

Abstract
γ‐Secretase complexes (GSECs) are multimeric membrane proteases involved in a variety of physiological processes and linked to Alzheimer's disease (AD). Presenilin (PSEN, catalytic subunit), Nicastrin (NCT), Presenilin Enhancer 2 (PEN‐2), and Anterior Pharynx Defective 1 (APH1) are the essential subunits of GSECs. Mutations in PSEN and the Amyloid Precursor Protein (APP) cause early‐onset AD. GSECs successively cut APP to generate amyloid‐β (Aβ) peptides of various lengths. AD‐causing mutations destabilize GSEC‐APP/Aβn interactions and thus enhance the production of longer Aβs, which elicit neurotoxic events underlying pathogenesis. Here, we investigated the molecular strategies that anchor GSEC and APP/Aβn during the sequential proteolysis. Our studies reveal that a direct interaction between NCT ectodomain and APP C99 influences the stability of GSEC‐Aβn assemblies and thereby modulates Aβ length. The data suggest a potential link between single‐nucleotide variants in NCSTN and AD risk. Furthermore, our work indicates that an extracellular interface between the protease (NCT, PSEN) and the substrate (APP) represents the target for compounds (GSMs) modulating Aβ length. Our findings may guide future rationale‐based drug discovery efforts.
Keywords: Alzheimer's disease, amyloid‐beta, Nicastrin, γ‐secretase, γ‐secretase modulators
Subject Categories: Molecular Biology of Disease; Neuroscience; Post-translational Modifications, Proteolysis & Proteomics
Introduction
γ‐Secretase intramembrane proteases (GSECs) play multifaceted roles in physiology and disease. Of great relevance is their critical involvement in Alzheimer's disease (AD) pathogenesis, where altered cleavage of the Amyloid Precursor Protein (APP; Fig 1A), and concomitant, relative increase in the generation of longer vs. shorter amyloid‐β (Aβ) peptides, has been proposed to underlie the disease (Selkoe & Hardy, 2016; Szaruga et al, 2017). GSEC activity is exerted by a group of heteromultimeric, membrane‐embedded complexes composed of Presenilin (PSEN1 or PSEN2; Li et al, 2000), Nicastrin (NCT) (Yu et al, 2000), Presenilin Enhancer 2 (PEN‐2; Francis et al, 2002), and Anterior Pharynx Defective 1 (APH1A or APH1B; Francis et al, 2002; Goutte et al, 2002) in a 1:1:1:1 stoichiometric ratio (Lazarov et al, 2006; Sato et al, 2007; Bai et al, 2015b). PSEN bears the catalytic center of the enzyme (De Strooper et al, 1998; Steiner et al, 1999; Wolfe et al, 1999; Ahn et al, 2010), while the other “non‐catalytic” subunits of the protease are required for the complex assembly, activation, structural stability/turnover, and trafficking (Thinakaran et al, 1996; Kim et al, 2003; Luo et al, 2003; Takasugi et al, 2003; Zhang et al, 2005). Once the quartet has been assembled, PSEN undergoes auto‐endoproteolytic cleavage, which yields an active pentameric protease complex, with the catalytic center located at the interface between the N‐ and C‐terminal fragments (NTF vs. CTF) of PSEN (De Strooper, 2003; Edbauer et al, 2003; Selkoe & Wolfe, 2007).
Figure 1. Schematic representation of APP processing and structural model (PDB: 5FN3) of the potential enzyme–substrate interface.
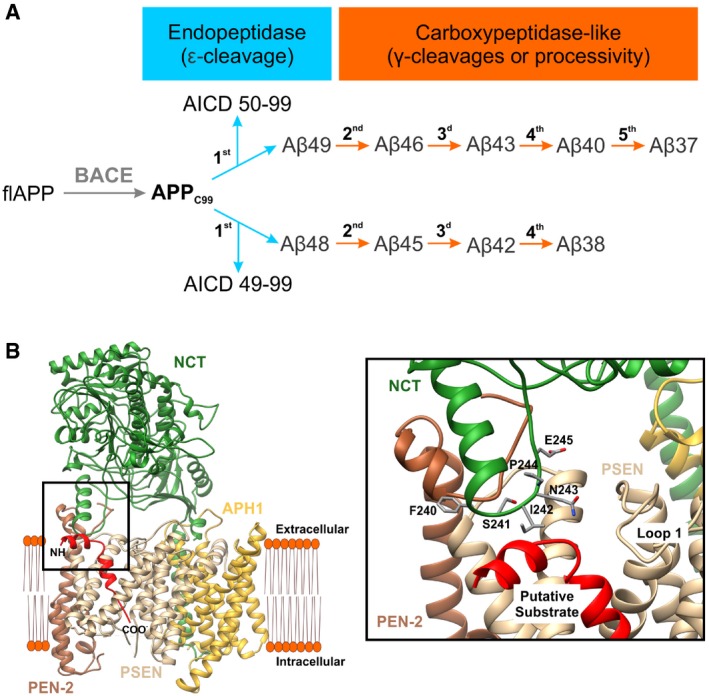
- In the amyloidogenic pathway, the ectodomain shedding of the full length APP (flAPP) by BACE yields the immediate GSEC substrate, APPC99. GSEC endopeptidase cleavage of APPC99 generates APP intracellular domain (AICD50‐99 or AICD49‐99) and a “de novo” generated Aβ substrate (Aβ48 or Aβ49, respectively). While the AICDs are released, the Aβ48 and Aβ49 are subjected to subsequent GSEC proteolysis (carboxypeptidase‐like γ‐cleavages).
- GSEC structure with co‐purifying helical peptide (PDB: 5FN3) is presented. PSEN1 is shown in light brown, PEN‐2 in dark brown, APH1A in gold, NCT in green, and the co‐purifying helical peptide in red. The topology of the helical peptide (putative GSEC substrate) is indicated by the amino and carboxyl terminal groups. The interface between the N‐terminal part of the helical peptide, the NCT ectodomain and PSEN1 is magnified, and NCT side chains are shown.
In addition to the proteolytic processing of APP, the cleavage of many other type I membrane proteins, such as Notch, ErbB4, and N‐Cadherin (reviewed in Haapasalo and Kovacs 2011), positions GSEC activity as a regulator of several key physiological events during embryonic development, hematopoiesis, and normal functioning of the nervous and immune system, as well as of disease processes, such as cancer (Jurisch‐Yaksi et al, 2013; McCarthy et al, 2017). Interestingly, the GSEC substrates are remarkably different in their amino acid sequences and lack any obvious common structural features (Beel & Sanders, 2008; Lleó, 2008). In fact, the only known prerequisite for GSEC cleavage is the removal of protruding ectodomains (when present) from type I membrane proteins by protein sheddases (Struhl & Adachi, 2000; Kopan & Ilagan, 2004), a step that generates C‐terminal transmembrane domains (TMDs) with short ectodomains—the immediate GSEC substrates—that may only then undergo GSEC‐mediated proteolysis.
APPC99 is the most studied substrate of GSECs due to its genetic and biochemical link to AD pathogenesis. Extensive research has revealed a rather unique mechanism by which APPC99, the C‐terminal fragment generated by β‐secretase‐mediated APP ectodomain shedding, is sequentially cut by GSECs (Qi‐Takahara, 2005; Yagishita et al, 2008; Takami et al, 2009). The first GSEC‐mediated endopeptidase (ε) cleavage releases a soluble cytosolic fragment (APP intracellular domain, AICD) and generates a transmembrane fragment (Aβ48 or Aβ49), which is successively cut by carboxypeptidase‐like (γ) cleavages, along the following product lines: Aβ49 → Aβ46 → Aβ43 → Aβ40 → Aβ37 or Aβ48 → Aβ45 → Aβ42 → Aβ38 (Fig 1A). The release of an Aβn peptide into the luminal/extracellular environment ends the sequential process (Takami et al, 2009). Importantly, the position of the ε‐cleavage on APPC99 defines the type of Aβ products (product line), while the efficiency of the sequential γ‐cleavages (number of cuts per substrate molecule, so‐called GSEC processivity) determines the length of the N‐terminal Aβ products.
Of note, mutations in both GSEC (enzyme, E) and APP (substrate, S) lead to early‐onset familial AD (FAD; Goate et al, 1991; Sherrington et al, 1995). Importantly, disease‐causing mutations in PSEN1 and PSEN2 genes consistently impair the efficiency of the γ‐cleavages, shifting Aβ profiles toward the generation of longer amyloidogenic peptides (Chávez‐Gutiérrez et al, 2012; Fernandez et al, 2014; Moore et al, 2015; Veugelen et al, 2016). This so‐called “GSEC dysfunction” promotes—as demonstrated by the analysis of post‐mortem brain samples from FAD patients (Szaruga et al, 2015)—the production of longer Aβ≥42 peptides, which trigger neurotoxic events underlying AD (Karran & De Strooper, 2016; Selkoe & Hardy, 2016). Furthermore, many but not all FAD‐causing PSEN variants (Chávez‐Gutiérrez et al, 2012; Fernandez et al, 2014; Szaruga et al, 2015) impair the initial GSEC endopeptidase (ε‐) cleavage and thus may have detrimental effects on GSEC‐mediated signaling cascades.
Importantly, our recent studies (Szaruga et al, 2017) have shown that the relative stabilities of the GSEC‐APP/Aβn (E‐S) complexes, formed during the sequential cleavage of APPC99 by GSEC, control the efficiency of the sequential proteolysis and thereby define the length of Aβ products. In addition, they have demonstrated that pathogenic mutations in PSEN1 and in APP destabilize the GSEC‐APP/Aβn interactions, leading to the “premature” release of longer, more hydrophobic Aβs (Szaruga et al, 2017). Of note, a remarkable correlation between the magnitude of the mutant‐induced weakening of the stability of the E‐S complexes and the corresponding age of disease onset (Szaruga et al, 2017) places the generation of longer Aβ≥42 peptides central to AD pathogenesis. The novel link between the E‐S complex (structural) stability and AD pathogenicity prompted us to investigate the molecular mechanisms securing GSEC‐APP/Aβn interactions. Here, we applied a structure‐based functional approach to determine the molecular strategies that anchor APP/Aβn during GSEC‐mediated proteolysis. Our analyses reveal that a direct interaction between the NCT ectodomain and APPC99/Aβn modulates the stability of GSEC‐APP/Aβn interactions and therefore regulates Aβ product length. In fact, the data raise the intriguing possibility that AD‐causing or protective single‐nucleotide variants (SNVs) may exist in the NCSTN gene. Furthermore, our analysis implicates the extracellular interface formed between GSEC (NCT, PSEN) and APPC99 in the regulation of the response to small compounds, referred to as GSEC modulators (GSMs). Collectively, these insights may guide future drug discovery efforts and the development of innovative strategies targeting safely and efficiently the destabilized GSEC‐APP/Aβn assemblies in AD.
Results
Structural data reveal a potential direct, short‐distance interaction between NCT and the substrate
We departed from the PDB:5FN3 high‐resolution GSEC structure to investigate the structural determinants securing E‐S complexes and thus controlling Aβ length. We hypothesized that this structure of GSEC (Fig 1B) (Bai et al, 2015a), which contains a co‐purifying helical peptide (putative substrate), depicts the interaction between GSEC and a “de novo” generated long Aβ substrate, just before it engages into the next catalytic turnover. In our working model, originally presented in Szaruga et al (2017) and discussed in‐depth here, key interactions between GSEC and the extracellular part of the putative substrate play a critical role in the stabilization of the E‐S assemblies during the sequential proteolysis of Aβn substrates. Accordingly, we explored the presumed E‐S co‐structure to define interactions that could contribute directly to the regulation of the strength of the GSEC‐APP/Aβ interactions. Interestingly, we found potential contact sites between the extracellular part of the helical co‐purifying peptide and the NCT ectodomain (amino acid (aa) residues 242–243 in human or 241–242 in mouse NCT (mNCT); please note that residue numbering used in this report corresponds to the sequence of mNCT unless specified otherwise; Fig 1B, zoom‐in) as well as with the first extracellular loop of PSEN1. While our recent analysis of FAD‐linked PSEN1 variants (Szaruga et al, 2017) has demonstrated the participation of the first extracellular loop of PSEN1 in the (de)stabilization of GSEC‐APP/Aβ interactions, the involvement of the ectodomain of NCT in the strength of E‐S interactions has not been shown before and, if true, would provide novel insights into the roles of the “non‐catalytic” GSEC subunits in the regulation of GSEC processivity of APP (Aβ length). Therefore, we investigated the contribution of NCT ectodomain to the stability of GSEC‐APP/Aβn interactions, which defines enzyme processivity.
