

Abstract
Transcription factors show rapid and reversible binding to chromatin in living cells, and transcription occurs in sporadic bursts, but how these phenomena are related is unknown. Using a combination of in vitro and in vivo single‐molecule imaging approaches, we directly correlated binding of the Gal4 transcription factor with the transcriptional bursting kinetics of the Gal4 target genes GAL3 and GAL10 in living yeast cells. We find that Gal4 dwell time sets the transcriptional burst size. Gal4 dwell time depends on the affinity of the binding site and is reduced by orders of magnitude by nucleosomes. Using a novel imaging platform called orbital tracking, we simultaneously tracked transcription factor binding and transcription at one locus, revealing the timing and correlation between Gal4 binding and transcription. Collectively, our data support a model in which multiple RNA polymerases initiate transcription during one burst as long as the transcription factor is bound to DNA, and bursts terminate upon transcription factor dissociation.
Keywords: binding dynamics, burst size, chromatin, stochastic, transcriptional activators
Subject Categories: Transcription
Introduction
During activation of transcription, transcriptional factors (TFs) bind to specific motif sequences in the promoters of genes and recruit factors such as chromatin regulators, coactivators, general transcription factors, and eventually RNA polymerase to initiate transcription. Understanding the molecular events that underlie gene activation requires knowledge about the kinetics of these processes. Studies of transcription dynamics in single cells have shown that some genes are constitutively transcribed with random initiation of single polymerases (1‐state model) (Zenklusen et al, 2008; Larson et al, 2011), whereas other genes are transcribed in short stochastic bursts of high transcriptional activity where several polymerases initiate, interspersed by periods of inactivity (2‐state model) (Golding et al, 2005; Chubb et al, 2006; Zenklusen et al, 2008; Bahar Halpern et al, 2015; Lenstra et al, 2015). Modulation of a gene's transcriptional activity can be done by changing the burst size (the number of polymerases in a burst), the burst duration, or the burst frequency (Bartman et al, 2016; Fukaya et al, 2016; Rodriguez et al, 2019), each with different effects on the cell‐to‐cell variability in a population (Raser & O'Shea, 2004).
Recent advances in imaging technologies allow for the direct measurement of TF binding dynamics at the single‐molecule level in living cells, providing insight into the search dynamics and mechanisms of gene activation (Elf et al, 2007; Tokunaga et al, 2008; Grimm et al, 2015; Liu & Tjian, 2018). Interpretation of the TF dynamics has been challenging, because the position and activity of target genes are often unknown, and target genes have different TF binding site affinities (Normanno et al, 2015). Target genes are also in different chromatin contexts, where the position or modification state of promoter nucleosomes can affect the accessibility of binding sites for TFs, although certain TFs, such as pioneer TFs, bind to DNA even in the context of a full or partially unwound nucleosome (Zaret & Mango, 2016). For example, at the GAL1/10 locus in budding yeast, the TF Gal4 binds to a partially unwrapped nucleosome with the help of the RSC remodeler (Floer et al, 2010). In addition to regulating accessibility, nucleosomes can significantly reduce the dwell time of transcriptional activators in vitro (Luo et al, 2014). However, how nucleosomes regulate TF binding dynamics in vivo is still mostly unexplored. In addition, the relationship between TF binding dynamics and the dynamics of transcription initiation is only starting to emerge (Larson et al, 2013; Senecal et al, 2014; Loffreda et al, 2017; Rullan et al, 2018).
The major difficulty in deciphering the causal relationships between TF dwell time, nucleosomes, and the dynamics of RNA synthesis comes from technical limitations. Although TF kinetics and RNA synthesis can be measured at single‐molecule resolution, it has been challenging to measure both simultaneously in the same cell at the active locus. Even if the location of target genes is known, the number of TF binding events at a specific locus would be limited, because of the high number of target genes and partial labeling of the TF population in single‐molecule imaging experiments (Liu & Tjian, 2018). Additionally, there is a mismatch in the timescale of TF binding (on the order of seconds) and transcription (on the order of minutes), and quantification of the transcription site intensity in a single plane is hampered by transcription site movement. Similarly, assessing the role of nucleosomes in regulating TF and transcription dynamics has proven difficult, since we lack in vivo tools to precisely control or visualize the position of nucleosomes around TF binding sites in single cells and observe their effect on TF binding dynamics.
Here, we used a combination of novel single‐molecule in vitro and in vivo imaging techniques to assay the interplay between TF dwell time, nucleosome binding and transcriptional bursting. We have developed a novel single‐molecule imaging platform to directly visualize both transcription binding dynamics and transcriptional output in the same cell in an endogenous setting. We exploited the characteristics of the galactose‐responsive genes in budding yeast, where the combination of low Gal4 expression and a small number of Gal4 target genes allows for the quantification of binding events to the specific target gene of interest. Moreover, we have employed an advanced 3D tracking technique to track the transcription site in 3D, which has allowed for the first time to directly correlate TF binding with transcription initiation kinetics at a single locus. We find that Gal4 precedes and overlaps with GAL10 transcription and that their fluctuations are coupled in time. Gal4 dwell time determines the transcription burst duration and depends on the affinity of the binding site. The burst duration is not modulated by galactose signaling, which instead regulates burst frequency. In addition, quantitative comparison of the in vivo Gal4 dwell time to the in vitro Gal4 binding rates in a nucleosomal context indicates that promoter nucleosomes reduce the Gal4 dwell time by orders of magnitude allowing for rapid Gal4 turnover. Overall, we show that TF binding to nucleosomal DNA is a key determinant of transcriptional burst duration, where multiple transcription initiation events can take place as long as the TF is associated with DNA.
Results
Mutations in the upstream activating sequence reduces burst size
Several often‐studied galactose‐responsive genes, such as GAL1 and GAL10, contain multiple UASs (upstream activating sequences), of which the spacing, configuration, and cooperativity may contribute to transcription. Therefore, to study the role of TF binding in bursting, we focused on the GAL3 gene, which contains a single Gal4 UAS in its promoter. Endogenous GAL3 transcription was visualized in live cells by the introduction of 14 PP7 repeats in the 5′ UTR of GAL3. Upon transcription, each PP7 repeat forms a stem loop that is bound by the PP7 bacteriophage coat proteins fused to a fluorescent protein. The PP7 loops did not affect GAL3 transcription levels, as the transcription site (TS) of the PP7‐tagged and non‐tagged allele in heterozygous diploid cells showed similar amount of nascent RNAs (Fig EV1A).
Figure EV1. Mutations in upstream activating sequence reduce burst size, but not burst frequency (related to Fig 1).
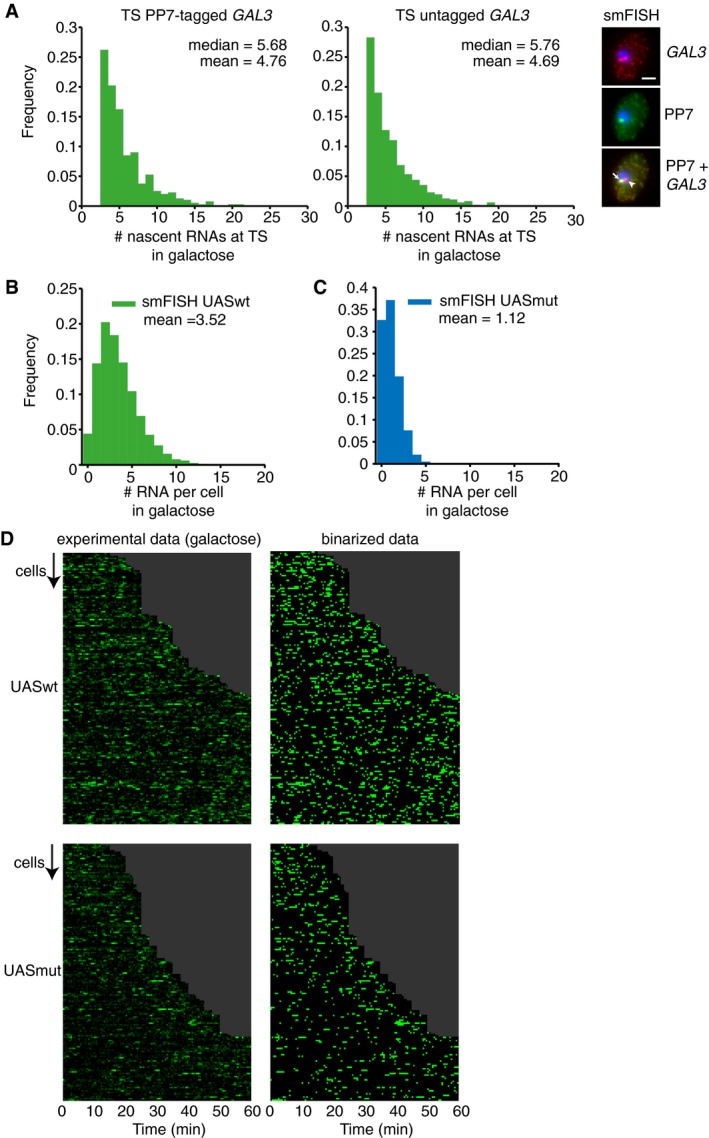
- GAL3 transcription site intensity of heterozygous diploids with and untagged and PP7‐tagged GAL3 allele, as measured by colocalization with the PP7 TS with smFISH. An example cell is shown on the right, with the PP7‐tagged GAL3 allele indicated with an arrow and the untagged allele indicated with an arrowhead. The dynamic range and distribution of the tagged and untagged allele are similar, indicating that the PP7‐tag does not affect transcription of GAL3. To prevent single RNAs from contributing to the TS distributions, TSs were defined as nuclear spots with 2.5‐fold the median intensity of cytoplasmic RNAs. n = 2,716 cells. Scale bar: 2 μm.
- Distribution of the number of RNAs per cell determined by smFISH. A strain with PP7‐tagged GAL3 driven by the UASwt GAL3 promoter was hybridized with PP7 probes. n = 11,839 cells.
- Same as (B) for a strain with UASmut GAL3 promoter. n = 4,845 cells.
- Heatmap of transcription site intensity of individual cells (rows) in galactose of one of three independent experiments. Left plots show experimental data for UASwt and UASmut at GAL3, right plots show binarized data after thresholding. In all three experiments combined, 324 cells were analyzed for UASwt and 250 cells for UASmut.
Live‐cell visualization of GAL3 transcription in galactose‐containing media revealed transcriptional bursts, with periods of high activity interspersed with periods of inactivity (Fig 1A, Movie EV1). The bursts were less frequent than for the previously measured GAL10 gene (Lenstra et al, 2015), with longer periods of gene inactivity. To determine on and off periods of transcription, a threshold was applied to intensity traces of the transcription site (Materials and Methods). In agreement with a two‐state model of bursting (Peccoud & Ycart, 1995), both the on and the off time are exponentially distributed (Fig 1B and C), with an average on time of 46.5 s ± 2.4 s and an average off time of 4.2 ± 0.2 min.
Figure 1. Mutations in upstream activating sequence (UAS) reduce burst size, but not burst frequency.
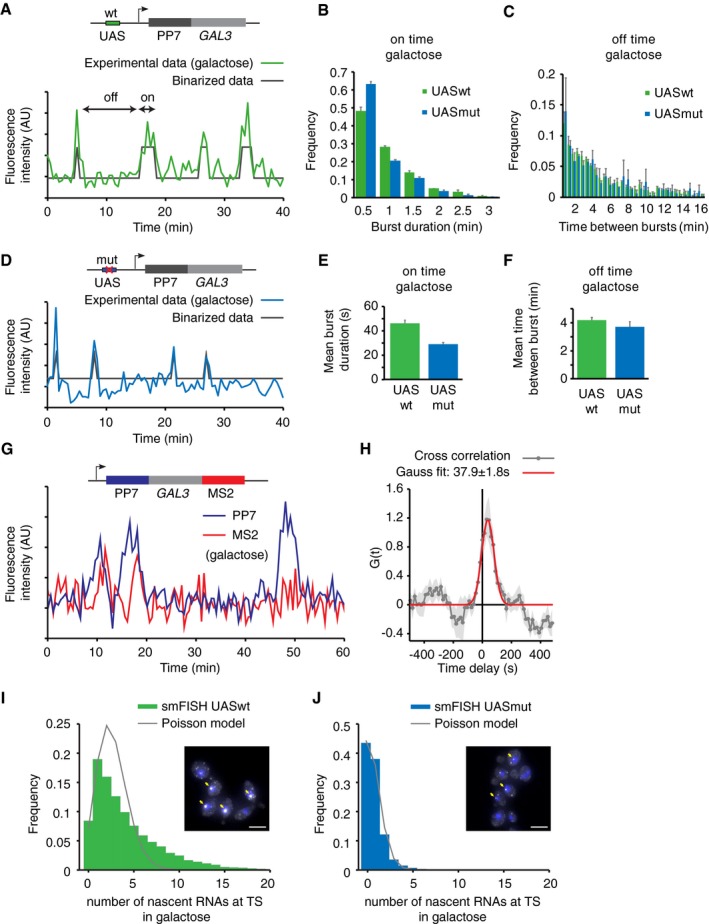
- Transcription at GAL3 is visualized by addition of PP7 loops at the 5′ of GAL3 in budding yeast. Example trace of the quantified fluorescence intensity of the transcription site. Traces are binarized to determine on (active) and off (inactive) times.
