

Abstract
We constructed a series of chimeric and mutant neuronal nicotinic acetylcholine receptor β subunits to map amino acid residues that determine sensitivity to competitive antagonists. The β2 and β4 subunits form pharmacologically distinct receptors when expressed in combination with the α3 subunit in Xenopus oocytes. At equipotent acetylcholine concentrations, α3β2 is 56-fold more sensitive to blockade by dihydro-β-erythroidine than is α3β4. The α3β2 combination is also sensitive to long-term blockade by neuronal bungarotoxin, whereas α3β4 is not. Pharmacological analysis of receptors formed by chimeric β subunits reveals that amino acid residues that determine both dihydro-β-erythroidine and neuronal bungarotoxin sensitivity are located within several sequence segments. The major determinant of sensitivity to both competitive antagonists is located between residues 54 and 63. A minor determinant of sensitivity to both antagonists lies between residues 1 and 54, whereas a minor determinant of NBT sensitivity lies between residues 74 and 80. Within region 54–63 of β2, mutant β2 subunits were used to identify threonine 59 as a residue critical in determining competitive antagonist sensitivity. Changing threonine 59 to lysine, as occurs in β4, causes a 9-fold decrease in dihydro-β-erythroidine sensitivity and a 71-fold decrease in neuronal bungarotoxin sensitivity. Changing polar threonine 59 to negatively charged aspartate causes a 2.5-fold increase in neuronal bungarotoxin sensitivity and has no effect on dihydro-β-erythroidine sensitivity.
Keywords: nicotinic receptor, neuronal, antagonists, mutant, chimera, neuronal bungarotoxin, dihydro-β-erythroidine
Nicotinic acetylcholine receptors (nAChRs) are found throughout the central and peripheral nervous systems, with 11 distinct genes encoding neuronal nAChR subunits (α2–α9, β2–β4) currently identified (Sargent, 1993; Elgoyhen et al., 1994). Functional neuronal nAChRs can be formed in Xenopusoocytes by expression of various combinations of these subunits (Duvoisin et al., 1989; Papke et al., 1989; Luetje et al., 1990b;Luetje and Patrick, 1991). Although these neuronal nAChR subunits are homologous with one another, each functional subunit combination is pharmacologically distinct. This may account for the diversity of neuronal nAChRs observed in vivo (Luetje et al., 1990a;Role, 1992; Sargent, 1993).
Identification of amino acid residues that are involved in forming the ligand-binding sites of nAChRs is essential to understanding how these receptors function. Affinity labeling experiments have identified several critical amino acid residues of the muscle-type α subunit (Kao et al., 1984; Dennis et al., 1988; Abramson et al., 1989; Galzi et al., 1990; Middleton and Cohen, 1991). Non-α subunits are also involved in forming the ligand-binding site. The ligand-binding sites of muscle nAChRs appear to be located at the interface between the α and γ subunits and between the α and δ subunits (Blount and Merlie, 1989; Pederson and Cohen, 1990; Czajkowski and Karlin, 1991;Middleton and Cohen, 1991). The ligand-binding sites of neuronal nAChRs appear to be formed in a similar manner, because both α and β subunits influence the pharmacological properties of these receptors (Luetje and Patrick, 1991). The residues identified by affinity labeling experiments, using Torpedo electric organ nAChRs, are highly conserved among muscle and neuronal nAChR subunits. Thus, these residues may form parts of the ligand-binding site common to all nAChRs, but cannot be responsible for the pharmacological diversity observed among nAChR subtypes.
An approach to identification of the amino acid residues of receptor subunits that confer differential pharmacological properties is to construct chimeras of pharmacologically distinct subunits. This approach has been used to identify several sequence segments of neuronal nAChR α subunits that affect sensitivity to agonists and the competitive antagonist neuronal bungarotoxin (NBT) (Luetje et al., 1993). Chimeric subunits have been used to identify regions of β2 and β4 that determine sensitivity to agonists (Figl et al., 1992; Cohen et al., 1995). This technique has also been used to localize the β subunit contribution to NBT sensitivity to the N-terminal 119 (Papke et al., 1993) or 80 (Wheeler et al., 1993) residues of β2.
We constructed a series of chimeric β subunits to more precisely identify regions of β subunits that determine competitive antagonist sensitivity. We used the structurally distinct competitive antagonists dihydro-β-erythroidine (DHβE) and NBT, which can distinguish between the α3β2 and α3β4 subunit combinations. Having identified residues 54–63 of β2 as containing the major determinant of competitive antagonist sensitivity, we then used a series of mutant β subunits to identify threonine 59 as the critical residue within this region.
MATERIALS AND METHODS
Materials.Xenopus laevis frogs were purchased from Nasco. RNA transcription kits were from Ambion. ACh, atropine, and 3-aminobenzoic acid ethyl ester were from Sigma (St. Louis, MO). Collagenase B was from Boehringer Mannheim (Indianapolis, IN). Sequenase 2.0 kits were from United States Biochemicals (Cleveland, OH). NBT was from Biotoxins. CloneAmp kits were from Gibco (Gaithersburg, MD). DHβE was a gift from Merck (Rahway, NJ).
