

Abstract
Longitudinal tracking of living cells is crucial to understand the mechanism of action and toxicity of cell-based therapeutics. To quantify the presence of administered cells in the host tissue without sacrifice of animals, labeling of the target cells with a nontoxic and stable contrast agent is a prerequisite. However, such long-term live cell tracking is currently limited by lacking fluorophores in the near-infrared (NIR) window with steady optical and physicochemical properties. Here we report, for the first time, the design of fixable cell tracking NIR fluorophores (CTNFs) with high optical properties, excellent cell permeation and retention, and high stability against chemical treatments. We demonstrated efficient cellular labeling and tracking of CTNFs using intraoperative optical fluorescence imaging by following the fate of NIR labeled cells from the time of injection into animals to ex vivo cellular analysis after resection of the target tissue. Due to the lipophilic cationicity and primary amine docking group, CTNF126 outperforms among the tested with rapid diffusion into the cytoplasmic membrane and sequestration inside the lysosomes, which prevents cellular efflux and improves cellular retention. Thus, CTNF126 will be useful to track cells in living organisms for the mechanism of action at the single cell level.
Keywords: Near-infrared fluorescence, fixable bioprobe, in vivo cell tracking, optical imaging, histological analysis
Graphical Abstract
CTNF126, a photostable lysosome targeting near-infrared fluorescent probe, tailored for comprehensive live cell tracing followed by in situ pathological analysis. The fluorophore exhibits high optical properties, i.e., extinction coefficient and quantum yield, excellent cell permeation, and lysosomal retention, which prevents efflux from the cell. In addition, CTNF126 can tolerate formaldehyde fixation and withstand harsh histopathological post-processing steps for further microscopic analysis.
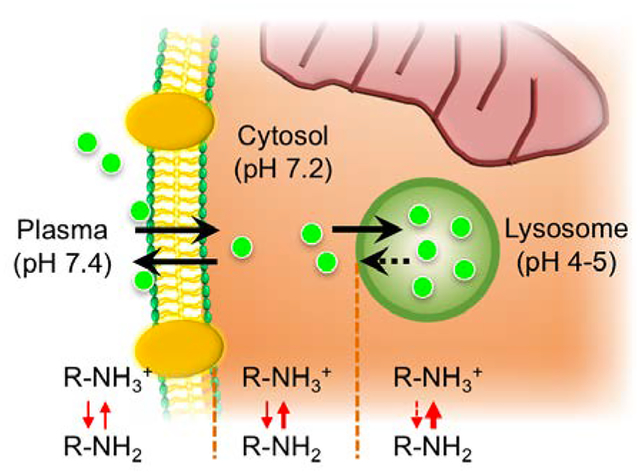
Recent advances in molecular imaging have provided insight for the in vivo behavior of transferred cells, aiding in both preclinical and clinical studies.[1–3] To extract data of biological function and activity from a single cell or from a population of cells, comprehensive and sequential imaging analysis from in vivo tracking to ex vivo pathophysiology is critical. However, it has been difficult to use fluorescence imaging techniques to visualize both the biological behavior of contrast agents in the body and molecular based consequences simultaneously because of the sensitivity of staining influenced by washing, fixation, and permeabilization on retention of fluorescence signal.[4] Therefore, it is important to improve the capability of tracers for real-time in vivo and ex vivo post-analysis in terms of binding specificity and chemical properties for better diagnostic interpretation.
General strategies for efficient biological imaging include the use of organic dyes,[5] inorganic nanoparticles,[6] and fluorescent proteins.[7] However, these often suffer from cytotoxicity and photobleaching as well as leakage of tracers and engulfment by neighboring cells, resulting in false-positives. Transfection of cells with fluorescent proteins, such as green fluorescent protein (GFP), allows longitudinal tracking of in vivo cell proliferation, but it can be difficult to reach the fluorescence density required for imaging cells in a short period of time.[8,9] To overcome these problems, different types of lipophilic organic fluorescent bioprobes have been developed to label subcellular components, such as cell membranes, lysosomes, and mitochondria.[10] However, tagging membrane can interfere with cellular mobility, and staining mitochondria can affect the membrane potential changes.[11] Lysosomes are digestive organelles with active endocytosis that absorb biomolecules to provide nutrition to the cells or degrade pathogens to protect the cells.[12] Therefore, staining lysosomes for the purpose of tracing cells can minimize potential alterations to the cellular activities.
