

Abstract
The majority of currently used therapeutics are small molecule-based and utilize occupancy-driven pharmacology as the mode of action (MOA), in which the protein function is modulated via temporary inhibition. New modalities that operate using alternative MOAs are essential for tapping into the “undruggable” proteome. The PROteolysis Targeting Chimera (PROTAC) technology provides an attractive new approach that utilizes an event-driven MOA. Small molecule-based heterobifunctional PROTACs modulate protein target levels by hijacking the ubiquitin-proteasome system to induce degradation of the target. Here, we address important milestones in the development of the PROTAC technology, as well as emphasize key findings from this previous year and highlight future directions of this promising drug discovery modality.
Introduction
The drug target landscape has undergone changes within the last decade. The focus has shifted from primarily focusing on traditional drug targets, such as G-protein coupled receptors (GPCRs), ion channels, and kinases towards including more challenging “undruggable” targets, which are nevertheless attractive from a biological perspective [1]. These targets generally include proteins without enzymatic function, such as transcription factors and scaffolding proteins [2]. Historically, drug targets with well-defined active sites suitable for accommodation of a small molecule have been the focus of pharmacological intervention. Therefore, established methods exist for drug development against these active site-containing targets and the discovery of traditional small molecule inhibitors [3]. As a result, the majority of drugs today are small molecule-based and primarily operate via occupancy-driven pharmacology as the mode of action (MOA) (Fig. 1a). Although successful, this MOA cannot be applied to all biological targets, especially those which lack enzymatic activity such as scaffolding proteins or proteins that function via protein-protein interaction (PPIs) [4]. Efficacy of drugs that operate via the occupancy-driven MOA is driven by retaining a high target occupancy where high drug doses are generally required, often leading to undesired side effects due to off-target binding associated with higher drug concentrations [5]. Furthermore, the development of resistance to inhibition/occupancy-driven therapeutics occurs in many disease indications such as cancer [6] and bacterial infections [7]. Consequently, efforts have been made to develop new drug classes, preferably with alternative MOAs in order to modulate non-traditional drug targets and combat resistance mechanisms. Hence, the drug class space has expanded to include new modalites such as nucleic acid-based therapeutics, modified peptides, recombinant proteins and monoclonal antibodies [1].
Figure 1.
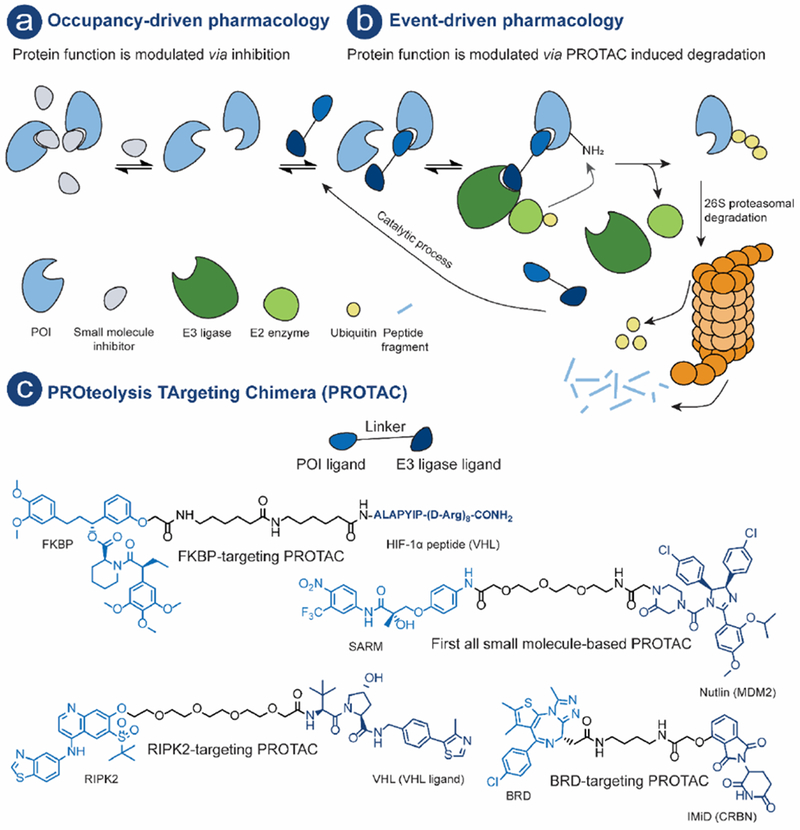
Schematic illustration of: a) Occupancy driven pharmacology – a small molecule-based drug, often an inhibitor, modulates protein function employing a non-catalytic MOA. b) Event-driven pharmacology (using PROTAC MOA as an example) – protein function is modulated by induced degradation. The PROTACs initiates a degradation cascade with POI ubiquitination followed by subsequent 26S proteasomal degradation of the POI. c) Schematic illustration of a PROTAC, POI ligand (royal blue) and an E3 ligand (dark blue), linker (black) and examples of PROTACs.
PROteolysis TArgeting Chimeras (PROTACs) have emerged as a new and promising modality utilizing an event-driven MOA, whereby protein levels are modulated by PROTAC-induced degradation [8,9] (Fig. 1b). A PROTAC is a hetero bifunctional molecule that consists of a protein of interest (POI) ligand and an E3 ubiquitin ligase (E3) recruiting ligand connected by a linker (Fig. 1c). PROTACs initiate a degradation cascade by forming a ternary complex with a POI and an E3, bringing the ubiquitination machinery in close proximity for subsequent POI ubiquitination. The polyubiquitinated POI is then recognized and degraded by the 26S proteasome. (Fig. 1b). The 26S proteasome is part of the ubiquitin-proteasome system (UPS) which is the primary mechanism used by eukaryotic cells to regulate protein levels [10,11]. In this review all compounds that meet the above definition will be referred to as PROTACs. Other names can however be found in the literature: e.g. specific and non-genetic IAP-dependent protein erasers (SNIPER); degrader; degronimids; PROteolysis TArgeting Peptide (PROTAP); Protein Degradation Probe (PDP).
This review covers the milestones of the PROTAC technology development, the current state of the technology with special focus on key findings from the previous year and highlights future directions of this promising approach for therapeutic discovery.
Past - PROTAC Development (2001-2016)
An overview of milestones for PROTAC development as well as protein targets that have successfully been modulated using the PROTAC technology is shown in Fig. 2. The first PROTAC, reported by the Crews and Deshaies laboratories in 2001, recruited the SCFβ-TRCP E3 to induce the degradation of methionine aminopeptidase-2 (MetAp-2) [33]. This first report was followed by PROTACs that induced degradation of the androgen (AR) and estrogen (ER) receptor, hence expanding the target scope [34]. Microinjection of these AR- and ER-targeting PROTACs demonstrated that they could function in an intact cell. The succeeding PROTACs used the HIF-1α peptide fragment bearing a cell-penetrating peptide sequence to recruit the Von Hippel-Lindau disease tumor-suppressor protein (VHL) E3 in intact cells without the need for microinjection (see Fig. 1c, FKBP-targeting PROTAC) [42]. Later, a shorter peptide fragment of HIF-1α was incorporated in a PROTAC targeting aryl hydrocarbon receptor nuclear translocator (ARNT) [39]. While these first generation PROTACs could induce specific degradation of their targets, they were active only in the low-micromolar range. Moreover, due to their peptidic character they suffered from poor cell permeability and hence low cellular activity which limited the PROTAC technology for development of novel therapeutics.
Figure 2.