Mutagenesis and construction of chimeric receptors. Chimeric and mutant subunits were constructed using PCR (Higuchi, 1990). Our notation for these subunits is to list the source of the N-terminal portion, followed by the residue number in the amino acid sequence in which the chimeric joint is made (numbering taken from the mature β2 subunit sequence), followed by the source of the C-terminal portion. For example, the chimeric subunit β4-204-β2 is composed of β4 sequence from the N terminus until residue 204, after which it is composed of β2 sequence. The β2 and β4 cDNAs in the Bluescript SK− vector were used as templates for PCR reactions. PCR products were subcloned into the pAMP1 vector using a CloneAmp kit (Gibco) or into the pCR-Script SK+vector (Stratagene, La Jolla, CA). To minimize the amount of PCR product in the final construct that would have to be sequenced, as much PCR product as possible was replaced with wild-type β2 or β4 sequence using existing restriction sites. Remaining sequence derived from PCR product was sequenced using Sequenase 2.0 (United States Biochemicals).
Injection of in vitro synthesized RNA intoXenopus oocytes.m7G(5′)ppp(5′)G-capped cRNA was synthesizedin vitro from linearized template DNA encoding the α3, β2, and β4 subunits, as well as the various chimeric and mutant subunits, using an Ambion mMessage mMachine kit. Mature X. laevis frogs were anesthetized by submersion in 0.1% 3-aminobenzoic acid ethyl ester, and oocytes were surgically removed. Follicle cells were removed by treatment with collagenase B for 2 hr at room temperature. Each oocyte was injected with 5–50 ng of cRNA in 50 nl of water and incubated at 19°C in modified Barth’s saline (88 mm NaCl, 1 mm KCl, 2.4 mm NaHCO3, 0.3 mm CaNO3, 0.41 mm CaCl2, 0.82 mm MgSO4, 100 μg/ml gentamicin, and 15 mm HEPES, pH 7.6) for 2–7 d. RNA transcripts encoding each subunit were injected into oocytes at a molar ratio of 1:1.
Electrophysiological recordings. Oocytes were perfused at room temperature (20–25°C), in a 300 μl chamber with perfusion solution (115 mm NaCl, 1.8 mm CaCl2, 2.5 mm KCl, 10 mm HEPES, pH 7.2, and 1.0 μm atropine). Perfusion was continuous at a rate of ∼20 ml/min. ACh was diluted in perfusion solution, and the oocytes were exposed to ACh for ∼10 sec using a solenoid valve. NBT sensitivity was tested by comparing ACh-induced current responses before and after the oocytes were incubated for 30 min in perfusion solution containing various concentrations of NBT and 100 μg/ml bovine serum albumin. Preincubation with NBT results in a slowly reversible competitive blockade of α3β2 but not α3β4 (Boulter et al., 1987; Duvoisin et al., 1989; Luetje et al., 1990b). DHβE sensitivity was tested by measuring the reduction of ACh-induced current responses when DHβE was coapplied with ACh. The response to ACh alone, before treatment with either NBT or DHβE, is taken as the control response. The ACh-induced response, after treatment with NBT or during coapplication with DHβE, is reported as a percent of the control response.
Current responses to agonist application were measured under two-electrode voltage clamp, at a holding potential of −70 mV, using a Knight Industrial Technologies voltage clamp unit. Micropipettes were filled with 3 m KCl and had resistances of 0.5–1.0 MΩ. Agonist-induced responses were captured, stored, and analyzed on a Macintosh IIci computer using a data acquisition program written with LabVIEW (National Instruments) and LIBI (University of Arizona) software (Luetje et al., 1993).
Dose–response and dose–inhibition data were fit with Passage II software by the nonlinear least-squares method. For dose–response data, we used the equation: current = maximum current/[1 + (EC50/[agonist])n], where n and EC50 represent the Hill coefficient and the agonist concentration producing half-maximal response, respectively. Rapid desensitization of these receptors can affect the accuracy of dose–response curves. However, this was found to account for only a small fraction of the difference in EC50 between α3β2 and α3β4 (Cohen et al., 1995). Rapid desensitization also makes the maximal response an unreliable standard with which to normalize data. For this reason, we normalized each response to the response to a low concentration of agonist. To compare and display results for different receptors, we then renormalized each value to the fit maximal response. For DHβE dose–inhibition data, we used the equation: current = maximum current/[1 + ([antagonist]/IC50)n], where n and IC50 represent the Hill coefficient and the antagonist concentration producing half-maximal inhibition, respectively. Fold differences between NBT dose–inhibition data for various receptors were determined by visual inspection of Figure 5B. Statistical significance was determined by using a two-sample t test after an F test to ensure equality of variance. For samples with unequal variance (p > 0.05), statistical significance was determined by using a two-samplet test for samples with unequal variance (Cochran’s method).
Fig. 5.