We focused on the design of fixable bioprobes to target lysosomes and trap inside the lysosomal membrane to eliminate efflux and washout after staining the cells. Frist, we developed heptamethine near-infrared (NIR) fluorophores for cellular tracking in living organisms (Scheme S1) because the NIR window (650–900 nm) is advantageous for bioimaging due to low tissue absorbance, low tissue scattering, and low autofluorescence compared with UV-visible region.[13] Next, a series of ionizable docking functional groups were substituted to the heptamethine cyanine imaging core to compare selective cellular retention and further docking in the subcellular component. Chloride (-Cl), carboxylate (-COOH), primary amine (-NH2), and secondary amine (-NH-) were introduced as a pendant domain on the mesocarbon of the heptamethine backbone (Fig. S1-S4). Chloro-substituted fluorophores were synthesized through salt condensation with Vilsmeier-Haack reagent to build the polymethine cyanine core (Scheme S1), and the phenoxide ions were conjugated to the meso-chlorine atom through the SRN1 displacement pathway. Because the protonated oxygen exhibits limited nucleophilicity, NaH was used to generate the phenoxide ion in situ. For the synthesis of fluorophore with a primary amine docking group (CTNF126), we initially protected tyramine hydrochloride using the Boc-group to ensure selective reactivity at the oxygen atom (Fig. 1a). After conjugation of the tyramine on the heptamethine cyanine, the Boc protection was removed to form the final structure of CTNF126 (see Supplementary Information for other chemicals).
Figure 1. Design of lysosome-targeted NIR fluorophores:

(a) Synthetic scheme of CTNF126 composed of two functional groups and (b) optical properties of three representative NIR fluorophores. Absorbance (Abs) and fluorescence (Fl) spectra were measured in FBS at 5 μM. (c) Longitudinal cell tracking process of NIR fluorophores and lysosomal sequestration of CTNF126. The thickness of red arrow represents ionization tendency.
Comparative absorption and fluorescence emission spectra were recorded for newly synthesized fluorophores, CTNF126, IR785, and CTNF103, and all showed stable optical properties in the NIR window (Fig. 1b). Table 1 summarizes the physiochemical and optical properties of newly synthesized NIR fluorophores. Extinction coefficient and fluorescence quantum yield values for all three were similar (ε >110,000 M−1cm−1; Φ > 23). However, CTNF126 has the highest molecular brightness in serum-containing media (44,660 M−1cm−1). In addition, all three fluorophores are lipophilic cations (logD > 3.0 at pH 7.4) which can readily diffuse across cell membrane by passive diffusion. However, only CTNF126 can becomes protonated (pKa > 6.0) in the acidic environment of lysosome (pH 4–5).[14] This shifts the equilibrium, favoring the ionized form that could limit the diffusion of CTNF126 back across the lysosomal membrane into the cytosolic space.[15] As a result, the protonated CTNF126 is retained inside the cell, which is necessary for bioprobe to be an inert cell tracer with enhanced photo-activity.
Table 1.
Physicochemical and optical properties of lipophilic NIR fluorescent fluorophores.