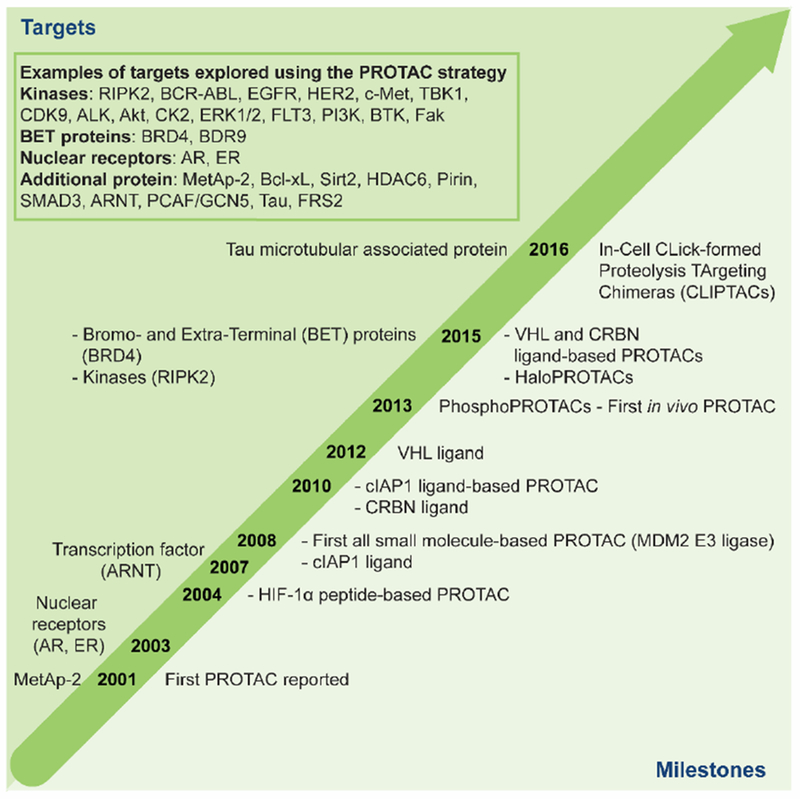
Time table describing the evolution of PROTACs (2001-2016): above time arrow (left): first time different protein or protein families were targeted using the PROTAC technology. Box: Example of targets that have been explored with the PROTAC technology [12–41]. Below time arrow (right): important milestones for the PROTAC technology development.
A significant advancement of the PROTAC technology was the identification of small molecule-based E3 recruiting ligands where the first all small molecule-based PROTAC was reported in 2008 [43]. The mouse double minute 2 homologue (MDM2) E3 was recruited by using a known MDM2-p53 PPI inhibitor, nutlin, as the E3 ligand[44]. The obtained PROTAC could induce degradation of AR (see Fig. 1c, First all small molecule-based PROTAC). Although this study demonstrated that developing cell permeable PROTACs is feasible, micromolar concentrations were required to induce the degradation of the AR [43]. Concurrently, bestatin methyl esters were found to bind to the cellular inhibitor of apoptosis protein 1 (cIAP1) and to promote its autoubiquitination and degradation [45]. The Hashimoto laboratory created the first PROTAC recruiting cIAP1 targeting cellular retinol- and retinoic acid-binding proteins (CRABP-I and II) for degradation using these bestatins [46]. Later, in 2012 high affinity peptidomimetic ligands for the VHL E3 were developed [47–49]. Further structure activity relationship (SAR) studies of the VHL ligand was later reported by the Ciulli laboratory which provided VHL ligands with improved physical chemical properties but similar affinities towards VHL [50–53]. During the same time, the E3 cereblon (CRBN) was identified as the molecular target of the immunomodulatory drugs (IMiDs), thalidomide, pomalidomide and lenalidomide [54–60]. Upon binding to CRBN, IMiDs were observed to promote the recruitment of neosubstrates, such as Ikaros, Aiolos and casein kinase 1A1 (CK1α), for ubiquitination and proteasomal degradation.
So far none of the obtained PROTACs had been characterized in vivo. In 2013, the PhosphoPROTACs, provided the first evidence that PROTACs function in vivo as these were able to inhibit tumor growth in murine models [41]. Furthermore, by taking advantage of the natural specificity of individual receptor tyrosine kinase (RTK) signaling pathways, the PhosphoPROTACs were able to distinguish between different RTK signaling pathways. Selectivity was obtained by incorporating different peptide sequencers as POI recruiting moiety known to be phosphorylated by a particular kinase. The activated PhosphoPROTAC was only obtained after co-treatment with a suitable growth factor, hence only the phosphorylated phosphoPROTAC has the ability to bind POI.
The utility of the small molecule-based VHL E3 ligand in facilitating targeted degradation was first demonstrated by its incorporation into HaloPROTACs [61]. These HaloPROTACs consisted of the VHL ligand as E3 recruiting moiety and a chloroalkane linker allowing for covalent attachment to HaloTag7 (HT7). HT7 is a modified bacterial dehalogenase that covalently binds to a chloroalkane [62]. The HaloPROTACs successfully recruited VHL to promote the degradation of ectopically expressed green fluorescent protein (GFP)-HT7 fusion protein. The most potent HaloPROTAC resulted in 90% maximum degradation (Dmax) of GFP-HT7, and displayed a low nanomolar DC50 (half-maximum degradation concentration, 19 nM) [61].
The access to all small molecule-based PROTAC compounds, with more drug-like properties, made it possible to generate highly potent cell permeable PROTACs. In 2015, the Crews laboratory, in collaboration with GSK, developed a receptor-interacting serine/threonine-protein kinase 2 (RIPK2)-targeting PROTAC (see Fig. 1c). Partially comprised of the small molecule VHL ligand, this PROTAC selectively induced RIPK2 degradation with low nanomolar cellular potency (DC50 = 1.4 nM) [12]. Further, mechanistic characterization using an in vitro ubiquitination assay demonstrated the catalytic nature of PROTACs. A negative control for the PROTAC which incorporates a stereoisomer of the VHL ligand that is unable to recruit VHL, was unable to reduce RIPK2 levels, demonstrating the E3 ligase-dependent mechanism for RIPK2 degradation. During the same timePROTACs targeting the bromodomain and extra-terminal (BRD/BET) family of epigenetic proteins using either CRBN or VHL E3 recruiting ligands were reported [63–65] (see Fig. 1c, for an example of a BRD-targeting PROTAC).
Further variations of PROTACs include, In-cell CLIck-formed Proteolysis TArgeting Chimeras (CLIPTACs) developed by Astex Pharmaceuticals [40]. CLIPTACs are PROTACs that are formed intracellularly by biocompatible reactions such as an inverse electron demand Diels–Alder reaction. Treating cells, sequentially with cell permeable tetrazine substituted thalidomide derivatives and trans-cyclo octene substituted POI ligand results in the formation of active PROTACs that successfully induced POI degradation (BRD4 and the extracellular signal–regulated kinases ERK1/2).
Altogether, these efforts exemplified the significance the PROTAC technology may impact on drug discovery.
Present
PROTAC Target Scope
To date, the most studied PROTAC targets belong to the BET [63–67] and kinase[68] families, the latter including the transmembrane RTKs [15,69] and a recent example lipid kinase, phosphatidylinositol 3-kinase (PI3K) [37]. Recent examples in addition to these protein families include, sirtuins (Sirt2) [31], histone deacetylases (HDACs) [36] and the epigenetic targets p300/CBP-associated factor (PCAF) and general control nonderepressible 5 (GCN5) [28]. During the last year, homo-PROTACs have been developed for auto-targeting of both VHL and CRBN [70,71]. Although most of the recently developed PROTACs utilize a small molecule-based E3 recruiting moiety, there are some examples that use the HIF-1α peptide as their E3 recruiting moiety for targeting proteins such as Smad3 [38], Akt [24], Tau [29,30] and Bcl-xL [25]. See figure 2, box, for summary of targets that have been explored using the PROTAC technology [12–41].