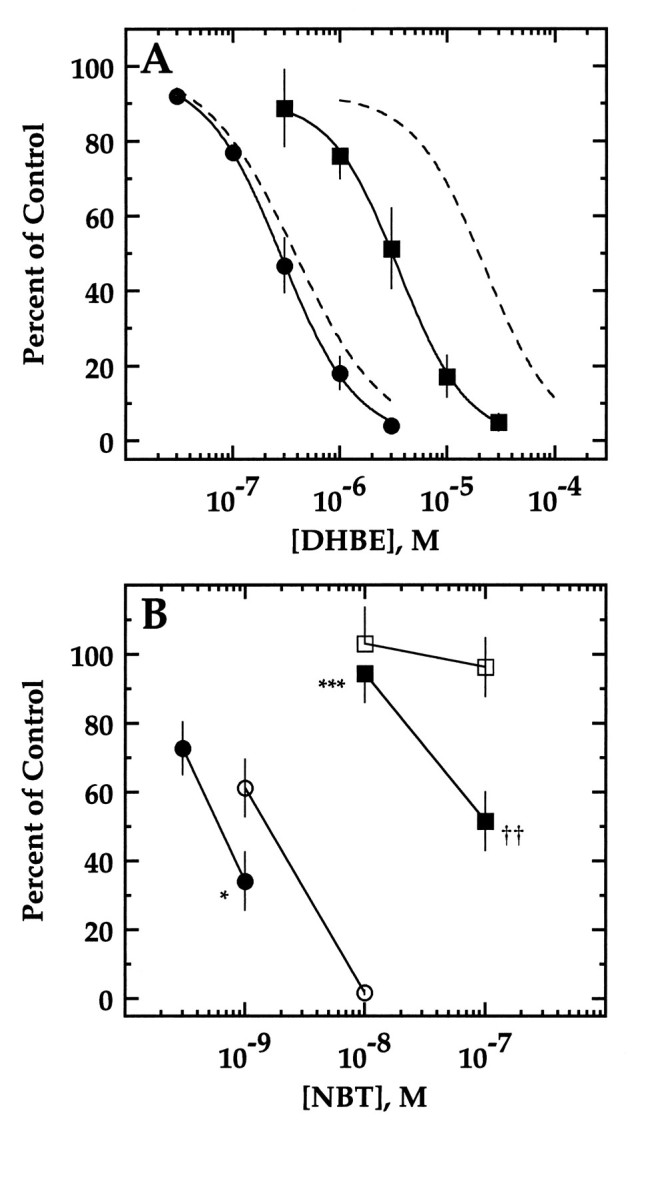
Effect of mutations of threonine 59 on DHβE and NBT sensitivity. A, DHβE sensitivity of α3β2,T59K (filled squares) and α3β2,T59D (filled circles). Current in response to an EC20 concentration of ACh in the presence of various concentrations of DHβE is presented as a percent of the response to ACh alone (mean ± SD of 3–6 oocytes). The lines are fits to a Hill equation (see Materials and Methods). IC50 values are 3.8 ± 0.9 μm for α3β2,T59K and 0.30 ± 0.07 μm for α3β2,T59D. Inhibition curves for α3β2 and α3β4 from Figure 2B are shown as dashed lines for reference. B, NBT sensitivity of α3β2,T59K (filled squares), α3β2,T59D (filled circles), α3β2 (open circles), and α3β4 (opensquares). Current in response to an ACh concentration at or below the EC50 after 30 min incubation with various concentrations of NBT is presented as a percentage of the response to ACh alone (mean ± SD of 3 separate oocytes). Significant differences from β2 are denoted by asterisks(*p < 0.02; ***p < 0.001). Significant differences from β4 are denoted by daggers(††p < 0.01). Some error bars are obscured by symbols.
RESULTS
The β2 and β4 subunits form receptors that are differentially sensitive to competitive antagonists
The β2 and β4 subunits each can form functional neuronal nAChRs when expressed in Xenopus oocytes in combination with α3 (Fig. 1). These two subunit combinations differ in their sensitivity to both DHβE and NBT. At equipotent agonist concentrations (see below), the α3β2 receptor is almost completely blocked by coapplication of 3 μm DHβE, whereas the α3β4 receptor is blocked only slightly. In addition, the α3β2 combination is completely blocked by 100 nm NBT, whereas the α3β4 is insensitive to this concentration of NBT. Because these competitive antagonists distinguish between the receptors based on the identity of the β subunits, they are useful probes to identify the structural basis for the contribution of the β subunit to competitive antagonist sensitivity.
Fig. 1.

α3β2 and α3β4 are pharmacologically distinct. Top traces, Current responses of an α3β2-expressing oocyte to 10 μm ACh alone and in combination with 3 μmDHβE (left), and current responses of a different α3β2-expressing oocyte to 1 μm ACh before and after 30 min incubation with 100 nmNBT (right).Bottom traces, Current responses of an α3β4-expressing oocyte to 100 μm ACh alone or in combination with 3 μmDHβE (left), and current responses of a different α3β4-expressing oocyte to 10 μm ACh before and after 30 min incubation with 100 nmNBT (right). ACh application of ∼10 sec is indicated by arrowheads. Scale bars: 150 nA, 10 sec.
