

Abstract
Immunotherapy using chimeric antigen receptor (CAR)‐engineered lymphocytes has shown impressive results in leukemia. However, for solid tumors such as colorectal cancer (CRC), new preclinical models are needed that allow to test CAR‐mediated cytotoxicity in a tissue‐like environment. Here, we developed a platform to study CAR cell cytotoxicity against 3‐dimensional (3D) patient‐derived colon organoids. Luciferase‐based measurement served as a quantitative read‐out for target cell viability. Additionally, we set up a confocal live imaging protocol to monitor effector cell recruitment and cytolytic activity at a single organoid level. As proof of principle, we demonstrated efficient targeting in diverse organoid models using CAR‐engineered NK‐92 cells directed toward a ubiquitous epithelial antigen (EPCAM). Tumor antigen‐specific cytotoxicity was studied with CAR‐NK‐92 cells targeting organoids expressing EGFRvIII, a neoantigen found in several cancers. Finally, we tested a novel CAR strategy targeting FRIZZLED receptors that show increased expression in a subgroup of CRC tumors. Here, comparative killing assays with normal organoids failed to show tumor‐specific activity. Taken together, we report a sensitive in vitro platform to evaluate CAR efficacy and tumor specificity in a personalized manner.
Keywords: CAR immunotherapy, colorectal cancer, cytotoxicity assays, natural killer cells, patient‐derived organoids
Subject Categories: Cancer, Immunology, Methods & Resources
Introduction
To increase the response rates to cancer immunotherapy, immune effector cells can be engineered to recognize tumor‐associated antigens (TAA) expressed on the surface of cancer cells. Introduction of chimeric antigen receptors (CAR) has shown impressive success in various hematological malignancies, and the use of autologous CAR‐engineered T cells in relapsed B‐cell acute lymphoblastic leukemia and relapsed large B‐cell non‐Hodgkin lymphoma was recently approved by the Food and Drug Administration (June & Sadelain, 2018). However, for solid cancers, CAR‐based therapies have faced several challenges: Most commonly, overexpressed tumor antigens show expression also in normal tissues, which may lead to serious side effects (Bonifant et al, 2016). Targeting cancer‐specific antigens, such as neoantigens, requires highly individualized approaches, and access to suitable preclinical models to test efficacy and safety. Clinical application of autologous CAR‐T cells is both labor‐ and cost‐intensive. The established natural killer (NK) cell line NK‐92 has been described as an alternative source for CAR‐engineered immune cells. The NK‐92 cell line was derived from a non‐Hodgkin's lymphoma (Gong et al, 1994) and displayed high non‐MHC‐restricted cytolytic activity in a number of preclinical studies (Suck et al, 2016). Together with the possibility of robust ex vivo expansion and engineering, NK‐92 cells may serve as a standardized platform for off‐the‐shelf CAR reagents (Zhang et al, 2017). Very recently, safety data from a phase I clinical trial with CD33‐specific CAR‐NK‐92 cells were reported (Tang et al, 2018). Another CAR‐NK‐92 cell product targeting the human epidermal growth factor receptor 2 (HER2) has entered a phase I clinical trial for the treatment of glioblastoma (NCT03383978; https://clinicaltrials.gov).
Metastatic colorectal carcinoma (CRC) accounts for the fourth most common cause of cancer‐related deaths worldwide (Ferlay et al, 2015). Conventional treatment options such as radiation and chemotherapy are often associated with severe side effects and disease recurrence. Hence, alternative and more personalized therapeutic strategies are urgently needed. Harnessing the enormous potential of the immune system appears an encouraging option to improve therapy outcomes in CRC patients. For microsatellite instable (MSI) tumors that are strongly immunogenic, checkpoint inhibition has shown a high clinical response rate (Le et al, 2015). However, microsatellite stable (MSS) tumors are refractory to immune checkpoint blockade, and patients with particularly poor prognosis show low leukocyte infiltration or reduced cytotoxic gene expression (Galon et al, 2006; Mlecnik et al, 2016) and may benefit from CAR‐based therapies. To develop effective CAR strategies, physiological preclinical models are required that recapitulate the individual tumor phenotype as well as the complex three‐dimensional (3D) tissue environment. The organoid culture system first described by Sato et al (2009, 2011) allows long‐term ex vivo expansion of gastrointestinal stem cells in a 3D extracellular matrix. The technology has been used to establish living biobanks of cancer and normal tissues that preserve the genetic and functional heterogeneity among CRC patients (van de Wetering et al, 2015; Fujii et al, 2016). Addition of stromal cells further allows to reconstruct the tissue microenvironment and to study immuno‐epithelial crosstalk (Farin et al, 2014; Rogoz et al, 2015; Nozaki et al, 2016; Noel et al, 2017). Moreover, tumor‐reactive T cells can be selectively expanded in co‐culture with tumor organoids from MSI patients (Dijkstra et al, 2018).
For quantitative characterization of cytotoxic responses to 3D organoids, there is an unmet need for standardized protocols. Monitoring of cell‐mediated killing is classically performed after single cell dispersal (Zaritskaya et al, 2010) that may result in spontaneous lysis due to sensitivity to enzymatic digest. In suspension, effector cell recruitment, motility, and interaction length cannot be addressed, which are key parameters for cytotoxicity in vivo (Weigelin et al, 2015; Halle et al, 2016). Current read‐outs for cell‐mediated cytotoxicity, such as flow cytometry‐based viability assays or enzyme release assays, only allow detection for a limited amount of time, which may not be sufficient to recapitulate the kinetics in a 3D environment. Finally, T‐cell responses (e.g., using PBMCs) show inherent donor variability and the directed antigens are unknown in most cases. In order to study cytotoxic responses to specific TAAs, we have taken advantage of the standardized CAR‐NK‐92 system. The aim was to establish a quantitative platform for CAR‐mediated cytotoxicity toward patient‐derived colon organoids. Endpoint and dynamic assays were developed to monitor cytotoxic responses in a 3D environment. We demonstrate targeting of universal antigens, neoantigens, and new candidate TAAs providing an informative platform for preclinical screening of efficacy and tumor cell specificity of CAR‐based strategies.
Results
Directing CAR cytotoxicity to human colon organoids
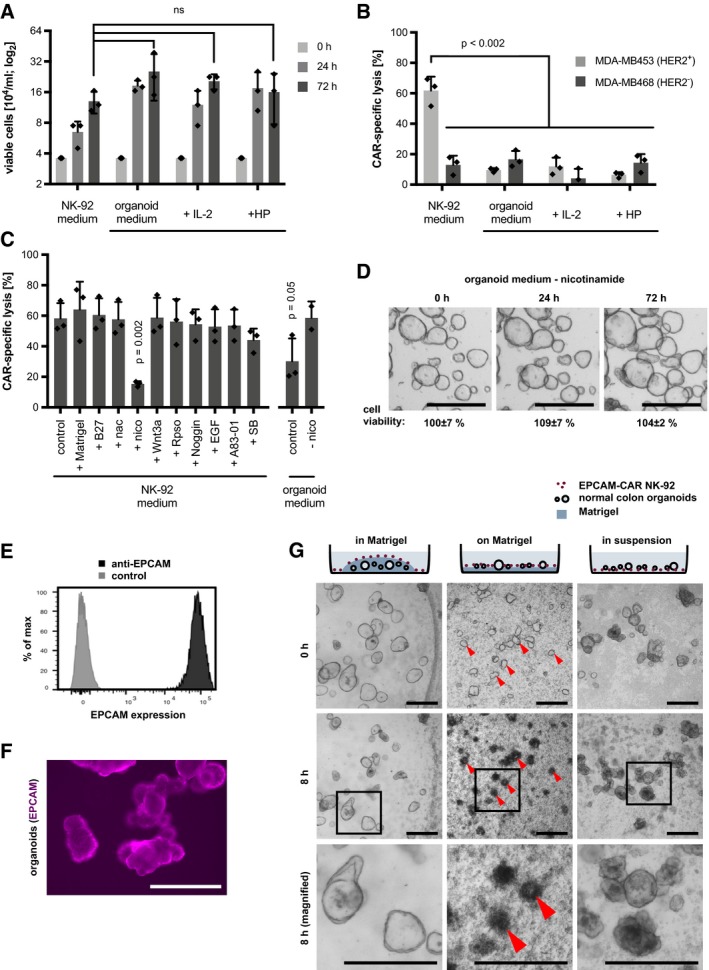
To establish a model for CAR targeting of organoids, we first tested the compatibility of NK‐92 cells with organoid culture medium. After 24 and 72 h of culture, we observed a comparable expansion as in the regular NK‐cell medium (Fig 1A), even in the absence of human serum and IL‐2 that are usually required for growth and activity. To test if the NK‐92 function is preserved under these conditions, a standard cytotoxicity assay was performed using HER2‐specific CAR‐NK‐92 cells that were co‐incubated with MDA‐MB453 (HER2‐positive) or MDA‐MB468 (HER2‐negative control) breast carcinoma cells as targets (Schönfeld et al, 2015). Specific lysis was strongly reduced in organoid medium even in the presence of human plasma or IL‐2 (Fig 1B). By testing all factors of the organoid medium individually, we identified nicotinamide as responsible for this inhibition (Fig 1C), a non‐essential factor as shown by cell viability assays (Fig 1D) that was subsequently omitted from the medium. To identify universal co‐culture conditions that allow effective killing of both normal and tumor human colon organoids, we used CAR‐NK‐92 cells that target EPCAM as a ubiquitously expressed epithelial surface antigen (Trzpis et al, 2007; Sahm et al, 2012; Fig 1E and F). Organoids were either embedded in Matrigel, on a thin layer of Matrigel or in suspension, followed by addition of EPCAM‐CAR NK‐92 cells or the control parental NK‐92 cells. Microscopic inspection showed that NK cells readily migrated on the surface but were incapable to penetrate the dense extracellular matrix (ECM). After 8 h, only culturing on a Matrigel‐coated layer but not in suspension resulted in a marked CAR‐induced lysis (Fig 1G). Our results suggest that co‐culture on an ECM layer can increase NK‐cell migration and/or stabilize the effector–target cell interaction and was therefore used for all subsequent experiments.
Figure 1. Organoid NK‐92 co‐culture model for efficient 3D killing assays.
-
AGrowth of NK‐92 cells in standard NK medium and organoid medium. 3.6 × 104 cells/ml were seeded, and organoid medium was supplemented with IL‐2 or human plasma (HP) followed by cell counting after 0, 24, 48, and 72 h. Mean ± SD on log2 scale, in n = 3 parallel cultures. ns: P > 0.05 (unpaired t‐test).
-
BReduced cytotoxic activity of CAR NK‐92 cells in organoid medium. FACS‐based cytotoxicity assay using HER2‐targeted CAR‐NK‐92 cells. Target cells were MDA‐MB453 (HER2+) and MDA‐MB468 (HER2−) breast carcinoma cells. Mean CAR‐specific lysis (± SD) was determined in triplicates. Significance was analyzed by unpaired t‐test.
-
CIdentification of nicotinamide (nico) as inhibitory factor in a standard cytotoxicity assay with MDA‐MB453 target cells. Each component of the organoid medium was added to the NK medium in a standard cytotoxicity assay (as in B). Mean CAR‐specific lysis (± SD) was determined in three independent experiments. Significant P‐values (compared to control) from unpaired t‐test analysis are shown. Nac: N‐acetylcysteine; SB: SB202190.
-
DUnaffected growth of normal colon organoids in nicotinamide‐free medium. Mean cell viability (± SD; in n = 3 wells; CellTiter‐Glo assay) compared to regular medium. The experiment was repeated twice independently. Scale bars: 500 μm.
-
E, FFlow cytometric and immunofluorescence microscopy analysis of EPCAM staining in normal colon organoids. Scale bar: 200 μm.