Mechanistic Considerations
There have been vast efforts made to understand the mechanism of PROTACs, and as of late the importance of ternary complex (POI-PROTAC-E3) formation has been a particular focus (Fig 3.).
Figure 3.
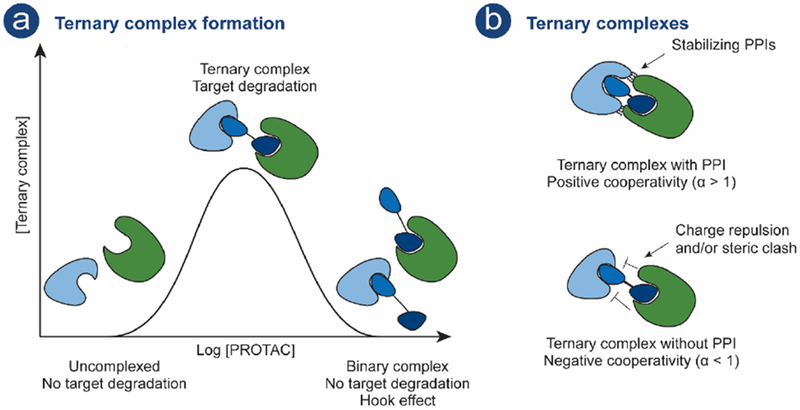
a) PROTAC-mediated ternary complex formation and Hook effect as a function of PROTAC concentration. PROTAC compound; POI ligand (royal blue), E3 ligand (dark blue) connected by a linker (black), POI (light blue), E3 (green). b) Top: Positive cooperativity is obtained when stabilizing PPIs are obtained between POI and E3. Bottom: Negative cooperativity is observed when charge repulsion and/or steric clashes is observed between POI and E3.
The mechanism of PROTAC-induced degradation is dependent on formation of a ternary complex enabling POI polyubiquitination and subsequent proteasomal degradation, as previously detailed (Fig. 1b). There are established mathematical models that describe ternary complex formation [72,73] which can be applied to the PROTAC-mediated ternary complex. These models predict a bell-shaped dependency on PROTAC concentration (Fig. 3a) [74]. For instance, at high PROTAC concentrations unproductive binary complexes may be observed and this phenomenon is referred to as the hook effect (Fig. 3a) [75]. Furthermore, favorable or repulsive interactions between the POI and E3 may affect ternary complex formation (Fig 3b). The term cooperativity (α) is used to describe these interactions where positive cooperativity (α > 1) occurs when stabilizing PPIs between the POI and E3 promote ternary complex formation. In contrast negative cooperativity (α < 1) occurs when interaction abrogate ternary complex formation. Positive cooperativity has been shown to minimize the extent of the hook effect, resulting in enhanced productive ternary complex formation [76].
The first crystal structure of a ternary complex, MZ1 (PROTAC) bound to the bromodomain of BRD4BD2 and to VHL was reported in 2017 by the Ciulli laboratory [77]. The crystal structure revealed contacts between BRD4 and VHL as well as interactions between the PROTAC linker and BRD4. Positive cooperativity, as evaluated by various biophysical methods, was shown to result in higher PROTAC potency and selectivity for induced degradation of individual BRD family members.
Recent studies have further shown that ternary complex formation might be more important in determining a productive PROTAC than the binary affinity of the POI ligand or the PROTAC for the target. A foretinib-based PROTAC, displaying low binary binding affinity towards the kinase p38α (Kd = 11 μM), could nonetheless potently induce its degradation (DC50 = 210 nM, Dmax = 91%) [78]. It was found that the PROTAC mediated a ternary complex formation with stabilizing PPIs between p38α and VHL which compensated for the low binary affinity and led to successful degradation. In line with this finding, high affinity POI ligand engagement alone was shown not to be sufficient to obtain potent PROTACs [79]. Despite incorporating a higher affinity ligand for BRD4 (I-BET726 (Kd = 4 nM)), a JQ1 (Kd = 100 nM) - containing PROTAC was more effective at promoting E3-mediated BRD4 degradation as evaluated by its ability to promote ternary complex formation through positive cooperativity [79].
There are, however, examples where cooperativity appears to be less important for efficient degradation [17,67]. Potent Bruton’s Tyrosine Kinase (BTK) (DC50 = 1–40 nM)- and BRD4 (DC50 = 5-50 nM)-targeting PROTACs, both of which recruit CRBN, showed very little to no cooperativity. Using crystal structures of PROTACs bound to both bromodomains of BRD4 (BD1 and BD2) and CRBN in combination with biochemical and cellular data, the Gray laboratory conduced investigations of the PROTAC mediated ternary complexes at a molecular level [67]. They found that different PROTAC linker lengths favored different ternary complex conformations in which BRD4 interacts with either the C-terminal or the N-terminal domain of CRBN.
Due to their catalytic nature, PROTACs have been described as “programmable essential activators” of ubiquitin ligase enzymes [80]. Programmable since PROTACs can conceivably be designed to target any POI; essential since no reaction, i.e. ubiquitination transfer, will occur in their absence; and finally, as activators since they mediate the ternary complex formation in a catalytic fashion. Hence, viewing PROTACs as activators can provide a framework for more robust PROTAC design.
In summary, the ternary complex formation is important to consider when developing PROTACs. The data discussed above suggest that determining binary POI-ligand affinity, or PROTAC affinity, alone may be insufficient to guide PROTAC design. Based on current data, the importance of cooperativity in the ternary complex formation differs between different E3s and probably also different POI, thereby making it difficult to determine general principles for ternary complex interaction on a molecular level. However, clearly PROTAC design should be aimed at obtaining a productive ternary complex.
PROTAC - Induced Degradation versus Inhibition
The PROTAC technology can provide enhanced selectivity beyond the POI ligand. Large efforts in drug discovery are directed towards identification of selective ligands. Although there are well-established strategies for the development of highly selective ligands - even for highly homologous protein families such as kinases, it still remains a major challenge [81].
Recently two lines of work highlight that the degradation MOA of PROTACs can provide enhanced selectivity among homologous targets compared to that provided by inhibition [27,78]. Both studies used the highly homologous kinase protein family for their investigations. Starting from the promiscuous kinase ligand, foretinib that binds around 130 kinases (determined by Discover X, kinome scan at 10 μM), the Crews laboratory demonstrated that the foretenib-based PROTAC has greater binary binding selectivity, as compared to foretinib and induced degradation of only a fraction of the kinases to which it binds [78]. Furthermore, depending on which E3 was recruited (VHL or CRBN) different degradation profiles were observed. The Gray laboratory also developed a promiscuous kinase-targeting PROTAC starting from a 2,4-diaminopyrimidine scaffold, which is a common motif in various kinase inhibitors [27]. Again, a difference in binary binding versus degradation profiles was observed: their 2,4-diamonpyrimidine-based PROTAC binds to 190 kinases but only induced degradation of 12 or 23 kinases in MOLM-14 or MOLT-4 cells, respectively. In summary, both studies concluded that PROTAC engagement by the POI does not correlate with degradation. Some kinases that displayed high binding affinity for the PROTAC were spared from degradation, while some low affinity binders were degraded e.g. p38α as already discussed above. Hence, these studies further demonstrate that the ternary complex formation is important to consider for PROTAC development. Enhanced degradation selectivity beyond the POI ligand was also observed when developing PROTACs targeting the TANK-binding kinase 1 (TBK1) [18]. Starting from an inhibitor that potently inhibits both TBK1 and the Inhibitor-κB kinase ε (IKKε), with IC50 of 1.3 nM and 8.7 nM, respectively, selective TBK1 PROTACs were developed. Interestingly, while the PROTAC selectively induced TBK1 degradation it retained inhibitory activity against both TBK1 and IKKε. Similar enhanced PROTAC selectivity has been observed for other targets including BRD4 [77], BCR-ABL [13] and more recent examples for PROTACs targeting HDACs and FMS Like Tyrosine kinase 3 (FLT-3) [36,69].
