzymes from widely divergent organisms all possess NAD+-dependent RNA 2′-phosphotransferase activity (Spinelli et al. 1998; Sawaya et al. 2005; Munir et al. 2018a,b).
A longstanding mystery surrounds the role that Tpt1 enzymes might play in taxa that have no fungal/plant-type RNA ligases. One possibility is that bacteria, archaea, or metazoa do have a capacity to install internal RNA 2′-PO4 groups, conceivably during RNA repair pathways that are as yet uncharacterized, and that such RNAs are potential substrates for Tpt1. An alternative scenario is that Tpt1 enzymes can catalyze reactions other than internal RNA 2′-PO4 removal, via their unique NAD+-dependent transferase mechanism.
Support for the latter scheme emerged from the recent findings that a subset of Tpt1 enzymes can transfer ADP-ribose from NAD+ to a 5′-monophosphate end of RNA or DNA to install a 5′-phospho-ADP-ribose cap structure (Munir et al. 2018b). Aeropyrum pernix Tpt1 (ApeTpt1) was particularly adept in this respect. Two other archaeal Tpt1s (from Pyrococcus horikoshii and Archaeoglobus fulgidis), one bacterial Tpt1 (from Clostridium thermocellum) and one fungal Tpt1 (from Chaetomium thermophilum) also exhibited 5′-phospho-ADP-ribose capping activity, albeit less effectively than ApeTpt1. In contrast, human Tpt1 and Runella slithyformis Tpt1 were inactive in 5′-phospho-ADP-ribose capping. The one-step synthesis of a 5′-phospho-ADP-ribosylated cap structure by a subset of Tpt1 enzymes (with no subsequent 5′-phosphotransferase step) extended the repertoire of the Tpt1 family and raised the prospect that Tpt1 enzymes might perform additional phospho-ADP-ribosylation reactions, with or without subsequent removal of the phosphate moiety.
The intrigue around Tpt1 was heightened by the 1.4 Å X-ray structure of Clostridium thermocellum Tpt1 (CthTpt1), produced as a recombinant protein in Escherichia coli and providentially crystallized with ADP-ribose-1″-phosphate in the NAD+ site (Banerjee et al. 2019). To our knowledge, there had been no prior description of ADP-ribose-1″-phosphate as a metabolite in E. coli. The presence of this molecule in the NAD+ site of CthTpt1 crystals is most plausibly explained by its genesis during CthTpt1 expression in vivo, e.g., via CthTpt1-catalyzed ADP-ribosylation of an endogenous bacterial phospho-substrate, followed by phosphoryl transfer to yield ADP-ribose-1″,2″-cyclic phosphate, which was hydrolyzed in situ to ADP-ribose-1″-phosphate. We construed this to be the first evidence that a bacterium has an endogenous substrate with which a Tpt1 enzyme can react. If the endogenous substrate for CthTpt1 is an RNA, it is either the case that (i) E. coli can form an RNA with an internal 2′-PO4 modification, via an as yet undefined pathway; or (ii) CthTpt1 can catalyze removal of an RNA terminal 2′-monophosphate or 3′-monophosphate moiety.
In the present study, we interrogate the reaction of Tpt1 enzymes with 5′ 32P-labeled pRNA2′p and pRNA3′p substrates. We report that CthTpt1 and ApeTpt1 catalyze the NAD+-dependent conversion of 2′-PO4 and 3′-PO4 ends to 2′-OH and 3′-OH ends, respectively, whereas Chaetomium Tpt1 selectively coverts a 2′-PO4 ends to a 2′-OH. In contrast, Runella slithyformis Tpt1 and human Tpt1 are not adept at either terminal phosphate removal reaction.
RESULTS
NAD+-dependent removal of RNA terminal 2′ and 3′ monophosphates by Clostridium thermocellulm Tpt1
Tpt1 proteins from diverse taxa displayed varying abilities to perform the recently discovered nucleic acid 5′-phospho-ADP-ribose capping reaction (Munir et al. 2018b). In order to test if the Tpt1 repertoire might embrace phosphoryl transfer reactions at the opposite end of the polynucleotide, we synthesized a pair of 10-mer RNA oligonucleotides with either a terminal 2′-PO4 or 3′-PO4 moiety. The 10-mers were 5′ 32P-labeled and then gel-purified for use in assaying terminal phosphate removal. Control reactions of the labeled pRNA2′p and pRNA3′p substrates with bacteriophage T4 polynucleotide 2′,3′ phosphatase (Das and Shuman 2013) resulted in quantitative removal of the 2′-PO4 or 3′-PO4 groups to yield 5′ 32P-labeled pRNA2′OH and pRNA3′OH products that migrated slower than the input substrate during denaturing PAGE (Fig. 1A, lanes T4). The instructive findings were that a 30 min reaction of 500 nM CthTpt1 with 200 nM pRNA2′p or pRNA3′p and 1 mM NAD+ resulted in the conversion of the input radiolabeled RNA into species that comigrated with the pRNA2′OH and pRNA3′OH products of the T4 enzyme-catalyzed 2′,3′ dephosphorylation reactions (Fig. 1A). Removal of the 2′-PO4 and 3′-PO4 groups by CthTpt1 depended on NAD+ (Fig. 1A). Additional slowly migrating NAD+-dependent reaction products were generated by CthTpt1, especially in the case of the pRNA3′p substrate (Fig. 1A). These species correspond to terminally phospho-ADP-ribosylated RNAs, as inferred from their susceptibility to post-reaction treatment with alkaline phosphatase (CIP; see below).
FIGURE 1.