- Histogram of GAL3 on time (burst duration) for cells with wt and mutated UAS, respectively, reveals shorter on times for mutated UAS. Errors indicate SE of three experiments, n = 324 cells UASwt, n = 250 cells UASmut.
- Histogram of GAL3 off times. Errors indicate SE of three experiments.
- Similar to (A) for cells were the UAS was mutated to a lower affinity UAS. Also see Fig EV1.
- Average on time for UASwt and UASmut from exponential fit. Errors indicate SD of fit.
- Average off time for UASwt and UASmut from exponential fit. Errors indicate SD of fit.
- Example traces of quantified fluorescence intensity at the transcription site for 5′PP7 and 3′MS2 at GAL3.
- Cross‐correlation of the PP7 and MS2 signals shows a peak at 37.9 s ± 1.8 s delay, n = 137 cells. Shaded area and errors indicate SEM.
- Distribution of number of nascent RNAs at the transcription site determined by smFISH. The distribution of UASwt does not fit a Poisson distribution (gray line), supporting that GAL3 is not constitutively transcribed, but is transcribed in bursts. Example image is shown, and yellow arrows indicate TSs. Scale bars: 5 μm. n = 11,839 cells.
- Same as (G) for cells with UASmut. The distribution of UASmut fits a Poisson distribution, indicating that GAL3‐UASmut is transcribed with random initiation of individual polymerases, similar to constitutive genes. n = 10,616 cells. See also Fig EV1B and C.
Source data are available online for this figure.
To investigate the role of Gal4 in determining this bursting pattern, the GAL3 UAS upstream of the PP7‐GAL3 gene was replaced with one of the GAL10 UAS sequences with a lower affinity (called UASmut; Fig 1D; Liang et al, 1996). Interestingly, analysis of the traces in this mutant revealed that the on time was significantly reduced from 46.5 s ± 2.4 s to 29.3 s ± 1.3 s, whereas the off times were similar (4.2 ± 0.2 min for UASwt and 3.7 ± 0.3 min for UASmut; Fig 1C–F). The on time of transcription (also referred to as burst duration) reflects the time that nascent RNA is visible at the site of transcription and includes the window of time when polymerases are loaded on the gene as well as the duration of elongation, termination, and release. We refer to the “active promoter time” as the interval over which RNA polymerases initiate at the promoter and the “burst size” as the number of polymerases which are loaded during this period. If post‐initiation processes are similar for both UASwt and UASmut, a reduced on time indicates that the active promoter time is reduced, likely resulting in fewer polymerases initiating during a burst for the UASmut.
To distinguish these different steps in the transcription cycle, we inserted the PP7 loops in the 5′ UTR of GAL3 and the orthogonal MS2 loops in the 3′ UTR of GAL3. As expected, dual‐color imaging of this PP7‐GAL3‐MS2 strain showed an increase in PP7 intensity followed by an increase in MS2, and a simultaneous drop of both signals (Fig 1G, Movie EV2). The peak in the PP7‐MS2 temporal cross‐correlation revealed that RNA polymerase takes on average 37.9 s ± 1.8 s to transcribe from the middle of the PP7 to the middle of the MS2 loops (Fig 1H), with an elongation rate of 64.5 ± 3.0 bp/s (3.9 ± 0.2 kb/min). Based on the length of the PP7‐GAL3 construct, the elongation time of this construct is calculated to be ~30 s. The average on time of 29.3 s ± 1.3 s in the UASmut construct thus suggests that the transcription on time is dominated by the elongation time with little time for loading of multiple polymerases. Therefore, the UASmut facilitates transcription of single RNAs during a burst with an active promoter time of a few seconds, whereas UASwt has a longer active promoter time of ~15 s.
To confirm that the burst size is reduced upon mutating the UAS, smFISH was performed on the UASwt and UASmut strains (Fig 1I and J). The distribution of nascent GAL3 RNA normalized to intensity of cytoplasmic RNAs shows that multiple RNAs are present at the TS in a GAL3 UASwt strain. In agreement with bursting transcription, the nascent transcript distribution does not fit to a Poisson model, which is the expected distribution for non‐bursting constitutive genes. In the strain with a UAS mutation, the number of nascent transcripts is reduced and fits well to a Poisson model. The UAS mutation thus results in transcription of single RNAs like a constitutively transcribed gene and results in loss of bursting at the locus. In summary, bursts of GAL3 transcription require high‐affinity binding of Gal4 to the promoter and reducing the affinity of Gal4 through a cis‐acting mutation in the UAS reduces the burst size.
Mutation in the UAS reduces dwell time of Gal4 on nucleosomal DNA in vitro
Since the in vivo experiments suggest the decrease in burst duration is due to a decrease in the affinity of Gal4 for the UAS, we performed a series of in vitro binding assays to directly compare Gal4 binding to the UASwt and UASmut sequences. First, we compared Gal4 binding affinity to the two sequences on naked DNA. In these experiments, Gal4 occupancy on naked DNA was measured with protein‐induced fluorescent enhancement (PIFE) (Hwang et al, 2011), where a Gal4 binding event enhances the fluorescence of a Cy3 fluorophore ~1.5‐fold (Fig 2A). Titrating Gal4 produces a binding curve that indicates stoichiometric binding to the UASwt sequence (Fig EV2A). Because of the stoichiometric binding, the relative affinity of Gal4 to the UASwt and UASmut sequences could be determined by titrating different concentrations of unlabeled competitor DNA. Wild‐type competitor DNA was more effective in competing with the labeled DNA for Gal4 binding than mutant competitor (IC50 4.0 ± 0.6 nM for UASwt versus 17.3 ± 3.5 nM for UASmut; Fig 2B), with a relative affinity change of 4.3‐fold (Fig 2C).
Figure 2. Mutations in UAS reduce residence time of Gal4 on nucleosomal DNA in vitro .
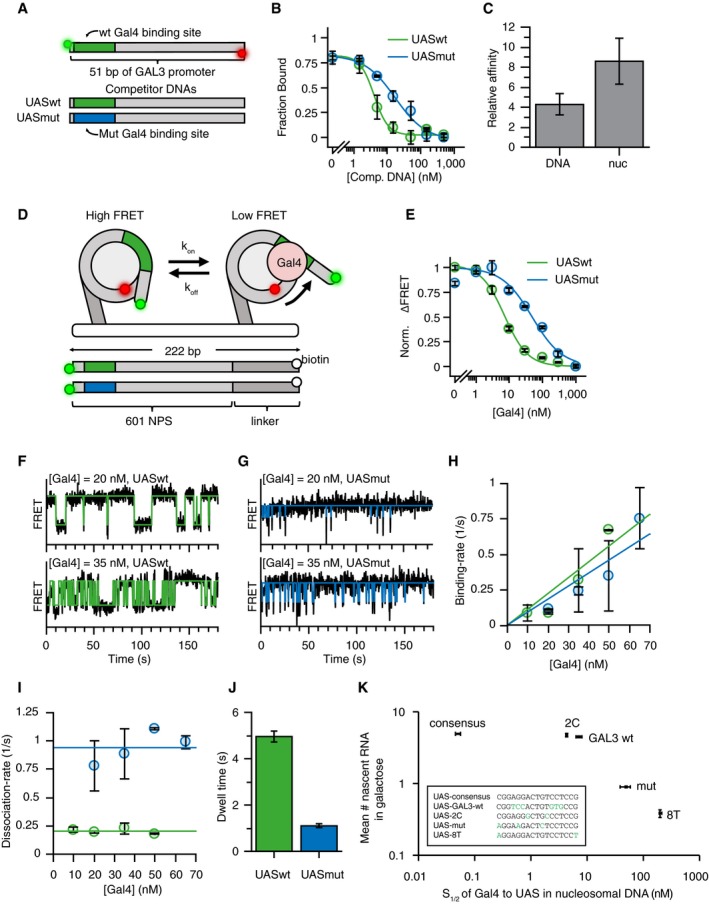
- Competitive binding experiments were performed to determine relative affinity of Gal4 to UASwt (green) and UASmut (blue) motifs. Gal4 occupancy on UASwt Cy3/Cy5 DNA was determined by measuring protein‐induced fluorescence enhancement (PIFE) on 51‐bp oligos containing either Gal4 UASwt or UASmut site 1 bp away from Cy3 fluorophore).
- Titrating unlabeled UASwt or UASmut competitor DNA reduces UASwt Cy3/Cy5 DNA occupancy. IC50UASwt = 4.0 ± 0.6 nM, IC50UASmut = 17.3 ± 3.5 nM. n = 3. Error bars indicate SD.
- Comparison between relative affinities of Gal4 UASwt versus UASmut in naked or nucleosomal DNA shows 4.3 ± 1.1 × difference in K D from naked DNA and 6.8 ± 1.7× from nucleosomal DNA. n = 3. Error bars indicate SD.
- Experimental setup for smFRET experiments to measure Gal4 binding at nucleosomal DNA. Gal4 binds to site 8 bp into nucleosome. A FRET pair in the entry/exit region provides readout on binding events (one fluorophore on DNA, one on histone). In the absence of Gal4, nucleosomes are in the high FRET state. A Gal4 binding event traps nucleosome in low FRET state.
- Gal4 affinity to nucleosomes containing UASwt or UASmut sequences gives S1/2 of 7.2 ± 0.8 nM and 48.9 ± 10.8 nM, respectively. n = 3. Error bars indicate SD.
- Example smFRET traces showing Gal4 binding to UASwt in nucleosomal DNA at two different Gal4 concentrations. States are determined using HMM fit.
- Same as (F) but for UASmut.
- Binding rates of Gal4 are concentration‐dependent, but are similar for UASwt and UASmut: kon UASwt = 0.011 ± 0.002/s/nM, kon UASmut = 0.009 ± 0.001/s/nM. n = 2 for all, except n = 4 for UASwt 10 nM Gal4 and n = 3 for UASwt 20 nM Gal4. Error bars indicate SD.
- The dissociation rate of Gal4 at UASwt is ˜5‐fold slower compared to the UASmut: koff UASwt = 0.20 ± 0.01/s, koff UASmut = 0.92 ± 0.05/s. n = 2 for all, except n = 4 for UASwt 10 nM Gal4 and n = 3 for UASwt 20 nM Gal4. Error bars indicate SD.
- Histogram of Gal4 dwell time to nucleosomal DNA containing UASwt and UASmut sequences. n = 11 for UASwt, n = 8 for UASmut. Error bars indicate SE.
- Scatter plot showing the average number of nascent RNA at TS (from smFISH) versus affinity of Gal4 to different UAS sequences in nucleosomal DNA (from in vitro measurements). In vivo transcription levels correlate with in vitro affinity of Gal4, but transcription saturates above wild‐type sequence. UAS sequences are shown in the box. For mean number of nascent RNA from smFISH, n = 2 for UASconsensus, UAS‐2C, UAS‐8T and n = 8 for UASwt, UASmut and errors indicate SEM. For in vitro affinity measurements, n = 3 and errors indicate SD.
Source data are available online for this figure.
Figure EV2. Mutations in UAS reduce residence time of Gal4 on nucleosomal DNA in vitro (related to Fig 2).
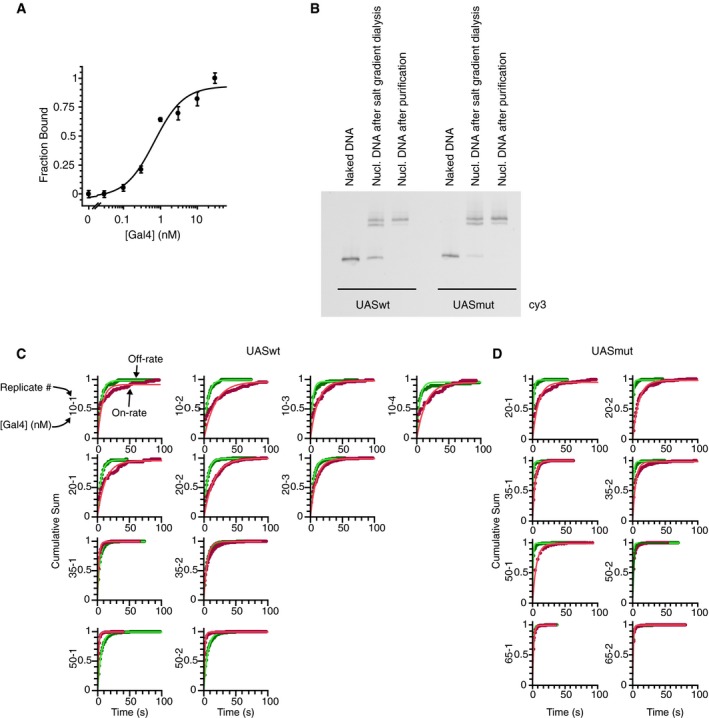
- Titration of Gal4 binding to the UASwt sequence where binding is measured through PIFE. With 500 pM DNA, K D = 680 ± 230 pM. Because the measured K D is essentially equal to the concentration of DNA used in this experiment, binding is considered to be stoichiometric and we assume the actual KD is much lower. In this experiment, ˜80% of DNA molecules are bound at [Gal4] = 5 nM. For the competition experiments to measure relative affinities between the UASwt and UASmut seq, 5 nM Gal4 was used. n = 3. Errors indicate SD.