Additionally, PROTAC linker length, as well as linker attachment point, can affect selectivity and degradation profiles. In a recent study, ternary complex conformation was found to be PROTAC linker-dependent for CRBN and BRD4 [67]. This suggests that PROTACs with the same E3 recruiting ligand and POI ligand can provide different selectivity profiles depending on differences in linker attachment points and linker chemical composition. Further, the linker length also influences degradation profiles: an increase in linker length of 3 atoms changes the degradation profile of a lapatinib-based PROTAC from one that targets both Epidermal Growth Factor Receptor (EGFR) and Human Epidermal growth factor Receptor 2 (HER2) into one that selectively degrades EGFR while HER2 levels are unchanged [15]. The fact that PROTAC technology can add a degree of selectivity above that of the parent POI ligand/inhibitor, both to target engagement and induced degradation, further strengthens its potential as a strategy to develop novel therapeutic agents.
During the past year, further studies showed that the event-driven PROTAC MOA can evade inhibitor resistance mechanisms, including target protein overexpression and resistance mutations.
A common resistance mechanism in response to therapeutic inhibition is the mutation of the target protein, e.g. mutations close to the inhibitor binding pocket that make inhibition less effective or ineffective. This is observed, for example, when ibrutinib is used for the treatment of chronic lymphocytic leukemia (CLL) [82] ), which is often dependent on the activity of BTK. Ibrutinib is an irreversible kinase inhibitor that covalently binds to a cysteine in the ATP binding pocket of BTK. More than 80% of all CLL patients develop resistance towards ibrutinib resulting from a cysteine-to-serine mutation (C481S). However, BTK-targeting PROTACs, developed from an ibrutinib derivative unable to covalently bind to BTK, can induce the degradation of both wildtype and mutant (C418S) BTK [16,83]. Additional recent studies have also reported PROTACs that successfully target BTK [17,27].
Furthermore, PROTACs can also evade other inhibition-derived resistance mechanisms [35]. Enzalutamide is an AR inhibitor used for the treatment of prostate cancer [84]. Resistance mechanisms which arise in response to enzalutamide treatment include AR mutations that turn enzalutamide into an agonist and/or the upregulation of the endogenous androgen ligand. Enzalutamide-derived PROTACs recruiting the VHL E3 can successfully induce degradation of various AR mutants for which inhibition is no longer efficacious, highlighting the event driven nature of these compounds. Additionally, these PROTACs were shown to outperform enzalutamide in antiproliferative and pro-apoptotic activity in AR overexpressing cells in the presence of synthetic androgen (R1881) [35].
Targeting kinases with PROTACs enables modulation of both scaffolding and enzymatic functions as compared to occupancy driven pharmacology that only affects enzymatic function [81]. This was demonstrated in a recent study, PROTACs targeting RTKs were able to target scaffolding functions as well as induce degradation of commonly observed RTK mutations that are resistant to kinase inhibitors [15]. PROTACs could successfully induce degradation of EGFR, HER2 and tyrosine-protein kinase Met (c-Met) including mutants of EGFR and c-Met. Depletion of these RTKs showed that downstream signaling was halted. Moreover, PROTAC-induced degradation showed a delay in kinome rewiring as compared to inhibition. This example also demonstrated for the first time that PROTACs are capable of inducing the degradation of transmembrane receptors. Similarly, PROTAC induced degradation of focal adhesion kinase (Fak) has also been demonstrated to affect scaffolding functions of Fak [85]. Induced degradation of Fak was shown to outcompete kinase activity inhibition both regarding Fak signaling as well as cell migration and invasion in human triple-negative breast cancer cells.
Also, a PROTAC targeting both PCAF and GCN5, which are closely related epigenetic proteins, was able to potently modulate expression of multiple inflammatory mediators in LPS-stimulated macrophages and dendric cells [28]. In contrast, inhibition of the bromodomain of PCAF and GCN5 with the POI ligand itself was insufficient to modulate the immunomodulatory function of these proteins.
To summarize, the PROTAC technology can escape resistance mechanisms that are difficult or impossible to modulate using the inhibitor MOA by offering means to modulate both enzymatic and non-enzymatic roles of proteins.
Present and Future Perspectives - Advancing the PROTAC Technology
Optimizing PROTAC Design, Synthesis and Evaluation
In order to accelerate and streamline PROTAC discovery, it is necessary to uncover PROTAC discovery principles and hence establish robust evaluation platforms [8] (Fig. 4a). To date, most of the reported PROTACs have been developed starting from a well-characterized ligand, often an inhibitor, as the POI recruiting moiety. The crystal structure of the POI ligand bound to its target and/or available POI ligand structure activity relationship (SAR) information has been used to guide PROTAC design, e.g. to identify linker attachment point on the POI ligand. In a recent example, PROTACs targeting the putative transcription factor regulator, pirin, were developed by using the physicochemical properties of the compounds to guide the PROTAC optimization. In only three iterations, selective and potent pirin-targeting PROTACs were obtained [32]. Computational protein-protein docking has been employed for rational design of highly a selective BRD4-targeting PROTAC [67].
Figure 4.
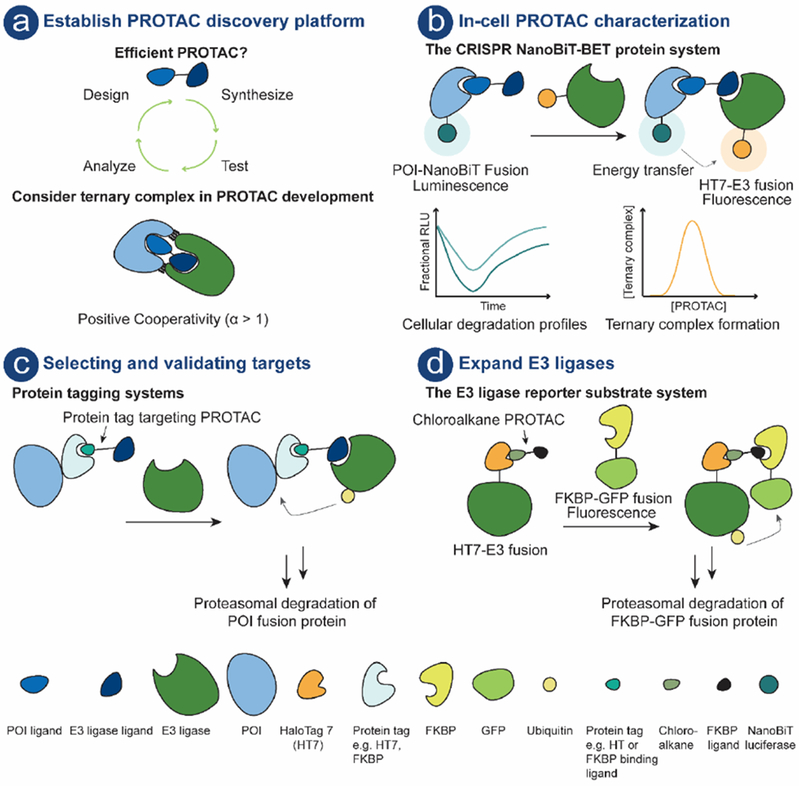
a) PROTAC compound; POI ligand (royal blue), E3 ligand (dark blue) connected by a linker (black). PROTAC platform with iterative design, synthesis, test and analyze cycle. b) Schematic illustration of the CRISPR NanoBiT-BET protein system [86]. This system enables PROTAC characterization in live-cells using endogenous protein levels. c) Protein tagging systems can be exploited for target selection and/or validation. The POI is fused to e.g. HT7 [61] or FKBP[87] and PROTAC mediated ternary complex formation results in polyubiquitination of the POI followed by proteasomal degradation. d) System for assessing E3 utility in the PROTAC technology [88]. Schematic illustration of the E3 reporter substrate system. E3 is fused to HT7, chloroalkane PROTAC covalently binds to HT7, and the recruitment of the reporter substrate (FKBP-GFP fusion protein) results in ternary complex, polyubiquitination and subsequent proteasomal degradation of the reporter substrate.