NAD+-dependent removal of RNA terminal 2′- and 3′-monophosphates by CthTpt1. (A) Tpt1 reaction mixtures (10 µL) containing 100 mM Tris-HCl, pH 7.5, 0.2 µM (2 pmol) 5′ 32P-labeled 10-mer pRNA2′p, or pRNA3′p substrates (shown at bottom), 1 mM NAD+ (where indicated by +), and 0.5 µM (5 pmol) CthTpt1 (where indicated by +) were incubated at 37°C for 30 min. T4 polynucleotide 2′,3′-phosphatase reaction mixtures (10 µL) containing 100 mM Tris-HCl, pH 7.5, 0.2 µM (2 pmol) 5′ 32P-labeled RNAs, 1 mM MgCl2, and 0.5 µM (5 pmol) T4 Pnkp were incubated at 37°C for 30 min. The products were analyzed by urea-PAGE and visualized by autoradiography. (B) Reaction mixtures (10 µL) containing 100 mM Tris-HCl, pH 7.5, 1 mM NAD+, 0.2 µM (2 pmol) 5′ 32P-labeled 10-mer pRNA2′p, or pRNA3′p substrates, and 0.5 µM (5 pmol) CthTpt1 (where indicated by +) were incubated at 37°C for 30 min. The reaction mixtures were heated at 65°C for 5 min and then either treated for 10 min at 37°C with 10 U of calf intestine alkaline phosphatase (CIP; from NEB) in 1× Cutsmart buffer (50 mM potassium acetate, 20 mM Tris-acetate, pH 7.9, 10 mM magnesium acetate, 100 µg/mL BSA, pH 7.9), or mock-incubated without phosphatase. The reactions were quenched with three volumes of cold 90% formamide, 50 mM EDTA, and the products were analyzed by urea-PAGE and visualized by autoradiography.
As expected, the 32P-labeled pRNA2′p and pRNA3′p substrates were eliminated by CIP, as were the predominant pRNA2′OH and pRNA3′OH products (Fig. 1B). In contrast, the most slowly migrating minor products were CIP-resistant, signifying that the 5′ 32P-labeled phosphate had been capped with ADP-ribose (Munir et al. 2018b). Because the mobility of the 5′-ADPR capped RNAs did not change after CIP treatment, we surmise that the original 2′-PO4 and 3′-PO4 groups were removed during the reaction with CthTpt1. The prominent labeled species in the pRNA3′p reaction that migrated between pRNA3′OH and ADPR-pRNAOH was effaced by CIP, suggesting that it corresponds to an anticipated pRNAp-ADPR intermediate in the terminal monophosphate removal reaction (Supplemental Fig. S1).
The temporal profile of the CthTpt1 reaction with the 5′ 32P-labeled pRNA2′p and pRNA3′p substrates is shown in Figure 2A,B. The kinetics of RNA terminal phosphate removal were quantified by plotting the sum of the pRNAOH and ADPR-pRNAOH species (expressed as the percent of total labeled RNA) as a function of reaction time (Fig. 2, OH). The kinetics of 5′ capping were evident as the sum of the ADPR-pRNAOH and ADPR-pRNAp species plotted as a function of time (Fig. 2, 5′-ADPR). Also quantified was the pRNAp-ADPR intermediate. Initial rates were determined by linear regression in Prism. The key findings were as follows. First, the rate of 2′-PO4 removal by CthTpt1 (2.94 ± 0.05 percent • min−1) was 2.4-fold faster than the rate of 3′-PO4 removal (1.24 ± 0.02 percent • min−1). Second, the pRNAp-ADPR intermediate accumulated to a significant extent during 3′-PO4 removal (up to 16% of total labeled RNA) and prior to the formation of the 3′-OH end product, but not during 2′-PO4 removal (1.6% of total RNA). This result suggests that transesterification of the ADPR ribose-O2″ to the 3′-PO4 of the pRNAp-ADPR intermediate was uniquely rate-limiting for 3′-PO4 removal. Third, the 2′ and 3′ terminal phosphate removal reactions were much more vigorous than the 5′-phospho-ADPR capping reaction, insofar as the extents of 2′-PO4 and 3′-PO4 removal after 45 min were 40-fold and 24-fold greater than the extent of 5′-capping, respectively.
FIGURE 2.

Temporal profile of RNA terminal 2′-PO4 and 3′-PO4 removal by CthTpt1. Reaction mixtures (100 µL) containing 100 mM Tris-HCl, pH 7.5, 1 mM NAD+, 0.2 µM (20 pmol) 5′ 32P-labeled 10-mer pRNA2′p, or pRNA3′p substrates (shown at bottom), and 0.5 µM (50 pmol) CthTpt1 were incubated at 37°C. Aliquots (10 µL) were withdrawn at the times specified and quenched immediately with three volumes of cold 90% formamide, 50 mM EDTA. The products were analyzed by urea-PAGE and visualized by autoradiography (A,B, left panels). The identities of the radiolabeled RNAs are indicated on the right. The kinetics of terminal phosphate removal were quantified in the right panels by plotting the sum of the pRNAOH and ADPR-pRNAOH species (expressed as the percent of total labeled RNA) as a function of reaction time (OH). The kinetics of 5′ capping were evident as the sum of the ADPR-pRNAOH and ADPR-pRNAp species plotted as a function of time (5′-ADPR). Also quantified was the pRNAp-ADPR intermediate. Each datum in the graphs is the average of three independent time course experiments ±SEM.