-
DCo‐culture strategies for analysis of CAR‐NK‐92‐mediated cytotoxicity. Organoids were embedded in Matrigel, or seeded on a Matrigel‐coated surface, or kept in suspension, followed by addition of EPCAM‐CAR NK‐92 cells (E:T ratio was 4:1). Schematic representation of experimental conditions (top) and morphological images after 0 and 8 h of co‐culture. Red arrows show examples of organoids that are efficiently lysed. In the magnified images (bottom row), abundant apoptotic bodies are present around the lysed organoids. Scale bars: 500 μm.
Quantitative analysis of CAR‐mediated cytotoxicity using a luciferase‐based 3D assay
For quantitative monitoring of CAR‐mediated cytotoxic activity against 3D organoids, we sought to develop assays that do not require single cell digestion. Firefly luciferase reporter has been described as a convenient read‐out of viable cells following cytotoxicity against 2D cell lines (Fu et al, 2010). To adapt this strategy, luciferase/GFP expression was introduced into organoids using lentiviral transduction, which allowed homogenous expression (Fig EV1A and B). By titration of organoids, linear and sensitive cell detection was confirmed (Fig EV1C). In addition, detergent‐induced cell lysis caused rapid solubilization of the cellular luciferase activity with < 20% residual signal detected in cellular debris, allowing accurate quantification of viable organoid cells.
Figure EV1. Luciferase activity as a quantitative read‐out for lysis‐resistant target cells.
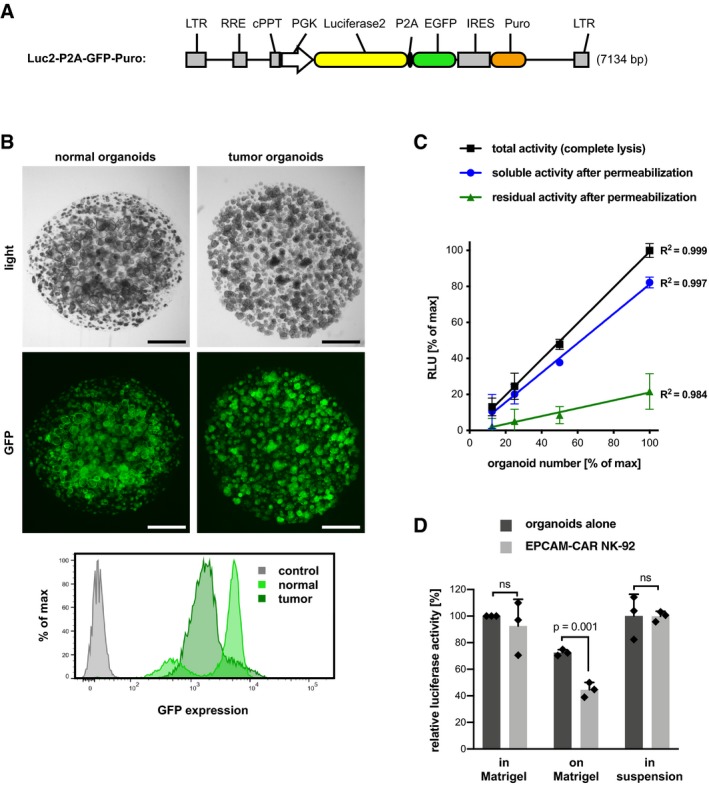
- Lentiviral construct to drive co‐expression of luciferase 2 and EGFP (separated by a P2A peptide). Stable expression is selectable by the puromycin resistance marker. Additional features are long‐terminal repeats (LTR), Rev response element (RRE), cPPT central polypurine tract, and the phosphoglycerate kinase (PGK) promoter.
- Patient‐derived normal and tumor colon organoids were lentivirally transduced to stably co‐express luciferase and GFP. Homogenous expression of GFP was observed. Scale bars: 1,000 μm. Bottom: Flow cytometric analysis of GFP expression. GFP‐negative organoids are shown as control.
- Linear detection of viable organoids. Luciferase activity (in relative light units, RLU) was quantified either after complete lysis (black line), or in the supernatant (blue line) or the remaining cell debris (green line) following cell permeabilization. Mean values (± SD) from n = 3 replicates. The coefficients of determination (R 2) are displayed. An organoid number of 100% corresponds to 1.25 × 105 epithelial cells. Note that residual signal after lysis was below 20% of total activity.
- Quantification of luciferase activity in different co‐culture settings (as in Fig 1G). Mean luciferase activity (± SD) from n = 3 replicates relative to growth in Matrigel. The presence of EPCAM‐CAR NK‐92 cells (E:T ratio was 1:1, for 8 h) reduces luciferase signal of normal organoids after seeding on a Matrigel‐coated surface. No reduction is detected after organoid seeding in Matrigel or in suspension. The experiment was repeated three times. Significance was analyzed by unpaired t‐test. Note that organoids were seeded on Matrigel 24 h before the start of co‐culture, which causes reduction of signal compared to standard culture.
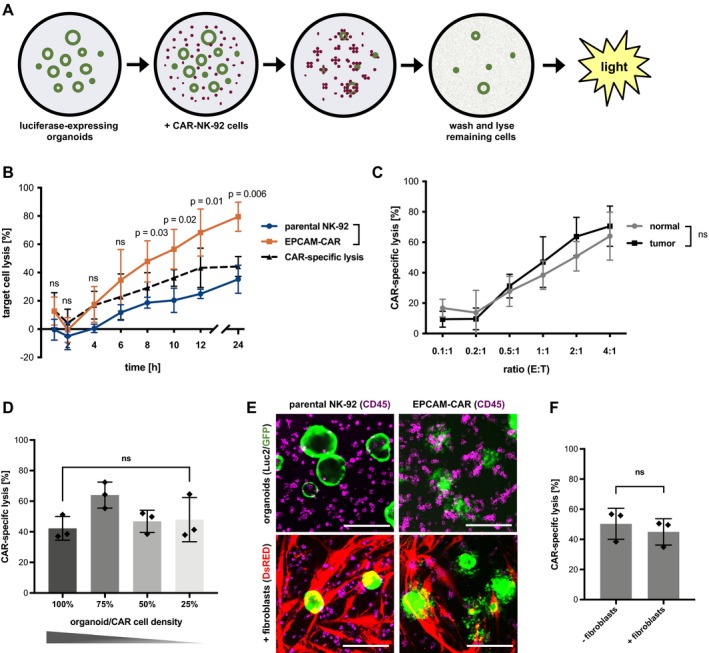
Using this assay, we tested the kinetics of cytotoxic activity of EPCAM‐CAR NK‐92 cells toward patient‐derived normal and tumor organoids (Fig 2A). Luciferase activity was significantly reduced only after co‐culture with organoids on a Matrigel‐coated layer, but not with organoids embedded in Matrigel or in suspension (Fig EV1D), confirming our morphological observations above (Fig 1G). After an initial refractory period of 2 h, a steady increase in cytotoxicity could be observed for both parental and CAR‐engineered NK‐92 cells (Fig 2B). However, target cell lysis was significantly increased in the case of EPCAM‐directed effector cells, where after 24 h, ~80% lysis was achieved. CAR‐specific lysis was determined by subtraction of cell lysis by parental NK‐92 cells and reached ~40% after 12 h. In order to determine the optimal effector‐to‐target (E:T) ratio, the absolute organoid cell number was first determined in an image‐based manner (Appendix Fig S1). Increasing cytotoxic efficiency could be observed at higher E:T ratios (Fig 2C) that were equally effective against normal and tumor organoids (which expressed similar levels of EPCAM). Next, the organoid and NK number was varied under constant E:T ratio to test whether the response is sensitive to the absolute cell densities. We found that the density had no major impact on the efficiency of killing, indicating that CAR cell recruitment can occur even at reduced effector cell density (Fig 2D). Moreover, we could achieve non‐perturbed lysis also in a tissue‐like environment, where organoids were seeded on a layer of colon fibroblasts, indicating that presence of EPCAM‐negative fibroblasts does not interfere with CAR cell recruitment (Fig 2E and F). Together, our results demonstrate that luciferase‐based measurement can serve as flexible read‐out for cytotoxicity in advanced tumor models.
Figure 2. Luciferase‐based quantification of CAR‐mediated cytotoxicity toward normal and tumor organoids.
-
ASchematic workflow. Luciferase‐expressing organoids allow quantification of residual cells after CAR‐mediated killing.
-
BKinetic analysis of EPCAM‐directed CAR‐NK‐92 cytotoxicity toward normal colon organoids. Remaining luciferase activity, normalized to organoids cultured alone, is shown as “target cell lysis”. Mean values (± SD) from n = 3 independent experiments. E:T ratio was 2:1. Statistical significance was analyzed by unpaired t‐test.
-
CEPCAM‐CAR‐specific lysis of normal and tumor organoids at different E:T ratios. Mean values (± SD) from n = 3 independent experiments after 8 h. ns: P > 0.05 (unpaired t‐test).
-
DInfluence of organoid/CAR cell density. At a constant E:T ratio of 1:1, decreasing numbers of organoids/NK‐92 cells were co‐cultured. EPCAM‐CAR‐specific lysis was measured in triplicates after 8 h (mean ± SD), and the experiment was replicated twice. ns: P > 0.05 (unpaired t‐test).
-
E, FEPCAM‐CAR targeting of organoids in the presence of human colonic primary fibroblasts. E:T ratio was 1:1. (E) Fluorescence microscopy analysis of luciferase/GFP‐expressing organoids (green); anti‐CD45‐APC‐labeled NK‐92 cells (magenta) and dsRED‐expressing fibroblasts (red) after 8 h. Scale bars: 200 μm. (F) Luciferase‐based cytotoxicity assay. Unaffected CAR‐specific lysis in the presence and absence of fibroblasts (measured in triplicates; mean ± SD). ns: P > 0.05 (unpaired t‐test).
Dynamic monitoring of CAR‐NK‐92 cytotoxicity by live‐cell imaging
To study cytotoxic responses in real time and at cellular resolution, we set up a confocal live‐cell imaging protocol. Colon organoids stably expressing GFP were cultured alone or together with EPCAM‐CAR cells or parental NK‐92 cells that were labeled by anti‐CD45 staining. Spinning‐disk microscopy in multiwell format allowed stable 3D imaging during 10 h and at 6‐min intervals (Fig 3A). From maximum image projection data, automatic image analysis was performed (see Appendix Fig S2) in organoids cultured alone (Movies EV1 and EV2), together with parental NK‐92 cells (Movies EV3 and EV4) or with EPCAM‐CAR cells (Movies EV5 and EV6). The loss of fluorescent organoid area over time was quantified as a measure of target cell killing (Figs 3B and C, and EV2A). To confirm a linear detection of target cell area, organoids were seeded at different concentrations and analyzed before and 8 h after addition of CAR or parental NK‐92 cells (Fig EV2B). Average GFP+ area from multiple imaging positions showed a significant and progressive decline after addition of EPCAM‐specific NK‐92 but not parental NK‐92 cells or organoids alone (Fig 3D). CAR‐specific lysis was evident after 2 h, reaching a level of ~40% after 10 h. Side‐by‐side comparison of image‐ and luciferase‐based read‐out showed very similar results, thus corroborating our protocols (Fig 3E). To study NK‐92 recruitment to the organoids, a second image analysis workflow was established that allows time‐resolved measurement of NK density in an area extending 50 μm around each organoid (see Appendix Fig S2). We detected a progressive recruitment of CAR NK‐92 cells that reached saturation after 2 h (Fig 3F), paralleling the observed kinetics of lysis. Parental NK‐92 cells were also enriched to the organoids compared to the overall density, albeit at a lower level than CAR cells (Fig 3F). These results suggest that parental NK‐92 cells do not achieve sufficiently high concentration or stable interaction with target cells to exert damage. To address if the organoid size has an impact on the killing efficiency, organoids were categorized by their initial size into three groups followed by tracking on a single organoid level (Fig EV2C–E). Comparison of the rate of relative area loss showed that small organoids are more effectively attacked (Fig EV2F), while larger organoids require longer time periods for lysis. For larger organoids, a stronger correlation between average of CAR‐NK density and relative area loss was found, indicating that a prolonged period of high local effector density is required to achieve complete lysis (Fig 3G).