Furthermore, PROTACs are often screened for activity by measuring protein levels using end-point techniques – primarily immunoblotting and more recently mass spectroscopy. These techniques are however, currently not amenable for high-throughput screeningand provide limited information to guide PROTAC structural optimization. As a result, the design and optimization of PROTACs are largely empirical making the iterative design, synthesis, test and analyze cycle (Fig. 4a) of PROTACs very laborious and time consuming.
Ternary complex binding data as well as ternary complex structural information would provide valuable information, given its recognized importance for determining PROTAC efficacy. Currently, there are only a few crystal structures reported of a PROTAC mediated ternary complex [67,77]. Methods to study ternary complex include Time-Resolved Fluorescence Energy Transfer (TR-FRET), AlphaLISA, Surface Plasmon Resonance (SPR) and Isothermal Titration Calorimetry (ITC) [74,89]. Although useful for ternary complex formation studies these methods do not recapitulate the full ubiquitin–proteasome system required for POI degradation.
In recent work by Promega, a real time live-cell system for PROTAC evaluation that enables both characterization of efficacy and MOA has been developed (Fig. 4b) [86]. The system combines CRISPR/Cas9 endogenous tagging and luminescent technology to kinetically measure target protein levels in live-cells. Furthermore, when this technology was combined with Bioluminescence Resonance Energy Transfer (NanoBRET), it enabled kinetic measurements of intracellular protein interactions along the degradation pathway such as ternary complex formation, ubiquitination, and PROTAC target engagement. This system may be valuable in particular when investigating non-traditional drug targets that lack enzymatic activity and/or down-stream signaling.
Expanding the E3 Space
There are more than 600 E3s predicted in the human genome but as of yet, only a few have been validated or exploited for PROTAC development [90]: specifically VHL, CRBN, MDM2, and cIAP1, which all have small molecule ligands, have been the E3 of choice. Peptide ligands for E3s have also been used with some success – the very first PROTAC recruited ß-TRCP by use of a peptide ligand; and a more recent PROTAC that contains a Keap1-binding peptide was able to recruit the Keap1 E3 to successfully induce degradation of the Tau protein [30,33]. Although these E3s have proven successful, some of them have limitations that need to be considered during PROTAC development. First, the originally used cIAP1 ligands suffered from specificity issues and caused cIAP1 degradation which made the utility of this E3 somewhat self-limiting [91,92]. Fortunately, more recent reports suggest that careful selection of the ligand for recruiting cIAP1 can largely circumvent autoubiquitination and hence self-degradation of cIAP1 [93]. Recently it was also reported that PROTACs that recruit the CRBN E3 can simultaneously mediate recruitment of neosubstrates, as similarly observed with IMiDs [94,95]. The simultaneous recruitment of neo-substrates may limit its utility in the PROTAC technology. Consequently, it is important to control for potential CRBN-mediated off-targets effects. Immunoblotting and negative control compounds, unable to recruit the POI, have been previously been employed. [16][28]. It would however be more thorough to perform global proteomic studies to control for yet unknown neo-substrates. The inherent activity of the E3 ligand can however be exploited in the PROTAC design, as illustrated in a recent example [96]. A BRD4-targeting PROTAC utilizing a nutlin derivate as E3 ligand, hence recruiting MDM2, showed dual MOA - induced degradation of BRD4 as well as upregulation of the tumor suppressor p53, providing a synergistic antiproliferative effect in several cancer cell lines. This example also illustrates that MDM2 can be utilized in the PROTAC technology to provide PROTACs with low nanomolar potency.
To further advance the PROTAC technology, efforts to identify additional E3s that can be utilized are needed. A cell-based E3 reporter system to evaluate which E3s can be recruited for induced protein degradation (Fig. 4d) was recently reported [88]. Six different E3s, representing the three major E3 classes were investigated. These were fused to HT7, and a FKBPF36V-GFP fusion was used as a degradation reporter substrate for a FK506 “bump” ligand-based HaloPROTAC. Two out of six E3s (Parkin and β-TRCP) showed potential for effective PROTAC recruitment. As previously mentioned, β-TRCP has been used as an recruiting E3 [33,34] but Parkin offers a novel example that could be exploited as a potential recruiting E3.
Selecting and Validating Targets
Target selection is one of the most important decisions in a drug discovery program [97]. Furthermore, there is a high probability that not all biological targets will be receptive to protein degradation and, as discussed above, PROTAC development can be a very time-consuming process. Therefore, early validation of selected POI targets would be very valuable.
In a recent paper, a promiscuous kinase-targeting PROTAC was used in a chemoproteomic approach to evaluate degradability of kinases [27]. Several well-characterized kinase therapeutic targets were identified as sensitive to PROTAC-induced degradation. Similar studies may be useful for other protein families before initiating PROTAC development. Furthermore, protein labeling systems including HaloTag (HT) [98,99] and His[100] tagged POI can also be utilized for determining target degradability and concomitantly enable target validation (Fig. 4c). This system has been employed with HaloPROTACs recruiting either VHL [61] or cIAP1 [98,99]. Currently, Promega offers around 9000 commercially available plasmids for HT-POI fusion proteins making this system readily accessible [101]. However, it should be noted that this system provides overexpressed levels of HT-POI which has been shown to affects normal protein turnover and homeostasis [86]. This may be overcome using CRISPR/Cas9 genome editing to obtain endogenous levels of HT tagged POIs. This has been done in a recent example, there CRISPR-mediated knock-in of FKBP12F36V provided endogenous expressed FKBP12F36V-POI fusion protein [87]. This system known as dTAG (Fig. 4c) can be used for early target validation. It is a cell-based system that enables immediate and selective control of POIs using CRBN-recruiting PROTACs. The dTAG approach was successfully employed to different FKBP12F36V tagged POIs including, BRD4, MYC and KRas. Additionally, it has been used in a separate study to evaluate PROTAC cell permeability, which is often a concern given their high molecular weight [27].
Targeting the Undruggable Proteome - Chemically Convert Binders to Potent PROTACs
So far most of the developed PROTACs target proteins are nonetheless part of the druggable proteome. The PROTAC technology has the potential to develop molecules capable of modulating challenging non-traditional drug targets. Since PROTAC technology only requires binders that temporarily mediate ternary complex formation, low affinity POI ligands can be incorporated into a PROTAC. This was recently demonstrated with the p38α targeting PROTAC, already discussed above, which displayed low affinity (11 μM) but could potently induce degradation of p38α (DC50 = 210 nM, Dmax = 91%). This opens possibilities to explore targets which have been proven difficult to access using traditional, occupancy-based MOA therapeutics.
An illustrative example of converting a binder into a PROTAC is the development of pirin-targeting PROTACs [32]. Pirin belongs to the cupin superfamily of proteins and has no known enzymatic activity or known endogenous ligand. To further study this protein, a PROTAC was developed starting from a pirin ligand discovered from a cell-based phenotypic screen. The PROTAC could successfully induce pirin degradation and competition with the PROTAC and the pirin ligand confirmed binding of the ligand to pirin. The PROTAC and small molecule probes offer chemical tools to further study the unexplored pirin protein. This strategy could be useful for investigating uncharacterized targets in cases were traditional tools are unsuccessful.