The dependence of product formation during a 30 min reaction on the amount of input CthTpt1 is shown in Figure 3. The specific activities for terminal phosphate removal, calculated from the slope of the titration curves at subsaturating enzyme, were 0.19 pmol/pmol for pRNA2′p and 0.10 pmol/pmol for pRNA3′p. Only the pRNA3′p reaction led to the accumulation of a pRNAp-ADPR intermediate at subsaturating enzyme levels (Fig. 3). Thus, CthTpt1 is acting in a stoichiometric fashion as a terminal phosphatase with the 10-mer substrates and is approximately twofold more effective at a terminal 2′-PO4 versus a terminal 3′-PO4. The specific activity of CthTpt1 in 5′-phospho-ADPR capping (0.0024 pmol/pmol for pRNA2′p and 0.0021 pmol/pmol for pRNA3′p) was 80-fold less than its activity in terminal 2′-PO4 removal. In contrast, titration of CthTp1 for removal of an internal 2′-PO4, using a 5′ 32P-labeled 6-mer RNA substrate pCCAA2′pAUOH that mimics a tRNA splice junction (Munir et al. 2018a), indicated that it acted catalytically, generating 150 fmol of 2′-OH RNA product per fmol of input CthTpt1 (data not shown).
FIGURE 3.
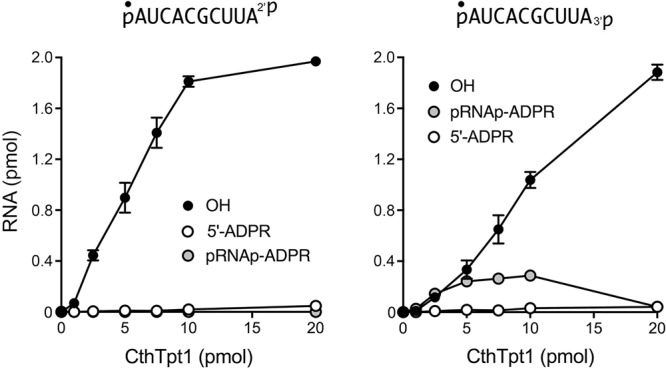
Dependence of 2′-PO4 and 3′-PO4 removal on CthTpt1 concentration. Tpt1 reaction mixtures (10 µL) containing 100 mM Tris-HCl, pH 7.5, 0.2 µM (2 pmol) 5′ 32P-labeled 10-mer pRNA2′p, or pRNA3′p substrates (shown at top), 1 mM NAD+, and CthTpt1 as specified were incubated at 37°C for 30 min. The products were analyzed by urea-PAGE. The extents of product formation, quantified as described in Figure 2, are plotted as a function of input enzyme. Each datum is the average of three independent titration experiments ±SEM.
RNA terminal 2′ and 3′ phosphatase activity of Aeropyrum pernix Tpt1
Reaction of 500 nM ApeTpt1 with 200 nM pRNA2′p or pRNA3′p and 1 mM NAD+ resulted in the conversion of the input radiolabeled RNA into two major products: pRNAOH and ADPR-pRNAOH (Fig. 4A). Product formation depended on NAD+. The kinetic profile of the ApeTpt1 reaction with the 5′ 32P-labeled pRNA2′p and pRNA3′p substrates is shown in Figure 5A,B and quantified as described above for CthTpt1. The identity of the labeled species formed at early times in the pRNA3′p reaction was verified by their sensitivity to CIP (see Fig. 4B), specifically insofar as the species designated as ADPR-pRNAp (which increased steadily at 1, 2, and 5 min) was converted by CIP into the more slowly migrating ADPR-pRNAOH capped RNA. As noted previously (Munir et al. 2018b), ApeTpt1 was more adept than CthTpt1 at 5′-phospho-ADPR capping (compare Figs. 2, 5). Nonetheless, the rate of 2′-PO4 removal by ApeTpt1 (12.8 ± 0.6 percent • min−1) was 6.7-fold faster than the rate of 5′ capping of the pRNA2′p substrate (1.91 ± 0.03 percent • min−1) (Fig. 5A). Similarly, the rate of 3′-PO4 removal by ApeTpt1 (5.01 ± 0.18 percent • min−1) was 2.8-fold faster than the rate of 5′ capping of the pRNA3′p substrate (1.82 ± 0.03 percent • min−1) (Fig. 5B). These data highlighted that ApeTpt1 was 2.6-fold faster at 2′-PO4 removal than 3′-PO4 removal.
FIGURE 4.

NAD+-dependent RNA terminal 2′-PO4 and 3′-PO4 removal by ApeTpt1. (A) Tpt1 reaction mixtures (10 µL) containing 100 mM Tris-HCl, pH 7.5, 0.2 µM (2 pmol) 5′ 32P-labeled 10-mer pRNA2′p, or pRNA3′p substrates (shown at bottom), 1 mM NAD+ (where indicated by +), and 0.5 µM (5 pmol) ApeTpt1 (where indicated by +) were incubated at 37°C for 30 min. T4 polynucleotide 2′,3′-phosphatase reactions were performed as described in Figure 1. The products were analyzed by urea-PAGE and visualized by autoradiography. The identities of the labeled RNAs are indicated on the right. (B) A reaction mixture (100 µL) containing 100 mM Tris-HCl, pH 7.5, 1 mM NAD+, 0.2 µM (20 pmol) 5′ 32P-labeled 10-mer pRNA3′p substrate (shown at bottom), and 0.5 µM (50 pmol) ApeTpt1 was incubated at 37°C. Aliquots (10 µL) were withdrawn at the times specified and quenched immediately with three volumes of cold 90% formamide, 50 mM EDTA. Duplicate aliquots were withdrawn and heated at 65°C for 5 min and then treated for 10 min at 37°C with CIP, as described in Figure 1. The products were analyzed by urea-PAGE and visualized by autoradiography. The identities of the labeled RNAs are indicated on the left. The conversion of ADPR-pRNAp to ADPR-pRNAOH by CIP is indicated on the right.