- Nucleosomes were reconstituted at a ratio of 1.25:1 of DNA:HO via salt gradient dialysis. Lanes 2 and 5 are nucleosome samples post salt gradient dialysis and before sucrose gradient purification. After dialysis, nucleosomes were loaded into 5–30% sucrose gradients and purified by centrifugation. Sucrose fractions containing nucleosomes were collected and concentrated. Lanes 3 and 6 are purified samples for UASwt and UASmut seq, respectively.
- Cumulative sum distribution for dwell times and unbound times for UASwt and UASmut single‐molecule experiments. Numbers indicate Gal4 concentration and replicate number.
In vivo, DNA is wrapped around nucleosomes. Previous measurements of Gal4 binding on naked versus nucleosomal DNA showed that nucleosomes reduce the dwell time of Gal4 ~1,000‐fold (Luo et al, 2014). Binding of Gal4 to the UAS sequences was therefore measured within a nucleosome by determining the change in energy transfer between two fluorophores positioned in the nucleosome entry/exit and DNA (Förster resonance energy transfer, FRET). Gal4 binding traps the nucleosome in a partially unwrapped state, which increases the distance between the fluorophores, reducing the FRET efficiency (Fig 2D). Similar to naked DNA, Gal4 has an 8.6‐fold higher affinity for the UASwt relative to the UASmut sequence (Fig 2C and E).
We then directly measured the binding and dissociation rates of Gal4 to both UAS sequences in a nucleosomal context by recording time‐resolved single‐molecule FRET trajectories between Gal4 and surface‐tethered nucleosomes (Fig 2F and G). As expected, the binding rate (frequency of binding) but not the dissociation rate (dwell time) is Gal4 concentration‐dependent (Fig 2H and I). The binding rates are similar for UASwt and UASmut, indicating that the frequency of binding is independent of the UAS sequence and likely regulated by the nucleosome unwrapping rate (Fig 2H). Interestingly, the UASmut shows a fivefold shorter dwell time than the wild type (4.96 s ± 0.25 for UASwt, 1.09 s ± 0.06 for UASmut; Fig 2I and J). A higher affinity binding site thus stabilizes Gal4 binding but does not influence the frequency of binding. This higher dwell time correlates with an increased burst size in vivo.
To generalize the relationship between binding affinity and transcription, we measured the affinity of five different sequences to nucleosomal DNA in vitro in comparison with the mean number of nascent RNAs in vivo, as determined by smFISH (Fig 2K). In agreement with our previous data, lower affinity binding correlates with lower nascent RNA production. However, binding sites with higher affinity than the wild‐type sequence did not increase nascent RNA output. This saturation suggests that either other regulatory factors limit the Gal4 dwell time in vivo or there is an inherent limit to the burst size even upon longer Gal4 binding.
To test whether in vitro measurements of Gal4 binding to nucleosomal DNA have in vivo relevance, we sought to determine the nucleosome occupancy near Gal4 binding sites in the presence or absence of galactose, i.e., induced and uninduced conditions. Specifically, we mapped stable and fragile nucleosomes by digestion with two different MNase concentrations (Kubik et al, 2015). Cells grown in galactose showed protected fragments of nucleosomal size (95–225 bp or 140–225 bp) around the Gal4 binding site in the GAL3 promoter (Figs 3 and EV3E). This MNase peak was only visible with low MNase digestion and only appeared after the two flanking stable nucleosomes moved upstream and downstream, which suggests the presence of a fragile partially unwrapped nucleosome around the Gal4 UAS (Kubik et al, 2015; Brahma & Henikoff, 2019). Fragile nucleosomes have also been reported around the GAL1‐10 UAS sites (Floer et al, 2010), and we find that fragile nucleosomes appear at many other galactose‐responsive genes (Fig EV3), strongly suggesting that Gal4 binds nucleosomal instead of naked DNA in vivo.
Figure 3. Gal4 binds at the edge of a fragile nucleosome in galactose in vivo .
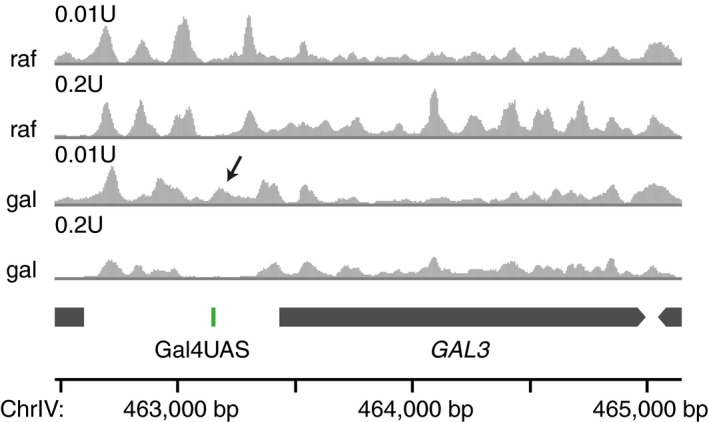
Profiles of nucleosome midpoint positions by MNase‐seq experiments. Samples were digested with the indicated MNase concentrations in both raffinose (raf) and galactose (gal) containing media. Midpoints of nucleosomes are smoothed by 31 bp. In galactose, the stable nucleosomes move away from the Gal4UAS, creating space for an additional fragile nucleosome (indicated by arrow).
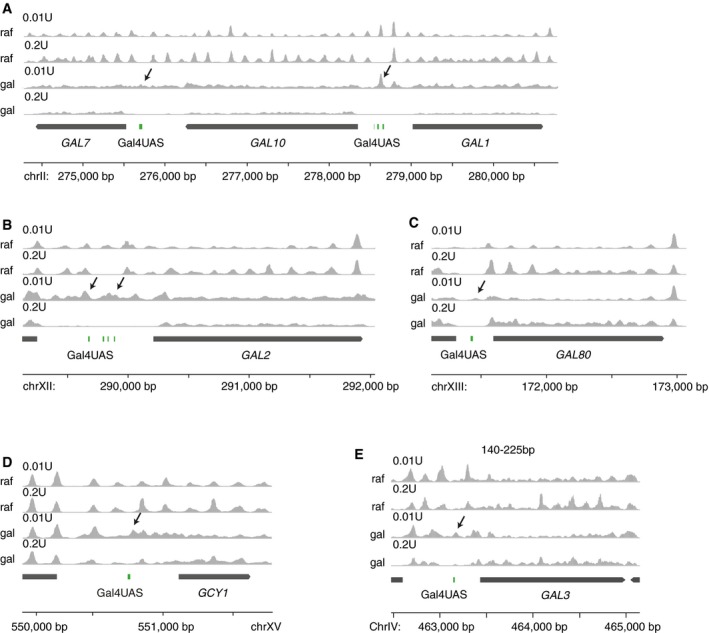
Figure EV3. Gal4 binds at the edge of a fragile nucleosome in galactose in vivo (related to Fig 3).
-
A–DGal4 binds at the edge of a fragile nucleosome in galactose at many galactose response genes in vivo. Profiles of nucleosome midpoint positions (95–225 bp) by MNase‐seq experiments at (A) GAL1, GAL10, GAL7, (B) GAL2, (C) GAL80, and (D) GCY1. Samples were digested with the indicated MNase concentrations in both raffinose (raf) and galactose (gal) containing media. Midpoints of nucleosomes are smoothed by 31 bp.
-
EProfile of nucleosome midpoint positions at GAL3 using a higher threshold (140–225 bp), which gives the same result at Fig 3.
Overall, in vitro and in vivo data indicate that Gal4 binding dynamics are correlated with RNA production, that longer binding supports larger burst sizes, that dwell time is shorter on nucleosome‐bound DNA, and that Gal4 binding sites contain fragile nucleosomes.
Gal4 dwell time in vivo shows two Gal4 binding populations
Our in vitro dwell‐time measurements indicate that Gal4 is bound to nucleosomal DNA on the order of seconds. Previous Gal4 binding measurements to naked DNA showed a 1,000‐fold longer dwell time (Luo et al, 2014), suggesting that nucleosomes may be required for rapid turnover of TFs in the nucleus. In living cells, many additional factors may also limit the dwell time of Gal4. We therefore used single‐molecule tracking (SMT) to analyze the Gal4 dwell time in vivo.
To visualize binding and diffusion of single molecules of Gal4, Gal4 was tagged with a HALO tag, which covalently binds bright synthetic dyes (Fig 4A). Addition of the HALO tag minimally affected its function, as Gal4‐HALO cells showed similar growth as wild type on galactose‐containing plates and minimal growth on LiCl2 plates (Fig EV4A). To support uptake of the synthetic dye, the gene encoding the Pdr5 transporter was also deleted (Ball et al, 2016). Lastly, 14 PP7 loops were added in the 5′UTR of GAL10 (Fig 4A), allowing the visualization of Gal4 binding kinetics specifically at one of its target genes. For this experiment, we used GAL10 instead of GAL3 because the GAL10 promoter contains four Gal4 UASs (instead of a single UAS for GAL3), which results in much higher transcriptional activity. Three GAL10 UASs are near consensus, and the fourth one is the UASmut sequence we used for GAL3. We reasoned that this may increase the chance of observing colocalization of TF binding at its target gene with respect to nascent RNA production.
Figure 4. Measurements of the Gal4 residence time in vivo shows two Gal4 populations, with long residence times colocalizing with target gene.
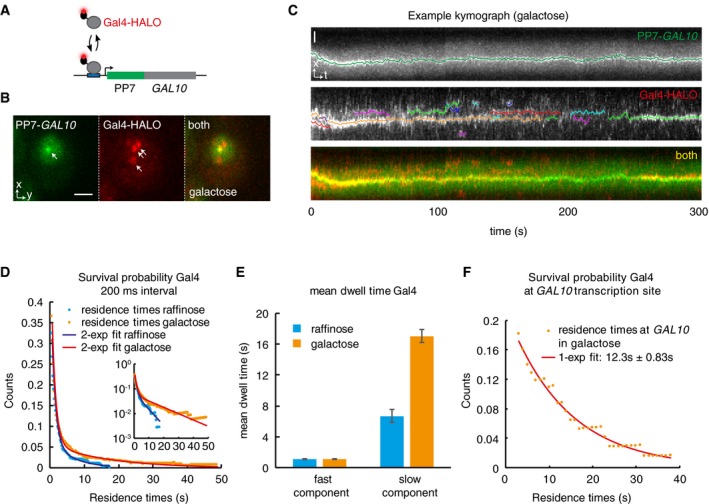
- Simultaneous imaging of Gal4 binding kinetics in vivo using single‐molecule tracking and RNA imaging of the GAL10 target gene. Gal4 is tagged with a HALO tag, which covalently binds to the dye JF646. Transcription of the target gene GAL10 is visualized by PP7 loops.
- Example image of a cell, showing the GAL10 TS (arrow, left panel), single molecules of Gal4 (arrows, middle panel), and an overlay (right panel). Scale bar: 2 μm.
- Example kymograph of a cell. Upper panel (PP7‐GAL10) shows the position of the TS over time. Middle panel (Gal4‐HALO) show tracks of Gal4. Data are in white, colored lines show analyzed tracks. Lower panel shows overlay, showing colocalization of some the Gal4 tracks to the GAL10 TS. Scale bar: 1 μm.
- Survival probability of the duration of Gal4 tracks (after displacement thresholding) at 200 ms interval from cells grown in raffinose (n = 258 tracks in 30 cells) or galactose (n = 346 tracks in 25 cells). Lines show bi‐exponential fit, indicating 2 Gal4 populations. Inset shows data in semi‐logarithmic plot.
- Residence time of the fast and slow components of the fits from (D). The slow component changes between conditions. Error bars indicate 95% CI.
- Survival probability of Gal4 residence times that colocalize with GAL10 TS (< 250 nm), showing that Gal4 with long residence time colocalizes with GAL10. Data were taken at 1s interval and show overlap between 267 Gal4 tracks and 83 GAL10 tracks, representing 458 bound molecules. Line shows exponential fit, with mean of 12.3 s ± 0.83 s. Errors indicate 95% CI.
Source data are available online for this figure.
Figure EV4. Measurements of the Gal4 residence time in vivo shows two Gal4 populations, with long residence times colocalizing with target gene (related to Fig 4).
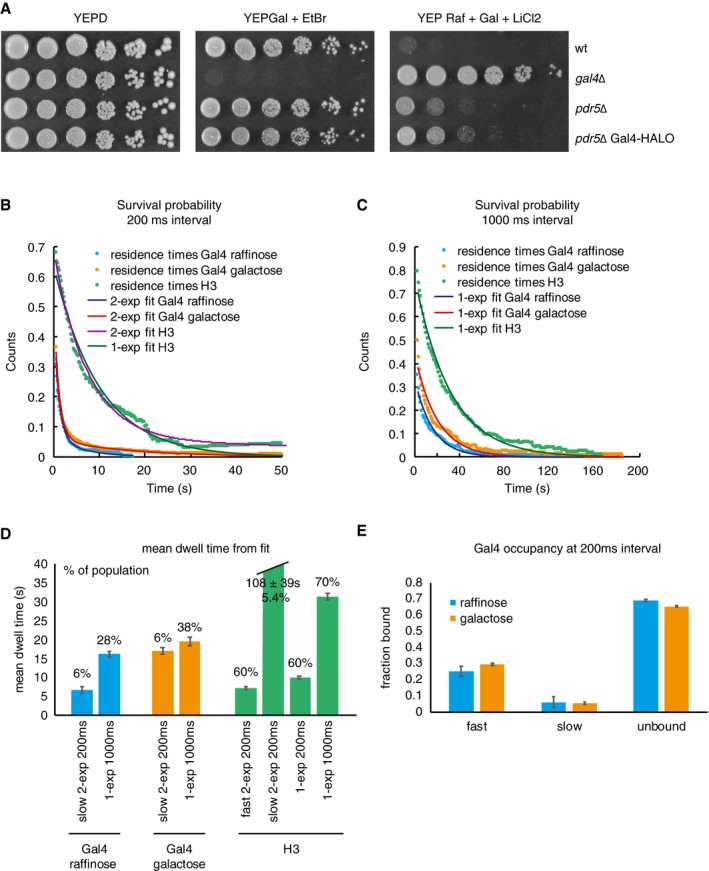
- Growth assay of wt, gal4∆, pdr5∆ and pdr5∆ Gal4‐HALO strains. Serial dilutions (fivefold) are shown after 3 days of growth on YEPD, YEPGal + EtBr, and YEP Raf + Gal + LiCl2.