Amino acid residues involved in determining competitive antagonist sensitivity are of particular interest because the competitive antagonist-binding sites of receptors are thought to overlap, at least partially, with the agonist-binding sites. DHβE has been shown to act competitively with ACh in ligand-binding experiments on rat brain homogenates (Williams and Robinson, 1984), and essential atomic groups of DHβE can superimpose with those of the agonist nicotine, suggesting that the two compounds can share a similar conformation (Sheridan et al., 1986). In addition, DHβE has been shown to be a purely competitive antagonist of the α7 neuronal nAChR exogenously expressed in Xenopus oocytes (Bertrand et al., 1992). We find that DHβE antagonism of both α3β2 and α3β4 can be overcome by increasing the ACh concentration, a result indicative of competitive antagonism (Table 1). NBT has also been shown to antagonize nAChRs in a competitive manner (Halvorsen and Berg, 1987).
Table 1.
DHβE antagonism can be overcome by increasing the acetylcholine concentration
| Receptor | [DHβE] (μm) | [ACh] (μm) | Percent of control |
|---|---|---|---|
| α3β2 | 1 | 10 | 21.5 ± 5.0 |
| α3β2 | 1 | 1000 | 96.1 ± 1.1 |
| α3β4 | 30 | 100 | 40.4 ± 9.9 |
| α3β4 | 30 | 10,000 | 95.9 ± 1.9 |
| α3β2,T59K | 10 | 30 | 17.2 ± 5.8 |
| α3β2,T59K | 10 | 3000 | 85.5 ± 1.8 |
ACh-induced current in the presence of DHβE is presented as a percentage of the control response to ACh alone (mean ± SD of 3–6 oocytes).
To evaluate accurately the degree of blockade by a competitive antagonist such as DHβE on α3β2 and α3β4, it is necessary that the ACh concentrations used be equipotent (i.e., at the same point on the dose–response curve) (Craig et al., 1993). For this reason, full dose–response curves for each subunit combination were constructed (Fig. 2A). The EC50 values for α3β2 and α3β4 were 70.8 ± 19.6 and 209.7 ± 40.7 μm, respectively (Table 2). The EC20 was chosen as an ACh test dose, because this is high enough to reliably yield useful current responses, but low enough to avoid extensive desensitization. In contrast to DHβE, incubation with NBT results in pseudoirreversible blockade; NBT slowly dissociates over a period of several hours. Because the postincubation ACh application only lasts for 10 sec, the ACh and NBT are not in direct competition. Determination of the percent blockade by NBT, therefore, is unrelated to the level of receptor activation by ACh.
Fig. 2.

A, Acetylcholine dose–response curves for α3β2 (circles) and α3β4 (squares). Symbols are the mean normalized responses ± SEM of three separate sets of oocytes, each set consisting of three to four separate oocytes. The lines are fits to a Hill equation (see Materials and Methods). EC50 and n values are 70.8 ± 19.6 μm and 0.74 ± 0.11 for α3β2, respectively, and 209.7 ± 40.7 μm and 1.56 ± 0.02 for α3β4, respectively. B, DHβE inhibition curves for α3β2 (circles) and α3β4 (squares). Increasing DHβE concentrations were coapplied with an EC20 ACh concentration of 100 μm for α3β4 and 10 μm for α3β2. The response in the presence of DHβE is reported as a percent of the response to ACh alone (mean ± SD of 3 oocytes). The lines are fits to a Hill equation (see Materials and Methods). IC50 values are 0.41 ± 0.17 μm for α3β2 and 23.1 ± 10.2 μm for α3β4. Some error bars are obscured by symbols.
Table 2.
EC50, Hill coefficient, and IC50values for receptors formed by wild-type, chimeric, and mutant β subunits
| Subunit | EC50(μm) | n | IC50(μm) |
|---|---|---|---|
| β2 | 70.8 ± 19.6 | 0.74 ± 0.11 | 0.41 ± 0.17 |
| β4 | 209.7 ± 40.7 | 1.56 ± 0.02 | 23.1 ± 10.2 |
| β4-54-β2 | 314.3 ± 108.8 | 0.90 ± 0.20 | – |
| β4-103-β2 | 433.3 ± 139.3 | 1.04 ± 0.16 | – |
| β4-133-β2 | 199.1 ± 72.4 | 1.66 ± 0.45 | – |
| β4-204-β2 | 518.6 ± 155.7 | 2.23 ± 0.30 | – |
| β2-54-β4 | 47.8 ± 45.3 | 0.84 ± 0.26 | – |
| β2-63-β4 | 72.7 ± 17.1 | 0.89 ± 0.02 | – |
| β2-74-β4 | 124.3 ± 96.8 | 0.87 ± 0.22 | – |
| β2-80-β4 | 40.0 ± 30.5 | 0.85 ± 0.12 | – |
| β2,N55S | 86.3 ± 47.5 | 0.57 ± 0.05 | – |
| β2,V56I | 67.9 ± 47.3 | 0.76 ± 0.10 | – |
| β2,T59K | 158.5 ± 114.1 | 0.92 ± 0.03 | 3.8 ± 0.9 |
| β2,E63T | 94.8 ± 26.1 | 0.76 ± 0.11 | – |
| β2,T59D | 76.2 ± 45.5 | 0.78 ± 0.07 | 0.30 ± 0.07 |
All subunits were functionally expressed in combination with α3. EC50 and n values, determined by fitting to a Hill equation (see Materials and Methods), are the mean ± SD of results from three to four separate oocytes, with the exception of β2 and β4, which are the mean ± SEM of results from three separate sets of oocytes, each consisting of three to four separate oocytes. IC50 values, determined by fitting to a Hill equation (see Materials and Methods), are the mean ± SD of results from three separate oocytes.