FIGURE 5.

Temporal profile of RNA terminal 2′-PO4 and 3′-PO4 removal by ApeTpt1. Reaction mixtures (100 µL) containing 100 mM Tris-HCl, pH 7.5, 1 mM NAD+, 0.2 µM (20 pmol) 5′ 32P-labeled 10-mer pRNA2′p, or pRNA3′p substrates (shown at bottom), and 0.5 µM (50 pmol) ApeTpt1 were incubated at 37°C. Aliquots (10 µL) were withdrawn at the times specified and quenched immediately with three volumes of cold 90% formamide, 50 mM EDTA. The products were analyzed by urea-PAGE and visualized by autoradiography (A,B, left panels). The identities of the radiolabeled RNAs are indicated. The temporal profiles of terminal phosphate removal, 5′ capping, and appearance of the pRNAp-ADPR intermediate were plotted in the graphs at right as described in Figure 2. Each datum in the graphs is the average of three independent time course experiments ±SEM.
The specific activities of ApeTpt1 for terminal phosphate removal, calculated from the slope of the titration curves at subsaturating enzyme, were 0.52 pmol/pmol for pRNA2′p and 0.23 pmol/pmol for pRNA3′p (not shown). The specific activity of ApeTpt1 in 5′-phospho-ADPR capping was 0.12 pmol/pmol for pRNA2′p and pRNA3′p. The specific activity of ApeTpt1 in removing an internal 2′-PO4, from the 6-mer RNA pCCAA2′pAUOH was 15 fmol/fmol (not shown). These results indicate ApeTpt1 is 10-fold less effective than CthTpt1 in the canonical internal 2′-phosphotransferase reaction but twofold more effective than CthTpt1 as a terminal 2′-phosphotransferase.
Chaetomium thermophilum is selective for NAD+-dependent removal of a terminal 2′-PO4
Reaction of 500 nM Chaetomium thermophilum Tpt1 (ChaetTpt1) with 200 nM pRNA2′p and 1 mM NAD+ resulted in 2′-PO4 removal to form a predominant pRNAOH product and a 5′ capped species ADPR-pRNAOH (Fig. 6A). In contrast, during a parallel reaction with the pRNA3′p substrate, ChaetTpt1 failed to remove the 3′-PO4, and instead generated a capped product ADPR-pRNAp that retained the original 3′-PO4. Treatment with CIP affirmed the product assignments, particularly the CIP-dependent conversion of ADPR-pRNA3′p to ADPR-pRNAOH (Fig. 6B). The kinetic profile of the ChaetTpt1 reaction with pRNA2′p is shown in Figure 6C and quantified in Figure 6D. The rate of 2′-PO4 removal (9.5 ± 0.5 percent • min−1) was 6.3-fold faster than the rate of 5′ capping (1.5 ± 0.04 percent • min−1). These results show that ChaetTpt1 is highly selective in its ability to ADP-ribosylate and remove a terminal RNA 2′-PO4 but not a 3′-PO4.
FIGURE 6.

NAD+-dependent RNA terminal 2′-PO4 removal by Chaetomium Tpt1. (A) Tpt1 reaction mixtures (10 µL) containing 100 mM Tris-HCl, pH 7.5, 0.2 µM (2 pmol) 5′ 32P-labeled 10-mer pRNA2′p, or pRNA3′p substrates (shown at bottom), 1 mM NAD+ (where indicated by +), and 0.5 µM (5 pmol) ChaetTpt1 (where indicated by +) were incubated at 37°C for 30 min. T4 polynucleotide 2′,3′-phosphatase reactions were performed as described in Figure 1. The products were analyzed by urea-PAGE and visualized by autoradiography. The identities of the radiolabeled RNAs are indicated on the left. (B) Reaction mixtures (10 µL) containing 100 mM Tris-HCl, pH 7.5, 1 mM NAD+, 0.2 µM (2 pmol) 5′ 32P-labeled 10-mer pRNA2′p, or pRNA3′p substrates, and 0.5 µM (5 pmol) ChaetTpt1 (where indicated by +) were incubated at 37°C for 30 min. The reaction mixtures were heated at 65°C for 5 min and then either treated for 10 min at 37°C with CIP or mock-incubated without CIP, as described in Figure 1. The products were analyzed by urea-PAGE and visualized by autoradiography. The conversion of ADPR-pRNAp to ADPR-pRNAOH by CIP is indicated on the right. (C) A reaction mixture (100 µL) containing 100 mM Tris-HCl, pH 7.5, 1 mM NAD+, 0.2 µM (20 pmol) 5′ 32P-labeled 10-mer pRNA2′p substrate (shown at bottom), and 0.5 µM (50 pmol) ChaetTpt1 was incubated at 37°C. Aliquots (10 µL) were withdrawn at the times specified and quenched immediately with three volumes of cold 90% formamide, 50 mM EDTA. The products were analyzed by urea-PAGE and visualized by autoradiography. The identities of the radiolabeled RNAs are indicated on the left. (D) The extents of terminal 2′-PO4 removal and 5′ capping calculated from kinetic assays in part C are plotted as a function of reaction time. Each datum is the average of three independent time course experiments ±SEM.