Progression to the Clinic
As mentioned above the drug class space has expanded to include alternative modalities. Even though the number of biologies, such as replacement proteins (e.g. insulin) or monoclonal antibodies (e.g. trastuzumab) [102], reaching the clinic have increased during recent years, the small molecule-based drugs will continue to contribute the most to the drug space [103]. An advantage with the small molecule-based PROTAC technology in contrast to biologies, is the cost of manufacturing. Small molecule-based drugs cost ~USD 700 per patient per year as compared to ~USD 15000 for biologies [66]. The cost of administration of a PROTAC may be even less than that of traditional small molecule drugs because of the catalytic MOA of the former, which may require lower or less frequent drug doses. Furthermore, an additional advantage of small molecule-based drugs include their cell permeability, hence their ability to modulate intracellular targets.
In the early stages of PROTAC development, their oral availability was questioned due to their non-adherence to the Lipinski’s Rule of Five [104]. In particular the high molecular weight of PROTACs was a concern regarding permeability. General principles for design of so-called beyond Rule of Five (bRo5) compounds are however evolving [105,106]. In fact, in 2017, Arvinas reported the first orally available PROTAC targeting AR [107]. PROTAC induced AR degradation was showed to be superior to inhibition in mouse xenograft studies, there 90% AR degradation was observed at a 1 mg/kg PO QD dose.
As already suggested, chemically converting failed small molecule-based drugs into PROTACs could give fast access to new therapeutics [8]. Abandoned drug discovery programs where high efficacy could not be reached using the inhibitor MOA may be especially attractive to explore, which is the major reason for failure in clinical development phases [108].
Conclusion
Drug discovery is expanding its repertoire and the PROTAC technology shows great potential as a new modality using an alternative MOA. Reports discussed here corroborate that the event-driven MOA exhibited by PROTACs offers several advantages over the occupancy-driven MOA. Key advancements from the last year include enhanced binding and degradation selectivity profiles towards homologous protein families and the ability to circumvent routine inhibition-derived resistance mechanisms. Additionally, the PROTAC technology shows potential for modulating targets where the inhibitor MOA is unsuccessful. Examples include PROTACs that target PCAF and GCN5 [28] , where inhibitor modulation has no effect, and where a binder was chemically converted to a PROTAC targeting pirin [32] underscores the PROTAC technology applicability. Furthermore, PROTAC development platforms are emerging that include methods for early target validation and systems that allow in cell characterization of ternary complex formation as well as POI degradation (Fig 4). It is clear that, established technologies for inhibitor discovery can be employed but should include ternary complex formation characterization as early as possible to guide PROTAC optimization. Since low affinity POI ligands can be utilized in PROTAC design, methodologies in fragment-based drug design (FBDD)[109] optimized for identification of low affinity ligands, in combination with ternary complex formation assays, may be useful for targeting non-traditional drug targets; especially for those were inhibition have been unsuccessful. Further, development of suitable computational tools that provide high throughput in silico characterization of ternary complexes would be useful for informing PROTAC design.
Since it has been demonstrated that the choice of E3 affects PROTAC degradation profile, efforts to expand the number of E3s that can be utilized are needed. Also, the characterization of tissue- and/or disease-specific E3s could potentially be utilized to further improve selectivity by developing PROTACs that are only active in a specific context, similar to phosphoPROTACs [41].
Further studies are needed to determine PROTAC efficiency in vivo, but as the first PROTACs are reaching clinical trials, this information will soon be available. It is an exciting time for the PROTAC technology and the knowledge about this highly promising strategy will improve over time, as PROTAC development principles are established and more POIs as well as E3s are explored.
Acknowledgement
We thank Doris Hellerschmied-Jelinek, Shanique Alabi, Stacey-Lynn Paiva, Daniel McQuaid, John Hines and George Burslem for helpful discussions and reading of the manuscript. MP gratefully acknowledges the Swedish Research Council (Vetenskapsrådet) for international postdoctoral funding (2016-00294). CMC gratefully acknowledges NIH (grant R35CA197589) for funding.
Biographies
Mariell Pettersson Biography
Dr. Mariell Pettersson is a postdoctoral fellow in Prof. Craig Crews’ group at Yale University funded by the Swedish Research Council. She obtained her PhD in 2015 from Gothenburg University (Gothenburg, Sweden). In her graduate work, in Prof. Morten Grøtlis group, she studied small-molecule and peptide-based inhibitors for disrupting the MDM2/p53 protein-protein interaction. Her current research is focused on expanding the PROTAC technology to target proteins that cannot be modulated by the traditional inhibition approach. Before joining Prof. Crews’ laboratory she was a senior scientist at a Sprint Bioscience AB (Stockholm, Sweden).
Craig Crews Biography
Prof. Craig Crews is the Lewis Cullman Professor of Molecular, Cellular and Developmental Biology and holds joint appointments in the departments of Chemistry and Pharmacology at Yale University. He graduated from the U. Virginia with a B.A. in Chemistry and received his Ph.D. from Harvard University in Biochemistry. On the faculty at Yale since 1995, his laboratory pioneered the use of small-molecules to control intracellular protein levels. In 2003, he co-founded Proteolix, whose proteasome inhibitor, Kyprolis™ received FDA approval for the treatment of multiple myeloma. Since Proteolix’s purchase by Onyx Pharmaceuticals in 2009, Prof. Crews has focused on a new ‘induced protein degradation’ drug development technology, PROTACs, which served as the founding IP for the New Haven-based biotech venture, Arvinas, Inc. In 2018 he received the Khorana Prize from the Royal Society of Chemistry, the 2018 Pierre Fabre Award for Therapeutic Innovation and was named an American Cancer Society Professor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
C.M.C. is founder, shareholder and consultant to Arvinas, Inc. In addition, his lab receives sponsored research support from Arvinas.