- Survival probability of the duration of Gal4 and H3 tracks (after displacement thresholding) at 200 ms interval from cells grown in raffinose or galactose. H3 is fit by a single and bi‐exponential, Gal4 by a bi‐exponential distribution, indicating 2 Gal4 populations. Gal4 raffinose: n = 258 tracks in 30 cells, Gal4 galactose: n = 346 tracks in 25 cells, H3: n = 175 tracks in 34 cells.
- Same as for (A) at 1,000 ms interval. Both H3 and Gal4 are fit to a single‐exponential fit. Gal4 raffinose: n = 153 tracks in 41 cells, Gal4 galactose: n = 238 tracks in 40 cells, H3: 213 tracks in 43 cells.
- Comparison of the mean residence times from data taken at 200 and 1,000 ms interval. The percentages above indicate the percentage of the population that the bar represents. H3 is a control, and is used to determine the longest measurable dwell time in our assay before we have bleaching effects. Although at 200 ms the 2‐exp fit of H3 shows that 5% of the molecules have a long dwell time of 108 s, the majority of H3 (60%) show a residence time of 7 s that is similar to Gal4. As expected, the average H3 dwell time (from 1‐exp fit) increases from 9.9 s at 200 ms to 31.2 s at 1,000 ms interval, indicating it is dominated by bleaching. Gal4 dwell time does not show the same increase between 200 and 1,000 ms interval, indicating that we are capturing the real dwell time. At 1,000 ms, we are well below the average dwell time of H3. Gal4 raffinose 200 ms: n = 258 tracks in 30 cells, Gal4 raffinose 1,000 ms: n = 153 tracks in 41 cells, Gal4 galactose 200 ms: n = 346 tracks in 25 cells, Gal4 galactose 1,000 ms: n = 238 tracks in 40 cells, H3 200 ms: n = 175 tracks in 34 cells, H3 1,000 ms: 213 tracks in 43 cells. Error bars indicate 95% CI.
- The fraction of fast‐ and slow‐bound Gal4 molecules is similar in raffinose and galactose. Gal4 raffinose: n = 258 tracks in 30 cells, Gal4 galactose: n = 346 tracks in 25 cells. Error bars indicate SD.
Cells were imaged with two colors simultaneously using HILO illumination (Highly Inclined and Laminated Optical sheet) to reduce out‐of‐focus fluorescence (Tokunaga et al, 2008). We observed single Gal4 molecules in the nucleus, some of which colocalize with the PP7‐GAL10 TS (Fig 4B, Movie EV3). The GAL10 TS and Gal4 molecules were tracked over time, as shown in the kymograph of Fig 4C. To determine whether Gal4 molecules were diffusing or bound to DNA, a distance threshold of 0.35 μm between frames was applied based on the mobility of histone H3 (HHT1‐HALO) molecules (Materials and Methods).
The survival probability of bound Gal4 molecules at all sites reveals binding of Gal4 on the order of several seconds, which is in the same dynamic range as the in vitro measurements on nucleosomal DNA (Fig 2J; Luo et al, 2014). The biphasic behavior in the semi‐log plot indicates two populations of Gal4 with different binding rates (inset, Fig 4D). Multiple distributions have previously been observed for other TFs and have been attributed to non‐specific and specific binding (Chen et al, 2014; Ball et al, 2016; Presman et al, 2017). In agreement with this interpretation, the core histone H3 only showed a longer binding population and no shorter binding population (Fig EV4B). A bi‐exponential fit of the Gal4 survival probability revealed that the fast binding fraction of ~1s has a similar dwell time in inactive and active conditions (raffinose and galactose, respectively; Fig 4E), supporting the idea that the fast binding is non‐specific. In contrast, the dwell time of the more stable binding fraction increases from 6.7 s ± 0.9 s to 17.1 s ± 0.8 s in active conditions. The fraction of fast‐ and slow‐bound Gal4 molecules is similar in both conditions (Fig EV4E). Since galactose‐responsive genes are inactive or lower transcribed in raffinose than in galactose, an increased dwell time suggests that longer binding is specific binding that is required to activate transcription.
Interpretation of TF dwell times in the complex milieu of the nucleus can be challenging since residence times are averaged across many binding sites that likely have different affinities. To obtain insight into the binding kinetics at a single target gene, we took advantage of the addition of PP7 RNA labeling for the GAL10 gene. When we focused on Gal4 binding events that specifically overlapped with the GAL10 transcription site in the presence of galactose, several longer binding events were observed, with a mean dwell time of 12.3 s ± 0.83 s (Fig 4F). Since alignment between channels may be imperfect, our stringent colocalization threshold of 250 nm may result in underestimation of the dwell time at GAL10. Regardless, the presence of longer binding events at a transcribing target gene indicates that more stable binding is associated with the activation of transcription.
The dwell time of TFs can be underestimated if molecules are bleached before they dissociate from the DNA. Histone H3 binds on much longer timescales and served as a control to determine the maximally measurable dwell time. Since experiments taken with 200 ms interval showed Gal4 dwell times similar to H3 dwell time, the same experiments were repeated with 1s interval. At this longer time interval, non‐specific binding was below the time resolution, and the survival probably followed a single‐exponential curve. Although the longer interval indeed resulted in an increase of the average H3 dwell time, Gal4 dwell time did not increase significantly (Fig EV4B–D). The measured average specific Gal4 dwell time (~17 s) is therefore unlikely to be the result of a technical artifact.
In summary, Gal4 shows two binding populations. Our data support a model where the short sub‐second binding is non‐specific binding of Gal4 in search for its target sites, and that the more stable several‐second binding is specific and productive binding to the promoters of target genes in order to activate transcription.
Gal4 binding correlates with transcription
Long Gal4 binding events (~12–17 s) which spatially colocalize with the GAL10 TS suggest that these binding events result in the production of nascent RNA. However, the SMT experiments visualize molecules in a single plane, and the three‐dimensional movement of the GAL10 TS out of the focal plane hampered quantification of the TS intensity. Using SMT, we were therefore unable to quantify if binding was followed by the production of RNA. Moreover, there is a dynamic mismatch, which poses a challenge for fluorescence visualization: Gal4 is bound on the order of 10 s, but bursts of transcription are separated by many minutes. To directly correlate Gal4 binding with RNA output, we imaged our dual‐tagged Gal4‐HALO and PP7‐GAL10 cells in a 3D orbital tracking microscope (Kis‐Petikova & Gratton, 2004; Levi et al, 2005a,b; Annibale & Gratton, 2015), which is able to track the GAL10 gene in 3D and simultaneously capture Gal4 binding. As a variation on conventional confocal scanning microscopy, an orbital tracking microscope makes circular orbits around an object of interest and uses a feedback loop to track the object in 3D with high temporal and spatial resolution (Fig 5A). Figure 5B shows an example trace of peaks of Gal4 binding, followed by increases in GAL10 transcription.
Figure 5. Real‐time correlation of Gal4 binding and transcription in vivo using orbital tracking.
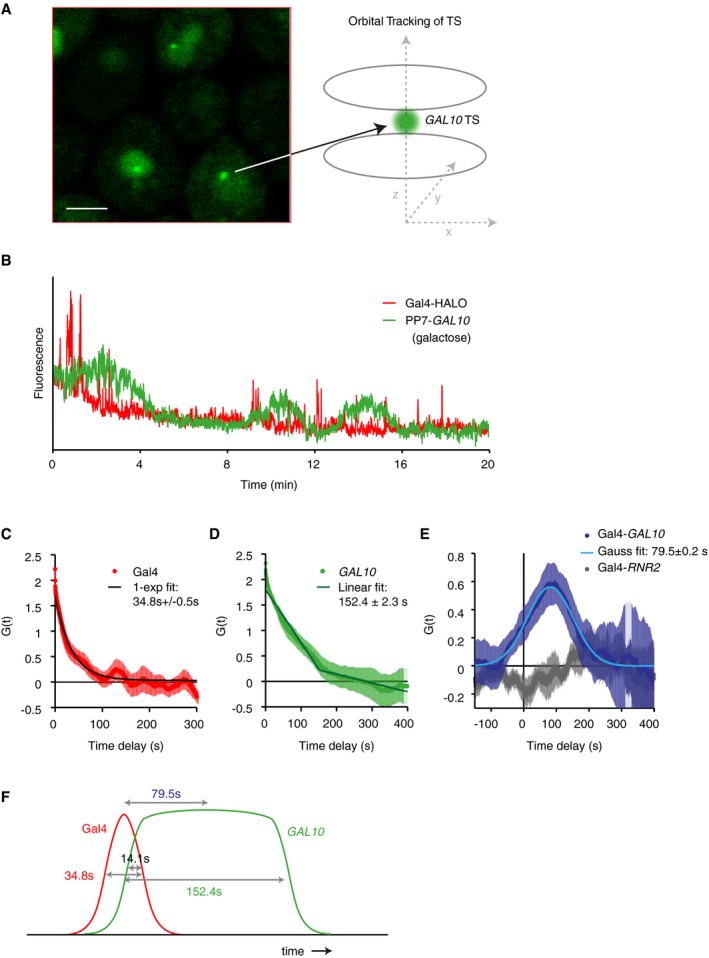
- Schematic of 3D orbital tracking of GAL10 TS. Scale bar: 2 μm.
- Example trace of Gal4 binding and GAL10 transcription in the same cell.
- Autocorrelation of Gal4. The exponential fit shows an average dwell time of 34.8 ± 0.5 s. n = 19 traces. Shaded area and errors indicate SEM.
- Autocorrelation of GAL10. The fit of 2 linear lines reveals a burst duration of 152.4 ± 2.3 s. n = 19 traces. Shaded area and errors indicate SEM.
- Cross‐correlations for Gal4‐GAL10 RNA (blue) and Gal4‐RNR2 RNA (gray, negative control). Asymmetry and positive temporal shift of the cross‐correlation function are consistent with a 79.5 ± 0.2 s delay between the middle of Gal4 and GAL10 signals (n = 19 traces), which is not seen in the negative control (n = 8 traces). Shaded areas and errors indicate SEM.
- Schematic of the different signal durations of Gal4 binding and GAL10 transcription. Gal4 binding overlaps with the GAL10 transcription for 14.1 s.
Source data are available online for this figure.
The autocorrelation function of Gal4 revealed a dwell time for Gal4 of 34.8 s ± 0.5 s (Fig 5C), which is longer than the 12‐s dwell time of the SMT experiments but is likely explained by an underestimation of the dwell time at GAL10 in the SMT from the stringent colocalization threshold. GAL10 transcription shows a duration of 152.4 s ± 2.3 s (Fig 5D), but it should be noted that the fit obtained by the intersect of two linear lines does not match the data perfectly and this measurement may be imprecise. Nevertheless, the resulting burst duration of 152.4 s for GAL10 TS is comparable to our previously reported burst duration (133 s) (Lenstra et al, 2015). The orbital tracking method is thus able to simultaneously capture both Gal4 binding and GAL10 transcription events.
With the ability to record TF binding and transcriptional activation at the same locus, we are able to measure the temporal sequence of events. The cross‐correlation between Gal4 and GAL10 revealed a positive peak at 79.5 s ± 0.2 s (Fig 5E), which indicates that fluctuations arising from Gal4 binding and GAL10 RNA production are temporally correlated and that Gal4 binding precedes the PP7‐GAL10 signal by ~80 s. Given the dwell time of Gal4 binding, the burst duration of GAL10, and the 80‐s delay between the middle of both signals, we calculated that Gal4 binding precedes RNA appearance, but that the nascent RNA levels already start rising during this dwell time with an overlap of 14.1 s (Fig 5F). The temporal correlation was specific, as the cross‐correlation peak disappeared in a negative control strain, where a non‐galactose‐responsive gene RNR2 was tracked together with Gal4 (Fig 5E). In addition, there was no positive correlation in a strain when no synthetic dye was added to the cells to label Gal4 (unpublished observations). The orbital tracking experiments thus indicate that the dynamics of Gal4 binding directly determines the GAL10 transcriptional bursting kinetics.