Via enzyme titrations, we determined that the specific activity of ChaetTpt1 for terminal 2′-PO4 removal was 0.84 pmol/pmol and that ChaetTpt1 was unable to remove a terminal 3′-PO4 (Fig. 7). The specific activity of ChaetTpt1 in 5′-phospho-ADPR capping was 0.15 pmol/pmol for pRNA2′p and 0.21 pmol/pmol for pRNA3′p (Fig. 7). The specific activity of ApeTpt1 in removing an internal 2′-PO4, from the 6-mer RNA pCCAA2′pAUOH was 86 fmol/fmol (not shown).
FIGURE 7.
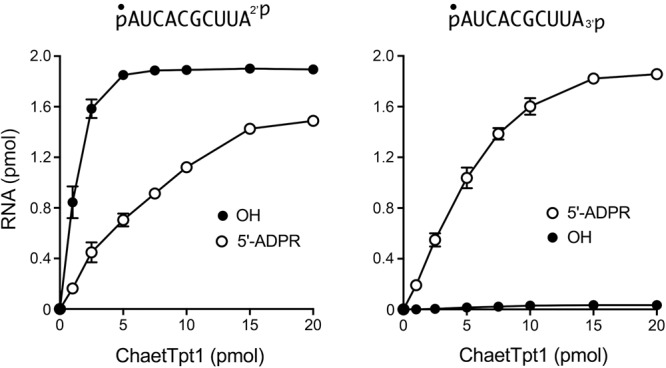
Dependence of 2′-PO4 removal on ChaetTpt1 concentration. Reaction mixtures (10 µL) containing 100 mM Tris-HCl (pH 7.5), 1 mM NAD+, 0.2 µM (2 pmol) 5′ 32P-labeled 10-mer pRNA2′p, or pRNA3′p substrates (shown at top), and ChaetTpt1 as specified were incubated at 37°C for 30 min. The reaction mixtures were analyzed by urea-PAGE. The extents of product formation are plotted as a function of input enzyme. Each datum is the average of three independent titration experiments ±SEM.
Runella slithyformis and human Tpt1 are inept at 2′ and 3′ terminal phosphate removal
Runella slithyformis Tpt1 (RslTpt1), which is among the biochemically best understood Tpt1 enzymes with respect to the canonical Tpt1 reaction (Munir et al. 2018a), and human Tpt1 (HsaTpt1) were both reported to be inactive in 5′-phospho-ADP-ribose capping (Munir et al. 2018b). Here we found that RslTpt1 and HsaTpt1 (at 500 nM concentration) were unreactive with 200 nM pRNA2′p or pRNA3′p and 1 mM NAD+ (Fig. 8A,B), the same conditions that were permissive for the terminal phosphatase activities of other Tpt1 orthologs. Control reactions performed in parallel showed that RslTpt1 and HsaTpt1 completely removed the internal 2′-PO4 from a 6-mer RNA mimetic of a tRNA splice junction (Fig. 8A,B).
FIGURE 8.

Runella and human Tpt1 are inept at RNA terminal 2′-PO4 and 3′-PO4 removal. Tpt1 reaction mixtures (10 µL) containing 100 mM Tris-HCl, pH 7.5, 0.2 µM (2 pmol) 5′ 32P-labeled 6-mer pCCAA2′pAUOH substrate, or 10-mer pRNA2′p or pRNA3′p substrates (shown at bottom), 1 mM NAD+ (where indicated by +), and 0.5 µM (5 pmol) RslTpt1 (A) or human Tpt1 (B) (where indicated by +) were incubated at 37°C for 30 min. T4 polynucleotide 2′,3′-phosphatase reactions were performed as in Figure 1. The products were analyzed by urea-PAGE.
DISCUSSION
The present study extends the catalytic repertoire of the Tpt1 enzyme family to include the NAD+-dependent conversion of RNA terminal 2′ and 3′ monophosphate ends to 2′-OH and 3′-OH ends, respectively. The salient finding here is that different Tpt1 enzymes vary in their capacity and positional specificity for terminal phosphate removal. To wit, the Clostridium and Aeropyrum Tpt1 proteins are active on 2′-PO4 and 3′-PO4 ends, with a 2.4- to 2.6-fold kinetic preference for the former. Chaetomium Tpt1 acts specifically on a terminal 2′-PO4 end and is virtually unreactive with a 3′-PO4. In contrast, Runella Tpt1 and human Tpt1 are ineffective in removing either a 2′-PO4 or 3′-PO4 end. A previous study of Saccharomyces cerevisiae Tpt1 using a 5′ 32P-labeled trinucleotide RNA substrate with a terminal 2′-PO4 (pAAA2′P) indicated that the yeast enzyme was extremely feeble at terminal 2′-PO4 removal, by a factor of 34,000 compared to its vigorous activity with a trinucleotide substrate with an internal 2′-PO4 (pApA2′PpA) (Steiger et al. 2001). In parallel studies with the E. coli Tpt1 ortholog KptA, Steiger et al. were unable to detect terminal 2′-PO4 removal from pAAA2′P.
Of the three Tpt1 enzymes that we find are active in terminal 2′-PO4 removal, ApeTpt1 was the fastest, followed by ChaetTpt1 and then CthTpt1. On a per enzyme basis, ChaetTpt1 has the highest specific activity in terminal 2′-PO4 removal, followed by ApeTpt1 and then CthTpt1. In each case, the rate and extent of the terminal 2′-PO4 phosphate removal reaction was greater than the rate and extent of 5′ ADPR capping of the RNA 5′-PO4 end. There is a rough correlation among the enzymes studied here between their capacities for terminal phosphate removal and 5′ ADPR capping, insofar as the Runella and human Tpt1 enzymes that are inept at terminal phosphate removal were the two enzymes found previously to be inactive in 5′ ADPR capping (Munir et al. 2018b). We do not see a clear connection between the taxonomy of the Tpt1 enzymes and an expanded catalytic repertoire. For example, whereas Tpt1 from the bacterium C. thermocellum is adept at terminal phosphate removal, the ortholog from the bacterium R. slithyformis is not.