References
- 1.Eric V et al. (2017) New Modalities for Challenging Targets in Drug Discovery. Angew. Chem., Int. Ed. 56, 10294–10323 [DOI] [PubMed] [Google Scholar]
- 2.Hopkins AL and Groom CR (2002) The druggable genome. Nat. Rev. Drug Discov. 1, 727–730 [DOI] [PubMed] [Google Scholar]
- 3.Valeur E and Jimonet P (2018) New Modalities, Technologies, and Partnerships in Probe and Lead Generation: Enabling a Mode-of-Action Centric Paradigm. J. Med. Chem 61, 9004–9029 [DOI] [PubMed] [Google Scholar]
- 4.Fuller JC et al. (2009) Predicting druggable binding sites at the protein–protein interface. Drug Discov. Today 14, 155–161 [DOI] [PubMed] [Google Scholar]
- 5.Adjei AA (2006) What Is the Right Dose? The Elusive Optimal Biologic Dose in Phase I Clinical Trials. J. Clin. Oncol 24, 4054–4055 [DOI] [PubMed] [Google Scholar]
- 6.Holohan C et al. (2013) Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 13, 714–726 [DOI] [PubMed] [Google Scholar]
- 7.Blair JMA et al. (2014) Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42–51 [DOI] [PubMed] [Google Scholar]
- 8.Churcher I (2018) Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones? J. Med. Chem 61, 444–452 [DOI] [PubMed] [Google Scholar]
- 9.Coleman KG and Crews CM (2018) Proteolysis-Targeting Chimeras: Harnessing the Ubiquitin-Proteasome System to Induce Degradation of Specific Target Proteins. Annu. Rev. Cancer Biol 2, 41–58 [Google Scholar]
- 10.Hochstrasser M (1995) Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr. Opin. Cell. Biol 7, 215–223 [DOI] [PubMed] [Google Scholar]
- 11.Navon A and Ciechanover A (2009) The 26 S Proteasome: From Basic Mechanisms to Drug Targeting. J. Biol. Chem 284, 33713–33718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bondeson DP et al. (2015) Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol 11, 611–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai AC et al. (2016) Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem., Int. Ed 55, 807–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demizu Y et al. (2016) Development of BCR-ABL degradation inducers via the conjugation of an imatinib derivative and a cIAP1 ligand. Bioorg. Med. Chem. Lett 26, 4865–4869 [DOI] [PubMed] [Google Scholar]
- 15.Burslem GM et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 25, 67–77.e63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buhimschi AD et al. (2018) Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 57, 3564–3575 [DOI] [PubMed] [Google Scholar]
- 17.Zorba A et al. (2018) Delineating the role of cooperativity in the design of potent PROTACs for BTK. PNAS 115, E7285–E7292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crew AP et al. (2018) Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J. Med. Chem 61, 583–598 [DOI] [PubMed] [Google Scholar]
- 19.Olson CM et al. (2017) Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol 14, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robb CM et al. (2017) Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem. Commun. 53, 7577–7580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bian J et al. (2018) Discovery of Wogonin-based PROTACs against CDK9 and capable of achieving antitumor activity. Bioorg. Chem. 81, 373–381 [DOI] [PubMed] [Google Scholar]
- 22.Zhang C et al. (2018) Proteolysis Targeting Chimeras (PROTACs) of Anaplastic Lymphoma Kinase (ALK). Eur. J. Med. Chem. 151, 304–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang CH et al. (2018) Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC). Biochem. Biophys. Res. Commun. 505, 542–547 [DOI] [PubMed] [Google Scholar]
- 24.Henning RK et al. (2016) Degradation of Akt using protein-catalyzed capture agents. J. Pept. Sci 22, 196–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng J et al. (2016) Proteolysis targeting peptide (PROTAP) strategy for protein ubiquitination and degradation. Biochem. Biophys. Res. Commun 470, 936–940 [DOI] [PubMed] [Google Scholar]
- 26.Chen H et al. (2018) Chemically induced degradation of CK2 by proteolysis targeting chimeras based on a ubiquitin–proteasome pathway. Bioorg. Chem 81, 536–544 [DOI] [PubMed] [Google Scholar]
- 27.Huang H-T et al. (2018) A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader. Cell Chem. Biol 25, 88–99.e86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bassi ZI et al. (2018) Modulating PCAF/GCN5 Immune Cell Function through a PROTAC Approach. ACS Chem. Biol 13, 2862–2867 [DOI] [PubMed] [Google Scholar]
- 29.Chu T-T et al. (2016) Specific Knockdown of Endogenous Tau Protein by Peptide-Directed Ubiquitin-Proteasome Degradation. Cell Chem. Biol 23, 453–461 [DOI] [PubMed] [Google Scholar]
- 30.Lu M et al. (2018) Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur. J. Med. Chem 146, 251–259 [DOI] [PubMed] [Google Scholar]
- 31.Schiedel M et al. (2018) Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem 61, 482–491 [DOI] [PubMed] [Google Scholar]
- 32.Chessum NEA et al. (2018) Demonstrating In-Cell Target Engagement Using a Pirin Protein Degradation Probe (CCT367766). J. Med. Chem 61, 918–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakamoto KM et al. (2001) Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. PNAS 98, 8554–8559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakamoto KM et al. (2003) Development of Protacs to Target Cancer-promoting Proteins for Ubiquitination and Degradation. Mol. Cell. Proteomics 2, 1350–1358 [DOI] [PubMed] [Google Scholar]
- 35.Salami J et al. (2018) Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol 1, 100–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang K et al. (2018) Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg. Med. Chem. Lett 28, 2493–2497 [DOI] [PubMed] [Google Scholar]
- 37.Li W et al. (2018) Phthalimide conjugations for the degradation of oncogenic PI3K. Eur. J. Med. Chem. 151, 237–247 [DOI] [PubMed] [Google Scholar]
- 38.Wang X et al. (2016) New strategy for renal fibrosis: Targeting Smad3 proteins for ubiquitination and degradation. Biochem. Pharmacol 116, 200–209 [DOI] [PubMed] [Google Scholar]
- 39.Lee H et al. (2007) Targeted Degradation of the Aryl Hydrocarbon Receptor by the PROTAC Approach: A Useful Chemical Genetic Tool. Chembiochem 8, 2058–2062 [DOI] [PubMed] [Google Scholar]
- 40.Lebraud H et al. (2016) Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci 2, 927–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hines J et al. (2013) Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. PNAS 110, 8942–8947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schneekloth JS et al. (2004) Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation. J. Am. Chem. Soc 126, 3748–3754 [DOI] [PubMed] [Google Scholar]
- 43.Schneekloth AR et al. (2008) Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett 18, 5904–5908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vassilev LT et al. (2004) In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 303, 844–848 [DOI] [PubMed] [Google Scholar]
- 45.Sekine K et al. (2008) Small Molecules Destabilize cIAP1 by Activating Auto-ubiquitylation. J. Biol. Chem 283, 8961–8968 [DOI] [PubMed] [Google Scholar]
- 46.Itoh Y et al. (2010) Protein Knockdown Using Methyl Bestatin–Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J. Am. Chem. Soc 132, 5820–5826 [DOI] [PubMed] [Google Scholar]
- 47.Buckley DL et al. (2012) Small-Molecule Inhibitors of the Interaction between the E3 Ligase VHL and HIF1α. Angew. Chem., Int. Ed 51, 11463–11467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buckley DL et al. (2012) Targeting the von Hippel–Lindau E3 Ubiquitin Ligase Using Small Molecules To Disrupt the VHL/HIF-1α Interaction. J. Am. Chem. Soc 134, 4465–4468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Molle I et al. (2012) Dissecting Fragment-Based Lead Discovery at the von Hippel-Lindau Protein:Hypoxia Inducible Factor 1α Protein-Protein Interface. Chem. Biol 19, 1300–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Testa A et al. (2018) 3-Fluoro-4-hydroxyprolines: Synthesis, Conformational Analysis, and Stereoselective Recognition by the VHL E3 Ubiquitin Ligase for Targeted Protein Degradation. J. Am. Chem. Soc 140, 9299–9313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soares P et al. (2018) Thioamide substitution to probe the hydroxyproline recognition of VHL ligands. Bioorg. Med. Chem 26, 2992–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soares P et al. (2018) Group-Based Optimization of Potent and Cell-Active Inhibitors of the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase: Structure–Activity Relationships Leading to the Chemical Probe (2S,4R)-1-((S)-2-(1-Cyanocyclopropanecarboxamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (VH298). J. Med. Chem. 61, 599–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frost J et al. (2016) Potent and selective chemical probe of hypoxic signalling downstream of HIF-α hydroxylation via VHL inhibition. Nat. Commun 7, 13312–13323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ito T et al. (2010) Identification of a Primary Target of Thalidomide Teratogenicity. Science 327, 1345–1350 [DOI] [PubMed] [Google Scholar]