Galactose signaling regulates burst frequency, but not burst duration
The repressor Gal80 can inhibit the activity of Gal4. In addition to the Gal4‐DNA binding dynamics, the binding dynamics of Gal80 to Gal4 may potentially contribute to bursting of the galactose‐responsive genes. We therefore checked whether bursting of the galactose‐responsive genes is affected by repression of Gal80 by comparing the nascent transcript distributions of GAL10 and GAL3 by smFISH in wt and gal80Δ cells (Fig EV5). In high galactose conditions (similar conditions as Figs 4 and 5), deletion of GAL80 does not change the transcriptional activity of either GAL10 or GAL3 (Fig EV5), indicating that Gal80 is inactive in these conditions. In agreement, previous findings indicate that Gal80 is fully dissociated from Gal4 in high galactose (Jiang et al, 2009). In full induction conditions, it is therefore highly unlikely that the Gal80‐Gal4 binding dynamics contributes to the control of bursting.
Figure EV5. Galactose signaling regulates transcription levels by modulating burst frequency but not burst duration (related to Fig 6).
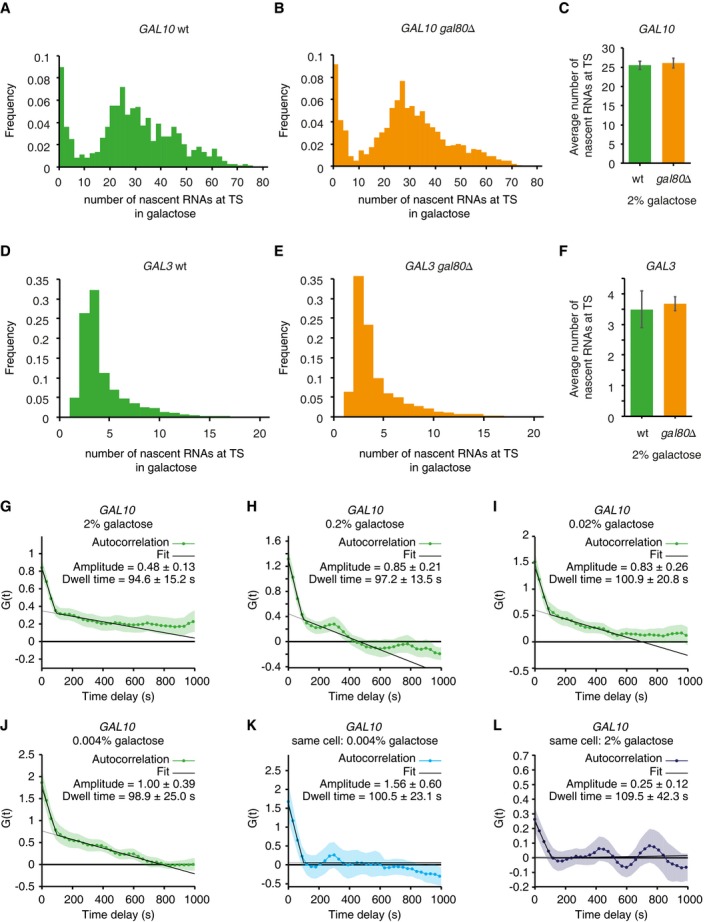
-
ADistribution of number of nascent RNAs at the GAL10 transcription site determined by smFISH in wild‐type cells grown in galactose.
-
BSame as (A) for gal80Δ cells.
-
CAverage number of nascent RNA at the GAL10 transcription site, determined by 8 smFISH experiments as in (A) for wt (n = 23,503 cells) and 4 experiments as in (B) for gal80Δ (n = 6,472 cells). Error bars indicate SEM.
-
DDistribution of number of nascent RNAs at the GAL3 transcription site determined by smFISH in wild‐type cells grown in galactose.
-
ESame as (D) for gal80Δ cells.
-
FAverage number of nascent RNA at the GAL3 transcription site, as determined by 3 smFISH experiments as in (D) for wt (n = 12,410 cells) and 4 experiments as in (E) for gal80Δ (n = 29,022 cells). Error bars indicate SEM.
-
G–JAutocorrelation of GAL10 TS intensity traces from cells after induction with media containing (G) 2% galactose, n = 86 cells (H) 0.2% galactose, n = 79 cells (I) 0.02% galactose, n = 84 cells (J) 0.004% galactose, n = 53 cells, corrected for non‐steady state effects (Lenstra et al, 2015). Dwell times and amplitudes are represented as bar graphs in Fig 6D and E. Shaded areas and errors indicate SEM.
-
K, LAutocorrelation of same‐cell dose response of GAL10 TS intensity traces from cells grown in (K) 0.004% galactose, and (L) after addition of 2% galactose (n = 13 cells), corrected for non‐steady state effects (Lenstra et al, 2015). Dwell times and amplitudes are represented as bar graphs in Fig 6G and H. Shaded areas and errors indicate SEM.
Repression by Gal80 may become more important in lower galactose concentrations. We therefore asked whether the burst duration and burst frequency are regulated in different galactose conditions. Due to the positive and negative feedback loops in the signaling pathway, the galactose‐responsive genes are known to display a bimodal dose response (Acar et al, 2005; Venturelli et al, 2012), where the fraction of induced cells increases in higher doses of galactose. However, whether the induced cells in different doses of galactose also show different bursting properties is unexplored.
We again focused on GAL10 transcription to determine burst duration and burst frequency at different galactose levels, because of its higher dynamic range as compared to GAL3 (Fig EV5). Traces of the transcription site intensity showed continuous bursting after the gene was induced (Fig 6A). For lower doses of galactose, the onset of the first burst of GAL10 transcription was delayed (Fig 6B). Since the off periods were very short and a new burst often started before the previous burst had finished, a threshold to determine the transcription on and off time would result in underestimation of the frequency and overestimation of the burst duration. The autocorrelation of the fluorescence intensity traces was therefore used to extract the burst duration (Figs 6C and EV5). In full induction conditions (2% galactose), the burst duration of ~95 s (Figs 6D and EV5) determined from autocorrelation was shorter than the burst duration of ~150 s measured previously with the orbital tracking experiments. The reasons for this discrepancy are not known at present but may be due to the imprecise fit of the orbital tracking data, or slightly different bleaching rates and/or signal‐to‐background ratios between the orbital tracking and time‐lapse imaging methods. Bleaching or differential background during the acquisition can result in fluctuations in the data at longer timescales, which contribute to the slower decaying linear component of the autocorrelation, and may influence the determination of the burst duration. Nevertheless, across four different galactose concentrations, the burst duration is invariant (Figs 6D and EV5), suggesting that the number of initiating polymerases per burst is the same within the resolution of our measurement. Changes in the autocorrelation amplitude also allow for extraction of relative frequencies (Fig 6C). In higher galactose concentration, cells show a higher burst frequency (lower amplitude; Figs 6E and EV5), indicating that bursting of GAL10 is modulated by galactose at the frequency level.
Figure 6. Galactose signaling regulates transcription levels by modulating burst frequency but not burst duration.
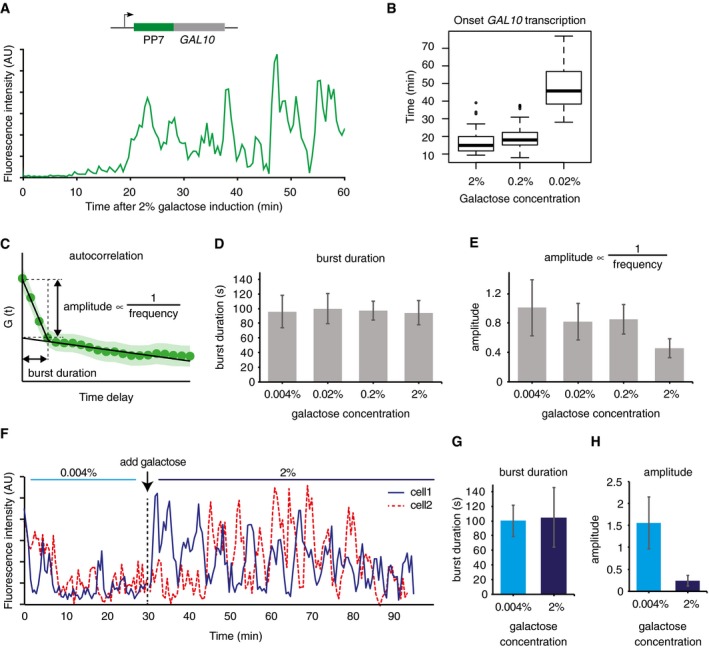
-
AExample trace of PP7‐GAL10 transcription in 2% galactose.
-
BBoxplot of the onset of the first transcription event in different doses of galactose. The box indicates quartiles, the horizontal line inside the box indicates the median, and the whiskers indicate 1.5 times the interquartile range of the box. Data points outside the whiskers are indicated with circles (2%: n = 86 cells, 0.2%: n = 79 cells, 0.02%: n = 84 cells).
-
CThe autocorrelation was used to interpret the burst duration and burst frequency changes. Burst duration is measured by the intersect of the two linear fits. When burst duration is constant, the amplitude is inversely related to the burst frequency.
-
DBurst duration from autocorrelation is constant across four different galactose concentrations (2%: n = 86 cells, 0.2%: n = 79 cells, 0.02%: n = 84 cells, 0.004%: n = 53 cells). Error bars indicate SEM.
-
EAmplitude of autocorrelation decreases (burst frequency increases) at higher doses of galactose (2%: n = 86 cells, 0.2%: n = 79 cells, 0.02%: n = 84 cells, 0.004%: n = 53 cells). Error bars indicate SEM.
-
FExample traces of two single cells exposed to two different doses of galactose.
-
G, HSame‐cell dose response shows same burst duration (G) but lower amplitude (H) at higher galactose concentration, indicating that galactose levels regulate burst frequency (n = 13 cells). Error bars indicate SEM.
Source data are available online for this figure.
The burst frequency changes of GAL10 are about twofold. However, the variability between cells is substantial (Fig 6E), making it challenging to compare bursting changes between different conditions. To circumvent this limitation and unambiguously measure transcriptional bursting changes in response to galactose, transcription was monitored in real time as the galactose concentration is increased during the experiment (Fig 6F). This single‐cell dose response is not affected by global cell‐to‐cell differences, allowing for direct measurements of the bursting changes in the same cell in two different doses of galactose. Again, the burst duration is similar in both concentrations, but the burst frequency clearly increases after increasing the galactose concentration (Figs 6F–H and EV5). In summary, we observe no change in burst duration in different galactose concentrations, indicating that the burst duration is not actively regulated by Gal80 or other components of the galactose‐sensing pathway. Instead, the galactose network regulates transcription levels by modulating the burst frequency and the onset of transcription.
Discussion
In this study, we have used a combination of in vivo and in vitro single‐molecule imaging approaches to determine the interplay between TF dwell time, nucleosomes, and the burst size of a gene. The direct visualization of Gal4 binding and GAL10 transcription reveals for the first time the timing and correlation of TF binding and target gene expression at single‐molecule resolution (Fig 5). The observed temporal coupling indicates that the dynamics of TF binding directly determines transcriptional bursting kinetics. Based on our results, we propose a model where multiple RNA polymerases are recruited to form a burst of transcription during the time that a TF is bound to the promoter. The burst terminates when the TF dissociates from the DNA (Fig 7).
Figure 7. Schematic of the interplay between transcriptional bursting, nucleosomes, and TF dynamics.
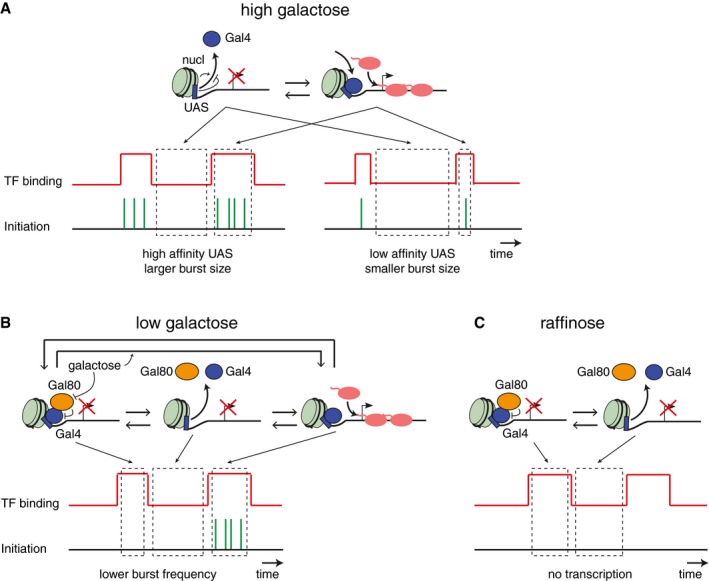
- In full induction conditions (high galactose), bursting of the GAL genes is determined by the binding dynamics of Gal4. A longer Gal4 dwell time results in a larger burst size. The Gal4 dwell time is determined by the affinity of the UAS and is significantly reduced by fragile nucleosomes.
- In lower galactose concentration, Gal80 represses Gal4 activity and transcription initiation during some bursts, resulting in a lower burst frequency. When Gal4 is not repressed, the burst duration is similar to full induction conditions. We propose that galactose concentration determines the availability of active Gal4.
- In inactive conditions, Gal4 still binds DNA, but is fully repressed by Gal80.