NCT ectodomain—aa 241‐244—regulates GSEC processivity of APPC99
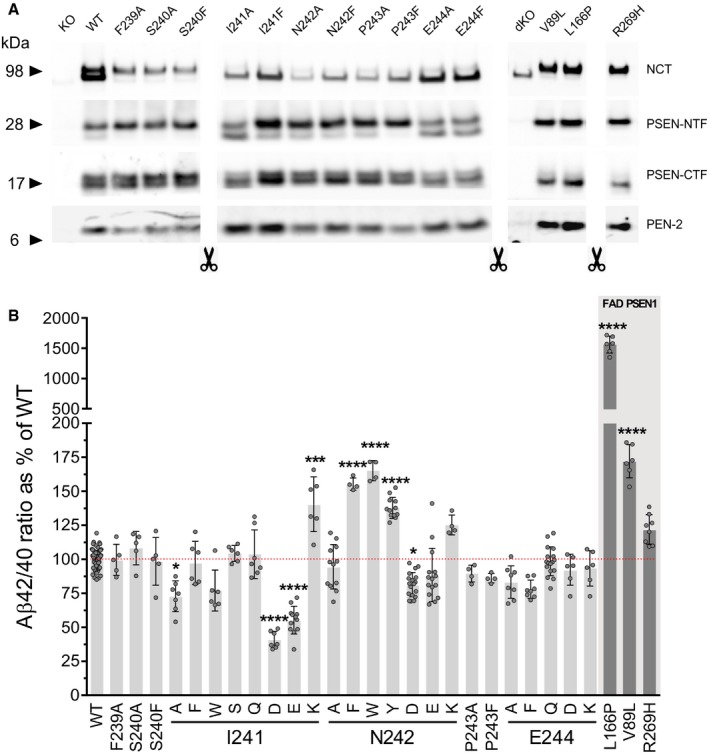
To challenge the potential involvement of the NCT‐241‐242 region in the regulation of the GSEC processivity of APPC99, we performed Ala/Phe mutagenic scanning of the region of interest and of the two neighboring amino acids on each side (NCT‐F239A/F, NCT‐S240A/F, NCT‐I241A/F, NCT‐N242A/F, NCT‐P243A/F and NCT‐E244A/F), followed by functional evaluation of the GSEC complexes in cell‐based assays. To this end, we used NCT knock‐out (KO) mouse embryonic fibroblasts (MEFs; Li et al, 2003) stably expressing either wild‐type or mutant NCT and transiently expressing wild‐type APPC99 substrate. Importantly, SDS–PAGE/Western blot analysis showed that all mutants restored the assembly, maturation and activity of GSEC, as indicated by the levels of mature NCT and PEN‐2 as well as the endoproteolytic generation of PSEN1‐NTF and PSEN1‐CTF fragments (Fig EV1A). To determine the effects of the tested mutant NCTs on GSEC processivity of APP, we quantified secreted Aβ38, Aβ40 and Aβ42 peptides by multi‐spot enzyme‐linked immunosorbent assay (MSD‐ELISA) and Aβ43 by standard ELISA. In line with our previous studies, Aβ43 was produced at low levels relative to the other (Aβ38/Aβ40/Aβ42) peptides in wild‐type or mild destabilizing (such as FAD‐linked V89L and R269H PSEN1) conditions (Veugelen et al, 2016; Fig EV2). We, therefore, used the Aβ (38 + 40)/42 ratio (product/substrate ratio of the 4th GSEC catalytic turnover) to estimate the overall processivity of GSEC toward APP. Importantly, we included the FAD‐linked PSEN1‐L166P, PSEN1‐V89L and PSEN1‐R269H mutant cell lines (Psen1 −/− /Psen2 −/− MEFs (Nyabi et al, 2002) stably expressing respective PSEN1 mutants) transiently expressing APPC99 as reference samples in the analyses. The PSEN1 pathogenic variants were selected based on their differential location in the PSEN1 structure and distinct clinical ages of disease onsets (24, 48.6 and 55 years, respectively; Cruts et al, 2012). In line with the reported detrimental effects of the FAD‐linked PSEN1 mutations on GSEC processivity of APP (Chàvez‐Gutièrrez et al, 2012; Fernandez et al, 2014; Veugelen et al, 2016), all tested pathogenic mutant GSECs lowered the Aβ (38 + 40)/42 ratio relative to the wild‐type enzyme. The results thus validate the use of the Aβ (38 + 40)/42 ratio for the detection of changes in the sequential GSEC‐mediated cleavage of APP.
Figure EV1. NCT ectodomain regulates the Aβ 42/40 ratio.
- Representative SDS–PAGE/Western blot analysis of CHAPSO‐solubilized membrane proteins from Ncstn −/− or Psen1 −/− /Psen2 −/− MEF cell lines stably expressing WT or mutant mouse NCT/human PSEN1 subunits (shown in Fig 2A). The presence of mature, glycosylated NCT, N‐terminal, and C‐terminal fragments of the endoproteolyzed PSEN1 and PEN‐2, compared to NCT knock‐out (KO) or PSEN1/2 double KO (dKO) cells, indicates that WT and mutants reconstitute GSEC complexes. Arrowheads indicate the position of molecular weight markers.
- Aβ peptides present in the extracellular media of cultured WT or mutant mNCT/hPSEN1 MEF cell lines, transiently expressing APPC99 substrate, were quantified by ELISA, and the Aβ 42/40 ratio were determined and plotted as % of the WT condition. The data are represented as mean ± SD, N ≥ 4 independent experiments. One‐way ANOVA and Dunnett's post hoc test were used to determine the statistical significance; ***P > 0.001 and ****P > 0.0001 (compared to WT condition). F(DFn, DFd): F(31, 290) = 873,6.
Source data are available online for this figure.
Figure EV2. Secreted Aβ profiles in cell‐based thermoactivity assays.
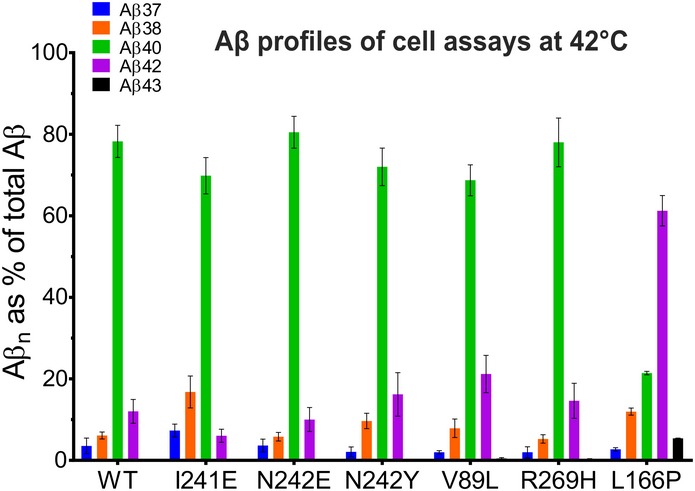
Aβ profiles (% contribution of individual Aβ37, Aβ38, Aβ40, Aβ42, and Aβ43 peptides to the total Aβ levels (Aβ37 + 38 + 40 + 42 + 43)) in the conditioned medium collected from WT or mutant (NCT) MEF cells lines co‐expressing WT or mutant APPC99 substrates, subjected to temperature challenge, were assayed by ELISA. The contribution of the Aβ43 peptides to the secreted Aβ pool is < 1% for the WT or mildly destabilizing FAD mutants (V89L and R269H). Data are represented as mean ± SD, N ≥ 3.
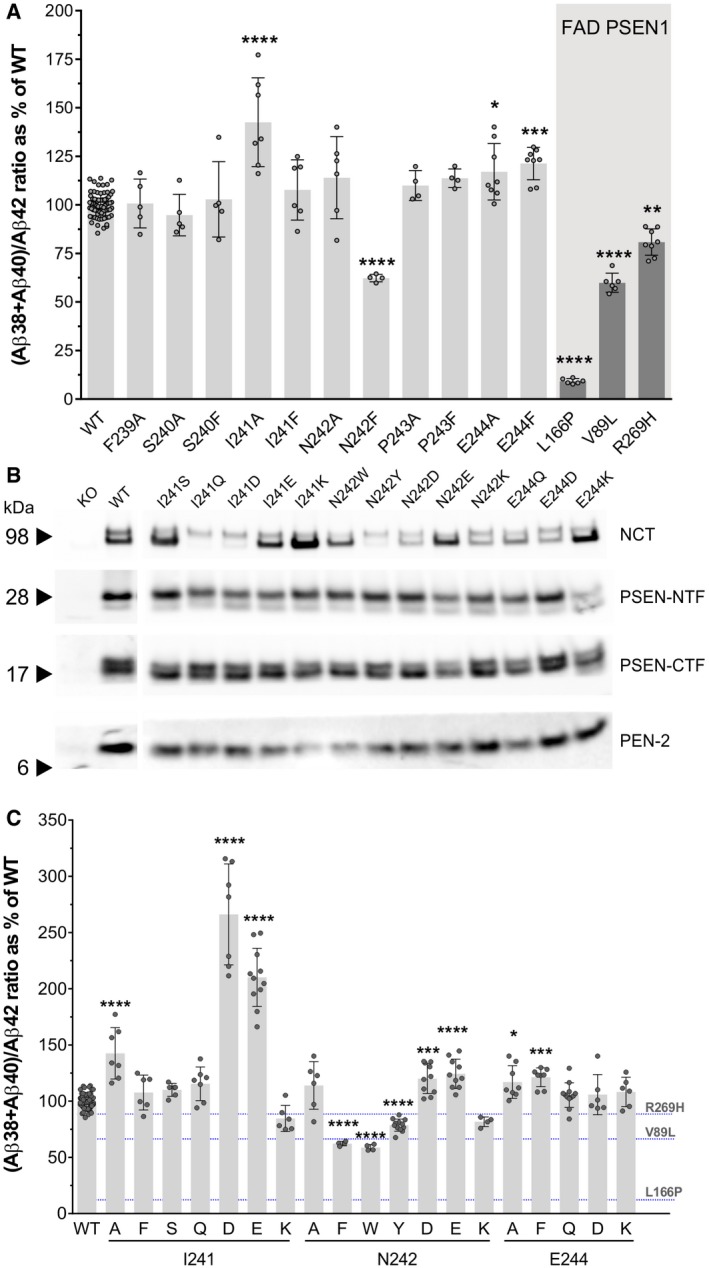
With regard to NCT, Ala/Phe substitutions of aa NCT‐I241, NCT‐N242 and NCT‐E244 significantly altered the Aβ (38 + 40)/42 ratio, supporting the involvement of NCT ectodomain in the regulation of the sequential GSEC cleavage of APP (Fig 2A). Interestingly, substitutions at these positions shifted enzyme processivity in both directions, with the NCT‐I241A substitution, and to a lesser extent the E244A and E244F mutants, enhancing the efficiency of the sequential GSEC cleavages on APP, and the NCT‐N242F mutation hindering the successive proteolysis. Notably, the NCT‐N242F substitution reduced GSEC processivity of APP to the levels similar to those exhibited by the FAD pathogenic PSEN1‐V89L variant (Fig 2A).
Figure 2. NCT ectodomain regulates GSEC processivity toward APPC 99 .
- Ala/Phe substitutions of aa 239‐244 were introduced in the NCT ectodomain to evaluate the role of this region in the regulation of GSEC processivity. Aβ38, Aβ40 and Aβ42 levels present in the conditioned medium collected from KO NCT MEF cells rescued with WT or respective mutant NCT GSECs and transiently expressing with APPC99 were quantified by ELISA. Aβ(38 + 40)/42 ratio was calculated to determine GSEC processivity toward APPC99. DKO PSEN1/PSEN2 MEFs rescued with the indicated FAD PSEN1 mutant and transduced with APPC99 were used as references.
- Representative SDS–PAGE/Western blot analysis of CHAPSO‐solubilized membranes from KO NCT MEF cell lines stably expressing WT or mutant NCT subunits. The presence of mature, glycosylated NCT, N‐terminal and C‐terminal fragments of the endoproteolyzed PSEN1 and PEN‐2 (compared to NCT knock‐out (KO) cells) indicates that WT and mutated NCTs reconstitute GSEC complexes. Arrowheads indicate the position of molecular weight markers.
- Quantification of the effects of the indicated substitutions of aa 241, 242 and 244 in NCT on GSEC processivity of APPC99, estimated as indicated above by the Aβ(38 + 40)/42 ratio. Fig EV1B shows the corresponding Aβ42/40 ratios.
Based on these data, we selected positions NCT‐241, NCT‐242 and NCT‐244 for further analysis. In the second round of site‐directed mutagenesis, we replaced the hydrophobic isoleucine at the position 241 with polar aa of small–medium size (Ser and Gln) or with differentially charged side chains (Asp, Glu and Lys). As shown in Fig 2B, all generated mutants reconstituted active GSEC complexes. Our analysis revealed that the introduction of negatively charged residues (Asp or Glu) resulted in a (mean ± SD) 2.66 ± 0.45 and 2.1 ± 0.26 fold elevation in the Aβ (38 + 40)/42 ratio, respectively (Fig 2C), while other tested mutations at this position did not significantly alter this ratio. Furthermore, the differential effects of the I241E vs. I241Q mutants on the Aβ (38 + 40)/42 ratio suggest that a charge–charge interaction underlies the activation of the sequential GSEC‐mediated proteolysis observed for the NCT I241E substitution. To test whether the introduction of a negative charge next to I241 would also increase GSEC processivity, we mutated NCT‐N242 to Asp or Glu. To investigate further the role of position N242 on GSEC processivity of APP, we introduced bulky residues with polar (Tyr, Trp) or charged (Lys) side chains at this position, generated mutant cell lines, and tested them in cell‐based GSEC activity assays. The analysis showed that all mutants at position 242 rescue GSEC complex formation (Fig 2B) and that introduction of a bulky aromatic group at position NCT‐242 is detrimental for GSEC processivity (Fig 2C). Furthermore, it revealed that the NCT‐N242E/D mutations, in contrast to Asp/Glu residues at position 241, result in mild increments in the Aβ (38 + 40)/42 ratio, indicating that a charge–charge interaction with activating effects on GSEC processivity of APP is optimally established with negatively charged NCT residues at position 241. Of note, mutagenesis of the E244 residue did not reveal any additional substitutions affecting GSEC processivity in a significant manner. In conclusion, the data demonstrate that residues 241, 242, and to a lesser extent 244, in NCT ectodomain contribute to the mechanism(s) governing the efficiency of the successive GSEC‐mediated cleavage on APP and thus modulate the length of Aβ product peptides.