Figure 3. Continuous imaging‐based monitoring of cytotoxic activity toward organoids.
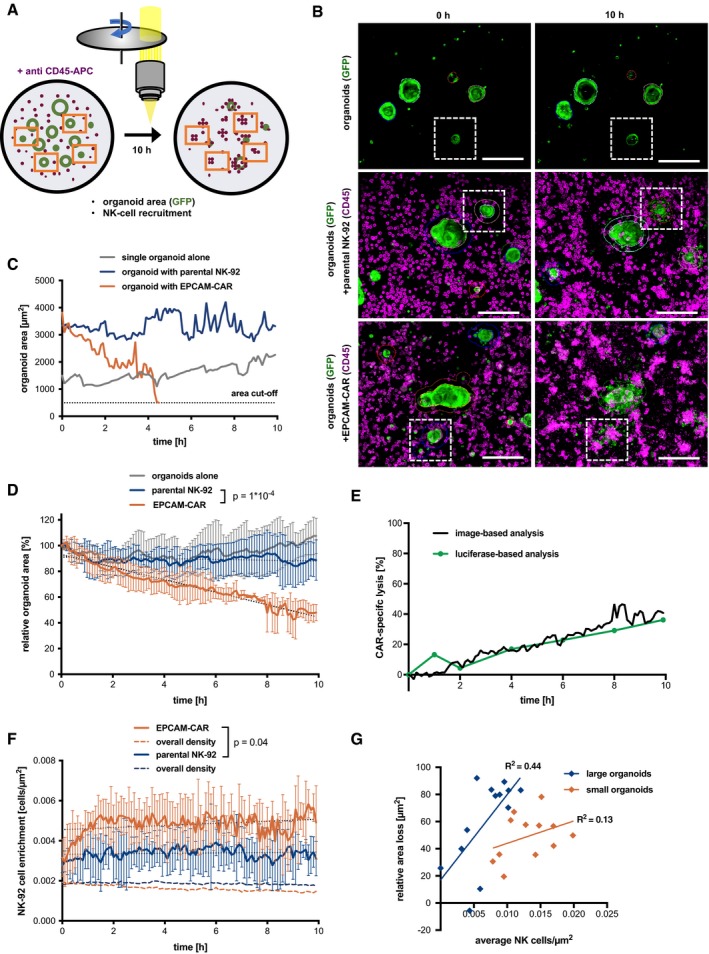
- Live imaging strategy using spinning‐disk microscopy.
- Exemplary data with GFP‐expressing organoids (green) and anti‐CD45‐APC‐labeled NK‐92 cells (magenta) at 0 and 10 h of co‐culture. Maximum intensity projections are shown. Organoid outlines are automatically detected; white boxes indicate organoids tracked in (C). Scale bars: 200 μm. See also Fig EV2 and Movies EV1, EV2 EV3 EV4 EV5 and EV6.
- Single organoid tracking (area) during CAR‐mediated killing.
- Quantitative monitoring of cytotoxicity by loss of GFP+ organoid area. Mean values from n = 4 imaging positions (± SD) are shown relative to the area detected at t = 0. Slopes were compared by linear regression; statistically significant differences were observed (ANCOVA).
- Comparative kinetics of CAR‐specific lysis detected by image‐based analysis (black line) and luciferase‐based measurement (green line; data are from Fig 2B).
- Time‐resolved recruitment of NK‐92 cells to target organoids. NK‐cell density was automatically determined in a 50 μm surrounding region (see Appendix Fig S4A). Mean ± SD from n = 4 imaging positions. Hatched lines show the NK‐92 density on the entire image. Slopes were compared by linear regression; significant differences were observed (ANCOVA).
- Correlation of area loss and NK‐cell density on a single organoid level. Organoids were classified as small (area < 2,000 μm2; n = 12) and large (area > 2,000 μm2; n = 13), respectively. Maximum relative area loss and average NK density for the entire co‐culture period were plotted, and coefficients of determination (R 2) are shown. Note that large organoids contribute mostly to overall recruitment in (F).
Figure EV2. Imaging‐based quantification and tracking of individual organoids.
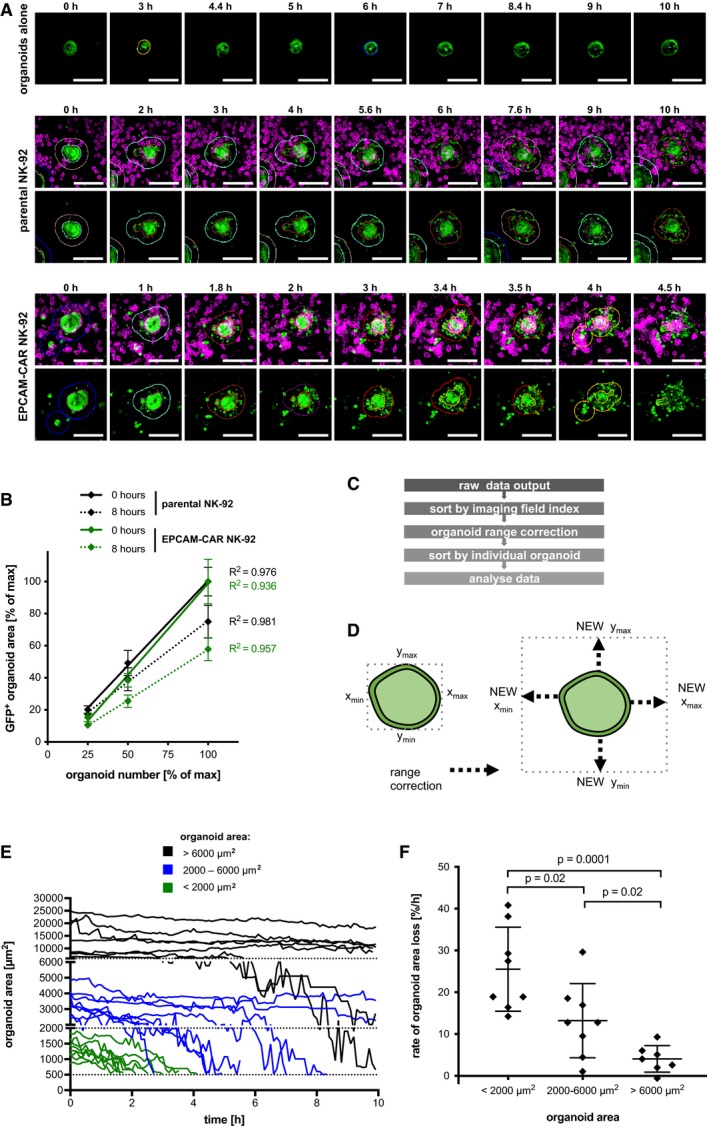
- Kinetic imaging data of single organoids (see Fig 3B and C). GFP‐positive organoids cultured alone or with parental NK‐92 cells or EPCAM‐CAR cells. NK‐92 cells were labeled with anti‐CD45‐APC. Maximum intensity projections of merged channels and the single GFP channel are shown. Organoid areas detected by the analysis software and expanded areas for determining NK‐cell recruitment are marked in color. Scale bars: 100 μm.
- Linear detection of organoid area. Organoids were seeded on Matrigel layer at different concentrations and co‐cultured with parental NK‐92 cells or EPCAM‐CAR cells. For each concentration, the organoid area was automatically quantified in 110 imaging fields before (0 h) or after (8 h) addition of NK‐92 cells. The entire imaging region was subdivided into n = 4 quadrants, and the relative organoid area is shown as mean (± SD). E:T ratio was kept constant at 4:1. 50% seeding density corresponds to the standard density. Coefficients of determination (R 2) are shown.
- Workflow for extracting single organoid data. Refer to methods for further details.
- Range correction for tracking of organoids that may grow or move during the analysis. The coordinates of each organoid at t = 0 were expanded.
- Single organoid tracking in co‐culture with EPCAM‐CAR cells (data from Fig 3). Normal organoids were classified as large (area > 6,000 μm2; n = 7), medium (area 2,000–6,000 μm2; n = 8), or small (area < 2,000 μm2; n = 8). The lower detection cut‐off was set at 500 μm2.
- Rate of relative area loss for single organoids of distinct sizes (analysis of data shown in E). Average rate (± SD; measured in n = 7–8 independent organoids) is shown. Statistical significance was analyzed by unpaired t‐test (ns: P > 0.05).
CAR targeting of tumor neoantigens
In order to study a tumor‐specific CAR response, we engineered organoids by lentiviral transduction to express a neoantigen, the epidermal growth factor receptor (EGFR) variant III (vIII) that can be found on several solid cancer types (Gan et al, 2013). Genomic deletion of exons 2–7 of the EGFR gene and amplification of the locus results in expression of truncated, constitutively active EGFRvIII (Fig EV3A). Although organoids were selected for stable expression by antibiotic resistance, we could not detect the EGFRvIII protein by immunoblot analysis (Fig EV3B and C). However, by RT‐PCR analysis transgene expression was confirmed arguing for protein expression below the immunoblot detection limit (Fig EV3D). By luciferase measurement, we detected potent and highly specific activity of EGFRvIII‐CAR NK‐92 cells toward EGFRvIII‐transduced organoids but not control organoids (Fig 4A and B). These results indicate that even low levels of neoantigen expression render the organoids highly susceptible for lysis.
Figure EV3. Generation and characterization of EGFRvIII neoantigen expressing organoids.
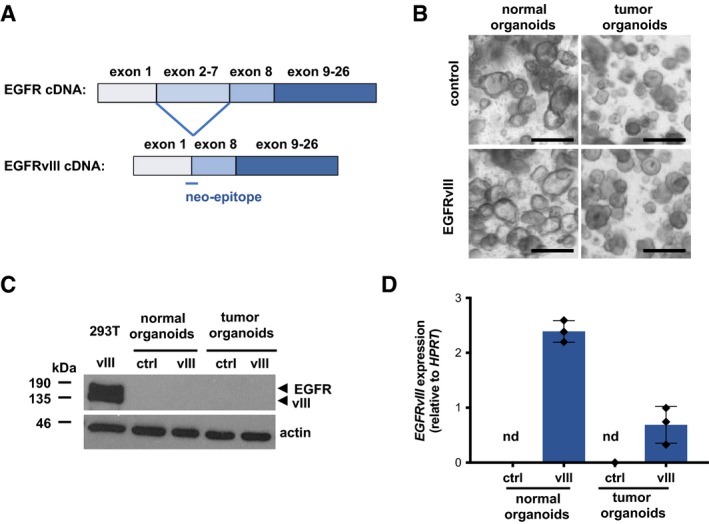
- Schematic representations of wild‐type EGFR and EGFRvIII cDNA lacking exons 2–7.
- Morphology of control and EGFRvIII‐expressing normal and tumor organoids. Scale bars: 250 μm.
- Immunoblot analysis of organoid total cell lysates with anti‐EGFR antibody. Actin served as loading control. Lysate of HEK‐293T cells transiently transfected with EGFRvIII lentiviral transfer vector served as positive control.
- qRT–PCR analysis of EGFRvIII expression (DNase I‐treated organoid total RNA). Average transgene expression (± SD) from n = 3 independent experiments (each measured in triplicates) is shown relative to HPRT expression level. nd: no expression detected in control organoids.