Moreover, our data show that the Gal4 dwell time depends on the affinity of the binding site (Fig 7A). Mutating the UAS promoter sequence (UASmut) reduces the Gal4 dwell time (Fig 2) and the RNA burst size (Fig 1) but does not affect the Gal4 binding frequency nor the burst frequency. Gal4 dwell time in vivo also appears to be considerably reduced by fragile promoter nucleosomes. The in vivo average dwell time of 17 s (Fig 4) is highly similar to the Gal4 in vitro dwell time at nucleosomal DNA (5 s, Fig 2), which is remarkable given that many more factors could influence Gal4 binding. In contrast, the Gal4 in vitro dwell time at naked DNA is 3 orders of magnitude more stable (Luo et al, 2014), indicating that addition of a nucleosome reduces the dwell time of Gal4 substantially. Indeed, Gal4 is able to bind to nucleosomal templates (Taylor et al, 1991), and at both the GAL3 and GAL10 promoter, the Gal4 binding sites appear to be covered by a fragile nucleosome in activating conditions (Fig 3; Floer et al, 2010). Nucleosomes thus not only serve to prevent TF binding, but also appear to play a novel role in limiting the dwell time of TFs. The presence of highly dynamic fragile nucleosomes in many yeast promoters (Kubik et al, 2015; Brahma & Henikoff, 2019) and at many galactose‐responsive genes (Fig EV3) suggests that nucleosomes may be generally required for rapid TF turnover. This mechanism of regulation may only apply to specific TF subclasses that can bind to partially unwrapped nucleosomal DNA, since previous studies in other systems have reported that the position of promoter nucleosomes affects the transcriptional burst frequency (Brown et al, 2013; Dadiani et al, 2013), likely by regulating DNA accessibility and TF binding frequency.
Transcriptional analysis at different galactose doses shows that burst duration is constant in different galactose concentrations (Fig 6). Since the burst duration is coupled to the Gal4 dwell time, the unchanged burst duration along the dose response suggests that Gal4 dwell time is likely not regulated by the galactose signaling network. Instead, galactose regulates the burst frequency (Fig 7B). Burst frequency regulation by upstream signaling networks has been observed previously in other systems and is often accompanied by increased transcription factor concentrations (Rodriguez et al, 2019). Absolute Gal4 levels do not increase in galactose, but our data are consistent with a model where the signaling network regulates the effective concentration of active Gal4 (Fig 7B). In inactive conditions, Gal4 is still bound to the promoter but repressed by the repressor Gal80 (Fig 7C) (Sellick et al, 2008), and increasing the galactose concentration could result in dose‐dependent unmasking of the Gal4 activation domain by Gal80 (Fig 7B).
We observe a reduced dwell time of Gal4 in raffinose versus galactose conditions. In raffinose, galactose‐responsive genes are either lowly transcribed (GAL80 and GAL3) or not transcribed (GAL10, GAL1, and GAL7), and transcription levels are increased in galactose. Even at inactive genes in raffinose, the promoter is bound by Gal4, but inactivated by Gal80. In addition to repressing transcriptional activation by Gal4, the reduced Gal4 dwell time in raffinose (Fig 4E) suggest that Gal80 may possibly act to reduce Gal4 dwell time on DNA. Alternatively, the difference in dwell time of Gal4 could be a result of post‐translational modifications (Loffreda et al, 2017), such as Gal4 phosphorylation or ubiquitination (Sellick et al, 2008). It will be interesting to uncover which other factors regulate the dwell time of Gal4.
Live‐cell tracking of individual Gal4 molecules by SMT indicates that several‐second binding of Gal4 is associated with the active transcription site (Fig 4). Dynamic association of Gal4 to chromatin has also been shown by previous competition ChIP experiments (Collins et al, 2009). However, the timescale accessible by ChIP‐based approaches is on the order of minutes, whereas SMT experiments report dwell times in the order of seconds. Indeed, the in vivo dwell‐time measurements by single‐molecule imaging of TFs are limited by the lifetime of the fluorophore (Liu & Tjian, 2018). If molecules bleach before they dissociate from the DNA, the dwell time can be underestimated or a long‐lived binding population can even be entirely missed. Two lines of evidence argue against this scenario for Gal4. First, increasing the frame interval did not increase the dwell‐time measurements for Gal4 (Fig EV4). Second, the positive cross‐correlation between Gal4 and GAL10 RNA (Fig 5) excludes the possibility that Gal4 would be bound stably to the promoter (on the order of minutes) and be independent from RNA output. Even if the in vivo dwell time of Gal4 is underestimated, the peak in the cross‐correlation between Gal4 and GAL10 would not be consistent with a model where several‐minute binding would be required for RNA production.
Recent studies have proposed a model where clustering or phase separation of the transcription machinery is important for driving transcription (Cisse et al, 2013; Hnisz et al, 2017; Lu et al, 2018). Although we do not exclude this possibility, it is important to note that the kinetics of our data can completely be described by the binding kinetics of individual TFs to single target genes, without need to invoke liquid droplets of multi‐molecular assemblies of multiple target genes. The correlation between the in vitro dwell time and in vivo transcriptional burst duration, and the observed similarity between the in vitro and in vivo dwell time of single Gal4 molecules, suggest that clustering does not contribute greatly to the binding kinetics of Gal4.
The connection between binding site affinity and burst size implies that the burst size of a gene is encoded in the genome. Large transcriptional burst sizes are associated with higher transcriptional noise (Raser & O'Shea, 2004; Raj et al, 2006). As a consequence, TF binding sites in the gene promoter do not only encode information on the level of transcription, but also on the associated cell‐to‐cell variability. We speculate that the depletion of consensus binding sites and the enrichment of clusters of weaker affinity sites in the genome are perhaps a way to regulate transcription levels without creating large burst sizes (Farley et al, 2015). On the other hand, the burst size may also be inherently limited for some genes, as introducing a stronger affinity binding site at GAL3 did not increase transcription levels (Fig 2K), although the explanation for this phenomenon is still unclear. Perhaps other regulatory factors limit the Gal4 dwell time in vivo, or alternatively, the stability of other factors may become limiting when Gal4 dwell time is increased.
In summary, we have used advanced single‐molecule microscopy experiments of the galactose system to show how endogenous TFs and nucleosomes regulate bursting kinetics. Whether the mechanism we propose also applies to other transcription networks remains to be determined. Overall, our results provide a framework for interpreting the fluctuations of TF binding and RNA production that have implications for understanding noise in gene expression.
Materials and Methods
Yeast strains and plasmids
Haploid or diploid yeast was transformed with a PCR product containing the PP7 loop cassette and loxP‐kanMX‐loxP. The kanMX marker was removed with CRE recombinase (pTL014). Coat protein was expressed from plasmids (pTL041, pTL092, pTL093) or was integrated (pTL147, pTL164) (Wosika et al, 2016). To introduce UAS mutations or the MS2 loop cassette, a PCR product/single‐stranded oligo and plasmid‐expressing Cas9 and a guide RNA (Laughery et al, 2015) were transformed. Strains, plasmids, and oligos used to construct the strains are listed in Tables EV1, EV2 and EV3.
Microscopy of transcription and image processing
Cells were imaged at mid‐log (OD 0.2–0.4) on coverslip with 2% agarose pads at 30°C. Imaging of transcription was performed on custom‐built wide‐field microscopes, consisting of an AxioObserver inverted microscope (Zeiss), a 100× NA 1.46 objective, an Evolve 512 EMCCD camera (Photometrics) or a sCMOS ORCA Flash 4v3 (Hamamatsu), a Tokai Hit stage incubator (INUB‐LPS) or a UNO Top stage incubator (OKOlab) at 30°C, and laser excitation at 488 (Excelsior, Spectra Physics) or LED excitation at 470/24 and 550/15 (SpectraX, Lumencor). Wide‐field images were recorded at 30 s interval for PP7‐GAL3 and PP7‐GAL10, and at 15 s interval for PP7‐GAL3‐MS2, with 9 z‐stacks (Δz 0.5 μm) and 150 ms exposure using Micro‐Manager software.
For image analysis, maximum intensity projections were computed. The intensity of the TS was calculated for each color separately by fitting a 2D Gaussian mask after local background subtraction as described previously (Coulon et al, 2014) using custom Python software. To determine the on and off periods, a threshold was applied to background subtracted traces of 2.5–3 times the standard deviations of the background. This number was chosen to reliably distinguish on and off periods from background levels at the single transcript level. Auto‐ and cross‐correlation functions were computed and averaged as described previously (Coulon et al, 2014; Lenstra et al, 2015). The burst duration is given by the intersection of the fast and slow linear components.
Single‐molecule FISH
Yeast cultures were grown to early mid‐log, fixed with 5% PFA (Electron Microscopy Sciences, 15714‐S) for 20 min, washed three times with buffer B (1.2 M sorbitol and 100 mM potassium phosphate buffer pH 7.5), permeabilized with 300 U of lyticase (Sigma‐Aldrich, L2524‐25KU), and washed with buffer B. Cells were immobilized on poly‐l‐lysine‐coated coverslip and permeabilized with 70% ethanol overnight. Coverslips were hybridized for 4 h at 37°C with hybridization buffer containing 10% dextran sulfate, 10% formamide, 2xSSC, and 5 pmol probe. For FISH targeting the repeats, four PP7 probes labeled with Cy3 were targeted to the loops (Table EV3). For FISH targeting GAL3, 48 probes labeled with Quasar 570 were used (Table EV3). Coverslips were washed 2× for 30 min with 10% formamide, 2xSSC at 37°C, 1× with 2xSSC, and 1× for 5 min with 1× PBS at room temperature. Coverslips were mounted on microscope slides using ProLong Gold mounting media with DAPI (Thermo Fisher, P36934). Cells were imaged on an AxioObserver inverted microscope (Zeiss) with LED illumination (SpectraX, Lumencor) and a sCMOS ORCA Flash 4v3 (Hamamatsu). Spots were localized in maximum intensity projected imaged using custom Python software by fitting to a 2D Gaussian mask with local background subtraction. Cell and nuclear outlines were determined with Cell Profiler (Carpenter et al, 2006). TS were defined as the brightest nuclear spot and were normalized to the median fluorescent intensity of cytoplasmic RNAs. The TS distribution was compared with a Poisson distribution. Colocalization of PP7 and GAL3 probes was determined with a maximum spot distance of 1 pixel.
Preparation of DNA, nucleosomes, and Gal4 for affinity measurements in vitro
To prepare DNA molecules for DNA binding experiments, Cy3 or Cy5 oligonucleotides were annealed by mixing at an equal‐molar ratio and purified on a Gen‐Pak FAX Anion exchange Column (Waters, WAT015490). To prepare DNA molecules for nucleosome experiments, oligonucleotides for PCR were labeled with Cy3 NHS ester (GE Healthcare) at an amino group at their 5′‐ends. Oligonucleotides were then purified by HPLC with 218TP C18 reverse‐phase column (Hichrom, 218TP54). DNA molecules were prepared by PCR from a plasmid containing the 601 nucleosome positioning sequence (NPS) with a UASwt or UASmut Gal4 binding site positioned 8 bp into the nucleosome. Following PCR amplification, DNA molecules were purified using a MonoQ column (GE Healthcare). All DNA sequences can be found in Table EV3.
Human recombinant histones were expressed and purified as previously described (Luger et al, 1999). Mutation H3 (C110A) was introduced by site‐directed mutagenesis (Agilent). The histone octamer was refolded by adding each of the histones together at a ratio of 1.1:1.1:1:1 (H2A:H2B:H3:H4) and purifying as previously described (Luger et al, 1999). H2A(K119C) containing HO was labeled with Cy5‐maleimide (GE Healthcare) as previously described Gibson et al (2016).
Nucleosomes containing Cy3‐labeled DNA and purified Cy5‐labeled histone octamer were reconstituted through double dialysis. DNA and octamer were mixed at a ratio of 1.25:1 DNA:HO in 0.5× TE pH 8 with 1 mM benzamidine hydrochloride (BZA) and 2M NaCl in a volume of 50 μl. The mixture was loaded into a dialysis chamber and placed into a large dialysis bag containing 80 ml of 0.5× TE pH 8, 1 mM BZA, and 2M NaCl and dialyzed extensively against 0.5× TE pH 8 with 1 mM BZA at 4C. Dialyzed nucleosomes were loaded onto 5–30% sucrose gradients and purified by centrifugation on an Optima L‐90 K Ultracentrifuge (Beckman Coulter) with a SW‐41 rotor. Sucrose fractions containing nucleosomes were collected, concentrated, and stored in 5× TE pH 8 on ice.
To prepare Gal4, amino acids 1–147 of Gal4 were expressed from plasmid pET28a containing Gal4‐1‐147 in E. coli Rosetta (DE3)pLysS cells (Millipore, 70956) by inducing with 1 mM IPTG + 10 μM ZnAc for 3 h. Cells were harvested by centrifugation and resuspended at 50 ml per 1 l starting culture in buffer A (50 mM Tris pH 8.0, 200 mM NaCl, 1 mM DTT, 10 μM ZnAc, 1 mM phenylmethanesulfonyl fluoride (PMSF), 20 μg/ml leupeptin, and 20 μg/ml pepstatin) and stored at −80°C. The cells were lysed by sonication and clarified by centrifugation, loaded onto a 5 ml HisTrap HP Ni‐NTA column (GE Healthcare, 17524801), equilibrated with buffer B (25 mM Tris pH 7.5, 200 mM NaCl, 0.2% Tween‐20, 10 mM imidazole, 20 μM ZnAc, 1 mM DTT, 1 mM PMSF), and eluted with elution buffer (25 mM Tris pH 7.5, 200 mM NaCl, 0.2% Tween‐20, 200 mM imidazole, 20 μM ZnAc, 1 mM DTT, 1 mM PMSF). Fractions containing Gal4 were dialyzed into buffer C (25 mM Tris pH 7.5 200 mM NaCl, 20 μM ZnAc, 1 mM DTT, 1 mM PMSF) and purified with a TSKgel SP‐5PW cation exchange column (Tosoh, 07161). Fractions containing Gal4 were concentrated using Amicon 10K filters (Millipore, UFC201024) and stored in buffer D (HEPES pH 7.5, 200 mM NaCl, 10% glycerol, 20 μM ZnAc, 1 mM DTT, 1 mM PMSF).