At the EC20 (10 μm for α3β2, 100 μm for α3β4) for each receptor, responses were measured in the presence of increasing DHβE concentrations (Fig. 2B). The IC50values for α3β2 and α3β4 were 0.41 ± 0.17 and 23.1 ± 10.2 μm, respectively. These inhibition curves were used to select 3 μm DHβE as a concentration that differentiates between the receptors. At 3 μm DHβE, there is relatively little blockade of the α3β4 receptor (86.9 ± 5.1% of control), whereas most of the α3β2 response is eliminated (10.0 ± 4.6% of control). An NBT concentration of 100 nm was chosen to differentiate between the receptors, because this concentration blocks α3β2 almost completely (3.4 ± 1.6% of control) and has little effect of α3β4 (96.3 ± 8.4% of control).
Sequence segment 54–63 of β subunits contains a major determinant of DHβE and NBT sensitivity
We constructed a series of chimeric β subunits to determine which sequence segments are responsible for differences in competitive antagonist sensitivity. Sections of one β subunit were substituted with homologous sections from the other β subunit. In Figure3, A and B, the chimeras contain an N-terminal section of β4 connected to a C-terminal section of β2. In Figure 3, C and D, the chimeras contain an N-terminal section of β2 connected to a C-terminal section of β4. Each of these chimeras was then expressed in Xenopusoocytes, in combination with α3, and full dose–response curves were obtained to determine the EC20 for ACh. The degree of blockade of an EC20 ACh response by 3 μm DHβE was then determined (Fig.3A,C). The degree of blockade by 100 nm NBT was also determined (Fig.3B,D).
Fig. 3.

DHβE and NBT sensitivity of receptors formed by chimeric β subunits. A, DHβE sensitivity of receptors formed by each of a series of chimeric subunits in which increasingly larger portions of the N-terminal end of β2 were replaced by the corresponding portion of β4. Current in response to an EC20 concentration of ACh in the presence of 3 μm DHβE is presented as a percent of the response to ACh alone (mean ± SD of 3–4 separate oocytes).B, NBT sensitivity of receptors formed by the chimeras inA. Current in response to an ACh concentration at or below the EC50 after 30 min incubation with 100 nm NBT is presented as a percentage of the response to ACh alone (mean ± SD of 3–4 separate oocytes, except for β4, which is mean ± SEM of 3 separate sets of oocytes, each set consisting of 3–4 separate oocytes). C, DHβE sensitivity of receptors formed by each of a series of chimeric subunits in which increasingly larger portions of β4 were replaced by the corresponding portion of β2. Current in response to an EC20concentration of ACh in the presence of 3 μmDHβE is presented as a percent of the response to ACh alone (mean ± SD of 3–4 separate oocytes). D, NBT sensitivity of receptors formed by the chimeras in C. Current in response to an ACh concentration at or below the EC50after 30 min incubation with 100 nm NBT is presented as a percentage of the response to ACh alone (mean ± SD of 3–4 separate oocytes, except for β2-54-β4, which is mean ± SEM of 3 separate sets of oocytes, each set consisting of 3–4 separate oocytes). Significant differences from β2 are denoted byasterisks (*p < 0.05; **p < 0.01; ***p < 0.001). Significant differences from β4 are denoted by daggers (†p < 0.05;†††p < 0.001). Some error bars are too small to appear.
Substitution of the first 54 N-terminal residues of β2 with the corresponding section of β4 (β4-54-β2) resulted in a subunit that formed receptors that were slightly, but significantly, less sensitive to 3 μm DHβE and 100 nmNBT than were receptors formed by wild-type β2. This intermediate sensitivity between that of wild-type β2- and β4-containing receptors suggests that a minor determinant of DHβE and NBT sensitivity is located within the first 54 N-terminal amino acids of the β subunit. A chimeric subunit in which the first 103 N-terminal residues of β2 were replaced with β4 sequence (β4-103-β2) formed receptors as insensitive to 3 μm DHβE and 100 nm NBT as were receptors formed by wild-type β4. This suggests that the section of the β subunit responsible for DHβE and NBT sensitivity is located within the first 103 N-terminal amino acids, with the segment 54–103 containing the major determinant. Chimeric subunits in which the first 133 or 204 N-terminal residues of β2 were replaced with β4 sequence (β4-133-β2, β4-204-β2) also formed receptors with DHβE and NBT sensitivities comparable to that of wild-type β4-containing receptors.