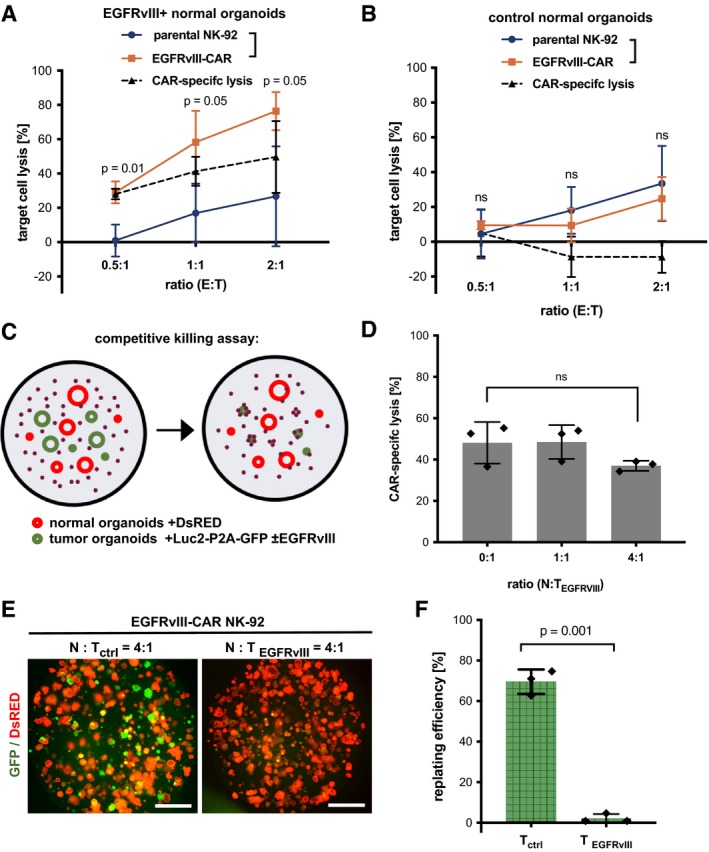
Figure 4. CAR‐mediated cytotoxicity against tumor organoids expressing the EGFRvIII neoantigen.
-
A, BLuciferase‐based quantification of target cell lysis of EGFRvIII‐expressing (A) and control (B) normal organoids by parental NK‐92 and EGFRvIII‐CAR cells after 8 h at different E:T ratios. Values are mean (± SD) from n = 3 independent experiments. Statistical significance was analyzed by unpaired t‐test (ns: P > 0.05).
-
CExperimental setup for competitive killing assays.
-
DCell killing at different normal (N) to tumor organoid ratios (TEGFRvIII). The amount of EGFRvIII‐expressing tumor organoids was kept constant (E:T ratio was 5:1). Luciferase‐based quantification of CAR‐specific lysis. Values are mean (± SD) from n = 3 technical replicates; the experiment was replicated twice. ns: P > 0.05 (unpaired t‐test).
-
E, FReplating assay to analyze the presence of viable tumor cells after prolonged treatment with EGFRvIII‐CAR NK‐92 cells. Normal DsRED‐expressing organoids (N) were mixed with luciferase/GFP‐expressing EGFRvIII‐negative (Tctrl) or EGFRvIII‐positive (TEGFRvIII) tumor organoids at a ratio of 4:1, followed by 48‐h co‐culture (E:T ratio 5:1) and organoid replating. (E) Fluorescence microscopy images after 4 days are shown. Scale bars: 1,000 μm. (F) Quantification of replating efficiency. The mean percentage of GFP‐positive organoids (± SD), relative to the starting ratio, was determined from fluorescent images in n = 3 independent wells. Statistical significance was analyzed by unpaired t‐test.
To recapitulate tumor‐specific targeting in vivo, we performed competitive co‐cultures in the presence of normal organoids (Fig 4C). Tumor organoids expressing EGFRvIII (luciferase and GFP‐positive) were co‐cultured with healthy colon organoids (luciferase‐negative and DsRED‐positive). No significant differences in CAR‐specific lysis were detected in the absence or presence of various amounts of normal organoids, demonstrating effective recruitment (Fig 4D). To test whether prolonged co‐culture could eliminate tumor cells but preserve viability of normal organoids, control normal and EGFRvIII‐expressing tumor organoids were seeded in a ratio of 4:1 and cultured with CAR cells for 48 h, followed by replating and colony‐formation assays of the remaining viable organoids. Fluorescent microscopic analysis of organoid numbers showed that after targeting, EGFRvIII‐expressing tumor organoids were reduced by 98%, while control (EGFRvIII‐negative) organoids only showed a reduction of 29% (Fig 4E and F). These results indicate that the organoid model allows testing of CAR efficiency and specificity in a complex and competitive microenvironment.
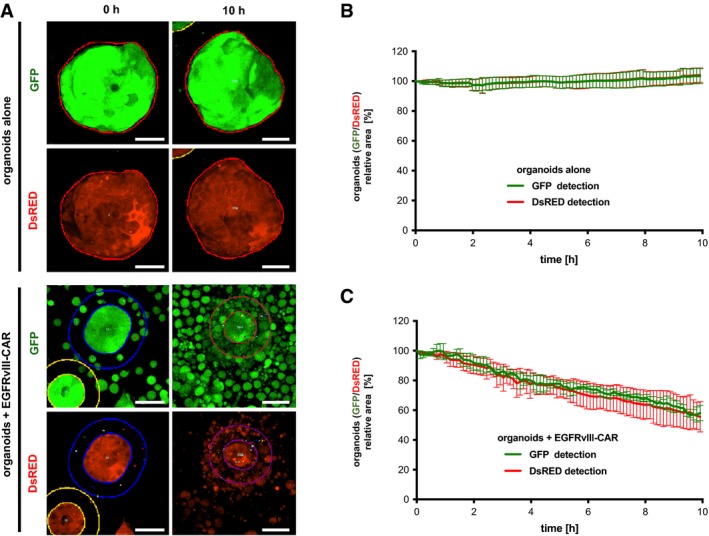
To dynamically visualize the tumor‐specific activity, live imaging was performed. Control experiments using GFP/DsRED double‐positive organoids showed that the image analysis workflow allows similar quantification of organoid area by detection of GFP or DsRED (Fig EV4). GFP expression in EGFRvIII‐CAR NK‐92 cells did not interfere with the analysis, indicating that the algorithm effectively excludes small structures (single cells or apoptotic bodies). Subsequently, co‐cultures of normal organoids (DsRED‐positive) and EGFRvIII‐expressing tumor organoids (GFP‐positive) were monitored alone (Fig 5A and Movie EV7) or incubated with parental NK‐92 (Movie EV8) or EGFRvIII‐CAR NK‐92 cells (Movie EV9). While specific loss of GFP‐positive area was readily observed, the DsRED‐positive area remained constant during the 10 h imaging interval (Fig 5B–D), indicating that CAR‐mediated killing does not induce bystander toxicity to normal tissue lacking the cognate surface antigen. Furthermore, specific recruitment of CAR cells was detected toward the GFP‐positive tumor cells but not to the adjacent DsRED‐positive normal cells (Fig 5E and F), demonstrating the potential of 3D live imaging to monitor on‐ and off‐target toxicity in a parallel fashion.
Figure EV4. Image‐based detection of organoid cytotoxicity with different fluorescent reporters.
-
AImaging data of GFP+ and DsRED+ organoids at 0 and 10 h of monoculture or co‐culture with GFP+ EGFRvIII‐CAR NK‐92 cells (E:T ratio was 2:1). EGFRvIII‐positive tumor organoids were used. Maximum intensity projection images are shown. Automatic organoid detection (thin lines) was performed with similar settings for both reporters (see Appendix Fig S2; the outer lines denote the dilated area for analysis of cell recruitment). Scale bars: 50 μm.
-
B, CQuantitative monitoring of organoid area during monoculture (B) or co‐culture with GFP+ EGFRvIII‐CAR NK‐92 cells (C). Experiments were performed as in (A). Mean values from n = 4 imaging positions (± SD) are shown relative to the area detected at t = 0. Note that comparable areas are measured for GFP and DsRED channels and that the presence of GFP+ EGFRvIII‐CAR NK‐92 only minimally affects the detected organoid area.
Figure 5. Dynamic analysis of CAR‐mediated cytotoxicity against tumor organoids.
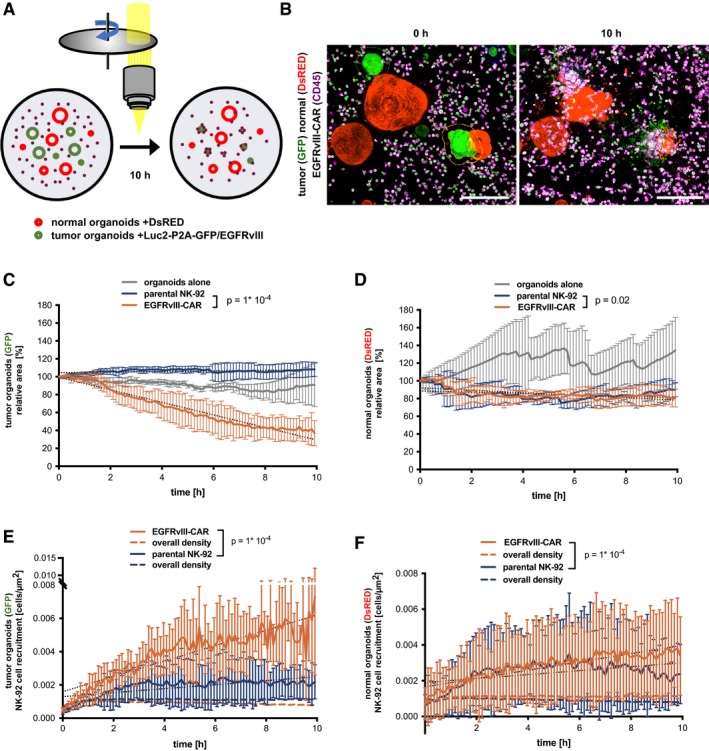
-
ALive imaging setup for competitive killing assays (normal to tumor ratio was 1:1, and E:T ratio was 3:1).
-
BExemplary data with DsRED‐expressing normal organoids (red), GFP‐expressing EGFRvIII‐positive tumor organoids (green), and anti‐CD45‐APC‐labeled EGFRvIII‐CAR NK‐92 cells (magenta) at 0 and 10 h of co‐culture. Maximum intensity projections are shown. Outlines of tumor organoids are automatically detected. Scale bars: 200 μm. See also Movies EV7, EV8 and EV9. Note that EGFRvIII‐CAR NK‐92 cells are also GFP+, but which does not perturb the analysis (see Fig EV4).
-
C, DQuantitative monitoring of cytotoxicity against EGFRvIII‐positive target cells (C; GFP+ area) or against bystander normal cells (D; DsRED+ area). The same data were analyzed in (C and D). Mean values from n = 4 imaging positions (± SD) are shown relative to the area detected at t = 0. Slopes were compared by linear regression; significant differences were observed (ANCOVA).
-
E, FTime‐resolved recruitment of EGFRvIII‐CAR and parental NK‐92 cells to EGFRvIII‐positive target cells (E) or bystander normal cells (F). Same data were analyzed in (C and D). Mean (± SD) from n = 4 imaging positions. NK‐cell density was automatically determined in a region of 50 μm surrounding each organoid. Hatched lines show the NK‐92 density on the entire image. Slopes were compared by linear regression; significant differences were observed (ANCOVA).