Our results here highlight RNA 2′-PO4 and 3′-PO4 ends as plausible endogenous substrates with which CthTpt1 reacted to form the ADP-ribose-1″-phosphate ligand retained in the NAD+ site in the CthTpt1 crystal structure (Banerjee et al. 2019). This is a more parsimonious scenario than invoking a hypothetical E. coli pathway for the formation of an internal RNA 2′-PO4. However, we cannot at this point exclude the possibility that CthTpt1 reacts with a nonnucleic acid phospho-substrate in E. coli.
It will be of interest to understand why certain Tpt1 enzymes react with noncanonical phospho-substrates whereas others do not. The structure of CthTpt1 as a product complex mimetic with ADP-ribose-1″-phosphate in the NAD+ site and pAp in the RNA site revealed that the essential active site amino acid chains (an Arg-His-Arg-Arg tetrad) form an elaborate network of contacts to the 1″-phosphate (equivalent to the splice junction 2′-PO4 prior to its transfer to NAD+) and the vicinal 3′-PO4 of the splice junction (Banerjee et al. 2019). The inference from the CthTpt1 structure that the junction 2′-PO4 and 3′-PO4 are the most critical determinants of substrate recognition accords with the biochemical insights of Steiger et al. (2001) who showed that an RNA trinucleotide with a gem-diphospho 2′-PO4,3′-PO4 terminus sufficed for efficient 2′-phosphotransferase activity of S. cerevisiae Tpt1. With this in mind, it is relatively straightforward to imagine that Tpt1 might react with a terminal 2′-PO4 via the same substrate binding mode seen in the CthTpt1 crystal—in which the terminal nucleobase makes a π-cation stack on an arginine and the 2′-PO4 is coordinated by two catalytic arginines—except that the 3′-PO4 is missing. We envision that (i) the absence of the 3′-PO4 contacts accounts (in large part) for the lower specific activity of CthTpt1 at a terminal 2′-PO4 versus an internal 2′-PO4; and (ii) the Runella and human Tpt1 enzymes that do not remove a terminal 2′-PO4 are more stringently reliant on contacts to a vicinal 3′-PO4 (and perhaps to 3′-flanking ribonucleotides) than are the Tpt1s than can execute the terminal 2′-phosphotransferase reaction. It would appear that said stringency is exerted during the first step of the Tpt1 reaction pathway, insofar as we do not detect RNA terminal 2′-phospho-ADP-ribosylation by RslTpt1 or human Tpt1.
Accounting for the ability of CthTpt1 and ApeTpt1 to remove a terminal 3′-PO4 in light of the CthTpt1 crystal structure requires some conformational gymastics at the terminal ribonucleotide, which would need to rotate about one or more bonds to place the 3′-PO4 in roughly the same position vis-à-vis NAD+ normally occupied by the 2′-PO4. The accumulation of a terminal 3′-phospho-ADP-ribosylated RNA intermediate during the CthTpt1 3′-phosphotransferase reaction (Fig. 2B) suggests that the geometry of the 3′-p-ADPR adduct is not optimal for the ensuing transesterification step in which the ADPR ribose O2″ attacks the 3′-phosphodiester and displaces the RNA 3′-OH. To our knowledge, there has been no prior description of enzymatic ADP-ribosylation of an RNA 3′-phosphate end. Transfer of an ADP-ribose from NAD+ to a DNA 3′-monophosphate end by human PARP1-E998Q and PARP3 was reported recently (Munner and Ahel 2017; Zarkovic et al. 2018).
In sum, our results here fortify the theme that ADP-ribosyl transfer to a phosphorylated substrate is the unifying mechanistic feature of Tpt1-catalyzed reactions, which are not limited to the canonical activity of Tpt1 healing the 2′-PO4, 3′–5′ phosphodiester RNA splice junction formed during fungal and plant tRNA splicing. An enticing extrapolation of this idea is that some Tpt1 enzymes might catalyze NAD+-dependent phosphoryl transfer reactions with nonnucleic acid phospho-substrates and that interrogation of such reactions in vitro might illuminate what Tpt1 is doing in its manifold biological niches.
MATERIALS AND METHODS
Recombinant Tpt1 proteins
Tpt1 enzymes from Clostridium thermocellum, Aeropyrum pernix, Homo sapiens, Runella slithyformis, and Chaetomium thermophilum were produced in E. coli and purified as described previously (Munir et al. 2018a,b).
Solid phase synthesis of RNA oligonucleotides with 2′-PO4 and 3′-PO4 termini
Oligonucleotide syntheses were carried out using an ABI 3400 DNA synthesizer (Applied Biosystems) on either a Unylinker or 3′-Phosphate-ON (ChemGenes) solid support at a 1 µmol scale. Conventional 2′-tert-butyl-dimethylsilyl (TBDMS) ribonucleoside and 2′-acetyl levulinyl (ALE) ribonucleoside phosphoramidites (0.15 M in MeCN) (ChemGenes) were used. For phosphitylation, bis-cyanoethyl,-N,N-diisopropyl-phosphoramidite (0.20 in MeCN) was used. Phosphoramidites were dissolved in MeCN and activated with 5-ethylthio-1H-tetrazole (0.25 M in MeCN). Capping was carried out by the simultaneous delivery of acetic anhydride in pyridine/THF and N-methylimidazole (16% in THF) and contacting the solid support for 6 sec. Oxidation of the phosphite triester intermediates to the phosphate triesters was affected with 0.1 M iodine in pyridine/H2O/THF (20 sec); a solution of 3% trichloroacetic acid in THF, delivered over 1.8 min, was used to deprotect DMTr groups. For 2′-phosphate-containing substrates, a solution of anhydrous TEA/MeCN (2:3 v/v) was used to remove cyanoethyl phosphate protecting groups, while a 0.5 M solution of hydrazine hydrate in pyridine/AcOH (3:2 v/v) was used to remove 2′-ALE protecting groups. All oligonucleotides were deprotected and cleaved from the solid support using a 29% aqueous ammonia/ethanol (3:1 v/v) solution. TBDMS groups were removed using TREAT-HF. Crude oligonucleotides were purified via HPLC and characterized by LC-MS.