Residues 241‐242 in NCT ectodomain regulate the stability of GSEC‐APP/Aβ interactions
We then investigated whether the observed effects of mutations at positions NCT‐I241 and NCT‐N242 on GSEC processivity of APP are mediated by changes in the stability of GSEC‐APP/Aβ complexes. To this end, we evaluated temperature‐mediated destabilization of GSEC‐APP/Aβ interactions in wild‐type and mutant NCT MEF cell lines, transiently expressing APPC99 substrate, cultured at 42°C. Importantly, we first determined that prolonged incubation at 42°C does not affect cell viability of wild‐type MEFs (Fig 3A) and that the Aβ (38 + 40)/42 ratio was sensitive to detect temperature‐induced changes in the stability of wild‐type and FAD‐linked PSEN1 L166P, V89L and R269H GSEC‐APP/Aβ assemblies. Clearly, the elevated temperature significantly affected the efficiency of the 4th APP cleavage mediated by the wild‐type enzyme, as demonstrated by the Aβ (38 + 40)/42 ratio (95% CI: 78–86% relative to 37°C, indicated with a blue box), and further enhanced the proven E‐S destabilizing effects (Szaruga et al, 2017) of the FAD‐linked PSEN1 mutants (Fig 2C vs. 3B, Aβ (38 + 40)/42 ratio (mean ± SD) = 9.2 ± 1.3%, 59.9 ± 4.9%, and 80.8 ± 6.8% at 37°C vs. 7.3 ± 0.66%, 41.9 ± 6.1%, and 70.4 ± 4.9% at 42°C for PSEN1 L166P, V89L and R269H mutants, respectively). These data validate the use of the cell‐based GSEC thermoactivity assay to assess the (de)stabilizing nature of structural variants in the GSEC complex on the APPC99 sequential proteolysis. Thus, we employed this assay to evaluate the potential stabilizing or destabilizing effects of mutant NCT‐I241E, NCT‐N242E or NCT‐N242Y on E‐S interactions. The cell‐based thermoactivity assay revealed that the NCT‐I241E and NCT‐N242E mutants counteract the detrimental effect of temperature on the Aβ (38 + 40)/42 ratio, indicating that their effects on GSEC processivity are mediated by the stabilization of GSEC‐APP/Aβ interactions (Fig 3B). On the contrary, the NCT‐N242Y substitution, similar to FAD‐linked PSEN1 mutations, enhanced the detrimental effect of temperature on Aβ (38 + 40)/42 ratio. These results demonstrate that the aa residues 241–242 within the NCT ectodomain influence Aβ generation by modulating the strength of GSEC‐APP/Aβ interactions.
Figure 3. The NCT ectodomain contributes to the stability of GSEC‐APP/Aβn complexes and accordingly modulates Aβ product length.
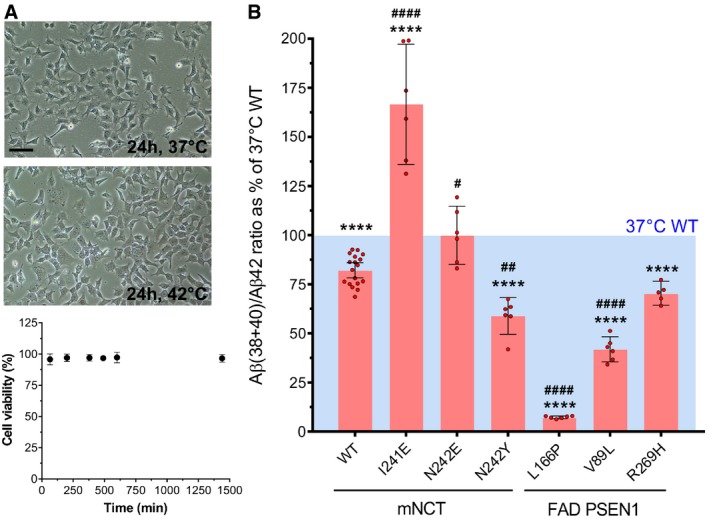
- (Top) Representative bright‐field microscopy images show MEF cells stably expressing WT or NCT mutant GSECs after 24‐h incubation at 37°C and 42°C. 25 μm scale bar is shown. (Bottom) Cell viability determined at the indicated time points is not affected by prolonged (≥ 24 h) incubation at 42°C. Data are presented as mean ± SD of 4 replicates.
- Relative GSEC‐APP/Aβn stabilities were assessed in cell‐based GSEC thermoactivity assays, which evaluate changes in GSEC processivity (Aβ(38 + 40)/42 ratio) upon incubation at 42°C for 24 h, relative to 37°C. Aβ peptides in the conditioned medium collected from the indicated MEF cell lines (transiently expressing APPC99) were quantified and the Aβ(38 + 40)/42 ratios determined. The data are shown as mean ± 95% CI, as % of the WT GSEC cell line incubated at 37°C for 24 h, N = 6 independent experiments. One‐way ANOVA followed by Dunnett's post hoc test in comparison with WT was used to determine the statistical significance (F(DFn, DFd): F(6, 46) = 100); ****P > 0.0001 compared to WT at 37°C, and # P > 0.05, ## P > 0.01 and #### P > 0.0001 compared to WT at 42°C. Note: Aβ profiles (including Aβ43 levels) normalized to total Aβ quantified in the cell‐based thermoactivity assays with the different mNCT WT/mutants are shown in Fig EV2.
NCT residue 241 establishes a direct, short‐distance interaction with the APP/Aβ substrate
To gain insights into the structural basis underlying the (de)stabilizing effects of NCT ectodomain on GSEC‐APP/Aβ interactions, we performed molecular dynamics (MD) simulations with the APP‐derived Aβ49 substrate bound to human GSEC as the co‐purifying peptide in the PDB:5FN3 structure (Fig 4A and I). Interestingly, the simulations suggested that human hNCT‐I242 (I241 in mNCT) and APPC99‐K28 are in close proximity to each other (mean Cα‐Cα distances of approx. 10 Å during the 3 μs sampling time). A negatively charged residue present at position hNCT‐242 induced the formation of a salt bridge between hNCT‐I242E (GSEC) and APPC99‐K28 in 34% of all sampled frames, while in 24% of all observations, the two side chains were bridged by a water molecule (Fig 4A, II and III). Subsequently performed post‐processing free energy calculations using molecular mechanics Poisson–Boltzmann surface area (MMPBSA) method (Genheden & Ryde, 2015) indicated that hNCT‐E242 indeed stabilizes GSEC‐APP/Aβ interactions by 2.90 ± 0.13 kcal/mol (mean ± SEM, with N = 1,500). Furthermore, the simulations supported the importance of the interactions between APP ectodomain and the first extracellular loop of PSEN1 (residues 109–118), a region reported previously to be involved in the substrate recognition (Takagi‐Niidome et al, 2015) and mutated in FAD.
Figure 4. Direct, short‐distance interaction between positions hNCT‐242 and APP/Aβ‐28 regulates GSEC processivity toward APPC 99 .
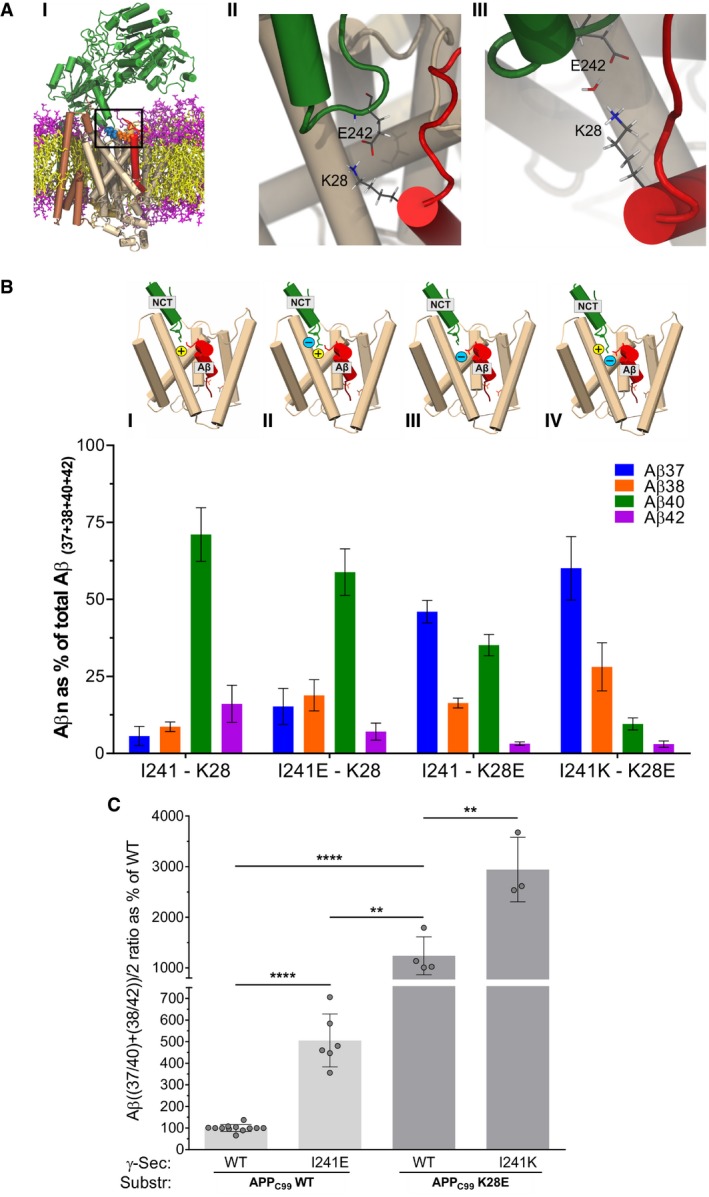
- Molecular dynamics simulations superimposing the cryo‐EM‐determined GSEC structure (PDB: 5FN3) with an NMR Aβ structure (PDB: 1IYT) on the co‐purifying helical peptide (I) present hNCT‐I242 and APPC99‐K28 at the NCT ectodomain–APP interface, and modeling of the NCT I242E mutant suggests a direct (II) or indirect (bridged by H2O, III) interaction between the NCT ‐E242 and the APPC99 ‐K28 side chains.
- Aβ profiles (% contribution of individual Aβ37, Aβ38, Aβ40 and Aβ42 peptides to the total Aβ levels (Aβ37 + 38 + 40 + 42)) in the conditioned medium collected from WT or mutant (NCT) MEF cells lines co‐expressing WT or mutant APPC99 substrates were assayed by ELISA. Schematic models of the tested GSEC‐APP complexes are shown.
- To account for the increased production of Aβ37 and Aβ38 at the expense of Aβ40 and Aβ42 in the tested conditions, the ratio of ((Aβ 37/40) + (Aβ 38/42))/2 was determined and used to estimate GSEC processivity of APPC99. All the data are presented as mean ± SD, N ≥ 3; unpaired, two‐tailed Student's t‐test was used to determine the statistical significance. **P > 0.01 and ****P > 0.0001. Calculated T‐ and Df values were as follows: T = 11,63, Df = 16 (I241‐K28 vs I241E‐K28); T = 11,37, Df = 14 (I241‐K28 vs. I241‐K28E); T = 4,572, Df = 8 (I241E‐K28 vs I241‐K28E); T = 4,497, Df = 5 (I241‐K28E vs. I241K‐K28E); and T = 2,561, Df = 7.
Based on the theoretical analysis, we speculated that the formation of a salt bridge between positions 241 in mouse NCT and K28 in APPC99 could explain the increments in the GSEC processivity of APP observed for the NCT‐I241E/D mutants (Figs 2C and 4B, I vs. II). To challenge this possibility experimentally, we “inverted” the putative salt bridge between NCT‐I241 and APPC99‐K28. If our hypothesis were correct, the “inverted” salt bridge (NCT‐I241K and NCT‐K28E) would also enhance GSEC processivity. Of note, previous studies have shown that K28 in APPC99/Aβ critically regulates Aβ length by an unknown mechanism (Ren et al, 2007; Kukar et al, 2011; Ousson et al, 2013; Jung et al, 2014). Accordingly, the single K28E APPC99 mutation shifted Aβ profiles (generated by the wild‐type GSEC) toward the generation of shorter Aβ peptides, relative to the wild‐type substrate (Fig 4B, I vs. III). Yet, the inclusion of the “inverted” salt bridge (I241K–K28E) further enhanced GSEC processivity of APPC99 (Fig 4B, III vs. IV), supporting a stabilizing role of the ionic interaction between NCT and APP/Aβ substrates.
ELISA analysis of Aβ profiles revealed a strong enhancement of the GSEC‐mediated generation of Aβ37 and Aβ38, at the expense of their precursors Aβ40 and Aβ42 in mutant NCT MEFs expressing mutant APPC99‐K28E substrates. Therefore, we included the contribution of the Aβ37 peptide in the estimation of GSEC processivity of APP by calculating the ((Aβ 37/40) + (Aβ 38/42)) ratio. The results clearly showed that the GSEC processivity of the mutant APPC99‐K28E was further enhanced when a positive charge was introduced at the position NCT‐241 (I241K) (Fig 4C, NCT‐WT–APPC99‐K28E vs. NCT‐I241K–APPC99‐K28E). This additive effect provides experimental evidence in support of a site‐specific interaction between NCT‐I241 and APPC99‐K28, with functional consequences on the efficiency of the sequential GSEC proteolysis.
To further verify the interaction between the aa at the positions 241 in NCT and 28 in APPC99, we performed chemical cross‐linking experiments using the heterobifunctional cross‐linker N‐β‐maleimidopropyl‐oxysuccinimide ester (BMPS) that presents amine‐ and thiol‐reactive groups connected by a 5.9 Å spacer arm (Fig 5A). To this end, we generated mutant NCT‐I241C and stably expressed it in the Ncstn −/− MEFs. SDS–PAGE/Western blot analysis of the GSEC complexes in the generated cell line showed the presence of the mature, fully glycosylated NCT as well as PEN‐2 and an efficient endoproteolytic generation of PSEN1‐NTF and PSEN1‐CTF, relative to the wild‐type condition (Fig EV3A), indicating that the mutant NCT‐I241C rescues GSEC complex formation and activity. Next, we expressed transiently the wild‐type APPC99 substrate in the wild‐type and the NCT‐I241C cell lines. In addition, we included non‐transduced cell lines as controls. To enhance the formation of the E‐S complexes and prevent their dissociation due to proteolysis, we treated the cells overnight with 1 μM GSEC inhibitor X, a transition state analog that halts substrate turnover but allows substrate binding to GSEC (Shearman et al, 2000). Then, we harvested the cells, prepared total membrane fractions, and subjected these fractions to a 2‐step cross‐linking protocol using BMPS. To evaluate the cross‐linking products, we detergent‐extracted membrane proteins and performed SDS–PAGE/Western blot analyses using 82E1 anti‐APPC99 (binds to the N‐terminus of APPC99/Aβ) and anti‐NCT antibodies (Fig 5B). Immunoblotting with the anti‐APPC99 antibody confirmed the expression of the substrate in the transduced cells (Fig 5B, lanes 3 and 4, bottom panel) and revealed the presence of a higher molecular weight band (~100 kDa) exclusively in the cross‐linked extracts prepared from the NCT‐I241C cell line overexpressing the APPC99 substrate (Fig 5B, lane 4 vs. 3, top panel). Further analysis with an anti‐NCT antibody showed that the electrophoretic mobility of the “high molecular weight” APPC99/Aβ‐positive band (cross‐linked NCT‐APPC99/Aβ complex) is slightly shifted toward a higher molecular mass, relative to the mobility of the mature, glycosylated NCT (Figs 5C and EV3C). The observed shift supports the cross‐linking of the wild‐type APP/Aβ substrate and mature mutant NCT‐I241C and furthermore indicates that the Cys241 in NCT is at least within 5.9 Å distance from a free amine group on the substrate, likely K28 in APPC99.