Evaluating CAR targeting of FRIZZLED receptors as rationale for colon cancer therapy
Next aim was to exploit this platform to test new CAR strategies targeting endogenously expressed TAAs. Recently, in a subset of CRC cases the loss of RNF43/ZNRF3 has been reported (Giannakis et al, 2014) that function together with LGR4/5 as R‐spondin receptors. The absence of R‐spondin activates their function as the E3‐ligases for Wnt‐receptor FRIZZLED (FZD). This results in FZD internalization and subsequent degradation (Fig 6A), while RNF43/ZNRF3‐deficient cells show persistently increased FZD surface levels and WNT pathway activity (Hao et al, 2012; Koo et al, 2012). Although in the majority of CRC patients WNT activation is caused by downstream mutations affecting the tumor suppressor Adenoma polyposis coli (APC), FZD overexpression induced by RNF43/ZNRF3 mutations might create a therapeutic vulnerability (Koo et al, 2015). Accordingly, the neutralizing pan‐FZD monoclonal antibody (OMP‐18R5) has shown efficacy in preclinical models (Gurney et al, 2012). Based on this monoclonal antibody sequence, we generated a novel CAR encompassing an OMP‐18R5‐derived single‐chain fragment variable (scFv) fused via a flexible hinge region and a transmembrane domain to a composite CD28‐CD3ζ signaling domain. The CAR was introduced into NK‐92 cells by lentiviral transduction (Appendix Fig S3A). Efficient, CAR‐dependent activity was confirmed using FZD5‐overexpressing MDA‐MB453 cells (Appendix Fig S3B). We subsequently took advantage of small intestinal mouse organoids deficient for Apc or double knock‐out (DKO) for Rnf43/Znrf3 that we have previously characterized (Farin et al, 2016) and that were transduced with luciferase/GFP lentiviral vector. Co‐cultures were performed in the presence or absence of R‐spondin with FZD‐CAR cells and parental NK‐92 cells for 8 h, and lysis was quantified by luminescence measurements (Fig 6B and C). A high CAR‐specific lysis was found, but neither in the presence nor in the absence of R‐spondin, any noticeable differences in the targeting between wild‐type, Apc‐KO, and Rnf43/Znrf3‐DKO organoids were detected. Also at reduced E:T ratios, we could not observe more pronounced cytotoxicity toward Rnf43/Znrf3‐DKO organoids (Fig 6D). Although we could not directly associate our results to the mouse Frizzled surface level due to the low abundance of the endogenous receptors, these results indicate that therapeutic targeting could result in off‐tumor cytotoxicity against normal colonic tissue.
Figure 6. Evaluation of Frizzled as a target for CAR therapy using a Rnf43/Znrf3‐deficient mouse organoid model.
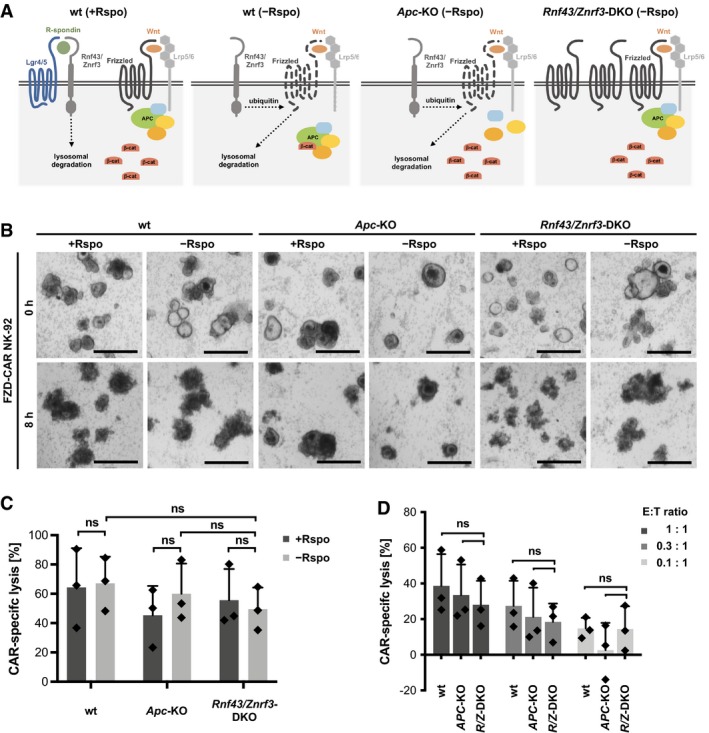
- Schematic model showing differential Frizzled levels and dependence on R‐spondin (Rspo) in normal organoids (wt) and APC‐ and Rnf43/Znrf3‐deficient cells.
- Light microscopic observation of normal and mutant organoids after (FRIZZLED) FZD‐CAR NK‐92‐mediated cytolysis in the presence or absence of R‐spondin (for 1 day before NK‐92 cell addition). Representative images of CAR‐targeted mouse organoids after 8 h of co‐culture are shown. Scale bars: 500 μm.
- Luciferase‐based quantification of FZD‐CAR‐specific killing in the presence and absence of R‐spondin. The E:T ratio was 1:1, and incubation time was 8 h. Values are mean (± SD) from n = 3 independent experiments. Statistical significance was analyzed by unpaired t‐test (ns: P > 0.05).
- FZD‐CAR‐specific killing at reduced E:T ratios. Experiments in the absence of R‐spondin and after 8 h of co‐culture. Values are mean (± SD) from n = 3 independent experiments. Statistical significance was analyzed by unpaired t‐test (ns: P > 0.05).
To further investigate the specificity of FZD‐CAR cells, we studied a panel of human organoids from primary CRC (CRC#1‐4). RNF43‐deficient tumor organoids exhibit a reported sensitivity to the Wnt‐secretion inhibitor IWP‐2 (van de Wetering et al, 2015), indicating their dependence on FZD signaling. By titration experiments, we identified CRC#4 as highly IWP‐2 sensitive (Fig EV5A). Sanger sequencing revealed an RNF43 frameshift mutation in this sample (Appendix Fig S4), while in the IWP‐2‐resistant lines, CRC#1‐3 instead damaging mutations in the APC mutation cluster region were detected. After stable transduction with luciferase/GFP, we measured the cytotoxic activity of parental NK‐92, FZD‐CAR, and EPCAM‐CAR cells toward normal and CRC#1‐4 organoids (Fig EV5B). Compared to the parental NK‐92, both CAR cells showed a uniformly high activity against all lines. The activity of EPCAM‐CAR cells could be associated with a uniform EPCAM expression level (Fig EV5C), arguing that the both CAR strategies result in non‐tumor‐specific activity also against normal epithelia of human origin.
Figure EV5. Evaluation of FZD‐CAR NK‐92 cells for targeting of human RNF43‐deficient CRC organoids.
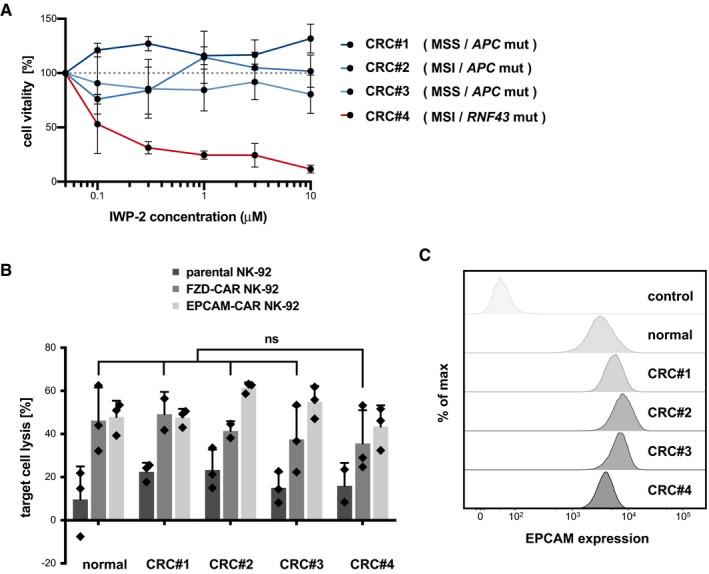
- IWP‐2 toxicity assay in different human CRC organoid lines. Cells were seeded in complete medium (– Wnt). Mean cell viability (± SD; in n = 3 wells; CellTiter‐Glo assay) after 6 days compared to culture in regular medium. The experiment was repeated twice independently. RNF43 and APC genomic loci were analyzed by Sanger sequencing (see Appendix Fig S4). The status of microsatellite instability/stability (MSI/MSS) and the presence of mutations are noted. RNF43 mutant organoids (CRC#4) show increased IWP‐2 sensitivity, indicating endogenous Wnt‐FZD signaling. WT organoids are dependent on exogenous Wnt and were not tested in this assay.
- Luciferase‐based quantification of cytotoxicity of parental, FZD‐CAR, and EPCAM‐CAR NK‐92 cells against normal and CRC organoid lines. Experiments were performed in the absence of R‐spondin. Mean target cell lysis (± SD; in n = 3 wells) after 8 h of co‐culture is shown. The E:T ratio was 2:1. Statistical significance was analyzed by unpaired t‐test (ns: P > 0.05). Note that the RNF43‐deficient organoids (CRC#4) show no increased sensitivity toward FZD‐CAR NK‐92 cells. The experiment was replicated three times independently.
- Flow cytometric analysis with anti‐EPCAM antibody shows similar EPCAM surface level on normal and CRC organoid lines. Unstained cells are shown as controls.
Discussion
For solid cancers, there is an unmet need for preclinical models to identify and select suitable target antigens for CAR therapy. Here, we describe a platform for assessment of CAR‐NK‐92‐mediated activity against patient‐derived organoids in a 3D environment. Compared to previous in vitro and in vivo models, the assays described here allow more physiological analysis of effector cell recruitment and cytotoxicity on a single organoid level. Using a panel of standardized CAR‐NK‐92 cells, this could facilitate rapid and individualized testing of therapy efficacy targeting various TAAs. In addition, potential unwanted toxicity to normal epithelia can be readily addressed. Adaptation of this technology may also help to improve CAR‐T strategies for CRC and other solid cancer entities.
Our results demonstrated that CAR‐NK‐92 cytotoxicity can be efficiently directed against tumor organoids even in a heterogeneous cellular microenvironment and at low levels of TAA expression. Moreover, in long‐term cultures near‐quantitative eradication of tumor cells was achieved in the absence of collateral damage to tumor antigen‐negative cells. However, given that strictly tumor‐specific antigens are not available in most cases, this potency may also result in serious toxicity. On‐target/off‐tumor toxicity can cause severe and life‐threatening side effects (Bonifant et al, 2016). Here, the organoid CAR platform may be useful for assessment of tumor cell specificity at an early stage. This may help to prioritize more laborious models such as humanized mice (Pennell et al, 2018) that however remain indispensible for toxicity testing in vivo that can include off‐cancer activity toward multiple organs. As an example, we generated and tested a novel CAR‐based on a therapeutic antibody (OMP‐18R5) that effectively blocks FZD receptors (Gurney et al, 2012). This antibody had been successfully tested in vitro and in xenograft studies leading to clinical trials (NCT01345201, NCT02005315, NCT01957007, NCT01973309). For patients with RNF43 and ZNRF3 mutations, FZD stabilization has been described, providing a therapeutic rationale (Giannakis et al, 2014; Koo et al, 2015). However, using isogenic wild‐type, Apc‐KO, and RnfF43/Znrf3‐DKO organoid lines, we could not identify a therapeutic window for the OMP‐18R5‐based CAR approach, arguing against this strategy.
Current in vitro models cannot fully capture the cellular processes that are relevant for immunotherapy of solid tumors. Most important restrictions are the availability of quantitative read‐outs for cytotoxicity during prolonged periods and a co‐culture setup that preserves a 3D tissue‐like environment. Here, we established robust assays to monitor cytotoxicity against organoids either by luciferase‐based endpoint measurement, which allows high‐throughput testing, or by live microscopy for continuous and cell‐resolved analysis. We observed that recruitment of effector cells is not limiting. Rather the E:T ratio was critical for effective killing, which could be observed on a single organoid level. We noted that killing efficiency varies between organoids of different sizes. While smaller organoids are lysed more rapidly, they only account for a minor fraction of all cells. The slower kinetics observed for larger organoids might more closely reflect the CAR response against solid tumor masses in vivo. In a tissue context, competition with neighboring tumor cells may reduce the local E:T ratio and decrease damage by prolonging the interval between NK‐cell contacts or reducing the membrane surface area that can be attacked. The possibility to trace responses in 3D thus allows to identify rate‐limiting processes to mechanistically improve CAR strategies.