Chimeric subunits were also constructed containing an N-terminal section of β2 connected to a C-terminal section of β4 (Fig.3C,D). Substituting the first 54 N-terminal residues of β2 into β4 (β2-54-β4) failed to increase the DHβE or NBT sensitivity of receptors formed by this chimera beyond that of receptors formed by wild-type β4. This result is consistent with the minor nature of the determinant between residues 1 and 54 (Fig. 3A,B). Substituting the first 63 N-terminal residues of β2 into β4 (β2-63-β4) resulted in a chimera that formed receptors as sensitive to blockade by 3 μm DHβE as receptors formed by β2. Substitution of the first 74 or 80 N-terminal residues of β2 into β4 also resulted in blockade by 3 μmDHβE that was not significantly different than blockade of α3β2. Receptors formed by β2-63-β4 and β2-74-β4 were slightly, but significantly, less sensitive to blockade by 100 nm NBT than were receptors formed by β2, whereas the β2-80-β4 chimera formed receptors with a sensitivity to 100 nm NBT indistinguishable from that of receptors formed by β2.
Taken together, the DHβE and NBT sensitivities of receptors formed by these chimeras (Fig. 3A–D) indicate that the major determinant for sensitivity to both competitive antagonists is within the amino acid segment from 54 to 63. This can be seen most clearly by considering the chimeras β4-54-β2 and β2-63-β4. Both β4-54-β2, which contains β2 sequence from residue 54 to the C terminus, and β2-63-β4, which contains β2 sequence from the N terminus to residue 63, form receptors nearly as sensitive to DHβE and NBT blockade as receptors formed by wild-type β2. The only β2 sequence common to these two chimeras is segment 54–63. In addition to this major determinant, a minor determinant of sensitivity to both antagonists is between 1 and 54, whereas a minor determinant of NBT sensitivity only may be within segment 74–80.
Threonine 59 of β2 is critical to both DHβE and NBT sensitivity
We examined sequence segment 54–63 in more detail by changing individual amino acid residues. The β2 sequence differs from β4 at only four residues within this region (Fig.4A). We changed each of these residues individually from what occurs in β2 to what occurs in β4 and then determined the DHβE and NBT sensitivity of receptors formed by these mutants (Fig. 4B,C). Changing threonine 59 of β2 to lysine (T59K) resulted in a significant loss in sensitivity to DHβE and NBT when compared to wild-type β2-containing receptors. Mutation V56I had a small, but significant, effect on both DHβE and NBT sensitivity. Mutation N55S had a small, but significant, effect on DHβE sensitivity, but no effect on NBT sensitivity, whereas mutation E63T had no effect on sensitivity to either antagonist.
Fig. 4.

Threonine 59 of β2 is critical to both DHβE and NBT sensitivity. A, Alignment of β2 and β4 sequences within segment 54–63. Residues that differ are denoted by solid circles. Tryptophan 57 is starred. B, DHβE sensitivity of receptors formed by each of a series of mutant β2 subunits. Current in response to an EC20 concentration of ACh in the presence of 3 μm DHβE is presented as a percent of the response to ACh alone (mean ± SD of 3 separate oocytes).C, NBT sensitivity of receptors formed by the β2 mutants in B. Current in response to an ACh concentration at or below the EC50 after 30 min incubation with 100 nm NBT is presented as a percentage of the response to ACh alone (mean ± SD of 3 separate oocytes, except for β4, which is mean ± SEM of three separate sets of oocytes, each set consisting of 3–4 separate oocytes). Significant differences from β2 are denoted by asterisks (*p < 0.05; **p < 0.01; ***p < 0.001). Significant differences from β4 are denoted by daggers(††p < 0.01;†††p < 0.001). Some error bars are too small to appear.
In Figure 5 we examined in more detail the degree to which mutation T59K affected DHβE and NBT sensitivity. The DHβE inhibition curve for receptors formed by β2,T59K is shifted to the right of the wild-type β2 curve approximately ninefold, accounting for about half of the difference between β2- and β4-containing receptors (Fig. 5A). Block of β2,T59K-containing receptors by DHβE remains competitive because an increased ACh concentration is able to overcome blockade (Table 1). Residue 59, together with residues 55 and 56, is responsible for most of the difference in DHβE sensitivity between β2 and β4. This can be seen most clearly by considering that blockade of β2- and β4-containing receptors by 3 μmDHβE differs by ∼77 percentage points. Block of mutants β2,N55S, β2,V56I, and β2,T59K by DHβE differs from block of β2 by 10, 9, and 41 percentage points, respectively, accounting for 60 percentage points. The remaining difference between block of β2- and β4-containing receptors (17 percentage points) is completely accounted for by segment 1–54, because block of β4-54-β2 receptors differs from that of β2 receptors by 19 percentage points. Threonine 59 also accounts for a substantial portion of the NBT sensitivity difference between β2- and β4-containing receptors (Fig. 5B). Receptors formed by β2,T59K were ∼71-fold less sensitive to NBT than β2-containing receptors. Although we know that α3β4 is at least 100-fold less sensitive to NBT than α3β2 (Fig. 5B), we are unable to determine exactly how much less sensitive α3β4 is attributable to a lack of a sufficient quantity of NBT. The remaining difference in NBT sensitivity between β2- and β4-containing receptors can be accounted for by the slight contribution from V56 and the minor determinants within regions 1–54 and 74–80.