Figure 5. Short‐distance interaction between NCT 241 and APPC 99 K28.
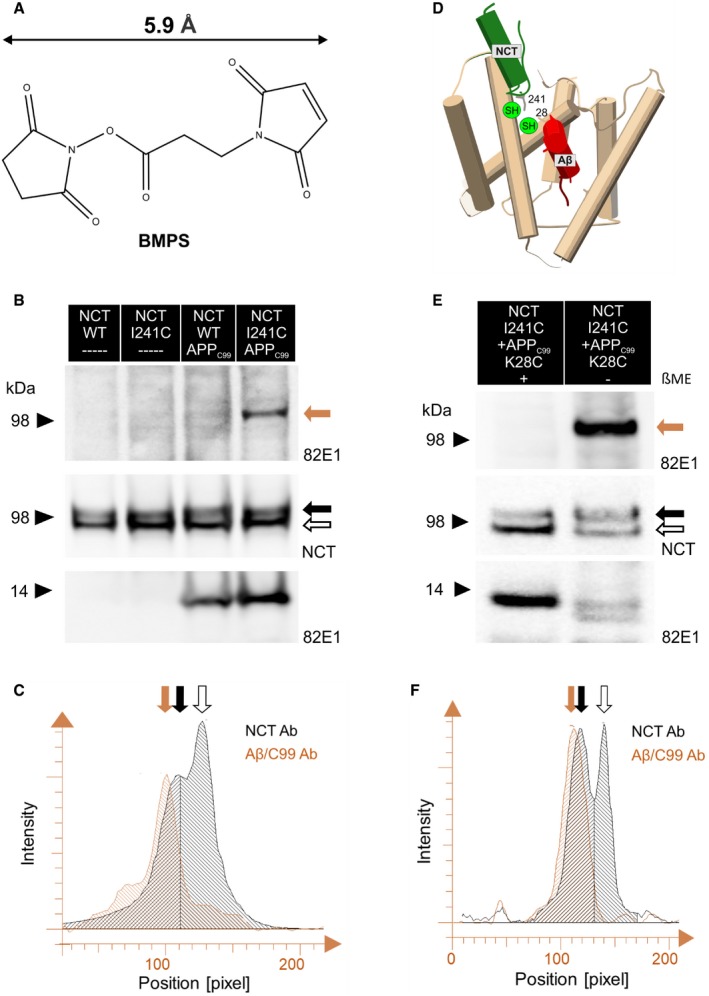
- Chemical structure of the BMPS hetero‐bifunctional (amine‐ and thiol‐reactive groups) cross‐linker.
- Introduction of Cys at position 241 in NCT allows chemical cross‐linking of NCT and APPC99 with BMPS. Representative SDS–PAGE/Western blot shows cross‐linked NCT‐APPC99, NCT and APPC99 bands in detergent‐extracted membrane proteins from BMPS cross‐linker‐treated membranes prepared from MEF cell lines expressing the indicated WT or mutant NCT GSEC complexes together with APPC99 or in the absence of the substrate. Orange, black and white arrows indicate the NCT‐APPC99/Aβ cross‐linked, mature NCT and immature NCT bands, respectively. Arrowheads indicate the position of molecular weight markers.
- Analysis of the Western blot density profiles in panel (B) shows that co‐migration of NCT (orange arrow) with APPC99 shifts the cross‐linked band toward a higher molecular weight than the observed for mature NCT (black arrow). Immature NCT is indicated with an open (white) arrow. Full‐range molecular weight density profiles are shown in Fig EV3C.
- Schematic representation of the GSEC‐APPC99 complex modified at NCT‐I241 and APP‐K28 by the introduction of Cys substitutions.
- Representative SDS–PAGE/Western blot presents NCT‐APPC99, NCT and APPC99 expression in detergent‐extracted membranes prepared from MEF cell lines co‐expressing the mutant NCT‐I241C GSEC complex with the APPC99‐K28C substrate. As negative control, the same sample was supplemented with β‐mercaptoethanol (BME) to reduce all disulfide bridges. A full‐range molecular weight Western blot is shown in Fig EV3B. Orange, black and white arrows indicate the NCT‐APPC99/Aβ cross‐linked, mature NCT and immature NCT bands, respectively. Arrowheads indicate the position of molecular weight markers.
- Analysis of the integrated density profiles of the respective Western blot bands supports the spontaneous formation of a disulfide bond between NCT‐I241C and APPC99 K28C (mature NCT and immature NCT are indicated with black and white arrows, respectively). The filled orange arrow points at the high molecular weight APPC99/Aβ density (cross‐linked NCT‐APPC99/Aβ band). Note that peak amplitudes for the cross‐linked NCT‐APPC99 and the mature NCT band were normalized to similar values for clarity purposes. Full‐range molecular weight density profiles are shown in Fig EV3D.
Source data are available online for this figure.
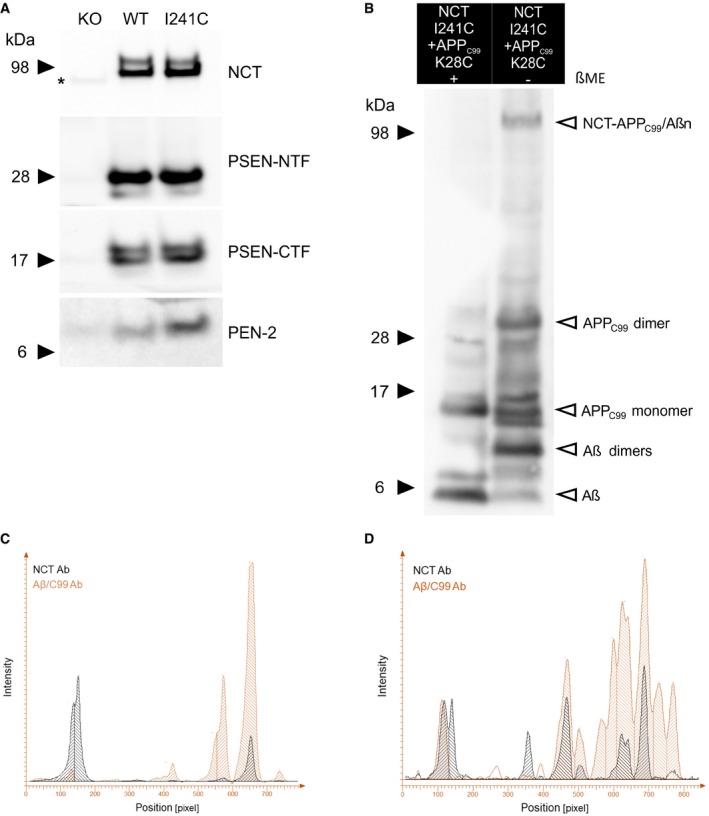
Figure EV3. Cross‐linking data demonstrate a short‐distance interaction between NCT 241 and APPC 99 K28.
-
ARepresentative SDS–PAGE/Western blot analysis of CHAPSO‐solubilized membrane proteins extracted from Ncstn−/− MEF cell lines stably expressing WT or mutant mNCT‐I241C subunits (shown in Fig 5). The presence of mature NCT, N‐terminal, and C‐terminal fragments of the endoproteolyzed PSEN1 as well as PEN‐2, compared to NCT knock‐out (KO) cells, indicates that WT and the I241C mutant reconstitute GSEC complexes. *Non‐specific band. Arrowheads indicate the position of molecular weight markers.
-
BRepresentative SDS–PAGE/Western blot presenting NCT‐APPC99/Aβ and APPC99 expression (as detected by 82E1 anti‐Aβ antibody) in detergent‐extracted membranes prepared from MEF cell lines co‐expressing respective NCT‐I241C mutant GSEC complex with APPC99‐K28C substrate. In the non‐reduced condition (‐ βME) bands corresponding to APPC99 and/or Aβ dimers are appearing, as indicated with arrowheads. To estimate the amount of substrate cross‐linked to GSEC (i.e., spontaneous disulfide bond formation between APPC99/Aβ and NCT), the NCT‐APPC99/Aβ band was quantified and normalized to “total APP/Aβ expression” (defined as the sum of the bands indicated with arrowheads). Mean efficiency ± SD = 15.4 ± 5.7%, N = 3 independent experiments. Arrowheads indicate the position of molecular weight markers.
- C, D
Source data are available online for this figure.
Based on the apparent short distance between NCT and APPC99, and to confirm the involvement of K28 in the direct interaction with NCT‐241, we tested the potential formation of a disulfide bond between these positions (Fig 5D). To this end, we expressed the APPC99‐K28C substrate in the NCT‐I241C cell line, prepared total membranes, and analyzed SDS‐extracted membrane proteins in SDS–PAGE/Western blot with anti‐APPC99/Aβ and anti‐NCT antibodies under reducing and non‐reducing conditions (Figs 5E and EV3D). The presence of a high molecular weight APPC99/Aβ‐positive band (~100 kDa) under non‐reducing conditions demonstrated the spontaneous formation of a disulfide bond between the NCT‐I241C and APPC99‐K28C mutants (Fig 5E and F). Quantification of the integrated densities of the high molecular weight band corresponding to the NCT‐APPC99/Aβ on the anti‐Aβ immunoblot revealed that ~15% (mean ± SD: 15.4 ± 5.7%, N = 3) of the substrate got cross‐linked to NCT (Fig EV3B). In conclusion, our analyses demonstrate that the direct, short‐distance interaction between NCT‐241 and APPC99‐28 modulates Aβ product profiles by stabilizing GSEC‐APP/Aβ interactions.
NCT ectodomain (residue 242) mediates the response to imidazole‐based GSEC modulators
Previous studies highlight K28 in APPC99 as a critical determinant of GSEC processivity of APP (Ren et al, 2007; Page et al, 2010; Kukar et al, 2011; Ousson et al, 2013; Jung et al, 2014) and show that, in addition to their effects on Aβ length, mutations at this position alter the response to GSMs (Page et al, 2010; Ousson et al, 2013; Jung et al, 2014), but the underlying mechanisms remain unknown. These intriguing findings and the short‐distance interaction between positions 28 in APPC99 and 241 in NCT prompted us to test the response of mutant (de)stabilizing NCT GSEC complexes to GSMs belonging to different classes (acidic vs imidazole‐based, Fig 6A). For this purpose, we transduced wild‐type and mutant (NCT‐I241E, NCT‐N242Y and NCT‐N242F) MEF cell lines with wild‐type APPC99, as described before, and treated the cell cultures with 0.3 μM, 1 μM GSM or vehicle control (0.1% DMSO) overnight. To evaluate the response of wild‐type and mutant GSEC complexes toward GSMs, we collected the cell media and quantified Aβ37, Aβ38, Aβ40 and Aβ42 levels by ELISA and determined Aβ profiles (Figs 6B and EV4A) as well as the Aβ37/Aβ40 and Aβ38/Aβ42 ratios (Fig EV4B and C). The results revealed that the acidic GSM activates exclusively the Aβ42 → Aβ38 cleavage (Fig 6C, P‐values for WT in columns 1 and 4), while the imidazole‐based GSMs II and III strongly activate both GSEC product lines, which is in agreement with previous findings (Page et al, 2010; Ousson et al, 2013). The GSMs II and III thus shifted Aβ profiles toward the production of the Aβ37 and Aβ38 peptides (blue and orange bars in Fig 6B, respectively), with concomitant reduction in the Aβ40 and Aβ42 species (green and purple bars in Fig 6B, respectively).
Figure 6. GSECs destabilized by the N242F and N242Y NCT mutations do not respond to imidazole‐based GSEC modulators.
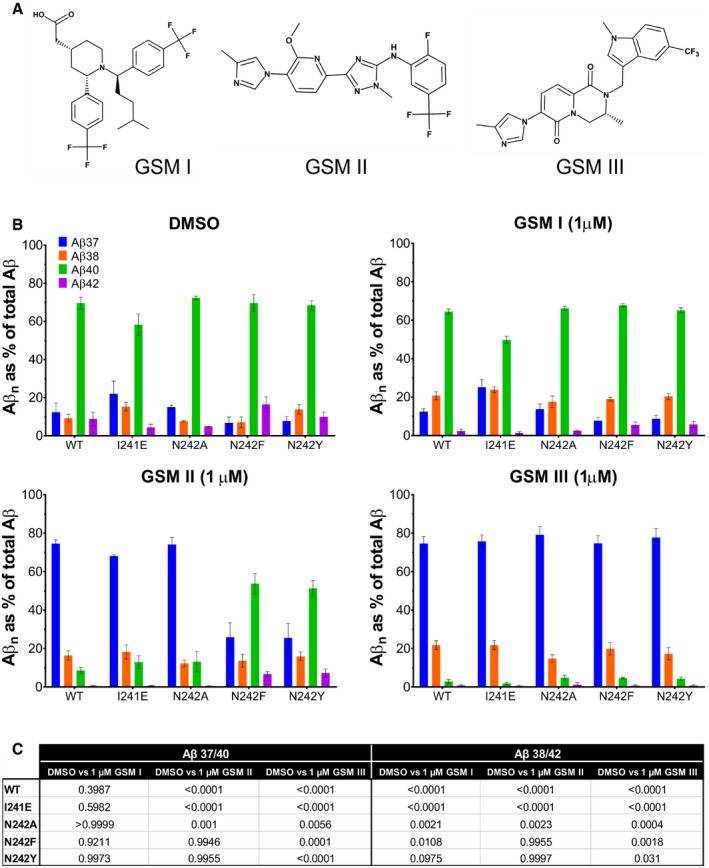
- Chemical structures of acidic GSEC modulator (GSM I) and imidazole‐based GSM II and GSM III (GSM B in Szaruga et al 2017).