- 55.Lopez-Girona A et al. (2012) Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 26, 2326–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fischer ES et al. (2014) Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krönke J et al. (2014) Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 343, 301–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petzold G et al. (2016) Structural basis of lenalidomide-induced CK1α degradation by the CRL4CRBN ubiquitin ligase. Nature 532, 127–130 [DOI] [PubMed] [Google Scholar]
- 59.Krönke J et al. (2015) Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 523, 183–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gandhi AK et al. (2014) Immunomodulatory agents lenalidomide and pomalidomide costimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4CRBN. Br. J. Haematol 164, 811–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buckley DL et al. (2015) HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 10, 1831–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ohana RF et al. (2009) HaloTag7: A genetically engineered tag that enhances bacterial expression of soluble proteins and improves protein purification. Protein Expr. Purif. 68, 110–120 [DOI] [PubMed] [Google Scholar]
- 63.Lu J et al. (2015) Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 22, 755–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Winter GE et al. (2015) Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zengerle M et al. (2015) Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol 10, 1770–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou B et al. (2018) Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J. Med. Chem 61, 462–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nowak RP et al. (2018) Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol 14, 706–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tan L and Gray NS (2018) When Kinases Meet PROTACs. Chin. J. Chem 36, 971–977 [Google Scholar]
- 69.Burslem GM et al. (2018) Enhancing Antiproliferative Activity and Selectivity of a FLT-3 Inhibitor by Proteolysis Targeting Chimera Conversion. J. Am. Chem. Soc 140, 16428–16432 [DOI] [PubMed] [Google Scholar]
- 70.Maniaci C et al. (2017) Homo-PROTACs: bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat. Commun 8, 830–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Steinebach C et al. (2018) Homo-PROTACs for the Chemical Knockdown of Cereblon. ACS Chem. Biol. 13, 2771–2782 [DOI] [PubMed] [Google Scholar]
- 72.Douglass EF et al. (2013) A Comprehensive Mathematical Model for Three-Body Binding Equilibria. J. Am. Chem. Soc 135, 6092–6099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lu C and Wang Z-X (2017) Quantitative Analysis of Ligand Induced Heterodimerization of Two Distinct Receptors. Anal. Chem 89, 6926–6930 [DOI] [PubMed] [Google Scholar]
- 74.Hughes Scott J. and Ciulli A. (2017) Molecular recognition of ternary complexes: a new dimension in the structure-guided design of chemical degraders. Essays Biochem. 61, 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Miles LEM (1975) Properties, variants, and applications of the immunoradiometric assay method. Res. Clin. Lab 5, 59–72 [DOI] [PubMed] [Google Scholar]
- 76.Roy RD et al. (2017) Cooperative binding mitigates the high-dose hook effect. BMC Syst. Biol 11, 74–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gadd MS et al. (2017) Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 13, 514–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bondeson DP et al. (2018) Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol 25, 78–87.e75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chan K-H et al. (2018) Impact of Target Warhead and Linkage Vector on Inducing Protein Degradation: Comparison of Bromodomain and Extra-Terminal (BET) Degraders Derived from Triazolodiazepine (JQ1) and Tetrahydroquinoline (I-BET726) BET Inhibitor Scaffolds. J. Med. Chem 61, 504–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fisher SL and Phillips AJ (2018) Targeted protein degradation and the enzymology of degraders. Curr. Opin. Chem. Biol 44, 47–55 [DOI] [PubMed] [Google Scholar]
- 81.Ferguson FM and Gray NS (2018) Kinase inhibitors: the road ahead. Nat. Rev. Drug Discov 17, 353–377 [DOI] [PubMed] [Google Scholar]
- 82.Woyach JA et al. (2014) Resistance Mechanisms for the Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. New. Engl. J. Med 370, 2286–2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sun Y et al. (2018) PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 28, 779–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sanford M (2013) Enzalutamide: A Review of Its Use in Metastatic, Castration-Resistant Prostate Cancer. Drugs 73, 1723–1732 [DOI] [PubMed] [Google Scholar]
- 85.Cromm PM et al. (2018) Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J. Am. Chem. Soc 140, 17019–17026 [DOI] [PubMed] [Google Scholar]
- 86.Riching KM et al. (2018) Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol 13, 2758–2770 [DOI] [PubMed] [Google Scholar]
- 87.Nabet B et al. (2018) The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol 14, 431–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ottis P et al. (2017) Assessing Different E3 Ligases for Small Molecule Induced Protein Ubiquitination and Degradation. ACS Chem. Biol 12, 2570–2578 [DOI] [PubMed] [Google Scholar]
- 89.Roy MJ et al. (2018) SPR-measured kinetics of PROTAC ternary complexes influence target degradation rate. bioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Clague MJ et al. (2015) The demographics of the ubiquitin system. Trends Cell Biol. 25, 417–426 [DOI] [PubMed] [Google Scholar]
- 91.Fulda S and Vucic D (2012) Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov 11, 109–124 [DOI] [PubMed] [Google Scholar]
- 92.Varfolomeev E et al. (2007) IAP Antagonists Induce Autoubiquitination of c-IAPs, NF-κB Activation, and TNFα-Dependent Apoptosis. Cell 131, 669–681 [DOI] [PubMed] [Google Scholar]
- 93.Ohoka N et al. (2017) In Vivo Knockdown of Pathogenic Proteins via Specific and Nongenetic Inhibitor of Apoptosis Protein (IAP)-dependent Protein Erasers (SNIPERs). J. Biol. Chem 292, 4556–4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ishoey M et al. (2018) Translation Termination Factor GSPT1 Is a Phenotypically Relevant Off-Target of Heterobifunctional Phthalimide Degraders. ACS Chem. Biol 13, 553–560 [DOI] [PubMed] [Google Scholar]
- 95.Matyskiela ME et al. (2018) SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Biol 14, 981–987 [DOI] [PubMed] [Google Scholar]
- 96.Hines J et al. (2018) MDM2-recruiting PROTAC Offers Superior, Synergistic Anti-proliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of p53. Cancer Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bunnage ME et al. (2015) Know your target, know your molecule. Nat. Chem. Biol 11, 368–372 [DOI] [PubMed] [Google Scholar]
- 98.Tomoshige S et al. (2015) Degradation of HaloTag-fused nuclear proteins using bestatin-HaloTag ligand hybrid molecules. Org. Biomol. Chem 13, 9746–9750 [DOI] [PubMed] [Google Scholar]
- 99.Tomoshige S et al. (2016) Efficient protein knockdown of HaloTag-fused proteins using hybrid molecules consisting of IAP antagonist and HaloTag ligand. Bioorg. Med. Chem 24, 3144–3148 [DOI] [PubMed] [Google Scholar]
- 100.Okitsu K et al. (2018) Development of a Small Hybrid Molecule That Mediates Degradation of His-Tag Fused Proteins. J. Med. Chem 61, 576–582 [DOI] [PubMed] [Google Scholar]
- 101.Promega. https://www.promega.com/products/pm/halotag-technology/kazusa-collection/?activeTab=1
- 102.Andrews L et al. (2015) A snapshot of biologic drug development:Challenges and opportunities. Human & Experimental Toxicology 34, 1279–1285 [DOI] [PubMed] [Google Scholar]
- 103.Campbell IB et al. (2018) Medicinal chemistry in drug discovery in big pharma: past, present and future. Drug Discov. Today 23, 219–234 [DOI] [PubMed] [Google Scholar]
- 104.Lipinski CA et al. (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews 23, 3–25 [DOI] [PubMed] [Google Scholar]
- 105.Doak BC et al. (2016) How Beyond Rule of 5 Drugs and Clinical Candidates Bind to Their Targets. J. Med. Chem 59, 2312–2327 [DOI] [PubMed] [Google Scholar]
- 106.DeGoey DA et al. (2018) Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection. J. Med. Chem 61, 2636–2651 [DOI] [PubMed] [Google Scholar]
- 107.Neklesa TK et al. (2017) An oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol. 35, 273–273 [Google Scholar]
- 108.Arrowsmith J and Miller P (2013) Phase II and Phase III attrition rates 2011–2012. Nat. Rev. Drug Discov 12, 569. [DOI] [PubMed] [Google Scholar]
- 109.Erlanson DA et al. (2016) Twenty years on: the impact of fragments on drug discovery. Nat. Rev. Drug Discov 15, 605–619 [DOI] [PubMed] [Google Scholar]