| Physicochemical property | CTNF126 | IR786 | CTNF103 |
|---|---|---|---|
| Formula | C42H50N3O | C32H36ClN2 | C41H45N2O3 |
| Molecular weight (Da) | 612.87 | 484.09 | 613.81 |
| LogD at pH 7.4 | 3.5 | 4.79 | 6.54 |
| Total charges at pH 7.4 | 1 | 1 | 0 |
| pKa, strongest basic | 9.81 | 1.92 | 4.05 |
| Topological polar surface area (Å2) | 41.5 | 6.25 | 52.78 |
| Hydrogen bonding acceptor(s) | 3 | 1 | 7 |
| Hydrogen bonding donor(s) | 1 | 0 | 1 |
| Optical property in media | CTNF126 | IR786 | CTNF103 |
| Absorbance maximum (λabs, nm) | 778 | 786 | 773 |
| Emission maximum (λem, nm) | 790 | 800 | 786 |
| Stokes shift (nm) | 12 | 14 | 13 |
| Extinction coefficient (ε, M−1cm−1) | 154,000 | 117,500 | 137,500 |
| Quantum yield at 770nm (Φ, %) | 29.0 | 23.0 | 29.0 |
| Molecular brightness (M−1cm−1) | 44,660 | 27,025 | 39,825 |
Figure 1c summarizes an innovative design of CTNF126 with two functional domains for ionizable docking and lipophilic imaging, respectively, that allows for avoidance of physical or biochemical interference of NIR bioprobes with cellular activities. The heptamethine cyanine core is used for the sequential imaging with high photostability and serum stability from in vivo to ex vivo using the FLARE imaging system.[16,17] Sufficient photostability in biological media is another essential property for prolonged live cell imaging.[5,6] As shown in Fig. S5, the photostability of CTNF126 was determined in warm serum, where all three fluorophores showed over 95% serum stability during 4 h post-incubation. Additionally, the structure provides appropriate lipophilicity allowing it to penetrate inside the live cell membrane. Rapid passive diffusion into the cell can be followed by selective lysosomal sequestration, and the fluorophores could be trapped inside the cells. Furthermore, the fixable property of CTNF126 allows retention in the tissue after resection and chemical treatment for histological analysis.
To compare the cellular permeability and retention, CTNF126, IR786, and CTNF103 were loaded into living PC3 cells individually, and their cellular behaviors were compared in various conditions (Fig. 2). All three lipophilic cations achieved saturation of loading into cells within 10 min, and with extracellular concentrations ranged from 0.5 to 10 μM. At these high concentrations, the tested fluorophores did not exhibit any evidence of toxicity (data not shown). In particular, no observed change in cellular morphology was observed, and neither was a decrease or increase in the rate of proliferation nor a change in cell plating efficiency. In addition, as shown in Figure 2a, the strong signal strength of CTNF126 was maintained in active proliferation of the cells after an additional 24 h incubation, while cells stained with IR786 and CTNF103 had virtually no intracellular retention signals due to the efflux of fluorophores. To show that CTNF126 is fixable, the cells were treated with 4% formaldehyde after staining and washed with 1% Tween 20 (Fig. 2b). Despite potential interaction of the chloride atom of IR786 with intracellular proteins, significantly reduced signal was observed after fixation and detergent washing (***P <0.05 for IR786; ***P <0.001 for CTNF103).[18] CTNF126, however, is fixed in place with no loss of signal after detergent washing. CTNF126 demonstrated high efficiency and rapid loading into live cells (Fig. S6), as well as exhibited excellent partitioning behavior and retention.
Figure 2. Physicochemical and optical stability of lysosome-targeted NIR fluorophores:

(a) Long-term cellular stability of CTNF126, IR786, and CTNF103 in live PC3 cells and (b) their stability after formaldehyde fixation and detergent washing was calculated (n = 5, mean ± s.d., *P <0.05; ***P <0.001). Scale bars = 25 μm.
To validate direct translation of CTNF126 for in vivo cell tracking, 1×106 PC3 cells stained with either CTNF126 or IR786 were suspended in the media solution and administered into syngeneic C57BL/6J mice intravenously. The mice were sacrificed at 5 min and 24 h post-injection, and the thoracic cavity was opened and imaged under real-time image guidance using the intraoperative optical imaging system (Fig. 3a). At an initial deposition, the cells with both CTNF126 and IR786 showed similar signal intensity from the lungs. However, after 24 h, only the cells stained with CTNF126 could be observed in the lungs. The signal intensity of the lung was attenuated at 24 h due to migration of the cells from the lung capillaries to the liver, which was traceable (i.e., increase in liver signal).[19] This in vivo cell tracking was emphasized by changing the injection method. As shown in Figure 3b, the cells orthotopically injected into the liver were found in the lung 6 h post-injection, which implies migration of the cells through hepatic vasculatures. NIR in vivo fluorescence imaging confirmed that prolonged lysosome sequestration of CTNF126 is the key to real-time visualization of tracing administrated cells. This mechanism differs from other lysosomal targeting and pH-sensitive probes where their purpose is to monitor metabolic cellular activities. In this case, bioprobes are effluxed from the targeted organelle in 1–2 h. Thus, the fixable property of CTNF126 inside the lysosome is unique from all other subcellular-targeted probes and has potential for clinical translation.