Arguably, CAR‐T‐ or CAR‐NK‐based strategies have not yet led to effective therapies in CRC, and one major challenge may be efficient cell administration. Low abundance of endogenous NK cells has been reported in primary colon cancer (Halama et al, 2011), and low lymphocyte infiltration (Galon et al, 2006) and/or an immunosuppressive environment correlate with poor prognosis and limit the benefit of checkpoint inhibition in CRC (Charoentong et al, 2017). TGF‐β‐mediated signaling in cancer‐associated fibroblasts has been identified as a critical mediator (Tauriello et al, 2018). While local CAR cell application may be challenging for primary CRC, our results show efficient effector cell recruitment and function in a tissue‐like context even in the presence of tumor‐derived fibroblasts. Transgenic expression of the chemokine receptor CCL9 could help to target cells to the gut (Chen et al, 2015). The limited penetration of NK cells into 3D Matrigel may serve as a model to study and improve immune cell infiltration into tissues. However, more relevant for treatment of metastatic CRC could be systemic administration or injection via the portal venous system to target disseminated tumor cells.
We recently showed that local CAR‐NK‐92 therapy can induce systemic endogenous anti‐tumor immunity in a glioblastoma model (Zhang et al, 2016, 2017). Thus, a combination of CAR‐NK‐92 cells with checkpoint inhibitors may further increase host T‐cell defenses. A second challenge will be to identify suitable target antigens for CAR therapy. Common neoepitopes have been described for MSI tumors only (Schwitalle et al, 2008; Inderberg et al, 2017). For the majority of non‐hypermutated tumors, differentially expressed surface molecules have to be targeted at the risk of off‐cancer toxicity. To limit such adverse reactions, new approaches have been developed such as the UniCAR system that allows precise control of active CAR dosage (Cartellieri et al, 2015) or the SynNotch technology that depends on combinatorial detection of multiple TAAs (Roybal et al, 2016). Testing in patient‐matched normal and tumor organoids may greatly assist to further adapt these strategies.
As a demonstration of the great potential of organoid systems for the development of cancer immunotherapies, tumor‐reactive T cells have recently been expanded from peripheral blood using co‐cultures of matched CRC organoids (Dijkstra et al, 2018). While this strategy was only successful in a fraction of HLA‐I‐positive tumors and only in MSI patients, future CAR‐T approaches might use organoids to selectively expand tumor‐reactive cells for a larger fraction of patients. With increasing access to organoids for other major cancer entities such as carcinomas of the breast, prostate, stomach, pancreas, and liver (Gao et al, 2014; Boj et al, 2015; Broutier et al, 2017; Sachs et al, 2018; Seidlitz et al, 2018), the co‐culture workflows established herein may provide a valuable tool for evaluating novel cancer immunotherapies also in a more general fashion.
Materials and Methods
Patient samples
CRC organoids were obtained from resected primary tumor tissues provided by the University Cancer Center Frankfurt (UCT). Normal human colon organoids and primary fibroblasts were derived from non‐pathological mucosa that was collected either during preemptive colonoscopy or from tumor‐adjacent normal colon after tumor resection. Written informed consent was obtained from all patients, and the study was approved by the Institutional Review Boards of the UCT and the Ethics Committee at the University Hospital Frankfurt (project number: SGI‐06‐2015).
Organoids and cell culture
Normal and tumor colon organoids were established and maintained as described previously (Sato et al, 2011; van de Wetering et al, 2015). Human organoids were embedded in growth factor reduced Matrigel (Corning), and normal human colon organoids were cultured in advanced DMEM/F12 supplemented with 50% Wnt3a‐conditioned medium (CM), 20% R‐spondin CM, 10% Noggin CM, 10 mM HEPES, 1× GlutaMax, 1× penicillin/streptomycin, 2% B27, 10 mM nicotinamide (Sigma‐Aldrich), 12.5 mM N‐acetylcystein (Sigma‐Aldrich), 500 nM A83‐01 (Tocris), 10 μM SB202190 (Biotrend), and 50 ng/ml human EGF (PeproTech). Unless stated otherwise, all media components were purchased from Life Technologies. Tumor organoids were grown in complete medium lacking Wnt3a CM. Generation and maintenance of Apc and Rnf43/Znrf3 mutant organoids from mouse small intestine was described previously (Farin et al, 2016). Normal mouse intestinal organoids were embedded in Reduced Growth Factor Basement Membrane extract (RGF BME 2; Cultrex; Amsbio) and grown in ENR medium containing advanced DMEM/F12 supplemented with 10% Noggin CM, 5% R‐spondin CM, 10 mM HEPES, 1× GlutaMax, 1× penicillin/streptomycin, 2% B27, 12.5 mM N‐acetylcystein, and 50 ng/ml human EGF, all as above. Apc and Rnf43/Znrf3 mutant organoids were cultured in medium lacking R‐spondin (EN medium). CM was prepared as described in Farin et al (2012). For each passage (every 5–7 days), organoids were dissociated by vigorous pipetting, washed from remaining matrix, pelleted, and seeded in fresh Matrigel/BME. 10 μM Y‐27632 (Selleckchem) was added to the culture medium after passage, and the medium was replaced every 2–3 days. For IWP‐2 titrations (Sigma‐Aldrich), organoids were seeded in a regular fashion in complete medium lacking Wnt3a CM in the presence of the indicated IWP‐2 concentrations and cultured for 6 days. Organoid viability was measured with CellTiter‐Glo® Assay (Promega).
Human primary fibroblasts were derived from homogenized colon tissues as described (Farin et al, 2012) and cultured on gelatin‐coated dishes in DMEM supplemented with 10% FCS, 1× penicillin/streptomycin and passaged weekly.
Human NK‐92 cells and their CAR‐engineered derivatives were maintained in X‐VIVO 10 medium (Lonza) supplemented with 5% heat‐inactivated human plasma (German Red Cross Blood Donation Service Baden‐Württemberg‐Hessen, Frankfurt, Germany) and 100 IU/ml IL‐2 (Proleukin; Novartis). EPCAM‐specific NK‐92 cells (NK‐92/31.28.z‐EGFP), HER2‐specific NK‐92 cells (NK‐92/5.28.z), and EGFRvIII‐targeting NK‐92 cells (NK‐92/MR‐1.1.28.z‐EGFP) were described previously (Sahm et al, 2012; Schönfeld et al, 2015; Genßler et al, 2016). A FZD‐specific CAR was designed based on a scFv fragment of the monoclonal antibody OMP‐18R5 (Gurney et al, 2012). A codon‐optimized CAR sequence encompassing an immunoglobulin heavy chain signal peptide, the OMP‐18R5 scFv fragment, a Myc‐tag, a CD8α hinge region, the transmembrane and intracellular domains of CD28, and the intracellular domain of CD3ζ was synthesized (GeneArt, Thermo Fisher Scientific) and inserted into the lentiviral transfer plasmid pSIEW upstream of IRES and EGFP sequences as previously described for other CAR constructs (Sahm et al, 2012). After transduction of NK‐92 cells, EGFP‐positive NK‐92 cells were enriched by flow cytometric cell sorting with a FACSAria Fusion fluorescence‐activated cell sorter (BD Biosciences). CAR surface expression in the obtained cell pool was confirmed by flow cytometric analysis with Alexa Fluor 647‐conjugated Myc‐tag‐specific antibody (clone 9E10; Santa Cruz Biotechnology) using a FACSCanto II flow cytometer (BD Biosciences). For all 3D cytotoxicity assays, sorted pools of the respective NK‐92 cells were used.
HEK293T, MDA‐MB453, and MDA‐MB468 cells were cultured in tissue culture‐treated dishes in DMEM supplemented with 10% FCS, 1× penicillin/streptomycin. Routine tests for mycoplasma were performed with negative results.
Lentiviral transduction
For lentiviral delivery of EGFRvIII, the encoding cDNA was PCR amplified from plasmid pLTR‐EGFRvIII (Hills et al, 1995), using Phusion High‐Fidelity DNA Polymerase (NEB). Primers were fwd: CAGGACCGGTTCTAGAGCGCTGCCACCATGCGACCCTCCGGGACG and rev: TTGTTGCGCCGGATCCTGCTCCAATAAATTCACTGCTTTG. The purified PCR fragment was inserted using the In‐Fusion HD Cloning kit (Clontech) into the lentiviral transfer plasmid lentiCas9‐Blast, a kind gift from Feng Zhang (Addgene plasmid # 52962; Sanjana et al, 2014), following BamHI and XbaI digests to excise the Cas9 ORF. For labeling cells with GFP and luciferase, the codon‐optimized luciferase reporter gene luc2 was amplified from plasmid pGL4.10[luc2] (Promega). Primers were fwd: GAACTAAACCGTCGACGCCACCATGGAAGACGCCAAAAACATAAAG and rev: CCATGGTGGCGTCGACTGGTCCAGGATTCTCTTCGACATCCCCTGCTTGTTTCAACAGGGAGAAGTTAGTGGCTCCGCTTCCGGACACGGCGATCTTTCCGCCCTTC, introducing a 3′ P2A sequence. The PCR fragment was inserted (as above) into the SalI site of pLV‐Pgk::GFP‐IRES‐Puro, 5′ of the GFP ORF. For labeling cells with DsRed, pLV‐LoxP‐DsRED‐STOP‐LoxP‐eGFP, a kind gift from Niels Geijsen (Addgene plasmid # 62732; D'Astolfo et al, 2015), was used. For FZD expression in MDA‐MB453 cells, the mouse FZD5 sequence together with a 5′ signal peptide sequence and a V5 epitope was amplified from pCDNA3.1‐V5‐FZD5 (kind gift of Madelon Maurice; Koo et al, 2012). Primer sequences were fwd: GAACTAAACCGTCGACGCCACCATGGTCCCGTGCACG and rev: CGCTTCCGGACTCGAGTACGTGCGACAGGGACACTTGC, ending with the last codon before stop. The PCR fragment was inserted (as above) into the SalI and XhoI sites of pLV‐Pgk::P2A‐DsRed‐IRES‐Puro lentivirus, 5′ and in frame with the P2A‐DsRED sequence. All constructs were sequence‐verified.
VSV‐G pseudotyped lentiviral particles were produced in HEK293T cells after polyethylenimine‐mediated transient transfection. Four days after transfection, the supernatant was collected and viral particles were concentrated by ultracentrifugation (20,000 g, 1 h, 4°C). Lentiviral transduction of organoids was performed as described (Koo et al, 2011) after enzymatic dissociation into single cells at 37°C for 5 min using StemPro Accutase (Life Technologies). Puromycin or blasticidin (1–2 μg/ml) was added to the culture medium after 3–4 days to select for transgene‐expressing cells. For lentiviral gene delivery to 2D cell lines and human primary fibroblasts, the adherent cells were incubated with viral particles for 8 h. Transgene‐expressing cells were selected as above.
Immunoblot analysis
Organoids were collected and separated from the Matrigel by mechanical shearing and were thoroughly washed. Cells were lysed using RIPA lysis buffer (150 mM NaCl, 50 mM Tris–HCl pH 7.6, 5 mM NaF, 5 mM β‐glycerophosphate, 1 mM EDTA, 1 mM Na‐orthovanadate, 1% Na‐deoxycholate, 1% NP‐40, supplemented with cOmplete protease inhibitor (Roche)) and additional sonification. After clearance by centrifugation, protein concentration was determined by standard Bradford assay. 20 μg of total proteins was loaded per lane for SDS–PAGE (10%) and subsequent transfer to a nitrocellulose membrane. Proteins were probed using the following antibodies: anti‐EGFR (clone D38B1; 1:1,000; Cell Signaling) and anti‐actin (polyclonal; 1:2,500; Sigma). Proteins were detected by chemiluminescence using species‐specific horseradish peroxidase‐labeled secondary antibodies (1:5,000).