The loss of DHβE and NBT sensitivity caused by the mutation T59K could be attributable to the change from the polar side chain of threonine to the positively charged side chain of lysine, or it could be attributable to the change in side chain volume. The identification of arginine 34 of NBT as a critical residue for neuronal nAChR blockade (Dewan et al., 1994; Fiordalisi et al., 1994) suggests that it is the introduction of the positively charged lysine that interferes NBT sensitivity. To explore this idea, we introduced a negative charge by changing this residue from threonine to aspartate (T59D). The DHβE and NBT sensitivity of receptors formed by β2,T59D is shown in Figure 5, A and B. The T59D mutation resulted in an increase in NBT sensitivity of ∼2.5 fold. The T59D mutation had no effect on DHβE sensitivity.
DISCUSSION
The neuronal nAChRs α3β2 and α3β4 differ in their sensitivity to the antagonists DHβE and NBT. Pharmacological analysis of a series of chimeric β subunits has allowed us to identify areas of the β subunits that determine sensitivity to these competitive antagonists. The major determinant of both DHβE and NBT sensitivity lies in sequence segment 54–63, with a minor determinant of sensitivity to both antagonists in region 1–54 and a minor determinant of NBT sensitivity in region 74–80. Within sequence segment 54–63, we identified threonine 59 of β2 as the critical residue. Changing this residue to lysine, as in β4, results in a 9-fold loss in DHβE sensitivity and a 71-fold loss in NBT sensitivity. Changing threonine 59 of β2 to aspartate, thus introducing a negative charge, caused a 2.5-fold increase in NBT sensitivity.
It has become clear recently that non-α subunits are involved in determining both the physical structure and pharmacological properties of the ligand-binding sites of nAChRs (Blount and Merlie, 1989;Duvoisin et al., 1989; Pederson and Cohen, 1990; Czajkowski and Karlin, 1991; Luetje and Patrick, 1991; Middleton and Cohen, 1991). Affinity labeling and mutagenesis studies of Torpedo electric organ and mammalian muscle nAChRs have identified amino acid residues of the γ and δ subunits that are associated with ligand binding (Cohen et al., 1992; Czajkowski et al., 1993; Sine, 1993; Fu and Sine, 1994). In covalent labeling experiments involving the competitive antagonistd-tubocurarine, Cohen et al. (1992) demonstrated incorporation of label onto a tryptophan residue of the γ and δ subunits (residue 55 and 57, respectively). This residue is conserved in the rat neuronal β2 and β4 subunits (position 57, Fig.4A), and thus appears to be a common feature of the ligand-binding sites of nAChRs. Interestingly, the homologous residue of neuronal α7 (tryptophan 54) is involved in determining sensitivity to both agonists and antagonists, leading to the proposal that α7 contributes both an “α component” and a “non-α component” when forming homo-oligomeric receptors (Corringer et al., 1995).
The conservation of this tryptophan among muscle and neuronal nAChRs means that this residue cannot be responsible for pharmacological differences between nAChR subtypes. It is the amino acid residues that differ among subunits that must be responsible for this diversity. We have identified such a residue, separated by only one residue from the conserved tryptophan, as the major determinant of differences in competitive antagonist sensitivity between β2- and β4-containing receptors. Changing this residue in β2 from threonine to what occurs in β4 (lysine) results in a substantial loss of both DHβE and NBT sensitivity. The change from threonine to lysine is a change in both the character (polar to negatively charged) and the size (55.7–101.5 Å3) of the side chain. Either or both of these properties might be responsible for the effect of changing this residue. Considering that arginine 34 of NBT has been identified as a critical residue involved in neuronal nAChR blockade (Dewan et al., 1994), we hypothesized that insertion of lysine at position 59 in β2 might be decreasing NBT sensitivity by electrostatic repulsion. If this were true, then introduction of a negative charge at this position might be expected to increase NBT sensitivity. Changing threonine 59 to aspartate does result, in fact, in an increase in NBT sensitivity (Fig. 5B).