Figure 3. In vivo tracking of lysosomal fluorophore-labeled cells using the intraoperative optical imaging system.

(a) CTNF126 and IR786-stained cells were trapped in the lung with strong fluorescent signal at 5 min post-intravenous injection. The cells labeled with CTNF126 were found in the liver as an evidence of cell migration from the lung capillaries at 24 h post-injection. (b) The cells injected into the liver orthotopically were found in the lung after 6 h, implicated liver-to-lung tracking of the cells. All NIR fluorescence images are identically normalized. Abbreviations used are: He, heart; In, intestine; Ki, kidney; Li, liver; Lu, lung. Scale bars = 3 mm.
One of the major challenges of fluorescence imaging in real-time cell tracking is insufficient thermal and biological stability of fluorophores, which limit continuous recording of the fate of cells after administration of stained cells.[20] Hematoxylin and eosin (H&E) is the gold standard for analyzing biopsy specimens in clinics, and cell tracking technology must be compatible to make a significant impact in the field.[21] However, H&E processing is extremely harsh, requiring exposure to high temperature of at least 58 °C for over 1 h and exposure to other organic solvents as strong as xylene during the tissue embedding and staining processes. There are currently no fixable cell-tracking NIR bioprobes that can withstand these punitive conditions.[4] CTNF126 is designed to be fixed covalently within the cell using formalin or paraformaldehyde by forming a cross-link methylene bridge, while retaining high stability.[22] To prove this, the same human tissue biopsy procedure used clinically was performed using cell pellets and lung tissue samples (Fig. 4). First, 1×107 PC3 cells stained with either CTNF126 or IR786 were collected using cell scraper and centrifuged at 1200 rpm for 10 min. The cell pellets were prepared into a paraffin block (Fig. S7) and underwent histological staining to evaluate the fluorescence retention or attenuation. As shown in Figure 4a, IR786 largely washed away during histological processing while CTNF126 was fixed in place and withstood high temperature and organic solvent washing. Next, the lung tissues were harvested after intravenous administration of the stained cells and performed the cryosection at a thickness of 10 μm followed by H&E staining. As expected, CTNF126 was resistant to the harsh processing and allowed further visualization of the cells while IR786 was mostly washed out from the stained cells localized in the lung tissue (Fig. 4b). This makes it possible to combine NIR fluorescence and H&E staining of the same slide, thus allowing us to locate a single cell on an H&E stained tissue slice while co-staining for differentiation markers [23].
Figure 4. Stability of histological processes, including formaldehyde fixation and H&E staining:

(a) Cell pellets mimicking the tissue structure and (b) the lung tissue sections harvested from C57BL/6 mice after intravenous administration of the cells labeled with lysosomal-targeted fluorophores. (c) Lysosomal sequestration of CTNF126 (left), LysoTracker (middle), and merged image of the two (right) in PC3 cells. All NIR fluorescence images have identical exposure and normalization. Scale bars = 25 μm.
To confirm the location of cellular uptake, PC3 cells were labeled with a series of NIR fluorophores. At a 2 μM concentration, IR786 and CTNF103 were found in the mitochondria (data not shown),[24] while CTNF126 targeted lysosomes confirmed by co-registration with LysoTracker (Fig. 4c). It is well known that lipophilic cations exhibit differential subcellular localization depending on concentrations: mitochondria uptake at low concentrations and endoplasmic reticulum (ER) accumulation at higher concentrations.[25,26] Next, to prove photostability in cells, CTNF126 and LysoTracker were dissolved in 100% serum at a concentration of 2 μM and incubated at 37 °C over 24 h (Fig. S8a). CTNF126 was stable in warm serum over the period of incubation (> 99%) without biological/chemical degradation (biodegradation) nor photo-induced degradation (photobleaching). Finally, the biological stability of CTNF126 was evaluated in PC3 cells, where CTNF126 showed significantly higher photostability compared with LysoTracker after 24 h incubation (Fig. S8b; n = 5, mean ± s.d., **P <0.01). This proves that CTNF126 is fixable inside lysosomes, which limits efflux elimination and stabilizes in the cell without degradation.