Ensemble PIFE measurements
Gal4 binding to its target site on a 51‐bp Cy3/5‐DNA was determined by protein‐induced fluorescence enhancement (PIFE) (Hwang et al, 2011), where Cy3 fluorescence increases upon protein binding. Fluorescence spectra were acquired with a Fluoromax4 fluorometer (Horiba) using an excitation wavelength of 510 nm. Gal4 affinity to DNA was measured by incubating 0.5 nM DNA with 0–30 nM Gal4 in 10 mM Tris–HCl pH 8, 130 mM NaCl, 10% glycerol, and 0.0075% v/v Tween‐20. From these experiments, we determined that ~80% of DNA molecules are bound by ~5 nM Gal4. Competition experiments were performed by incubating Cy3/5 DNA with 5 nM Gal4 and 0–500 nM unlabeled competitor DNA containing either the UASwt or UASmut Gal4 binding site for 30 min. For each titration, we determined the S1/2, the concentration at which the fluorescence signal decreases by 50%. With these measurements, we calculated the relative affinity of Gal4 to the UASwt and UASmut sequences (relative affinity = S1/2 UASmut competitor/S1/2 UASwt competitor). Fluorescence spectra were analyzed with Matlab to determine the change in Cy3 fluorescence.
Ensemble FRET measurements
Gal4 binding to its site within a nucleosome was detected through FRET. 0.5 nM nucleosomes were incubated for at least 5 min with 0–1,000 nM Gal4 in 10 mM Tris–HCl pH 8, 130 mM NaCl, 10% glycerol, and 0.0075% v/v Tween‐20. FRET efficiency was calculated using the RatioA method with a custom matlab program (Clegg, 1992).
Single‐molecule TIRF microscope
The smTIRF microscope was built on an inverted IX73‐inverted microscope (Olympus) as previously described (Roy et al, 2008). 532‐ and 638‐nm diode lasers (Crystal Lasers) were used for Cy3 and Cy5 excitation. The excitation beams were expanded and then focused through a quartz prism (Melles Griot) at the surface of the quartz flow cell. A 1.3 N.A. silicone immersion objective (Olympus) was used to collect fluorescence which was separately imaged onto an iXon3 EMCCD camera (Andor) with a custom‐built emission path containing bandpass filters and dichroic beam splitters (Chroma Tech). Each video was acquired using Micro‐Manager software.
Single‐molecule fluorescence measurements of Gal4 binding kinetics
Flow cells were functionalized as previously described (Kinz‐Thompson et al, 2013). Briefly, quartz microscope slides (Alfa Aesar) were sonicated in toluene and then ethanol, and then further cleaned in piranha solution (3:1 mixture of concentrated sulfuric acid to 50% hydrogen peroxide). Slides were washed in water and, once completely dry, incubated in 100 μM mPEG‐Si and biotin‐PEG‐Si (Laysan Bio) overnight in anhydrous toluene. Functionalized quartz slides and coverslips were assembled into microscope flow cells using parafilm to define channels. Before each experiment, the flow cell is treated sequentially with 1 mg/ml BSA, 40 μg/ml streptavidin, and biotin‐labeled nucleosomes.
Biotinylated nucleosomes were allowed to incubate in the flow cell at room temperature for 5 min and then washed out with imaging buffer containing the desired concentration of Gal4. The samples were first exposed to 638‐nm excitation to determine the location of Cy5 molecules and then 532 nm for FRET measurements. The imaging buffer for FRET experiments contained 10 mM Tris–HCl pH 8, 130 mM NaCl, 10% glycerol, 0.0075% v/v Tween‐20, 0.1 mg/ml BSA, 2 mM Trolox, 0.0115% v/v COT, 0.012% v/v NBA, 450 μg/ml glucose oxidase, and 22 μg/ml catalase.
Single‐molecule time series were fit to a two‐state step function by the hidden Markov method using vbFRET (Bronson et al, 2009). Idealized time series were further analyzed using custom‐written Matlab programs to determine the dwell‐time distributions of the TF‐bound and TF‐unbound states. Approximately 50% of FRETing traces fluctuated and were used in the analysis of FRET data. Dwell‐time and unbound‐time cumulative sum distributions were fit to single‐exponential distributions using matlab to obtain rate constants for the transitions between bound and unbound states.
MNase‐seq
Preparation and analysis of mono‐nucleosomal DNA was performed as described previously (de Jonge et al, 2017) with minor modifications. Briefly, cells were grown in SC + 2% raffinose or SC + 2% galactose from OD 0.3 to OD 1.0, fixed in 1% PFA, washed with 1 M sorbitol, treated with spheroplasting buffer (1M sorbitol, 1 mM β‐mercaptoethanol, 10 mg/ml zymolyase 100T (US biological, Z1004.250)) and washed twice with 1 M sorbitol. Spheroplasted cells were treated with 0.01171875 or 0.1875 U micrococcal nuclease (Sigma‐Aldrich, N5386‐200UN) in digestion buffer (1 M sorbitol, 50 mM NaCl, 10 mM Tris pH7.4, 5 mM MgCl2, 0.075% NP‐40, 1 mM β‐mercaptoethanol, 0.5 mM spermidine) at 37°C. After 45 min, reactions were terminated on ice with 25 mM EDTA and 0.5% SDS. Samples were treated with proteinase K for 1 h at 37°C and decrosslinked overnight at 65°C. Digested DNA was extracted with phenol/chloroform (PCI 15:14:1), precipitated with NH4‐Ac, and treated with 0.1 mg/ml RNaseA/T1. The extent of digestion was checked on a 3% agarose gel.
Sequencing was performed on one HiSeq2500 lane using Illumina TruSeq v4 chemistry. Paired‐end 125‐bp reads were aligned with Bowtie 2. Nucleosome dyads were found by taking the middle of each paired read of insert size between 95 and 225 bp (or 140 and 225 bp) and were smoothed with a 31‐bp window, as described in de Jonge et al (2017). Data are deposited in GEO with accession number: GSE116337.
Growth assay
Serial dilutions (fivefold) of BY4741, YTL219, YTL267, and YTL302 strains were spotted on YEP + 2% glucose, YEP + 2% galactose + 20 μg/μl ethidium bromide, and YEP + 2% raffinose + 2% galactose + 40 mM lithium chloride + 0.003% methionine. Growth was assessed after 3 days at 30°C.
Single‐molecule tracking of Gal4
Cells were grown in SC + 2% glucose overnight, washed in SC + 2% raffinose, and grown in SC + 2% raffinose for 4 h. After 2 h, cells were labeled with 5 nM JF646 (for Gal4) and 0.01 nM (for H3) (Grimm et al, 2015). Before imaging, cells were washed once and immobilized on coverslip with 2% agarose pads containing 2% raffinose or 2% galactose. SMT movies were acquired a custom‐built microscope, containing a Highly Inclined Laminated Optical (HILO) sheet illumination at 488 nm and 647 nm, a 100× 1.49 TIRF objective (Olympus), three EM‐CCD iXon Ultra 888 cameras (Andor), and an UNO stage top incubator at 30°C (OKOlab). Images captured simultaneously with dual‐color illumination at 488 and 647 nm for 30 ms at 200 ms or 1,000 ms interval.
Spots were tracked independently for the GAL10 transcription site and Gal4 channel using custom Matlab software based on MatTrack (Mazza et al, 2013). The labeling density is such that very few (< ~10) molecules are labeled in each nucleus, making it unlikely to have multiple labeled molecules in any one spot. Particle positions were fit with a 2D Gaussian mask, tracked using nearest‐neighbors approach, and manually verified. Only molecules that were tracked in more than 4 frames were considered to be bound. To determine whether a molecule was bound or diffusing, a threshold was used based on tracking of histone H3. Tracking of histone H3 (HHT1‐HALO) showed that 99% of single molecules had a frame‐to‐frame displacement of < 0.35 μm at 200 ms interval or < 0.37 μm at 1,000 ms interval. These maximum displacements were used to determine whether Gal4 particles are chromatin bound or diffusing. The cumulative distribution of dwell times of bound Gal4 molecules (survival probability plot) was corrected for bleaching by dividing the distribution by the decay of the number of particles found over time. To extract the average dwell times, the survival probability distribution was fit to a one‐ or two‐component exponential decay function.
To select for traces that colocalized with the GAL10 transcription site, images were registered using beads, and a 250 nm distance threshold was used to determine overlap. The survival probability plot was fit to a one‐component dwell exponential function.
Orbital tracking
Cells were prepared as for SMT experiments. Active TSs were identified at high laser power, but once orbital tracking of an TS was initiated, the power was reduced to reduce photobleaching. Orbital tracking was performed according to Annibale and Gratton (2015), Anzalone et al (2014). Tracking of transcription sites was achieved using two orbits of radius ~87 nm at a position 145 nm above the TS, followed by two orbits 145 nm below the TS, with 64 points per orbit and a pixel dwell time of 1024 μs per pixel. Each orbit had a duration of 65.5 ms with a total sampling time of 262 ms (3.8 Hz) sampling rate, allowing for the measurement of Gal4 and GAL10 RNA occupancy at high sampling rate for 10–20 min while keeping the transcription site in focus via the z‐piezo and active feedback from microscope software. If the spot disappears because the TS is inactive, the feedback loops wait at the same spot for the reappearance of signal.
Imaging and orbital tracking were performed on an ISS Alba FCS microscope (Champaign) with 488‐nm and 633‐nm excitation, two SPCM‐ARQH Avalanche Photodiodes (Pacer), a 1 MHz IOtech 3000 Data Acquisition card (Measurement Computing Corporation), and a Nano‐F25HS high‐speed z‐piezo (Mad City Labs) coupled to a Nikon Ti‐U inverted microscope with a CFI Plan Apochromat 60 × 1.2 NA water immersion objective (Nikon). Data acquisition was performed using SimFCS 3.0 software written by Enrico Gratton (Laboratory for Fluorescence Dynamics, University of California, Irvine). Bead alignment showed that the point spread function alignment in both channels was ~60 nm. When using one photon excitation for cross‐correlation spectroscopy, alignment of both lasers in 3D is critical to achieve success (Schwille et al, 1999). A 0.5–2× variable beam expander was therefore placed in front of the 488‐nm laser to achieve alignment in z due to chromatic aberration.
Fluorescence intensity appeared in carpet plots of angle versus time showing a Gaussian peak in the RNA channel with intermittent signal in the red channel assumed to be binding of fluorescently labeled Gal4 molecules. Sections of the fluorescence intensity traces were selected for active transcription by the appearance of signal above background in the carpet plots. Traces were background subtracted by the lower quartile of the signals. Data analysis was performed using custom software written in IDL (Harris Geospatial Solutions) and Python. Temporal correlation functions of the fluorescent signal were calculated as previously from ½ the average photon intensity over the 4‐orbital period of 262 ms directly from the DC component of the fluorescence intensity trace as shown in equation 18 of Kis‐Petikova and Gratton (2004). Correlation functions were averaged over 10–20 measurements. This resulted in very robust and reproducible correlation functions from which the dwell time of Gal4 molecules, GAL10 RNA and temporal relationships could be estimated. The GAL10 RNA autocorrelation is best fit with a linear model as reported in Larson et al (2011), Gal4 autocorrelation function is best fit using an exponential reaction dominant diffusion binding model (Digman & Gratton, 2009; Michelman‐Ribeiro et al, 2009) and the cross‐correlation is best fit to a shifted Gaussian model.
Author contributions
Conceptualization: TLL, MLF, DRL, and MGP; Investigation: BTD, AH, HPP, and TLL; Methodology and Software: DAB, MLF, and TLL; Writing—Original draft: BTD, MLF, and TLL; Writing—Review and Editing: DRL and MGP; Supervision; DRL, MGP, MLF, and TLL.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Movie EV1
Movie EV2
Movie EV3
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4B
Source Data for Figure 4C
Source Data for Figure 4D
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank L. Lavis for providing synthetic HALO dyes and G. Mehta, T.S. Karpova, V. Wosika, S. Pelet, J. Wisniewski, Y. Fondufe‐Mittendorf, K. Luger, J. Widom, for strains and plasmids. We thank T.S. Karpova for assistance with SMT experiments, Wim de Jonge for help with MNase‐seq experiments, Yi Luo for help with the ensemble in vitro FRET measurements, B. van Steensel for reading of the manuscript, and D. Stavreva and members of the D.R.L., M.G.P., M.L.F., and T.L.L. laboratories for helpful discussions. This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (D.R.L.), the Research Corporation for Science Advancement and the Gordon and Betty Moore Foundation (GBMF5263.10 M.L.F.), National Institutes of Health (1R15GM123446‐01 M.L.F., T32GM086252 B.T.D., R01GM121858 M.G.P, and R01GM121966 M.G.P.), the KWF Dutch Cancer Society (KWF 2012‐5394 T.L.L), the gravitation program CancerGenomiCs.nl from the Netherlands Organisation for Scientific Research (NWO, T.L.L.), and the European Research Council (ERC Starting Grant 755695 BURSTREG, T.L.L.).