- Aβ profiles (% contribution of individual Aβ37, Aβ38, Aβ40 and Aβ42 peptides to the total Aβ levels (Aβ37 + 38 + 40 + 42)) in the conditioned medium collected from vehicle (DMSO) or GSM‐treated (1 μM final concentration) MEF cells co‐expressing respective WT or mutant NCT GSEC with APPC99 were determined by ELISA. Data generated using 0.3 μM GSM‐treated MEF cells are shown in Fig EV4. Data are presented as mean ± SD, N ≥ 3 independent experiments.
- Aβ 37/40 and Aβ 38/42 ratios were calculated to evaluate the efficiency of the GSEC‐mediated cleavage of APPC99 (Fig EV4) and statistical analysis (P‐values) (two‐way ANOVA with Sidak's post hoc test) for the effect of GSMs on WT and mutant complexes is presented (response to GSM normalized to the corresponding vehicle condition).
Figure EV4. NCT ectodomain (N242) modulates the response toward imidazole‐based GSEC modulators.
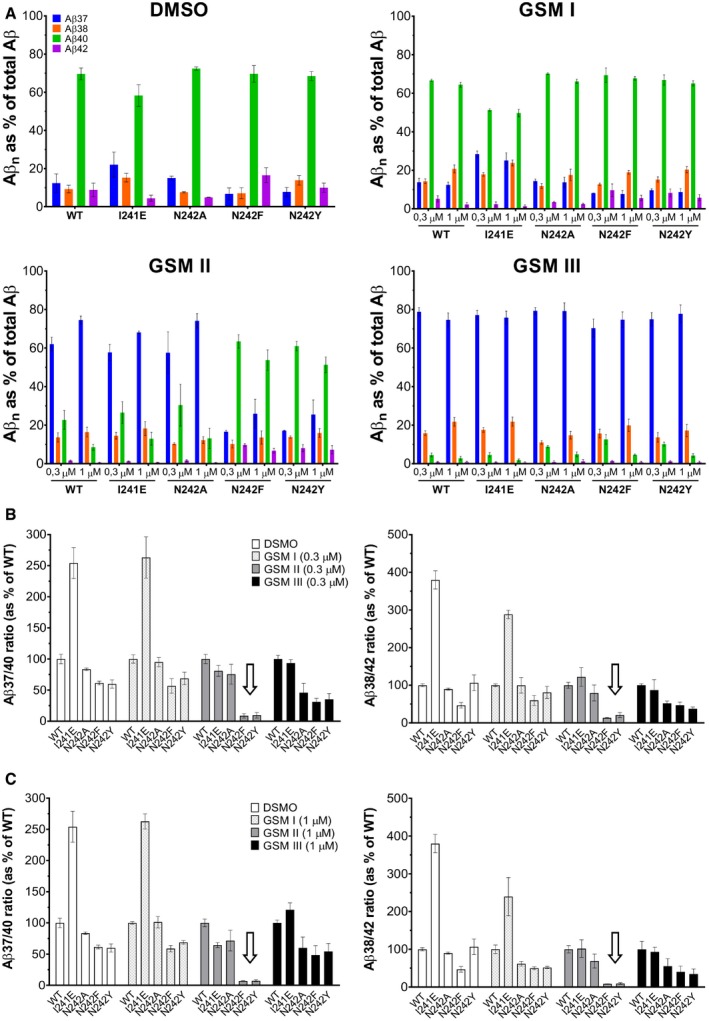
-
AAβ profiles generated by 0.3 and 1 μM GSM (I‐II‐III) or vehicle control (0.1% DMSO)‐treated mNCT mutant MEF cell lines (shown in Fig 6).
-
B, CAβ 37/Aβ40 and Aβ 38/Aβ42 ratios were quantified to evaluate the effect of GSM 0.3 μM (B) or 1 μM (C) treatment on the processivity of different mNCT mutant GSECs (shown in Fig 6). The Aβ ratios are normalized to the response of the wild‐type GSEC toward vehicle (0.1% DMSO condition) or GSM treatment. The arrows indicate the lack of response to GSM II of the N242F/Y substitutions.
Our analysis revealed that wild‐type as well as mutant GSEC complexes responded markedly to GSM I treatment (right upper panel in Fig 6B), with the exception of the NCT‐N242Y GSEC mutant which showed a partial response (Figs 6C and EV4, P‐values for N242Y in column 4). Most importantly, we observed that the NCT‐N242Y and NCT‐N242F mutant GSECs did not respond to GSM II, as indicated by the Aβ profiles (Fig 6B) showing that Aβ40 (green bar) remains the major Aβ peptide produced by these mutant cell lines in the presence of 1 μM GSM II. In fact, statistical analysis on the Aβ37/Aβ40 and Aβ38/Aβ42 ratios reveals no significant differences between the processivities of the NCT‐N242Y and NCT‐N242F mutant cell lines treated with vehicle (DMSO) or 1 μM GSM II (Fig 6C, P‐values for N242F and N242Y in columns 2 and 5). In contrast, the NCT‐N242A GSEC mutant responded similarly to WT toward GSM II (Fig 6B and C, P‐values for N242A in columns 2 and 5). All together, these data demonstrate that bulky residues at the NCT‐242 position not only impair the stability of GSEC‐APP/Aβ assembly (and therefore enzyme processivity, as shown in Fig 2C) but also have an impact on the activation of GSEC by the imidazole‐based GSM II. These results could be explained by the contribution of residue NCT‐242 to the binding pocket of GSM II or to its participation in the allosteric mechanisms triggered upon GSEC‐GSM interaction. We then reasoned that analysis of the mutant responses to another imidazole‐based compound could enable to distinguish between these two scenarios. Interestingly, we found that the NCT‐N242Y and NCT‐N242F mutant GSECs did respond to GSM III, as indicated by the relative increments in Aβ37 (blue bars) in the right lower panel of Fig 6B and the significant changes in the Aβ 37/40 and Aβ 38/42 ratios for DMSO‐ vs. 1 μM GSM III‐treated mutant cell lines (Fig 6C, P‐values for N242F and N242Y in columns 3 and 6). These findings thus demonstrate that the mechanisms underlying GSEC processivity of APP remain intact in the mutant NCT‐N242F and NCT‐N242Y protease complexes, and point toward an altered binding of imidazole‐based GSM II to NCT‐N242Y and NCT‐N242F GSECs. Furthermore, our studies also reveal a reduced response of the NCT‐N242Y, NCT‐N242F and NCT‐N242A mutant GSEC complexes to GSM III in the presence of 0.3 μM GSM III compared to the wild‐type protease (Fig EV4). In conclusion, the differential responses of GSEC complexes bearing NCT‐N242A, NCT‐N242Y and NCT‐N242F mutants toward imidazole‐based GSM II and III (Fig 6B and C) suggest partially distinct binding sites for these compounds. Our studies therefore assign a direct and active role to the ectodomain of NCT in the GSEC modulation by GSMs.
Discussion
We have recently demonstrated that the stability of GSEC‐APP/Aβn substrate interactions controls the length of Aβ product peptides and thereby affects the risk for AD. Notably, our previous findings show that FAD‐causing mutations destabilize GSEC‐APP/Aβ (E‐S) complexes and the magnitudes of their detrimental effects remarkably correlate with age of disease onset (Szaruga et al, 2017). The novel pathogenic link motivated us to investigate the intermolecular interactions that secure GSEC‐Aβn assemblies during the sequential GSEC‐mediated proteolysis. We departed from the high‐resolution structure of GSEC in complex with a co‐purifying helical peptide (PDB:5FN3, Bai et al 2015a). We hypothesize that this structure depicts the interaction between GSEC and the fragment generated after the initial proteolytic cleavage of the transmembrane domain of the substrate (de novo generated long Aβ from APPC99), just before it engages into the next catalytic turnover. The putative substrate occupies the substrate binding site, but in contrast to APPC83 (Zhou et al, 2019), only the most N‐terminal part of its transmembrane domain remains in a helical structure, while its unstructured C‐terminal part extends along the substrate binding channel to reach the active site. We actually proposed that the first endopeptidase‐mediated backbone break exerts a strong destabilizing effect on the helical structure of APPC99, leading to this configuration. In our model, further unwinding of the N‐terminal helix of the substrate must occur upon each GSEC cut, in order to provide the length of the substrate to fill the S1′–S3′ enzyme pockets (Bolduc et al, 2016b) during the stepwise catalysis. The sequential unwinding of the N‐terminal helix progressively destabilizes the E‐S assembly, increasing the probability of its dissociation and consequent release of Aβn. This “unwinding model”—originally proposed in Szaruga et al (2017)—suggests that the interactions established between the N‐terminal helical structure of the substrate and the protease “anchor” the E‐S complex and thereby define the length of the N‐terminal products (Aβ from APP). Interestingly, our analysis identified a potential contact site between the ectodomains of NCT (aa residues 241–242 in mNCT) and the co‐purifying peptide (putative substrate). Accordingly, we hypothesized that the potential “NCT‐APP/Aβ” interface could directly contribute to the stability of E‐S interactions and thereby modulate the length of Aβ products. Note that the established role of the non‐catalytic subunits (NCT, PEN‐2 and APH1) of the protease in PSEN/GSEC function and turnover (Gertsik et al, 2015; Carroll & Li, 2016) supports their contribution to the stability of GSEC‐APP/Aβ complexes.
Our studies revealed differential contributions of residues 241–242 in NCT to GSEC‐Aβn interactions, implicating for the first time the ectodomain of NCT in the mechanisms controlling Aβ length. Interestingly, we showed that aromatic substitutions at the position NCT‐242 (N242F/W/Y) significantly reduce GSEC processivity of APP to the levels similar to those exhibited by FAD pathogenic PSEN1 variants (Figs 2C and EV1B), while the presence of negatively charged residues at the position NCT‐241 promotes the production of shorter Aβ peptides. Further, cell‐based GSEC thermoactivity analysis demonstrated that the mutant NCT induced shifts in Aβ profiles by influencing the stability of GSEC‐APP/Aβn interactions (Fig 7).
Figure 7. Proposed model for GSEC‐mediated cleavage of APP and its modulation by GSMs.
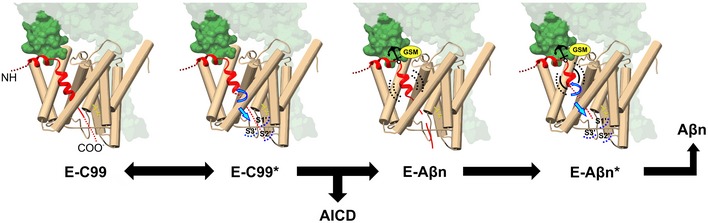
Once the E‐S complex is formed, the first endopeptidase cut strongly destabilizes the helical transmembrane domain of APPC99, leading to the unwinding of the most C‐terminal helical part of the Aβ substrate and providing the length to the “de novo” generated Aβ substrate to reach the active site (model originally proposed in Szaruga et al (2017)). The further unwinding of the Aβ substrate with each sequential cleavage stretches the substrate and provides the length to fill the catalytic pockets but weakens the GSEC‐Aβn interaction, until the eventual E‐S dissociation triggers Aβ release. Here, we propose that the extracellular interface that includes NCT (241/242), APP (K28) and the first extracellular loop of PSEN1 anchors GSEC‐APP/Aβn complexes during the sequential proteolytic mechanism, and the E‐S interface is the target of selected imidazole‐based GSMs.
The theoretical GSEC‐Aβ model (developed here) pointed toward the existence of a positively charged residue in the APP substrate and suggested a direct interaction between positions I242 in hNCT and K28 in APPC99, which was confirmed in cross‐linking experiments. Of remark, the spontaneous formation of a disulfide bond between NCT and APP (when Cys residues were introduced at positions 241 in mNCT and 28 in APPC99) defined it as a short‐distance interaction surrounded by an aqueous environment, and thus likely located in the extracellular/luminal environment (Rehder & Borges, 2010).
During the preparation of this report, the co‐structures of the GSEC with the Notch or APPC83 substrate became available (Yang et al, 2018; Zhou et al, 2019). Interestingly, the GSEC–Notch complex shows an extracellular interface between the ectodomains of the NCT subunit and Notch, with the NCT‐241–242 aa located at the E‐S interface (Fig EV5A). The GSEC‐APPC83 complex shows the APP‐K12 residue (corresponding to K28 in APPC99) in the proximity to the NCT‐241 residue; however, since the interface in the GSEC‐APPC83 structure is not fully resolved, it is possible that the cross‐linking of GSEC (PSEN1‐Q112C) to the APPC83 V8C substrate affected this region in the atomic model (Fig EV5B). The functional data presented here, together with the novel GSEC–substrate co‐structures (Yang et al, 2018; Zhou et al, 2019), suggest a conserved mechanism for the stabilization of GSEC–substrate interactions and may support the involvement of NCT ectodomain in the substrate recognition by the GSEC complex, by stabilizing the initial GSEC–substrate assembly. Of note, the ectodomain of NCT was initially proposed to act as a substrate receptor in the protease, with the anionic NCT‐E322 residue and the free N‐terminal amine group of the substrate interacting (Shah et al, 2005). However, the model has been challenged by biochemical studies (Chávez‐Gutiérrez et al, 2008; Zhao et al, 2010) and the recent E‐S structural data finally disproved it. The currently accepted model for NCT function assigns to its ectodomain a rather passive role in the “substrate selection”, in which it restricts—by steric hindrance—the access of substrates presenting large ectodomains (Bolduc et al, 2016a). Our data provide compelling evidence for additional, novel implications of the NCT ectodomain, i.e., its active role in the regulation of the GSEC processivity of APP and the modulation of the response to GSEC modulators (GSMs). Previous reports have shown that APP‐K28 mutations exert profound effects on Aβ generation via an unknown mechanism (Ren et al, 2007; Kukar et al, 2011) and alter the response to GSMs (Page et al, 2010; Ousson et al, 2013; Jung et al, 2014). Here, we demonstrate that the effects of K28 on APP processing are explained in part by its interaction with the ectodomain of NCT.