In conclusion, a novel NIR bioprobe CTNF126 was synthesized with excellent optical properties (i.e., high extinction coefficient, quantum yield, and photostability) for the purpose of sequential cell tracking from the body to microscopy. CTNF126 can withstand all thermal and chemical steps in histological tissue processing while retaining stable NIR fluorescence. We proved that CTNF126 is lysosome fixable with outstanding physicochemical and optical properties in serum containing media as well as organic solvents, enabling tracing of single cells for determining the mechanism of various diseases in the body. The lipophilic cation structure allows cellular membrane permeation, and the primary amine is used for rapid sequestration followed by efficient intracellular fixation by formalin through reductive amination. This ionizable amine group also reduces the risk of efflux of NIR fluorophores from the cells. Therefore, CTNF126 will be useful in staining various types of living organisms to gain direct evidence of their in vivo and ex vivo fate at the single cell level.
Experimental Section
Chemicals and syntheses
All chemicals and solvents were of American Chemical Society grade or HPLC purity. All chemicals were purchased from Fisher Scientific (Pittsburgh, PA), Sigma-Aldrich (St. Louis, MO), and Acros Organics (Pittsburgh, PA), unless noted otherwise. LysoTracker™ was obtained from Molecular Probes (Eugene, OR), and IR786 was purchased from Sigma-Aldrich. For synthesis of CTNF126 and CTNF103, heptamethine fluorophores were synthesized first, and they were assorted with side groups. Chloro-substituted dyes were synthesized as previously reported, through salt condensation with Vilsmeier-Haack reagent to build the heptamethine cyanine core.[16,17,27,28] Then, substitution of different ionizable docking functional groups (phenoxide ions) were made on the meso-chlorine atom of the heptamethine cyanine core to compare selective cellular retention of each fluorophore and further docking in the subcellular component (Scheme S1). The final compounds were analyzed by 1H NMR, 13C NMR, and HRMS (See Supplementary Information for detailed chemical syntheses and analysis).
Measurement of optical properties
Absorbance and fluorescence were measured using fiberoptic HR2000 (200–1100 nm) spectrometers (Ocean Optics, Dunedin, FL). Fluorescence excitation was provided by a 5 mW, 655 nm laser diode (Opcom Inc., Xiamen, China) coupled through a 300 mm core diameter, NA 0.22 fiber (Fiberguide Industries, Stirling, NJ). All optical measurements were made in serum-containing media. 5 μM of each fluorophore solution was prepared in 10% FBS solutions and incubated at 37 °C for 4 h to evaluate their photostability and physicochemical stability. In silico calculations of physicochemical properties such as molecular weight, charge, distribution coefficient (logD at pH 7.4), pKa, refractivity, topological polar surface area (TPSA), H-bond donors/acceptors, and rotatable bonds were calculated using JChem calculator plugins (ChemAxon, Budapest, Hungary). Data plotting was performed using Prism version 4.0a software (GraphPad, San Diego, CA) and Microsoft Excel (Redmond, WA).
Live cell labeling and in vitro imaging
Human prostate cancer cells (PC3 cells) were seeded into 24-well plates (5×104 cells per well) and incubated at 37 °C in humidified 5% CO2 incubator in DMEM containing 10% fetal bovine serum (FBS) and 1% penicillin streptomycin for 2 days. After washing twice with media solution, CTNF126, IR786, and CTNF103 were added to each well at a concentration of 2 μM and incubated for 30 min at 37 °C. To improve image contrast, cells were washed three times with media solution prior to imaging. The images were acquired either right after the triple wash, or right after additional washing with 1% Tween 20 to compare fluorescent signal reduction among the contrast agents. The live cell imaging was performed using Nikon TE2000 epifluorescence microscope equipped with a 75W Xenon light source and an Orca-ER (Hamamatsu, Bridgewater, NJ) camera. The filter set (Chroma Technology, Brattleboro, VT) composed of 710±25 nm excitation filter, 785 dichroic mirror, and 810±20 nm emission filter was used to detect all NIR fluorophores. These cells were observed again after 2 days to compare long-term stability.