Quantitative RT–PCR analysis
Total RNA was extracted using the Macherey‐Nagel NucleoSpin RNA Kit according to the manufacturer's instructions. For cDNA synthesis, random hexamers and M‐MLV Reverse Transcriptase enzyme (Promega) were used after DNase I digest. Relative gene expression was measured using Power SYBR® Green PCR Master Mix (Thermo Fisher Scientific) on a StepOneTM Real‐Time PCR instrument (Applied Biosystems). EGFRvIII transgene expression was studied using the following primers fwd: CAGAATACCTAAGGGTCGCG and rev: ATACAAGTCAGGTTGCCAGC. Quantification was performed using the dCT method, and for normalization, HPRT expression was studied using the following primers fwd: GCTGAGGATTTGGAAAGGGT and rev: CATCTCGAGCAAGACGTTCA.
Organoid genotyping
Genomic DNA was extracted using the DNeasy Blood & Tissue kit (Qiagen) and amplified using Phusion High‐Fidelity DNA Polymerase (NEB). Primers for the human APC locus were fwd: AGCTCCATCCAAGTTCTGCAC and rev: CTTGCCACAGGTGGAGGTAAT (1132 bp product) and for the human RNF43 locus were fwd: GTGGATGTGGTTCCAGGGG and rev: CCTGGGACCCTCTCGATCTT (1,244 bp product). DNA fragments were sequenced by Sanger sequencing (LightRun, Eurofins Genomics) using internal and external primers.
Immunostaining
Organoids were collected in chilled Organoid Harvesting Solution (Biozol) and incubated for 15 min on ice to completely dissolve remaining Matrigel. Organoids were pelleted and incubated with allophycocyanin (APC)‐coupled EPCAM‐specific antibody (clone HEA125; 1:300; Miltenyi Biotec) on ice for 1 h for EPCAM surface staining. After washing thoroughly, organoids were resuspended in complete medium and imaged using the CQ1 Confocal Quantitative Image Cytometer (Yokogawa).
Flow cytometry
Organoids were collected and enzymatically dissociated into single cells at 37°C for 5 min using StemPro Accutase. Cells were pelleted, resuspended in FACS buffer (PBS, 2% FBS, 2 mM EDTA), and filtered through a 0.40‐μm strainer. Suspensions of equivalent cell numbers were incubated with VioBlue‐coupled anti‐EPCAM (clone REA764; 1:150; Miltenyi Biotec) antibody for 30 min on ice. For live‐cell gating, eFluor780 (1:500; Invitrogen) was added. Surface antigen expression or GFP was measured on a FACSCanto II flow cytometer (BD Biosciences) and analyzed using FACSDiva software (BD Biosciences) and FlowJo v10 software.
NK‐92 cytotoxicity assays
Cytotoxicity assays with single cell suspensions of cell lines were performed as described (Sahm et al, 2012). To study the impact of the organoid media, each factor was added to the NK medium at 1x concentration (as above). Killing experiments were excluded if the CAR‐specific lysis in positive controls was < 30% and the experiment was repeated using a freshly recovered cryovial of CAR‐NK‐92 cells. If organoid/Matrigel layer accidently detached during the washing steps, the respective wells were marked and excluded from the analysis.
For organoid cytotoxicity assays, organoids were seeded on a Matrigel layer or on a layer of primary human colon fibroblasts and incubated with the respective NK‐92 cells as follows. 48‐well standard culture plates with a growth area of 1 cm2 (Greiner Bio‐One) were first moistened using culture medium. Subsequently, each well was evenly covered with 35 μl undiluted Matrigel which was allowed to solidify overnight at RT. Alternatively, human primary fibroblasts were seeded on gelatin‐coated 48‐well plates at a confluency of 80–90% and cultured for 1 day. Confluent organoids were collected, mechanically sheared, pelleted, washed, and seeded at a split ratio of 1:2.5. Organoids were resuspended in 150 μl of the respective culture medium supplemented with 10 μM of Y‐27632 per assay replicate. The organoid suspension was carefully added to the center of the Matrigel‐ or fibroblast‐covered wells, respectively. Organoids were grown for 24 h before addition of NK‐92 cells in 500 μl of medium without Y‐27632. For standard cytotoxicity assays, ~105 organoids cells (determined as described below) were seeded per well of the 48‐well plate (1 cm2), which was defined as organoid density of 100%.
NK‐92 cells and their CAR‐engineered derivatives were pelleted, washed, and resuspended in co‐culture medium as indicated below. Cells were counted using a hemocytometer, and the required number of cells in a total volume of 500 μl (or 1 ml for long‐term co‐cultures) per well of a 48‐well plate was co‐incubated with target cells for the desired time period at 37°C. The following co‐culture media were used: For human organoids, the medium contained complete normal or tumor medium (as above) lacking nicotinamide. 0.01 μM Prostaglandin E2 (PGE2; R&D Systems) was added to induce a cystic phenotype. For mouse small intestinal organoids, the respective culture media were used (as above). For luciferase‐based quantification, the supernatant was removed after co‐culture and each well was washed with pre‐warmed culture medium. Subsequently, 500 μl luciferase assay reagent composed of 10% One‐Glo EX Reagent (Promega) in lysis buffer (50 mM NaCl, 50 mM Tris–HCl pH 7.4, 1% Triton X‐100) was added to each well and incubated at room temperature for 15 min protected from light. Lysates were collected and stored at −80°C until measurement. 100 μl of each sample was transferred to an opaque 96‐well microtiter plate and measured using a LUMIstar Galaxy Luminometer (BMG LABTECH). Target cell lysis was defined as the difference between luciferase activity of organoids cultured alone and residual luciferase activity after co‐culture. CAR‐specify lysis was defined as the difference between target cell lysis by CAR‐NK‐92 cells and parental NK‐92 cells.
Image‐based counting of organoid cell number
To determine the exact epithelial cell count seeded per well of the 48‐well plate, the same amount of organoids was seeded on Matrigel‐coated 24‐well Imaging Plates CG (Miltenyi Biotec) and grown for 24 h. Cells were fixed (chilled 4% paraformaldehyde, 20 min) and permeabilized (1% Triton X‐100, 5 min), and nuclei were stained using NucBlue Fixed Cell ReadyProbes (Thermo Fisher Scientific) following the manufacturer's instructions. For each well, DAPI image stacks of entire organoids (4 μm z‐distance, 20–35 steps) were acquired in 60–90 randomly selected areas at 20× magnification (each corresponding to 0.64 mm2) using the CQ1 Confocal Quantitative Image Cytometer. The z‐stacks were analyzed using the application “Spheroid Structure” included in the CQ1 Software. Briefly, after smoothening and local thresholding, nuclei were detected as objects and expanded in all three dimensions for segmentation (see Appendix Fig S1). The mean number of nuclei per image region was extrapolated to the total area per well.
Live‐cell imaging‐based NK‐92 cytotoxicity assay
For all live‐cell imaging experiments, black‐walled glass bottom 24‐well Imaging Plates CG (Miltenyi Biotec) with a growth area of 1.36 cm2 were used. Moistened wells were covered with a layer of 50 μl undiluted Matrigel and allowed to solidify overnight at RT. For seeding, organoids stably expressing green or red fluorescent protein (GFP; DsRED) were seeded (as above) in a total volume of 500 μl per well and grown for 24 h before addition of NK‐92 cells. NK‐92 cells (prepared as above) were resuspended in a total volume of 1 ml co‐culture medium per well and stained by addition of APC‐conjugated anti‐CD45 (clone 2D1; 1:150; Invitrogen). All live‐cell imaging experiments were performed at 37°C, 5% CO2, and 20% humidity. Fluorescence images were acquired at identical positions in intervals of 6–7 min at 20× magnification using the CQ1 Confocal Quantitative Image Cytometer. Per position 13 z‐stacks with a step size of 4.2 μm were acquired (50 μm total z‐range). Per condition four positions were imaged using identical settings.
Image processing and analysis
Image analysis was done using the CQ1 software (Yokogawa). The exact procedure and software settings are shown in Appendix Fig S2. Image stacks were processed to maximum intensity projections (MIP) for further analysis. A custom algorithm was designed to analyze the loss of organoid area (as a measure for target cell killing) and the recruitment of NK‐92 cells to their targets. For detection of organoids, GFP/DsRED images were smoothened and binarized by local thresholding that was simultaneously adjusted for an entire well/condition. Edges of binarized objects were eroded to avoid detection of noise (e.g., apoptotic bodies). Only objects with an area greater than the threshold of 500 μm2 were defined as organoids. For measurement of effector cell recruitment, the detected organoids were expanded by addition of a belt area with a constant diameter of 50 μm (“dilation circle”), and NK‐92 cells within the dilated organoid area were counted. For this, the CD45 signals were smoothened and binarized by local thresholding. The area of binarized objects was eroded, and objects were reduced to points. A minimum distance to neighboring points was defined (0.1 μm) to avoid double counting of cells. All points detected within the dilated organoid area were counted as recruited effector cells. In order to measure the background density of NK‐92 cells (total number of NK‐92 cells per imaging frame), the same algorithm was applied to the total image).
The .csv files generated by the CQ1 software were imported to MS Excel 2016 for further analysis. For tracking of organoid area loss and NK‐92 cell recruitment over time for individual organoids, a macro was designed (Fig EV2C). Raw data included the organoid coordinates, the organoid area, and NK‐92 cell count. For each imaging position, the coordinates (x min, x max, y min, y max) served as unique identifier for each organoid. The initial range was expanded (NEW x min, NEW x max, NEW y min, NEW y max; see Fig EV2D) to track organoids that change size or position over the imaging interval. After filtering for the new coordinates, each organoid was assigned a unique number.
Statistical analysis
All statistical analysis was performed using GraphPad Prism 7.0d software. Data are presented as mean, and all error bars show standard deviation (SD). Statistical significance was evaluated with two‐tailed, unpaired Student's t‐test. For multiple t‐tests, unequal SD was assumed and P‐values were filtered using the desired FDR = 1% and the two‐stage method by Benjamini, Krieger, and Yekutieli. Differences were considered as not significant (ns) when P > 0.05. The built‐in linear regression was used to calculate correlation coefficients (R 2) and to determine whether slopes are significantly different (ANCOVA test).
Author contributions
TES, MHPG, CZ, and JR performed the experiments. CZ, MHM, BEM, JR, and TD provided technical support. TES, MHPG, and HFF analyzed the data. HFF and WSW conceived the study, supervised the experiments, and obtained funding. TES and HFF drafted the manuscript. All authors discussed the results and commented on the manuscript.
Conflict of interest
C.Z. and W.S.W. are named as inventors on patents and patent applications in the field of cancer immunotherapy owned by Georg‐Speyer‐Haus. The other authors declare no potential conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Movie EV9
Review Process File
Acknowledgements
We thank Lisa Sevenich for excellent help with live imaging experiments, Hans Clevers and Calvin Kuo for Wnt3a, Noggin, and R‐spondin producing cell lines, Stefan Stein for assistance with cell sorting, Thomas Brunner and Marcus Groettrup for scientific comments, and Constantin Menche and Barnabas Irmer for support with the sequencing analysis. The UCT biobank is acknowledged for support with collection of patient tissues. Grant Support: H.F.F. has been supported by DKTK junior researcher group funding and by the LOEWE Center Frankfurt Cancer Institute (FCI) funded by the Hessen State Ministry for Higher Education, Research and the Arts [III L 5 ‐ 519/03/03.001 ‐ (0015)].