The 3′-PO4 10-mer 5′-AUCACGCUUA3′-P was synthesized on 3′-Phosphate-ON lcaa CPG 500 Å solid support following standard 3′ to 5′ solid phase synthesis. The use of this solid support ensures a phosphate moiety remains on the 3′ terminus of the RNA upon cleavage from the support. The 2′-PO4 10-mer 5′-AUCACGCUUA2′-P was synthesized on Universal Unylinker lcaa CPG 500 Å solid support by first coupling a 5′-DMTr-2′-ALE adenosine 3′-O-cyanoethyl (CE) phosphoramidite (0.15 M in MeCN) for 15 min. Subsequently, to remove the cyanoethyl phosphodiester protecting group, anhydrous TEA/MeCN (2:3 v/v) solution was passed through the solid support (20 min, repeated 3×). This step ensures that the phosphate linkages vicinal to the 2′-ALE group are in the diester form, thus preventing chain cleavage in the ensuing removal of the ALE group (Lackey et al. 2009). After washing with MeCN (20 min) and drying the support over an argon stream (10 min), the synthesis columns were temporarily removed from the synthesizer and dried in vacuo for 30 min. To remove the 2′-ALE groups, the columns were returned to the synthesizer and a freshly prepared solution of 0.5 M hydrazine hydrate in pyridine/AcOH (3:2 v/v) was flowed through the columns (20 sec flow + 3.75 min sleep, repeated 4×). After washing (MeCN, 10 min) and drying (Ar gas, 10 min), the solid supports were dried again in vacuo (30 min). To phosphitylate at the newly exposed 2′-OH, bis-cyanoethyl-N,N-diisopropyl-phosphoramidite (0.20 M in MeCN) was coupled for 30 min, and then further oxidized using 0.1 M iodine in pyridine/H2O/THF (20 sec). To complete the oligonucleotide, standard 3′ to 5′ synthesis was continued on the 5′ terminus of the growing oligonucleotide using the sequence 5′-AUCACGCUU to yield the desired 10-mer oligonucleotides.
Deprotection and cleavage of oligonucleotides from the solid support was achieved by treatment with 1 mL of cold 29% aqueous ammonia/ethanol (3:1, v/v) for 48 h at room temperature. Samples were centrifuged and the supernatant was transferred to a clean 1.5 mL Eppendorf tube and vented for 30 min, chilled on dry ice, and evaporated to dryness. Removal of the 2′-silyl protecting groups was achieved by treatment with a 300 µL solution of NMP/Et3N/TREAT-HF (3:4:6, v/v) for 90 min at 65°C, followed by quenching with 3 M NaOAc buffer (50 µL; pH 5.5) and precipitation of the crude oligonucleotide from cold butanol (1 mL, −20°C). Samples were chilled on dry ice for 30 min and then centrifuged. After removing the supernatant, the remaining pellet (containing RNA) was evaporated to dryness, taken up in autoclaved milliQ water (1 mL), filtered, and quantitated by UV spectroscopy.
Crude oligonucleotides were purified by ion exchange (IE) HPLC using a Waters Protein-Pak DEAE 5PW anion exchange column (21.5 × 150 mm). A mobile phase of 1 M aqueous LiClO4 in milli-Q water was used for analysis and purification of the RNA (0%–24% LiClO4 over 30 min, 4 mL/min, 60°C). Following collection of the desired peaks, fractions were combined and excess LiClO4 salts were removed using Gel Pak 2.5 size exclusion columns (Glen Research).
Purified oligonucleotides were characterized by electrospray ionization-mass spectrometry (the HPLC elution profiles and MS analyses are shown in Supplemental Fig. S2) and quantitated by UV spectroscopy. Extinction coefficients were determined using the IDT OligoAnalyzer tool (www.idtdna.com/analyzer/Applications/OligoAnalyzer).
5′ 32P-labeled oligonucleotide substrates
A synthetic 6-mer RNA oligonucleotide 5′-CCAA2′PAU containing an internal 2′-PO4 (Munir et al. 2018a) and the two synthetic 10-mer RNAs with 2′-PO4 or 3′-PO4 termini were 5′ 32P-labeled by reaction with phosphatase-dead T4 polynucleotide kinase (Pnkp-D167N) in the presence of [γ32P]ATP. The reactions were quenched with 90% formamide, 50 mM EDTA, 0.01% xylene cyanol, and the radiolabeled RNAs were purified by electrophoresis through a 40-cm 20% polyacrylamide gel containing 7 M urea in 45 mM Tris-borate, 1 mM EDTA. The radiolabeled oligonucleotides were eluted from excised gel slices, recovered by ethanol precipitation, and resuspended in 10 mM Tris-HCl, pH 6.8, 1 mM EDTA (TE) and stored at −20°C.