In vivo cell tracking and histological analysis
Animals were housed in an AAALAC-certified facility and were studied under the supervision of BIDMC IACUC in accordance with the approved institutional protocol (#057–2014). 6-week-old C57BL/6 mice (male; 20–25 g) were purchased from Charles River Laboratories (Wilmington, MA). Animals were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine intraperitoneally (Webster Veterinary, Fort Devens, MA). B16F10 cells were stained with 2 μM of CTNF126 or IR786 for 30 min at 37 °C, and detached with cell scraper. 1×106 cells per mouse were injected intravenously into the mice. Animals were sacrificed 5 min and 24 h post-injection of the cells, respectively.
The lung cavity was opened to observe injected cells in the lung using our custom-built intraoperative NIR imaging system, which equipped 760 nm excitation light (3.6 mWcm−2) and white light (400–650 nm) at 5500 lux. Color and NIR fluorescence images were acquired simultaneously with custom software at rates of up to 15 Hz over a field of view with a diameter of 15 cm. The lungs were resected and embedded in Tissue-Tek O.C.T. compound (Sakura Fineteck, Torrance, CA).
The tissues were then cryosectioned at 10 μm interval and stained with hematoxylin and eosin (H&E) for further visualization with the customized TE2000 NIR fluorescence microscope. For validation of fluorescent signal retention during histological analysis, we preceded a standard method using cell pellets. PC3 pellets were seeded onto 100 × 15 mm petri dishes and incubated at 37 °C in humidified 5% CO2 incubator in the media solution until the cell confluency reached 90%. 1×107 cells (~ 5 petri dishes) were stained with CTNF126 or IR786 at a concentration of 2 μM and incubated for 30 min at 37 °C. After washing with media solution, the cells were collected using cell scraper and centrifuged at 1200 rpm for 10 min. After the same histological preparation was conducted, the sectioned slides were imaged using the microscope using 20X objective.
Quantitation and Statistical Analysis
Fluorescent intensity (FI) of a region of interest (ROI) over various tissues was quantified using Image J software (NIH, Bethesda, MD). Signal-to-background ratio (SBR) is FI of ROI / background (BG) intensity. Results were presented as mean (n = 5) ± standard deviation (s.d.). Statistical analysis was performed using a one-way ANOVA between multiple groups. A P value of less than 0.05 was considered significant (*P <0.05; **P <0.01; ***P <0.001).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. John V. Frangioni for many helpful discussions and experimental support and Ivey Choi for manuscript editing. This study was supported by the NIH grants #R01-EB022230 and #R21CA223270, and the Marine Biotechnology Program #20170263 funded by the Ministry of Oceans and Fisheries, South Korea.
Contributor Information
G. Kate Park, Gordon Center for Medical Imaging, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA; Interdisciplinary Program in Bioengineering and School of Chemical and Biological Engineering, Seoul National University, Seoul 151-742, South Korea.
Jeong Heon Lee, Gordon Center for Medical Imaging, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA.
Andrew Levitz, Department of Chemistry, Georgia State University, Atlanta, GA 30303, USA.
Georges El Fakhri, Gordon Center for Medical Imaging, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA.
Nathaniel S. Hwang, Interdisciplinary Program in Bioengineering and School of Chemical and Biological Engineering, Seoul National University, Seoul 151-742, South Korea
Maged Henary, Department of Chemistry, Georgia State University, Atlanta, GA 30303, USA.
Hak Soo Choi, Gordon Center for Medical Imaging, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA.