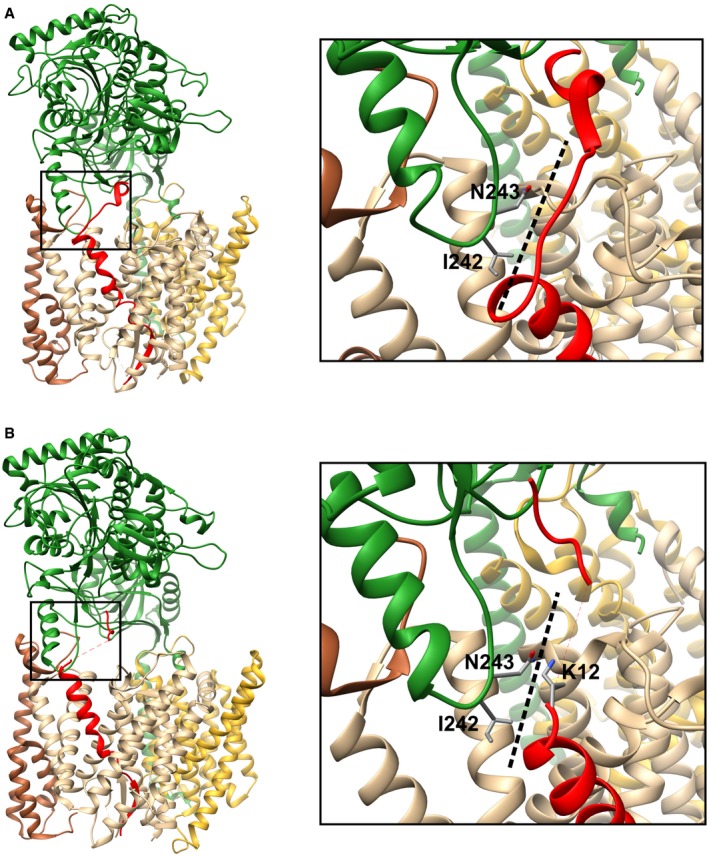
Figure EV5. NCT‐APP interface highlighted in the 6IDF and 6IYC GSEC–substrate co‐structures.
-
A, BGSEC co‐structure with Notch (PDB:6IDF) (A) or APPC83 (PDB:6IYC) (B) substrate is presented. PSEN1 is shown in light brown, PEN‐2 in dark brown, APH1A in gold, NCT in green, and Notch/APPC83 in red. The side chains of the NCT‐I241 and NCT‐N242 (together with K12 (corresponding to K28 in APPC99) in the APPC83 structure) are shown, and the interface between Notch/APPC83, NCT ectodomain, and PSEN1 is magnified and marked with a dashed line.
The proven direct, short‐distance interaction between K28 in APPC99 and position 241 in NCT ectodomain (Fig 5) and their shared implication in the regulation of GSEC processivity motivated us to investigate the effects of the (de)stabilizing mutations at positions NCT‐241/242 on the GSEC response to GSMs. Remarkably, our data revealed that the substitutions in NCT‐242 that destabilize GSEC‐APP/Aβ interactions (N242F and N242Y, as demonstrated in Fig 3) also had strong deleterious effects on the response to imidazole‐based GSM II in both production pathways (> 90% reduction for the mutant NCT‐N242F and NCT‐N242Y cell lines at 1 μM GSM II relative to the wild‐type GSEC, Fig EV4). Clearly, GSM II promoted the conversion of Aβ40 into Aβ37 and Aβ42 into Aβ38 by the wild‐type protease but failed to engage the NCT‐N242F/Y mutant GSEC complexes on these specific cuts. The similar responses of the wild‐type and NCT‐N242A mutant GSECs indicate that bulky substitutions at position NCT‐242 not only destabilize GSEC‐APP/Aβ interactions, and accordingly change Aβ product profiles, but also impair drastically the response to GSM II. The results could be explained by the contribution of residue NCT‐242 to the formation of the binding pocket for GSM II or by its participation in the allosteric mechanisms triggered upon GSEC‐GSM interaction. In this regard, our studies revealed that the NCT‐N242F/Y mutant GSEC complexes largely respond to GSM III. These observations support the contribution of NCT to the formation of the binding pocket for imidazole‐based GSMs, rather than its participation in the mechanisms underlying the response. Of note, the binding site of another imidazole‐based GSM (ST2038, structurally similar to the GSM II) has been located to the first extracellular loop of PSEN‐NTF (Takeo et al, 2014), which according to structural data is in proximity to the identified NCT‐APP interface. Furthermore, the novel GSEC‐Notch/APPC83 co‐structures place the substrate in between the studied NCT region and the first extracellular loop of PSEN1 (Fig EV5) supporting our conclusions. Finally, a cross‐competition study between the imidazole‐based GSM (ST2038) and the acidic GSM I shows that pre‐treatment with GSM I substantially decreases the affinity labeling of the GSEC complex by the ST2038 GSM (Takeo et al, 2014), suggesting partially overlapping binding pockets for acidic and imidazole‐based GSMs. The lack of response of the mutant NCT‐N242Y to GSM I observed here supports a similar conclusion.
Of potential clinical significance, the marked impact of the aa substitutions within NCT ectodomain on APP processing suggests the potential existence of AD‐causing or protective single‐nucleotide variants (SNVs) in the NCSTN (i.e., NCT encoding) gene. The data specifically highlight the position 242 in NCT, where a single‐nucleotide change—N242Y substitution—lowers GSEC processivity to the levels associated with pathogenic PSEN1 mutations. Intriguingly, no AD pathogenic or protective mutations have been identified so far in NCT or in any of the non‐catalytic subunits of the GSEC complex. It should be noted that in clinical practice, genetic testing varies between different centers and generally involves testing specifically for known genetic causes of dementia syndromes (Koriath et al, 2018). However, a high proportion of the genetic risk for AD remains uncharacterized. Rare highly penetrant mutations in PSEN1, PSEN2 and APP are thought to account for only 5‐10% of all early‐onset AD (EOAD, onset < 65 y) cases, while the heritability of EOAD has been estimated to range from 92 to 100%, thus leaving 90–95% of the affected families with an unidentified genetic cause for the disease (Cacace et al, 2016). In late‐onset AD, which has a heritability of 70–80%, five new genetic risk loci have recently been identified, with pathway analysis implicating immunity, lipid metabolism, tau binding proteins and APP metabolism (Kunkle et al, 2019). Furthermore, the enrichment of rare variants in the APP metabolism in this study suggests that additional rare variants remain to be identified.
Collectively, the presented data demonstrate for the first time the existence of an extracellular NCT‐APP interface implicated in the modulation of the strength of GSEC‐APP/Aβ interactions (and thus Aβ length) as well as in the response of E‐S complexes to GSMs (E‐S stabilizers). Based on our original findings, the novel E‐S structural data (Yang et al, 2018; Zhou et al, 2019) and the literature, we propose that the extracellular GSEC (NCT‐PSEN)‐APP interface encompassing the aa region 241‐242 in NCT ectodomain, K28 in APP and the first extracellular loop of PSEN plays a critical role in the stabilization of GSEC‐APP/Aβn interactions and is involved in the binding of selected imidazole‐based GSMs. Notably, the idea of selectively targeting APP processing with substrate targeting compounds (Kukar et al, 2008) has been raised in the past but has been abandoned due to studies, showing that potent pharmaceutics modulating Aβ length bind rather to the protease complex. Our studies may conciliate these views.
Conclusions
This study demonstrates that NCT plays an active role in the regulation of the efficiency of the sequential GSEC cleavage of APP by establishing a direct interaction with the APP/Aβn substrate. The results of the cross‐linking experiments of NCT and APPC99 validate the position of the co‐purifying helical peptide in the PDB:5FN3 GSEC structure as the substrate binding site for APP, and thus put this co‐structure as a suitable model for the investigation of the structural–functional relationships existing within the GSEC‐Aβn complexes. Our results also suggest that an extracellular interface between GSEC and APP could be the target for GSMs (E‐S stabilizers). These novel insights may conciliate different views on the location of GSM binding pocket(s) and guide future efforts to develop safe therapies targeting GSEC‐APP/Aβ assemblies, i.e., the generation of GSEC stabilizing compounds (GSSCs). Finally, this study raises additional considerations with potential implications for the clinic, as the remarkable involvement of NCT in the regulation of the Aβ product length suggests a potential link between single‐nucleotide variants in NCSTN and AD risk.
Materials and Methods
Antibodies and compounds
Following antibodies were used in the study: anti‐FLAG M2 (F3165) from Sigma‐Aldrich, anti‐PSEN‐CTF (MAB5232) from Merck, anti‐human PSEN‐NTF (MAB1563) from Millipore, anti‐NCT (#612290) from BD Biosciences, horseradish peroxidase (HRP)‐conjugated anti‐mouse (#1721011) and anti‐rabbit IgG (#1721019) from Bio‐Rad, HRP‐conjugated anti‐rat IgG (#P0450) from Agilent and anti‐human amyloid‐beta (N) (82E1, #10323) from IBL. Anti‐mouse PSEN‐NTF (B19.3), PSEN‐PEN‐2 (B126.2) and PSEN‐APP‐CTF (B63) antibodies were made in‐house. Antibodies used in the ELISA and GSMs were obtained through collaboration with Janssen Pharmaceutica NV (Beerse, Belgium). The ELISA capture antibodies were JRD/Aβ37/3 for Aβ37, JRF AB038 for Aβ38, JRF/cAb40/28 for Aβ40 and JRF/cAb42/26 for Aβ42, and the detection antibody was JRF/AbN/25 raised against the N‐terminus of Aβ. Acid‐based GSM I (2‐[(1R,2S)‐1‐[4‐methyl‐1‐[4‐(trifluoromethyl)phenyl] pentyl]‐2‐[4‐(trifluoromethyl)phenyl]‐4‐piperidyl] acetic acid) and imidazole‐based GSM II (N‐[2‐fluoro‐5‐(trifluoromethyl)phenyl]‐5‐[3‐methoxy‐4‐(4‐methylimidazol‐1‐yl) phenyl]‐2‐methyl‐1,2,4‐triazol‐3‐amine) and GSM III ((3R)‐3‐methyl‐7‐(4‐methyl‐1H‐imidazol‐1‐yl)‐2‐{[1‐methyl‐5‐(trifluoromethyl)‐1H‐indol‐3‐yl]methyl}‐1H, 2H, 3H, 4H, 6H‐pyrido [1,2‐a] pyrazine‐1,6‐dione) were synthesized as described previously (Crump et al, 2011; Velter et al, 2014). GSEC inhibitor X (#565771) was purchased from Calbiochem.
Generation of stable cell lines
Ncstn −/− (Li et al, 2003) and Psen1 −/− Psen2 −/− (Nyabi et al, 2002) mouse embryonic fibroblasts (MEFs) were cultured in Dulbecco's modified Eagle's medium (DMEM)/F‐12 (Life Technologies) containing 10% fetal bovine serum (FBS) (Life Technologies). Ncstn −/− MEFs were transduced with retroviruses, carrying pMSCVpuro plasmids encoding respective wild‐type or mutant (F239A, S240A, S240F, I241A, I241C, I241D, I241E, I241S, I241F, I241K, I241Q, I241W, N242A, N242D, N242E, N242F, N242K, N242W, N242Y, P243A, P234F, E244A, E244D, E244F, E244K and E244Q) NCTs, using a replication‐defective recombinant retroviral expression system (Clontech). Human PSEN1 MEF cell lines were generated by rescuing PSEN expression in knock‐out Psen1 −/− Psen2 −/− MEFs with either wild‐type human or mutant PSEN1 (V89L, L166P, R269H) variants. Briefly, retroviruses were generated by co‐transfecting HEK293T cells with pMSCVpuro wild‐type or mutant NCT or human PSEN1‐encoding plasmids and the PIK helper plasmids, as reported previously (Chávez‐Gutiérrez et al, 2008). Viral particles were harvested 48 h post‐transfection and used to infect Ncstn −/− or Psen1 −/− Psen2 −/− fibroblasts plated at 30–40% confluency. Culture medium was supplemented with 5 μg/ml puromycin (Sigma‐Aldrich) to select clones stably expressing PSEN or NCT, which were further maintained in DMEM/F‐12, 10% FBS and 3 μg/ml puromycin. NCT and PSEN1 expression levels and the reconstitution of the functional GSEC complexes in the different cell lines were analyzed by SDS–PAGE/Western blot.
Transduction of MEF cell lines with adenoviruses carrying APP‐encoding plasmids for cell‐based activity assays
Mouse embryonic fibroblast cell lines were transduced with recombinant adenoviruses carrying plasmids encoding APPC99, as described previously (Chávez‐Gutiérrez et al, 2008). Briefly, MEF cells stably expressing either WT or mutants mNCT/hPSEN1 were plated at the density of 1 × 105 cells/well into 12‐well plates and 16 h later transduced with recombinant adenoviruses Ad5/CMV‐APP. Seven hours post‐transduction, the medium was refreshed with low‐serum medium (DMEM/F‐12 medium containing 0.2% serum). The cells were then incubated for 24 h at 37°C or 42°C, respectively, and the conditioned medium was collected for Aβ analysis (Szaruga et al, 2017). When indicated, the low‐serum culture medium was supplemented with 0.3 μM or 1 μM GSMs (GSM I, GSM II and GSM III) or vehicle control (0.1% DMSO final concentration).