The EMBO Journal (2019) 38: e100928
References
- Boj SF, Hwang C‐I, Baker LA, Chio IIC, Engle DD, Corbo V, Jager M, Ponz‐Sarvise M, Tiriac H, Spector MS et al (2015) Organoid models of human and mouse ductal pancreatic cancer. Cell 160: 324–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ (2016) Toxicity and management in CAR T‐cell therapy. Mol Ther Oncolytics 3: 16011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarró LM, Bradshaw CR, Allen GE, Arnes‐Benito R, Sidorova O, Gaspersz MP et al (2017) Human primary liver cancer‐derived organoid cultures for disease modeling and drug screening. Nat Med 23: 1424–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartellieri M, Loff S, von Bonin M, Bejestani EP, Ehninger A, Feldmann A, Koristka S, Arndt C, Ehninger G, Bachmann MP (2015) Unicar: a novel modular retargeting platform technology for CAR T cells. Blood 126: 5549–5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, Hackl H, Trajanoski Z (2017) Pan‐cancer immunogenomic analyses reveal genotype‐immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep 18: 248–262 [DOI] [PubMed] [Google Scholar]
- Chen HJ, Sun J, Huang Z, Hou H, Arcilla M, Rakhilin N, Joe DJ, Choi J, Gadamsetty P, Milsom J et al (2015) Comprehensive models of human primary and metastatic colorectal tumors in immunodeficient and immunocompetent mice by chemokine targeting. Nat Biotechnol 33: 656–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Astolfo DS, Pagliero RJ, Pras A, Karthaus WR, Clevers H, Prasad V, Lebbink RJ, Rehmann H, Geijsen N (2015) Efficient intracellular delivery of native proteins. Cell 161: 674–690 [DOI] [PubMed] [Google Scholar]
- Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, Slagter M, van der Velden DL, Kaing S, Kelderman S et al (2018) Generation of tumor‐reactive T cells by co‐culture of peripheral blood lymphocytes and tumor organoids. Cell 174: 1586–1598.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farin HF, Van Es JH, Clevers H (2012) Redundant sources of Wnt regulate intestinal stem cells and promote formation of Paneth cells. Gastroenterology 143: 1518–1529.e7 [DOI] [PubMed] [Google Scholar]
- Farin HF, Karthaus WR, Kujala P, Rakhshandehroo M, Schwank G, Vries RGJ, Kalkhoven E, Nieuwenhuis EES, Clevers H (2014) Paneth cell extrusion and release of antimicrobial products is directly controlled by immune cell–derived IFN‐γ. J Exp Med 211: 1393–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farin HF, Jordens I, Mosa MH, Basak O, Korving J, Tauriello DVF, de Punder K, Angers S, Peters PJ, Maurice MM et al (2016) Visualization of a short‐range Wnt gradient in the intestinal stem‐cell niche. Nature 530: 340–343 [DOI] [PubMed] [Google Scholar]
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136: E359–E386 [DOI] [PubMed] [Google Scholar]
- Fu X, Tao L, Rivera A, Williamson S, Song X‐T, Ahmed N, Zhang X (2010) A simple and sensitive method for measuring tumor‐specific T cell cytotoxicity. PLoS ONE 5: e11867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii M, Shimokawa M, Date S, Takano A, Matano M, Nanki K, Ohta Y, Toshimitsu K, Nakazato Y, Kawasaki K et al (2016) A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell 18: 827–838 [DOI] [PubMed] [Google Scholar]
- Galon J, Costes A, Sanchez‐Cabo F, Kirilovsky A, Mlecnik B, Lagorce‐Pagès C, Tosolini M, Camus M, Berger A, Wind P et al (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313: 1960–1964 [DOI] [PubMed] [Google Scholar]
- Gan HK, Cvrljevic AN, Johns TG (2013) The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J 280: 5350–5370 [DOI] [PubMed] [Google Scholar]
- Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK et al (2014) Organoid cultures derived from patients with advanced prostate cancer. Cell 159: 176–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genßler S, Burger MC, Zhang C, Oelsner S, Mildenberger I, Wagner M, Steinbach JP, Wels WS (2016) Dual targeting of glioblastoma with chimeric antigen receptor‐engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 5: e1119354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakis M, Hodis E, Jasmine M, Yamauchi M, Rosenbluh J, Cibulskis K, Saksena G, Lawrence MS, Qian ZR, Nishihara R et al (2014) RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet 46: 1264–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong JH, Maki G, Klingemann HG (1994) Characterization of a human cell line (NK‐92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 8: 652–658 [PubMed] [Google Scholar]
- Gurney A, Axelrod F, Bond CJ, Cain J, Chartier C, Donigan L, Fischer M, Chaudhari A, Ji M, Kapoun AM et al (2012) Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci USA 109: 11717–11722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, Koch M, Weitz J, Kloor M, Zoernig I et al (2011) Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Cancer Res 17: 678–689 [DOI] [PubMed] [Google Scholar]
- Halle S, Keyser KA, Stahl FR, Busche A, Marquardt A, Zheng X, Galla M, Heissmeyer V, Heller K, Boelter J et al (2016) In vivo killing capacity of cytotoxic T cells is limited and involves dynamic interactions and T cell cooperativity. Immunity 44: 233–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao H‐X, Xie Y, Zhang Y, Charlat O, Oster E, Avello M, Lei H, Mickanin C, Liu D, Ruffner H et al (2012) ZNRF3 promotes Wnt receptor turnover in an R‐spondin‐sensitive manner. Nature 485: 195–200 [DOI] [PubMed] [Google Scholar]
- Hills D, Rowlinson‐Busza G, Gullick WJ (1995) Specific targeting of a mutant, activated EGF receptor found in glioblastoma using a monoclonal antibody. Int J Cancer 63: 537–543 [DOI] [PubMed] [Google Scholar]
- Inderberg EM, Wälchli S, Myhre MR, Trachsel S, Almåsbak H, Kvalheim G, Gaudernack G (2017) T cell therapy targeting a public neoantigen in microsatellite instable colon cancer reduces in vivo tumor growth. Oncoimmunology 6: e1302631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- June CH, Sadelain M (2018) Chimeric antigen receptor therapy. N Engl J Med 379: 64–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo B‐K, Stange DE, Sato T, Karthaus W, Farin HF, Huch M, van Es JH, Clevers H (2011) Controlled gene expression in primary Lgr5 organoid cultures. Nat Methods 9: 81–83 [DOI] [PubMed] [Google Scholar]
- Koo B‐K, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, van Es JH, Mohammed S, Heck AJR, Maurice MM et al (2012) Tumour suppressor RNF43 is a stem‐cell E3 ligase that induces endocytosis of Wnt receptors. Nature 488: 665–669 [DOI] [PubMed] [Google Scholar]
- Koo B‐K, van Es JH, van den Born M, Clevers H (2015) Porcupine inhibitor suppresses paracrine Wnt‐driven growth of Rnf43;Znrf3‐mutant neoplasia. Proc Natl Acad Sci USA 112: 7548–7550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D et al (2015) PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med 372: 2509–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlecnik B, Bindea G, Kirilovsky A, Angell HK, Obenauf AC, Tosolini M, Church SE, Maby P, Vasaturo A, Angelova M et al (2016) The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci Transl Med 8: 327ra26 [DOI] [PubMed] [Google Scholar]
- Noel G, Baetz NW, Staab JF, Donowitz M, Kovbasnjuk O, Pasetti MF, Zachos NC (2017) A primary human macrophage‐enteroid co‐culture model to investigate mucosal gut physiology and host‐pathogen interactions. Sci Rep 7: 45270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki K, Mochizuki W, Matsumoto Y, Matsumoto T, Fukuda M, Mizutani T, Watanabe M, Nakamura T (2016) Co‐culture with intestinal epithelial organoids allows efficient expansion and motility analysis of intraepithelial lymphocytes. J Gastroenterol 51: 206–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennell CA, Barnum JL, McDonald‐Hyman CS, Panoskaltsis‐Mortari A, Riddle MJ, Xiong Z, Loschi M, Thangavelu G, Campbell HM, Storlie MD et al (2018) Human CD19‐targeted mouse T cells induce B cell aplasia and toxicity in human CD19 transgenic mice. Mol Ther 26: 1423–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogoz A, Reis BS, Karssemeijer RA, Mucida D (2015) A 3‐D enteroid‐based model to study T‐cell and epithelial cell interaction. J Immunol Methods 421: 89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, Lim WA (2016) Precision tumor recognition by T cells with combinatorial antigen‐sensing circuits. Cell 164: 770–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, Balgobind AV, Wind K, Gracanin A, Begthel H et al (2018) A living biobank of breast cancer organoids captures disease heterogeneity. Cell 172: 373–386.e10 [DOI] [PubMed] [Google Scholar]
- Sahm C, Schönfeld K, Wels WS (2012) Expression of IL‐15 in NK cells results in rapid enrichment and selective cytotoxicity of gene‐modified effectors that carry a tumor‐specific antigen receptor. Cancer Immunol Immunother 61: 1451–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F (2014) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods 11: 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ et al (2009) Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature 459: 262–265 [DOI] [PubMed] [Google Scholar]
- Sato T, Stange DE, Ferrante M, Vries RGJ, van Es JH, van den Brink S, van Houdt WJ, Pronk A, van Gorp J, Siersema PD et al (2011) Long‐term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology 141: 1762–1772 [DOI] [PubMed] [Google Scholar]
- Schönfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, Nowakowska P, Bönig H, Köhl U, Kloess S et al (2015) Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2‐specific chimeric antigen receptor. Mol Ther 23: 330–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwitalle Y, Kloor M, Eiermann S, Linnebacher M, Kienle P, Knaebel HP, Tariverdian M, Benner A, von Knebel Doeberitz M (2008) Immune response against frameshift‐induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology 134: 988–997 [DOI] [PubMed] [Google Scholar]
- Seidlitz T, Merker SR, Rothe A, Zakrzewski F, von Neubeck C, Grützmann K, Sommer U, Schweitzer C, Schölch S, Uhlemann H et al (2018) Human gastric cancer modelling using organoids. Gut 68: 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suck G, Odendahl M, Nowakowska P, Seidl C, Wels WS, Klingemann HG, Tonn T (2016) NK‐92: an “off‐the‐shelf therapeutic” for adoptive natural killer cell‐based cancer immunotherapy. Cancer Immunol Immunother 65: 485–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, Yin J, You F, Zhu M, Shen W et al (2018) First‐in‐man clinical trial of CAR NK‐92 cells: safety test of CD33‐CAR NK‐92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res 8: 1083–1089 [PMC free article] [PubMed] [Google Scholar]
- Tauriello DVF, Palomo‐Ponce S, Stork D, Berenguer‐Llergo A, Badia‐Ramentol J, Iglesias M, Sevillano M, Ibiza S, Cañellas A, Hernando‐Momblona X et al (2018) TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554: 538–543 [DOI] [PubMed] [Google Scholar]
- Trzpis M, McLaughlin PMJ, de Leij LMFH, Harmsen MC (2007) Epithelial cell adhesion molecule: more than a carcinoma marker and adhesion molecule. Am J Pathol 171: 386–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelin B, Bolaños E, Teijeira A, Martinez‐Forero I, Labiano S, Azpilikueta A, Morales‐Kastresana A, Quetglas JI, Wagena E, Sánchez‐Paulete AR et al (2015) Focusing and sustaining the antitumor CTL effector killer response by agonist anti‐CD137 mAb. Proc Natl Acad Sci USA 112: 7551–7556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor‐Weiner A, Kester L et al (2015) Prospective derivation of a living organoid biobank of colorectal cancer. Cell 161: 933–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaritskaya L, Shurin MR, Sayers TJ, Malyguine AM (2010) New flow cytometric assays for monitoring cell‐mediated cytotoxicity. Expert Rev Vaccines 9: 601–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Burger MC, Jennewein L, Genßler S, Schönfeld K, Zeiner P, Hattingen E, Harter PN, Mittelbronn M, Tonn T et al (2016) ErbB2/HER2‐specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst 108: djv375 [DOI] [PubMed] [Google Scholar]
- Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, Wels WS (2017) Chimeric antigen receptor‐engineered NK‐92 cells: an off‐the‐shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front Immunol 8: 533 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Movie EV9
Review Process File