REFERENCES
- [1].Naumova AV, Modo M, Moore A, Murry CE, Frank JA, Nat Biotechnol 2014, 32, 804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nguyen PK, Nag D, Wu JC, Adv Drug Deliv Rev 2010, 62, 1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Park GK, Hoseok s., Kim GS, Hwang NS, Choi HS, Appl Spectrosc Rev 2018, 53, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pagani FD, DerSimonian H, Zawadzka A, Wetzel K, Edge AS, Jacoby DB, Dinsmore JH, Wright S, Aretz TH, Eisen HJ, Aaronson KD, Journal of the American College of Cardiology 2003, 41, 879. [DOI] [PubMed] [Google Scholar]
- [5].Lee JH, Park G, Hong GH, Choi J, Choi HS, Quant Imaging Med Surg 2012, 2, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B., Bawendi MG, Frangioni JV, Nat Biotechnol 2007, 25, 1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Blasberg RG, Gelovani J, Mol Imaging 2002, 1, 280. [DOI] [PubMed] [Google Scholar]
- [8].Arnhold S, Lenartz D, Kruttwig K, Klinz FJ, Kolossov E, Hescheler J, Sturm V, Andressen C, Addicks K, J Neurosurg 2000, 93, 1026. [DOI] [PubMed] [Google Scholar]
- [9].Dong L, Zhang X, Yu C, Yu T, Liu S, Hou L, Fu L, Yi S, Chen W, Exp Ther Med 2013, 6, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hu F, Liu B, Org Biomol Chem 2016, 14, 9931. [DOI] [PubMed] [Google Scholar]
- [11].Frye LD, Edidin M, J Cell Sci 1970, 7, 319. [DOI] [PubMed] [Google Scholar]
- [12].De Duve C, Wattiaux R, Annu Rev Physiol 1966, 28, 435. [DOI] [PubMed] [Google Scholar]
- [13].Owens EA, Lee S, Choi J, Henary M, Choi HS, Wiley Interdiscip Rev Nanomed Nanobiotechnol 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lassailly F, Griessinger E, Bonnet D, Blood 2010, 115, 5347. [DOI] [PubMed] [Google Scholar]
- [15].Kazmi F, Hensley T, Pope C, Funk RS, Loewen GJ, Buckley DB, Parkinson A, Drug Metab Dispos 2013, 41, 897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Choi HS, Nasr K, Alyabyev S, Feith D, Lee JH, Kim SH, Ashitate Y, Hyun H, Patonay G, Strekowski L, Henary M, Frangioni JV, Angew Chem Int Ed Engl 2011, 50, 6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Njiojob CN, Owens EA, Narayana L, Hyun H, Choi HS, Henary M, J Med Chem 2015, 58, 2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yuan L, Wang L, Agrawalla BK, Park SJ, Zhu H, Sivaraman B, Peng J, Xu QH, Chang YT, J Am Chem Soc 2015, 137, 5930. [DOI] [PubMed] [Google Scholar]
- [19].Choi HS, Ashitate Y, Lee JH, Kim SH, Matsui A, Insin N, Bawendi MG, Semmler-Behnke M, Frangioni JV, Tsuda A, Nat Biotechnol 2010, 28, 1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kim SH, Park G, Hyun H, Lee JH, Ashitate Y, Choi J, Hong GH, Owens EA, Henary M, Choi HS, Biomed Mater 2013, 8, 014110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Weir C, Morel-Kopp MC, Gill A, Tinworth K, Ladd L, Hunyor SN, Ward C, Heart Lung Circ 2008, 17, 395. [DOI] [PubMed] [Google Scholar]
- [22].Gustavson KH, J Biol Chem 1947, 169, 531. [PubMed] [Google Scholar]
- [23].Inoue K, Gibbs SL, Liu F, Lee JH, Xie Y, Ashitate Y, Fujii H, Frangioni JV, Choi HS, J Nucl Med 2014, 55, 1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nakayama A, Bianco AC, Zhang CY, Lowell BB, Frangioni JV, Mol Imaging 2003, 2, 37. [DOI] [PubMed] [Google Scholar]
- [25].Chen LB, Methods Cell Biol 1989, 29, 103. [DOI] [PubMed] [Google Scholar]
- [26].Terasaki M, Song J, Wong JR, Weiss MJ, Chen LB, Cell 1984, 38, 101. [DOI] [PubMed] [Google Scholar]
- [27].Hyun H, Wada H, Bao K, Gravier J, Yadav Y, Laramie M, Henary M, Frangioni JV, Choi HS, Angew Chem Int Ed Engl 2014, 53, 10668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Owens EA, Hyun H, Kim SH, Lee JH, Park G, Ashitate Y, Choi J, Hong GH, Alyabyev S, Lee SJ, Khang G, Henary M, Choi HS, Biomed Mater 2013, 8, 014109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.