MEF electroporation with plasmids encoding wild‐type or mutant APPC99‐3xFLAG substrates
Mouse embryonic fibroblast cell lines were electroporated with 5 μg pSG5**APPC99‐3xFLAG‐WT or pSG5**APPC99‐3xFLAG‐K28E plasmids using the NEPA21 super electroporator transfection system (Nepagene). Freshly trypsinized cells were set to a concentration of 1.5 × 106 cells/ml in OPTI‐MEM (Thermo Fisher Scientific) and 100 μl of the cell suspension was mixed with 5 μg of DNA, and transferred to a Nepa 2 mm gap cuvette (#EC‐002S, Nepagene) for electroporation (poring pulse (voltage = 140V, length = 7.5 ms, interval = 50 ms, # of pulses = 2, decay rate = 10% and polarity = +); transfer pulse (voltage = 20V, length = 50 ms, interval = 50 ms, # of pulses = 5, decay rate = 40% and polarity = +/−)). The electroporated cells were mixed with 400 μl DMEM/F‐12 medium containing 10% FBS and 3 μg/ml puromycin immediately after the electroporation and plated into 24‐well plates. The medium was refreshed with low‐serum medium 12 h post‐electroporation and collected 36 h later for the Aβ quantification.
Quantification of Aβ production by (MSD) ELISA
To determine the GSEC processivity of APP, Aβ37, Aβ38, Aβ40, Aβ42 and Aβ43 levels in conditioned medium (cell‐based assays) were measured. To quantify the concentration of Aβ37, Aβ38, Aβ40 and Aβ42 peptides, Multi‐Spot 96‐well MSD‐ELISA plates pre‐coated with anti‐Aβ37‐, anti‐Aβ38‐, anti‐Aβ40‐ and anti‐Aβ42‐specific antibodies using the multiplex Meso Scale Discovery (MSD) technology were used. Non‐specific protein binding to the plates was blocked with 150 μl/well blocking buffer (PBS supplemented with 0.1% casein) for 1.5 h at room temperature (while shaking at 600 rpm), and the blocked plates were rinsed 5 times with 150 μl/well washing buffer (PBS supplemented with 0.05% Tween‐20). 25 μl of SULFO‐TAG JRF/AbN/25 detection antibody diluted in blocking buffer was mixed with 25 μl of standards (synthetic human Aβ1‐37, Aβ1‐38, Aβ1‐40 and Aβ1‐42 peptides at known concentrations) or 25 μl analyzed samples, both diluted in blocking buffer, and the mix (50 μl/well) was loaded on the plate for ELISA analysis. After overnight incubation at 4°C, the plates were rinsed 5 times with washing buffer and developed by the addition of 150 μl/well of the 2 × MSD Read Buffer T (Tris‐based buffer containing tripropylamine). The signals were read immediately on a Sector Imager 6000 (Meso Scale Discovery). To quantify the concentration of the Aβ43 peptide in cell‐based assays, conditioned medium samples were loaded on the ELISA plates pre‐coated with anti‐human Aβ (1‐43) rabbit IgG, supplied with the human amyloid‐β (1‐43) (FL) assay kit (IBL) and peptide levels were measured following the supplier's protocol.
Cross‐linking in MEFs
Mouse embryonic fibroblast cells stably expressing either wild type of I241C mutant mouse NCT were plated into 10 cm tissue culture dishes at the density of 1.5 × 106 cells/dish and transduced with recombinant adenovirus Ad5/CMV‐APP bearing human APPC99, as described above. 36 h post‐transduction, the cells were collected by scraping in ice‐cold PBS and total membranes prepared. To induce the cross‐linking between free sulfhydryl groups (i.e., cysteines), 30 μg membrane protein resuspended in 50 mM MES and 100 mM NaCl pH 6.5 was mixed with N‐β‐maleimidopropyl‐oxysuccinimide ester (BMPS, Thermo Fisher Scientific) at a final concentration of 10 μM and incubated for 2 h on ice. The excess of the cross‐linker was removed by washing the membrane pellets in buffer B. To promote the reaction with free amine groups, membranes were then resuspended in 100 mM HEPES and 100 mM NaCl pH 7.5 and incubated again on ice for 2 h. Cross‐linked membranes were pelleted by ultracentrifugation, solubilized in 4 × sample buffer (NuPAGE LDS, Thermo Fisher Scientific) supplemented with 4% β‐mercaptoethanol (BME) and analyzed by SDS–PAGE/Western blot.
To test for the potential formation of a disulfide bridge between NCT‐I241C and APP‐K28C mutants, NCT‐I241C MEF cell line was electroporated with 7.5 μg of pSG5**APP‐C99‐3xFLAG‐K28C plasmid per 1 × 106 cells. The medium was refreshed with low‐serum medium containing GSEC Inhibitor X (1 μM) 12 h post‐electroporation and cells were harvested 24 h later. Membrane fractions were prepared from the electroporated cells and resuspended in 50 mM MES and 100 mM NaCl pH 6.5. 30 μg protein (measured by Bradford protein assay) was resolved by SDS–PAGE under reducing (4% BME in sample buffer) or non‐reducing (no BME in sample buffer) conditions. The presence of the cross‐linked products was evaluated by Western blot.
Molecular dynamics simulations
The NCT‐I242E mutant molecular dynamics (MD) simulation conducted for this study was based on a 3000‐ns‐long simulation of the APP‐derived substrate Aβ49 in complex with WT (human) GSEC, part of a computational study of GSEC‐Aβn complexes (Hitzenberger & Zacharias, 2019). The structures were based on PDB entries 5FN2 (Bai et al, 2015a) (GSEC) and 2LP1 (Barrett et al, 2012) (APPC99). To make sure that the substrate remains in a conformation that is cleavable during the sampling, a transition state‐like geometry (Singh et al, 2009) at the interface between the cleavage (C99‐V46 and C99‐I47) and the active site (PSEN‐D257 and PSEN‐D385) was stabilized by the use of appropriate harmonic restraints with weak force constants (for details, cf. to the previous study; Hitzenberger & Zacharias, 2019). The complex of GSEC‐I242E and GSEC‐Aβ49 was placed in a bilayer of 301 POPC molecules and solvated in a 0.15 M KCl solution containing 53,829 water molecules. Periodic boundary conditions were employed, and the rectangular simulation box had a volume of approx. 2,200,000 Å3. Lipids, water and proteins were described by the Lipid14 (Dickson et al, 2014), the TIP3P (Mark & Nilsson, 2001) and AMBER14SB (Maier et al, 2015) force fields, respectively. The target temperature was set to 303.15K by the use of the Langevin thermostat (Goga et al, 2012) with a collision frequency of 1 ps−1. The desired pressure of 1.0 bar was maintained by the Monte Carlo barostat (Åqvist et al, 2004). Non‐bonded interactions were described up until a distance of 8 Å, and the particle mesh Ewald method (Darden et al, 1993) was used to describe long‐range effects. To allow for 4.0 fs time steps, the SHAKE algorithm (Ryckaert et al, 1977) and the hydrogen mass repartitioning method (Hopkins et al, 2015) were employed. The simulations were performed using the CUDA (Nickolls et al, 2008) version of PMEMD (Götz et al, 2012; Le Grand et al, 2013; Salomon‐Ferrer et al, 2013), which is part of the AMBER16 simulation package (Case et al, 2016). For evaluation, a 3000‐ns‐long simulation trajectory was generated. Trajectory analysis was carried out using CPPTRAJ (Roe & Cheatham, 2013) and VMD (Humphrey et al, 1996), which was also utilized to render the simulation snapshots. For free energy calculations, the molecular mechanics Poisson–Boltzmann surface area (MMPBSA) (Genheden & Ryde, 2015) post‐processing method as implemented in the MMPBSA.py program (Miller et al, 2012) has been employed. In order to account for the effects of the POPC bilayer, an implicit lipid model with a permittivity of ε = 2.0 has been used. The membrane slab was assumed to be 34 Å in thickness so that it covers the same region as the lipid tails in the explicit treatment. The head group region was assumed to be part of the aqueous layer (ε = 80.0) in order to ensure a realistic treatment of the electrostatics at the lipid–water interface where E242 and K28 are located (this region was populated by a high amount of water molecules—especially in the cavity region between NCT and PSEN, which is void of lipids). The implicit lipid slab was centered around the mean center of mass (COM) of the explicit bilayer in the simulation. The (internal) permittivity of the protein was assumed to be ε = 1.0 and the salt concentration was set to 0.15 M. In order to estimate the influence of the I242E mutation to ΔΔGbind, 1,500 frames of the simulation generated for this study were compared to the same amount of frames of the previously performed WT GSEC‐Aβ49 simulation (Hitzenberger & Zacharias, 2019). Due to the large system sizes and the enormous computational effort involved in estimating changes of entropy upon binding, only PBSA‐intrinsic solvation entropies have been considered.
Statistical analysis
All statistical analyses were performed using the GraphPad Prism 7 or 8 software. One‐way ANOVA with Dunnett's post hoc test was used to test the significance of the changes between groups unless indicated otherwise. In Fig 4, an unpaired, two‐tailed Student's t‐test was used to test the significance between tested groups. In Fig 6, two‐way ANOVA with Sidak's post hoc test was used to determine the response to the pharmacological treatments. P‐value < 0.05 was used as a pre‐determined threshold for statistical significance. All statistical analyses are described in the corresponding figure legends.
Author contributions
LC‐G designed the study, analyzed the data, supervised the research, and wrote the manuscript. DP performed experiments and analyzed the data. SL performed cloning and protein purification and assisted with experiments. KMZ analyzed data. NSR provided input on clinically related matters. MH and MZ performed modeling and molecular dynamics simulations studies. MM provided Aβ ELISA antibodies. FB provided GSM compounds.
Conflict of interest
D.P., M.H., S.L., K.M.Z., N.S.R, M.Z., and L.C.G. declare no competing interests. M.M. and F.B are employees of Janssen Pharmaceutica.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 5
Acknowledgements
This work was funded by the Stichting Alzheimer Onderzoek (SAO; S16013), the KU Leuven Methusalem Grant (METH/14/07) and the FWO (research project G0B2519N). D.P. is supported by a PhD fellowship from the FWO (SB/1S23819N). N.S.R. is supported by a University of London Chadburn Lectureship in Medicine. We thank Janssen Pharmaceutica for anti‐Aβ monoclonal antibodies and GSMs. We are grateful to Maria Szaruga for their critical reading of the manuscript and thank Michel Vande Kerckhove for helpful discussions.
The EMBO Journal (2019) 38: e101494
References
- Ahn K, Shelton CC, Tian Y, Zhang X, Gilchrist ML (2010) Activation and intrinsic γ ‐secretase activity of presenilin 1. Proc Natl Acad Sci USA 107: 21435–21440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Åqvist J, Wennerström P, Nervall M, Bjelic S, Brandsdal BO (2004) Molecular dynamics simulations of water and biomolecules with a Monte Carlo constant pressure algorithm. Chem Phys Lett 384: 288–294 [Google Scholar]
- Bai XC, Rajendra E, Yang G, Shi Y, Scheres SHW (2015a) Sampling the conformational space of the catalytic subunit of human γ‐secretase. Elife 4: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XC, Yan C, Yang G, Lu P, Ma D, Sun L, Zhou R, Scheres SHW, Shi Y (2015b) An atomic structure of human γ‐secretase. Nature 525: 212–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, Beel AJ, Sanders CR (2012) The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336: 1168–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beel AJ, Sanders CR (2008) Substrate specificity of γ‐secretase and other intramembrane proteases. Cell Mol Life Sci 65: 1311–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc DM, Montagna DR, Gu Y, Selkoe DJ, Wolfe MS (2016a) Nicastrin functions to sterically hinder γ‐secretase‐substrate interactions driven by substrate transmembrane domain. Proc Natl Acad Sci 113: E509–E518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc DM, Montagna DR, Seghers MC, Wolfe MS, Selkoe DJ (2016b) The amyloid‐beta forming tripeptide cleavage mechanism of γ‐Secretase. Elife 5: e17578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacace R, Sleegers K, Van Broeckhoven C (2016) Molecular genetics of early‐onset Alzheimer's disease revisited. Alzheimer's Dement 12: 733–748 [DOI] [PubMed] [Google Scholar]
- Carroll CM, Li YM (2016) Physiological and pathological roles of the γ‐secretase complex. Brain Res Bull 126: 199–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Betz R, Botello‐Smith W, Cerutti DS, Cheatham TE, Darden TA, Duke RE, TGiese TJ, Gohlke H, Goetz AW et al (2016) AMBER 2016. University of California, San Francisco.