The EMBO Journal (2019) 38: e100809
Contributor Information
Matthew L Ferguson, Email: mattferguson@boisestate.edu.
Tineke L Lenstra, Email: t.lenstra@nki.nl.
Data availability
The sequencing (MNase‐seq) data from this publication have been deposited to the GEO database https://www.ncbi.nlm.nih.gov/geo/ and assigned the identifier GSE116337. Microscopy data are available upon request.
References
- Acar M, Becskei A, van Oudenaarden A (2005) Enhancement of cellular memory by reducing stochastic transitions. Nature 435: 228–232 [DOI] [PubMed] [Google Scholar]
- Annibale P, Gratton E (2015) Single cell visualization of transcription kinetics variance of highly mobile identical genes using 3D nanoimaging. Sci Rep 5: 9258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone A, Annibale P, Gratton E (2014) 3D orbital tracking in a modified two‐photon microscope: an application to the tracking of intracellular vesicles. J Vis Exp 92: e51794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar Halpern K, Tanami S, Landen S, Chapal M, Szlak L, Hutzler A, Nizhberg A, Itzkovitz S (2015) Bursty gene expression in the intact mammalian liver. Mol Cell 58: 147–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball DA, Mehta GD, Salomon‐Kent R, Mazza D, Morisaki T, Mueller F, McNally JG, Karpova TS (2016) Single molecule tracking of Ace1p in Saccharomyces cerevisiae defines a characteristic residence time for non‐specific interactions of transcription factors with chromatin. Nucleic Acids Res 44: e160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartman CR, Hsu SC, Hsiung CC‐S, Raj A, Blobel GA (2016) Enhancer regulation of transcriptional bursting parameters revealed by forced chromatin looping. Mol Cell 62: 237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahma S, Henikoff S (2019) RSC‐associated subnucleosomes define MNase‐sensitive promoters in yeast. Mol Cell 73: 238–249.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronson JE, Fei J, Hofman JM, Gonzalez RL, Wiggins CH (2009) Learning rates and states from biophysical time series: a Bayesian approach to model selection and single‐molecule FRET data. Biophys J 97: 3196–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, Mao C, Falkovskaia E, Jurica MS, Boeger H (2013) Linking stochastic fluctuations in chromatin structure and gene expression. PLoS Biol 11: e1001621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J et al (2006) Cell Profiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7: R100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang Z, Li L, Chen B‐C, Revyakin A, Hajj B, Legant W, Dahan M, Lionnet T, Betzig E et al (2014) Single‐molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156: 1274–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubb JR, Trcek T, Shenoy SM, Singer RH (2006) Transcriptional pulsing of a developmental gene. Curr Biol 16: 1018–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisse II, Izeddin I, Causse SZ, Boudarene L, Senecal A, Muresan L, Dugast‐Darzacq C, Hajj B, Dahan M, Darzacq X (2013) Real‐time dynamics of RNA polymerase II clustering in live human cells. Science 341: 664–667 [DOI] [PubMed] [Google Scholar]
- Clegg RM (1992) Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol 211: 353–388 [DOI] [PubMed] [Google Scholar]
- Collins GA, Lipford JR, Deshaies RJ, Tansey WP (2009) Gal4 turnover and transcription activation. Nature 461: E7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon A, Ferguson ML, De Turris V, Palangat M, Chow CC, Larson DR (2014) Kinetic competition during the transcription cycle results in stochastic RNA processing. Elife 3: 03939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadiani M, van Dijk D, Segal B, Field Y, Ben‐Artzi G, Raveh‐Sadka T, Levo M, Kaplow I, Weinberger A, Segal E (2013) Two DNA‐encoded strategies for increasing expression with opposing effects on promoter dynamics and transcriptional noise. Genome Res 23: 966–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digman MA, Gratton E (2009) Analysis of diffusion and binding in cells using the RICS approach. Microsc Res Tech 72: 323–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elf J, Li G‐W, Xie XS (2007) Probing transcription factor dynamics at the single‐molecule level in a living cell. Science 316: 1191–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley EK, Olson KM, Zhang W, Brandt AJ, Rokhsar DS, Levine MS (2015) Suboptimization of developmental enhancers. Science 350: 325–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floer M, Wang X, Prabhu V, Berrozpe G, Narayan S, Spagna D, Alvarez D, Kendall J, Krasnitz A, Stepansky A et al (2010) A RSC/nucleosome complex determines chromatin architecture and facilitates activator binding. Cell 141: 407–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukaya T, Lim B, Levine M (2016) Enhancer control of transcriptional bursting. Cell 166: 358–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson MD, Brehove M, Luo Y, North J, Poirier MG (2016) Methods for investigating DNA accessibility with single nucleosomes. Methods Enzymol 581: 379–415 [DOI] [PubMed] [Google Scholar]
- Golding I, Paulsson J, Zawilski SM, Cox EC (2005) Real‐time kinetics of gene activity in individual bacteria. Cell 123: 1025–1036 [DOI] [PubMed] [Google Scholar]
- Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH et al (2015) A general method to improve fluorophores for live‐cell and single‐molecule microscopy. Nat Methods 12: 244–250, 3 p following 250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, Sharp PA (2017) A phase separation model for transcriptional control. Cell 169: 13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H, Kim H, Myong S (2011) Protein induced fluorescence enhancement as a single molecule assay with short distance sensitivity. Proc Natl Acad Sci USA 108: 7414–7418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Frey BR, Evans ML, Friel JC, Hopper JE (2009) Gene activation by dissociation of an inhibitor from a transcriptional activation domain. Mol Cell Biol 29: 5604–5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge WJ, O'Duibhir E, Lijnzaad P, van Leenen D, Groot Koerkamp MJ, Kemmeren P, Holstege FC (2017) Molecular mechanisms that distinguish TFIID housekeeping from regulatable SAGA promoters. EMBO J 36: 274–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinz‐Thompson CD, Palma M, Pulukkunat DK, Chenet D, Hone J, Wind SJ, Gonzalez RL (2013) Robustly passivated, gold nanoaperture arrays for single‐molecule fluorescence microscopy. ACS Nano 7: 8158–8166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kis‐Petikova K, Gratton E (2004) Distance measurement by circular scanning of the excitation beam in the two‐photon microscope. Microsc Res Tech 63: 34–49 [DOI] [PubMed] [Google Scholar]
- Kubik S, Bruzzone MJ, Jacquet P, Falcone J‐L, Rougemont J, Shore D (2015) Nucleosome stability distinguishes two different promoter types at all protein‐coding genes in yeast. Mol Cell 60: 422–434 [DOI] [PubMed] [Google Scholar]
- Larson DR, Zenklusen D, Wu B, Chao JA, Singer RH (2011) Real‐time observation of transcription initiation and elongation on an endogenous yeast gene. Science 332: 475–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DR, Fritzsch C, Sun L, Meng X, Lawrence DS, Singer RH (2013) Direct observation of frequency modulated transcription in single cells using light activation. Elife 2: e00750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughery MF, Hunter T, Brown A, Hoopes J, Ostbye T, Shumaker T, Wyrick JJ (2015) New vectors for simple and streamlined CRISPR‐Cas9 genome editing in Saccharomyces cerevisiae . Yeast 32: 711–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenstra TL, Coulon A, Chow CC, Larson DR (2015) Single‐molecule imaging reveals a switch between spurious and functional ncRNA transcription. Mol Cell 60: 597–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi V, Ruan Q, Gratton E (2005a) 3‐D particle tracking in a two‐photon microscope: application to the study of molecular dynamics in cells. Biophys J 88: 2919–2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi V, Ruan Q, Plutz M, Belmont AS, Gratton E (2005b) Chromatin dynamics in interphase cells revealed by tracking in a two‐photon excitation microscope. Biophys J 89: 4275–4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SD, Marmorstein R, Harrison SC, Ptashne M (1996) DNA sequence preferences of GAL4 and PPR1: how a subset of Zn2 Cys6 binuclear cluster proteins recognizes DNA. Mol Cell Biol 16: 3773–3780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Tjian R (2018) Visualizing transcription factor dynamics in living cells. J Cell Biol 217: 1181–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffreda A, Jacchetti E, Antunes S, Rainone P, Daniele T, Morisaki T, Bianchi ME, Tacchetti C, Mazza D (2017) Live‐cell p53 single‐molecule binding is modulated by C‐terminal acetylation and correlates with transcriptional activity. Nat Commun 8: 313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Yu D, Hansen AS, Ganguly S, Liu R, Heckert A, Darzacq X, Zhou Q (2018) Phase‐separation mechanism for C‐terminal hyperphosphorylation of RNA polymerase II. Nature 558: 318–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, Richmond TJ (1999) Preparation of nucleosome core particle from recombinant histones. Methods Enzymol 304: 3–19 [DOI] [PubMed] [Google Scholar]
- Luo Y, North JA, Rose SD, Poirier MG (2014) Nucleosomes accelerate transcription factor dissociation. Nucleic Acids Res 42: 3017–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazza D, Ganguly S, McNally JG (2013) Monitoring dynamic binding of chromatin proteins in vivo by single‐molecule tracking. Methods Mol Biol 1042: 117–137 [DOI] [PubMed] [Google Scholar]
- Michelman‐Ribeiro A, Mazza D, Rosales T, Stasevich TJ, Boukari H, Rishi V, Vinson C, Knutson JR, McNally JG (2009) Direct measurement of association and dissociation rates of DNA binding in live cells by fluorescence correlation spectroscopy. Biophys J 97: 337–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normanno D, Boudarène L, Dugast‐Darzacq C, Chen J, Richter C, Proux F, Bénichou O, Voituriez R, Darzacq X, Dahan M (2015) Probing the target search of DNA‐binding proteins in mammalian cells using TetR as model searcher. Nat Commun 6: 7357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peccoud J, Ycart B (1995) Markovian modeling of gene‐product synthesis. Theor Popul Biol 48: 222–234 [Google Scholar]
- Presman DM, Ball DA, Paakinaho V, Grimm JB, Lavis LD, Karpova TS, Hager GL (2017) Quantifying transcription factor binding dynamics at the single‐molecule level in live cells. Methods 123: 76–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S (2006) Stochastic mRNA synthesis in mammalian cells. PLoS Biol 4: e309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raser JM, O'Shea EK (2004) Control of stochasticity in eukaryotic gene expression. Science 304: 1811–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Ren G, Day CR, Zhao K, Chow CC, Larson DR (2019) Intrinsic dynamics of a human gene reveal the basis of expression heterogeneity. Cell 176: 213–226.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Hohng S, Ha T (2008) A practical guide to single‐molecule FRET. Nat Methods 5: 507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rullan M, Benzinger D, Schmidt GW, Milias‐Argeitis A, Khammash M (2018) An optogenetic platform for real‐time, single‐cell interrogation of stochastic transcriptional regulation. Mol Cell 70: 745–756.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwille P, Haupts U, Maiti S, Webb WW (1999) Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one‐ and two‐photon excitation. Biophys J 77: 2251–2265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellick CA, Campbell RN, Reece RJ (2008) Galactose metabolism in yeast‐structure and regulation of the leloir pathway enzymes and the genes encoding them. Int Rev Cell Mol Biol 269: 111–150 [DOI] [PubMed] [Google Scholar]
- Senecal A, Munsky B, Proux F, Ly N, Braye FE, Zimmer C, Mueller F, Darzacq X (2014) Transcription factors modulate c‐Fos transcriptional bursts. Cell Rep 8: 75–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor IC, Workman JL, Schuetz TJ, Kingston RE (1991) Facilitated binding of GAL4 and heat shock factor to nucleosomal templates: differential function of DNA‐binding domains. Genes Dev 5: 1285–1298 [DOI] [PubMed] [Google Scholar]
- Tokunaga M, Imamoto N, Sakata‐Sogawa K (2008) Highly inclined thin illumination enables clear single‐molecule imaging in cells. Nat Methods 5: 159–161 [DOI] [PubMed] [Google Scholar]
- Venturelli OS, El‐Samad H, Murray RM (2012) Synergistic dual positive feedback loops established by molecular sequestration generate robust bimodal response. Proc Natl Acad Sci USA 109: E3324–E3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wosika V, Durandau E, Varidel C, Aymoz D, Schmitt M, Pelet S (2016) New families of single integration vectors and gene tagging plasmids for genetic manipulations in budding yeast. Mol Genet Genomics 291: 2231–2240 [DOI] [PubMed] [Google Scholar]
- Zaret KS, Mango SE (2016) Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr Opin Genet Dev 37: 76–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenklusen D, Larson DR, Singer RH (2008) Single‐RNA counting reveals alternative modes of gene expression in yeast. Nat Struct Mol Biol 15: 1263–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Movie EV1
Movie EV2
Movie EV3
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4B
Source Data for Figure 4C
Source Data for Figure 4D
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
The sequencing (MNase‐seq) data from this publication have been deposited to the GEO database https://www.ncbi.nlm.nih.gov/geo/ and assigned the identifier GSE116337. Microscopy data are available upon request.