Assay of Tpt1 activity
Reaction mixtures containing 100 mM Tris-HCl (pH 7.5), 0.2 µM 5′ 32P-labeled RNA substrates, 1 mM NAD+, and Tpt1 as specified in the figure legends were incubated at 37°C. The reactions were quenched at the times specified in the figure legends by addition of three volumes of cold 90% formamide, 50 mM EDTA. The products were analyzed by electrophoresis (at 55 W constant power) through a 40-cm 20% polyacrylamide gel containing 7 M urea in 45 mM Tris-borate, 1 mM EDTA, and visualized by autoradiography and/or scanning the gel with a Fujifilm FLA-7000 imaging device. The products were quantified by analysis of the gel scans in ImageQuant.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the U.S. National Institutes of Health (National Institute of General Medical Sciences, R35-GM126945), the Geoffrey Beene Cancer Research Center, and the National Science and Engineering Council of Canada (Discovery grant to M.J.D.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.071142.119.
REFERENCES
- Banerjee A, Munir A, Abdullahu L, Damha MJ, Goldgur Y, Shuman S. 2019. Structure of tRNA splicing enzyme Tpt1 illuminates the mechanism of RNA 2′-PO4 recognition and ADP-ribosylation. Nat Commun 10: 218 10.1038/s41467-018-08211-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culver GM, McCraith SM, Zillman M, Kierzek R, Michaud N, LaReau RD, Turner DH, Phizicky EM. 1993. An NAD derivative produced during transfer RNA splicing: ADP-ribose 1″–2″ cyclic phosphate. Science 261: 206–208. 10.1126/science.8392224 [DOI] [PubMed] [Google Scholar]
- Culver GM, McCraith SM, Consaul SA, Stanford DR, Phizicky EM. 1997. A 2′-phosphotransferase implicated in tRNA splicing is essential in Saccharomyces cerevisiae. J Biol Chem 272: 13203–13210. 10.1074/jbc.272.20.13203 [DOI] [PubMed] [Google Scholar]
- Das U, Shuman S. 2013. Mechanism of RNA 2′,3′-cyclic phosphate end-healing by T4 polynucleotide kinase-phosphatase. Nucleic Acids Res 41: 355–365. 10.1093/nar/gks977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey JG, Mitra D, Somoza MM, Cerrina F, Damha MJ. 2009. Acetal levulinyl ester (ALE) groups for 2′-hydroxyl protection of ribonucleosides in the synthesis of oligoribonucleotides on glass and microarrays. J Am Chem Soc 131: 8496–8502. 10.1021/ja9002074 [DOI] [PubMed] [Google Scholar]
- McCraith SM, Phizicky EM. 1991. An enzyme from Saccharomyces cerevisiae uses NAD+ to transfer the splice junction 2′-phosphate from ligated tRNA to an acceptor molecule. J Biol Chem 266: 11986–11992. [PubMed] [Google Scholar]
- Munir A, Abdullahu L, Damha MJ, Shuman S. 2018a. Two-step mechanism and step-arrest mutants of Runella slithyformis NAD+-dependent tRNA 2′-phosphotransferase Tpt1. RNA 24: 1144–1157. 10.1261/rna.067165.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munir A, Banerjee A, Shuman S. 2018b. NAD+-dependent synthesis of a 5′-phospho-ADP-ribosylated RNA/DNA cap by RNA 2′-phosphotransferase Tpt1. Nucleic Acids Res 46: 9617–9624. 10.1093/nar/gky792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munner D, Ahel I. 2017. Reversible mono-ADP-ribosylation of DNA breaks. FEBS J 284: 4002–4016. 10.1111/febs.14297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popow J, Schlieffer A, Martinez J. 2012. Diversity and roles of (t)RNA ligases. Cell Mol Life Sci 69: 2657–2670. 10.1007/s00018-012-0944-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya R, Schwer B, Shuman S. 2005. Structure-function analysis of the yeast NAD+-dependent tRNA 2′-phosphotransferase Tpt1. RNA 11: 107–113. 10.1261/rna.7193705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli SL, Malik HS, Consaul SA, Phizicky EM. 1998. A functional homolog of a yeast tRNA splicing enzyme is conserved in higher eukaryotes and in Escherichia coli. Proc Natl Acad Sci 95: 14136–14141. 10.1073/pnas.95.24.14136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli SL, Kierzek R, Turner DH, Phizicky EM. 1999. Transient ADP-ribosylation of a 2′-phosphate implicated in its removal from ligated tRNA during splicing in yeast. J Biol Chem 274: 2637–2644. 10.1074/jbc.274.5.2637 [DOI] [PubMed] [Google Scholar]
- Steiger MA, Kierzek R, Turner DH, Phizicky EM. 2001. Substrate recognition by a yeast 2′-phosphotransferase involved in tRNA splicing and its Escherichia coli homolog. Biochemistry 40: 14098–14105. 10.1021/bi011388t [DOI] [PubMed] [Google Scholar]
- Steiger MA, Jackman JE, Phizicky EM. 2005. Analysis of 2′-phosphotransferase (Tpt1p) from Saccharomyces cerevisiae: evidence for a conserved two-step reaction mechanism. RNA 11: 99–106. 10.1261/rna.7194605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarkovic G, Belousova EA, Talhaoui I, Saint-Pierre C, Kutuzov MM, Matkarimov BT, Biard D, Gasparutto D, Lavrik OI, Ishchenko AA. 2018. Characterization of DNA ADP-ribosyltransferase activities of PARP2 and PARP3: new insights into DNA ADP-ribosylation. Nucleic Acids Res 46: 2417–2431. 10.1093/nar/gkx1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.