- Chávez‐Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H et al (2012) The mechanism of γ‐Secretase dysfunction in familial Alzheimer disease. EMBO J 31: 2261–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez‐Gutiérrez L, Tolia A, Maes E, Li T, Wong PC, De Strooper B (2008) Glu332 in the nicastrin ectodomain is essential for gamma‐secretase complex maturation but not for its activity. J Biol Chem 283: 20096–20105 [DOI] [PubMed] [Google Scholar]
- Crump CJ, Fish BA, Castro SV, Chau DM, Gertsik N, Ahn K, Stiff C, Pozdnyakov N, Bales KR, Johnson DS et al (2011) Piperidine acetic acid based γ‐secretase modulators directly bind to presenilin‐1. ACS Chem Neurosci 2: 705–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Theuns J, Van Broeckhoven C (2012) Locus‐specific mutation databases for neurodegenerative brain diseases. Hum Mutat 33: 1340–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J Chem Phys 98: 10089–10092 [Google Scholar]
- De Strooper B (2003) Aph‐1, Pen‐2, and Nicastrin with Presenilin generate an active gamma‐Secretase complex. Neuron 38: 9–12 [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F (1998) Deficiency of presenilin‐1 inhibits the normal cleavage of amyloid precursor protein. Nature 391: 387–390 [DOI] [PubMed] [Google Scholar]
- Dickson CJ, Madej BD, Skjevik ÅA, Betz RM, Teigen K, Gould IR, Walker RC (2014) Lipid14: the amber lipid force field. J Chem Theory Comput 10: 865–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C (2003) Reconstitution of γ ‐secretase activity. Nature Cell Biology 5: 486–489 [DOI] [PubMed] [Google Scholar]
- Fernandez MA, Klutkowski JA, Freret T, Wolfe MS (2014) Alzheimer presenilin‐1 mutations dramatically reduce trimming of long amyloid β‐peptides (Aβ) by γ‐secretase to increase 42‐to‐40‐residue Aβ. J Biol Chem 289: 31043–31052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC et al (2002) aph‐1 and pen‐2 are required for Notch pathway signaling, γ‐secretase cleavage of βAPP, and presenilin protein accumulation. Dev Cell 3: 85–97 [DOI] [PubMed] [Google Scholar]
- Genheden S, Ryde U (2015) The MM/PBSA and MM/GBSA methods to estimate ligand‐binding affinities. Expert Opin Drug Discov 10: 449–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertsik N, Chiu D, Li YM (2015) Complex regulation of γ‐secretase: from obligatory to modulatory subunits. Front Aging Neurosci 7: 342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goate A, Chartier‐Harlin M‐C, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 349: 704–706 [DOI] [PubMed] [Google Scholar]
- Goga N, Rzepiela AJ, De Vries AH, Marrink SJ, Berendsen HJC (2012) Efficient algorithms for langevin and DPD dynamics. J Chem Theory Comput 8: 3637–3649 [DOI] [PubMed] [Google Scholar]
- Götz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC (2012) Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. generalized born. J Chem Theory Comput 8: 1542–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutte C, Tsunozaki M, Hale VA, Priess JR (2002) APH‐1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci USA 99: 775–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapasalo A, Kovacs DM (2011) The many substrates of presenilin/γ‐secretase. J Alzheimers Dis 25: 3–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitzenberger M, Zacharias M (2019) Structural modeling of γ‑Secretase Aβn complex formation and substrate processing. ACS Chem Neurosci 10: 1826–1840 [DOI] [PubMed] [Google Scholar]
- Hopkins CW, Le Grand S, Walker RC, Roitberg AE (2015) Long‐time‐step molecular dynamics through hydrogen mass repartitioning. J Chem Theory Comput 11: 1864–1874 [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 7855: 33–38 [DOI] [PubMed] [Google Scholar]
- Jung JI, Ran Y, Cruz PE, Rosario AM, Ladd TB, Kukar TL, Koo EH, Felsenstein KM, Golde TE (2014) Complex relationships between substrate sequence and sensitivity to alterations in γ‐secretase processivity induced by γ‐secretase modulators. Biochemistry 53: 1947–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurisch‐Yaksi N, Sannerud R, Annaert W (2013) A fast growing spectrum of biological functions of γ‐secretase in development and disease. Biochim Biophys Acta Biomembr 1828: 2815–2827 [DOI] [PubMed] [Google Scholar]
- Karran E, De Strooper B (2016) The amyloid cascade hypothesis: are we poised for success or failure? J Neurochem 139: 237–252 [DOI] [PubMed] [Google Scholar]
- Kim S, Ikeuchi T, Yu C, Sisodia SS (2003) Regulated hyperaccumulation of presenilin‐1 and the “γ‐Secretase” complex. J Biol Chem 278: 33992–34002 [DOI] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX (2004) Gamma‐secretase: proteasome of the membrane? Nat Rev Mol Cell Biol 5: 499–504 [DOI] [PubMed] [Google Scholar]
- Koriath C, Kenny J, Adamson G, Druyeh R, Taylor W, Beck J, Quinn L, Mok TH, Dimitriadis A, Norsworthy P et al (2018) Predictors for a dementia gene mutation based on gene‐panel next‐ generation sequencing of a large dementia referral series. Mol Psychiatry. 10.1038/s41380-018-0224-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukar TL, Ladd TB, Bann MA, Fraering PC, Narlawar R, Maharvi GM, Healy B, Chapman R, Welzel AT, Price RW et al (2008) Substrate‐targeting γ‐secretase modulators. Nature 453: 925–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukar TL, Ladd TB, Robertson P, Pintchovski SA, Moore B, Bann MA, Ren Z, Jansen‐West K, Malphrus K, Eggert S et al (2011) Lysine 624 of the amyloid precursor protein (APP) is a critical determinant of amyloid β peptide length: support for a sequential model of γ‐secretase intramembrane proteolysis and regulation by the amyloid β precursor protein (APP) juxtamembrane region. J Biol Chem 286: 39804–39812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle BW, Grenier‐Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie‐Wolf A et al (2019) Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 51: 414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov VK, Fraering PC, Ye W, Wolfe MS, Selkoe DJ, Li H (2006) Electron microscopic structure of purified, active γ‐secretase reveals an aqueous intramembrane chamber and two pores. Proc Natl Acad Sci 103: 6889–6894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand S, Götz AW, Walker RC (2013) SPFP: speed without compromise – A mixed precision model for GPU accelerated molecular dynamics simulations. Comput Phys Commun 184: 374–380 [Google Scholar]
- Li YM, Lai MT, Xu M, Huang Q, DiMuzio‐Mower J, Sardana MK, Shi XP, Yin KC, Shafer JA, Gardell SJ (2000) Presenilin 1 is linked with gamma‐secretase activity in the detergent solubilized state. Proc Natl Acad Sci USA 97: 6138–6143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Ma G, Cai H, Price DL, Wong PC (2003) Nicastrin is required for assembly of presenilin/γ‐secretase complexes to mediate notch signaling and for processing and trafficking of β‐amyloid precursor protein in mammals. J Neurosci 23: 3272–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lleó A (2008) Activity of gamma‐secretase on substrates other than APP. Curr Top Med Chem 8: 9–16 [DOI] [PubMed] [Google Scholar]
- Luo WJ, Wang H, Li H, Kim BS, Shah S, Lee HJ, Thinakaran G, Kim TW, Yu G, Xu H (2003) PEN‐2 and APH‐1 coordinately regulate proteolytic processing of presenilin 1. J Biol Chem 278: 7850–7854 [DOI] [PubMed] [Google Scholar]
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 11: 3696–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark P, Nilsson L (2001) Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J Phys Chem A 105: 9954–9960 [Google Scholar]
- McCarthy AJ, Coleman‐Vaughan C, McCarthy JV (2017) Regulated intramembrane proteolysis: emergent role in cell signalling pathways. Biochem Soc Trans 45: 1185–1202 [DOI] [PubMed] [Google Scholar]
- Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE (2012) MMPBSA.py: an efficient program for end‐state free energy calculations. J Chem Theory Comput 8: 3314–3321 [DOI] [PubMed] [Google Scholar]
- Moore S, Evans LDB, Andersson T, Portelius E, Smith J, Dias TB, Saurat N, McGlade A, Kirwan P, Blennow K et al (2015) APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep 11: 689–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickolls J, Buck IAN, Garland M (2008) Scalable Parallel. In IEEE Hot Chips 20 Symposium (HCS), pp 40–53, Stanford, CA: [Google Scholar]
- Nyabi O, Pype S, Mercken M, Herreman A, Saftig P, Craessaerts K, Serneels L, Annaert W, de Strooper B (2002) No endogenous Aβ production in presenilin‐deficient fibroblasts. Nat Cell Biol 4: E164 [DOI] [PubMed] [Google Scholar]
- Ousson S, Saric A, Baguet A, Losberger C, Genoud S, Vilbois F, Permanne B, Hussain I, Beher D (2013) Substrate determinants in the C99 juxtamembrane domains differentially affect γ‐secretase cleavage specificity and modulator pharmacology. J Neurochem 125: 610–619 [DOI] [PubMed] [Google Scholar]
- Page RM, Gutsmiedl A, Fukumori A, Winkler E, Haass C, Steiner H (2010) β‐Amyloid precursor protein mutants respond to γ‐secretase modulators. J Biol Chem 285: 17798–17810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi‐Takahara Y (2005) Longer forms of amyloid‐β protein: implications for the mechanism of intramembrane cleavage by γ‐secretase. J Neurosci 25: 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehder DS, Borges CR (2010) Cysteine sulfenic acid as an intermediate in disulfide bond formation and nonenzymatic protein folding. Biochemistry 49: 7748–7755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Schenk D, Basi GS, Shapiro IP (2007) Amyloid β‐protein precursor juxtamembrane domain regulates specificity of γ‐secretase‐dependent cleavages. J Biol Chem 282: 35350–35360 [DOI] [PubMed] [Google Scholar]
- Roe DR, Cheatham TE (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput 9: 3084–3095 [DOI] [PubMed] [Google Scholar]
- Ryckaert JP, Ciccotti G, Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n‐alkanes. J Comput Phys 23: 327–341 [Google Scholar]
- Salomon‐Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC (2013) Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh ewald. J Chem Theory Comput 9: 3878–3888 [DOI] [PubMed] [Google Scholar]
- Sato T, Diehl TS, Narayanan S, Funamoto S, Ihara Y, De Strooper B, Steiner H, Haass C, Wolfe MS (2007) Active γ‐secretase complexes contain only one of each component. J Biol Chem 282: 33985–33993 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Wolfe MS (2007) Presenilin: running with scissors in the membrane. Cell 131: 215–221 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 8: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE III, Sudhof T, Yu G (2005) Nicastrin functions as a gamma‐secretase‐substrate receptor. Cell 122: 435–447 [DOI] [PubMed] [Google Scholar]
- Shearman MS, Beher D, Clarke EE, Lewis HD, Harrison T, Hunt P, Nadin A, Smith AL, Stevenson G, Castro JL (2000) L‐685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid β‐protein precursor γ‐secretase activity. Biochemistry 39: 8698–8704 [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K et al (1995) Cloning of a gene bearing missense mutations in early‐onset familial Alzheimer's disease. Nature 375: 754–760 [DOI] [PubMed] [Google Scholar]
- Singh R, Barman A, Prabhakar R (2009) Computational insights into aspartyl protease activity of presenilin 1 (ps1) generating alzheimer amyloid β‐peptides (aβ40 and aβ42). J Phys Chem B 113: 2990–2999 [DOI] [PubMed] [Google Scholar]
- Steiner H, Duff K, Capell A, Romig H, Grim MG, Lincoln S, Hardy J, Yu X, Picciano M, Fechteler K et al (1999) A loss of function mutation of presenilin‐2 interferes with amyloid β ‐peptide production and notch signaling. J Biol Chem 274: 28669–28673 [DOI] [PubMed] [Google Scholar]
- Struhl G, Adachi A (2000) Requirements for presenilin‐dependent cleavage of notch and other transmembrane proteins. Mol Cell 6: 625–636 [DOI] [PubMed] [Google Scholar]
- Szaruga M, Veugelen S, Benurwar M, Lismont S, Sepulveda‐Falla D, Lleo A, Ryan NSSSNS, Lashley T, Fox NCCC, Murayama S et al (2015) Qualitative changes in human gamma‐secretase underlie familial Alzheimer's disease. J Exp Med 212: 2003–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN et al (2017) Alzheimer's‐causing mutations shift Aβ length by destabilizing γ‐secretase‐Aβn interactions. Cell 170: 443–456.e14 [DOI] [PubMed] [Google Scholar]
- Takagi‐Niidome S, Sasaki T, Osawa S, Sato T, Morishima K, Cai T, Iwatsubo T, Tomita T (2015) Cooperative roles of hydrophilic loop 1 and the C‐terminus of presenilin 1 in the substrate‐gating mechanism of γ‐secretase. J Neurosci 35: 2646–2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima‐Kawashima M, Funamoto S, Ihara Y (2009) Gamma‐Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta‐carboxyl terminal fragment. J Neurosci 29: 13042–13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T (2003) The role of presenilin cofactors in the gamma‐secretase complex. Nature 422: 438–441 [DOI] [PubMed] [Google Scholar]
- Takeo K, Tanimura S, Shinoda T, Osawa S, Zahariev IK, Takegami N, Ishizuka‐Katsura Y, Shinya N, Takagi‐Niidome S, Tominaga A et al (2014) Allosteric regulation of γ‐secretase activity by a phenylimidazole‐type γ‐secretase modulator. Proc Natl Acad Sci 111: 10544–10549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M et al (1996) Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo . Neuron 17: 181–190 [DOI] [PubMed] [Google Scholar]
- Velter AI, Bischoff FP, Berthelot D, De Cleyn M, Oehlrich D, Jaroskova L, Macdonald G, Minne G, Pieters S, Rombouts F et al (2014) Anilinotriazoles as potent gamma secretase modulators. Bioorganic Med Chem Lett 24: 5805–5813 [DOI] [PubMed] [Google Scholar]
- Veugelen S, Saito T, Saido TC, Chávez‐Gutiérrez L, De Strooper B (2016) Familial Alzheimer's disease mutations in presenilin generate amyloidogenic Aβ peptide seeds. Neuron 90: 410–416 [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ (1999) Two transmembrane aspartates in presenilin‐1 required for presenelinen doproteolysis and gamma‐secretase activity. Nature 398: 513–517 [DOI] [PubMed] [Google Scholar]
- Yagishita S, Morishima‐Kawashima M, Ishiura S, Ihara Y (2008) Aβ46 is processed to Aβ40 and Aβ43, but not to Aβ42, in the low density membrane domains. J Biol Chem 283: 733–738 [DOI] [PubMed] [Google Scholar]
- Yang G, Zhou R, Zhou Q, Guo X, Yan C, Ke M, Lei J, Shi Y (2018) Structural basis of Notch recognition by human γ‐secretase. Nature 565: 192–197 [DOI] [PubMed] [Google Scholar]
- Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T et al (2000) Nicastrin modulates presenilin‐mediated notch/glp‐1 signal transduction and βAPP processing. Nature 407: 48–54 [DOI] [PubMed] [Google Scholar]
- Zhang YW, Luo WJ, Wang H, Lin P, Vetrivel KS, Liao F, Li F, Wong PC, Farquhar MG, Thinakaran G et al (2005) Nicastrin is critical for stability and trafficking but not association of other presenilin/gamma‐secretase components. J Biol Chem 280: 17020–17026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Liu Z, Ilagan MXG, Kopan R (2010) γ‐Secretase composed of PS1/Pen2/Aph1a can cleave notch and amyloid precursor protein in the absence of Nicastrin. J Neurosci 30: 1648–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yang G, Guo X, Zhou Q, Lei J, Shi Y (2019) Recognition of the amyloid precursor protein by human γ‐secretase. Science 363: eaaw0930 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 5