Construction and functional analysis of chimeric receptor subunits allows identification of structural differences that confer unique pharmacological properties. This methodology has been used to map determinants of both agonist and antagonist sensitivity on neuronal nAChR β subunits. Chimeric subunits have been used to identify the general region on β subunits that contributes to NBT sensitivity.Papke et al. (1993) showed that substitution of the first 119 amino acids of the β4 subunit with the corresponding section of β2 can confer NBT sensitivity onto the β4 subunit. The identity of the β subunit also influences the NBT sensitivity of the receptors α4β2 and α4β4. Wheeler et al. (1993) showed that a chimeric subunit composed of the N-terminal 80 residues of β2 followed by β4 sequence, formed receptors with α4 that were sensitive to NBT blockade. A series of β subunit chimeras, expressed in combination with the α3 subunit, has been used to identify the sequence segment 104–120 of β2 as important in determining sensitivity to the agonist cytisine (Figl et al., 1992; Cohen et al., 1995). Consistent with these reports, we find that receptors formed by α3 and the chimera β4–103-β2 have a cytisine sensitivity similar to that of receptors formed by wild-type α3β2 (data not shown).Cohen et al. (1995) also show that region 104–120 is responsible for part of the difference in EC50 for ACh, as well as for part of the difference in Hill slope, between α3β2 and α3β4.
Differential sensitivity to agonists may result from differences in affinity or efficacy, making it difficult to infer conclusions about the functional role of the sequence segment being mapped. Competitive antagonists are ideal probes, because they compete with agonist for a common binding site but do not activate the receptors; they have no efficacy. Because our determination of the DHβE sensitivity of each receptor is dependent on use of equipotent concentrations of ACh, it is important that differences in DHβE sensitivity not be an artifact of differences in ACh dose–response curve characteristics. The α3β2 and α3β4 ACh dose–response curves clearly differ in both EC50 and apparent Hill coefficient (Table 2). However, these differences can be taken into consideration when DHβE dissociation constants (Ki) for each receptor are calculated (Leff and Dougall, 1993). The resulting DHβEKi values of 0.21 and 32.4 μm (for α3β2 and α3β4, respectively) differ by ∼154-fold. Additional arguments against differences in DHβE sensitivity being artifactual are that DHβE sensitivity maps differently than the ACh EC50 and the apparent Hill coefficient (compare Fig. 3 and Table 2), and that use of both DHβE and NBT as probes has identified the same residue as a major determinant of competitive antagonist sensitivity. If DHβE antagonism is competitive, why do determinants of DHβE sensitivity and ACh EC50 differ? Although competitive antagonists do compete for a common binding site with agonists, their interactions with the binding site would not necessarily be coextensive with those of agonists. In fact, this would not be expected, because competitive antagonists differ from agonists by lacking efficacy.
Although the pseudoirreversibility of NBT blockade makes concerns over ACh dose–response curves irrelevant, NBT is a large peptide toxin and may block receptor activation by adventitiously occluding the ligand-binding site after binding elsewhere on the receptor. Therefore, mapping the areas of the receptor responsible for differential sensitivity to NBT may identify regions of uncertain significance. However, both DHβE and NBT sensitivity map similarly, identifying threonine 59 as a major determinant and region 1–54 as containing a minor determinant, supporting the view that NBT is a useful probe. The significance of region 74–80, containing an additional minor determinant of NBT sensitivity, is unclear. Another example of determinants of NBT sensitivity overlapping with those of small ligand sensitivity occurs on α subunits. The α2 subunit forms receptors with β2 that are insensitive to NBT and are more sensitive to nicotine than to ACh, whereas α3β2 receptors are blocked by NBT and are much less sensitive to nicotine than to ACh. Region 195–215 contains determinants of both properties. Within this region, the glutamine residue at position 198 of α3 (proline in α2) was shown to be an important determinant of both NBT and nicotine sensitivity (Luetje et al., 1993). Similar results regarding the role of glutamine 198 in determining nicotine sensitivity have been obtained recently using chicken neuronal nAChR α subunits (Hussy et al., 1994).
The exact physical role of residues identified in this and previous studies, that confer pharmacological differences among nAChR subtypes, remains unclear. In this study, we provide data consistent with a direct interaction between residue 59 of β2/β4 and NBT. Thus, these residues may be structural features of the binding site and may participate in the binding of ligand. Alternatively, these residues may not actually participate in binding of ligand, but impinge upon those that do, reshaping the site enough to alter ligand sensitivity. More extensive mutagenesis of identified residues will be required to distinguish between these possibilities. Particularly promising is the potential for incorporating unnatural amino acids at these sites (Nowak et al., 1995).
Footnotes
This work was supported by grants to C.W.L. from the National Institute on Drug Abuse (DA08102), the American Heart Association Florida Affiliate, and the Pharmaceutical Research and Manufacturers of America Foundation. C.W.L. is an Initial Investigator of the American Heart Association, Florida Affiliate. We thank Floyd Maddox for technical assistance.
Correspondence should be addressed to Dr. Charles W. Luetje, Department of Molecular and Cellular Pharmacology (R-189), University of Miami School of Medicine, P.O. Box 016189, Miami, FL 33101.
REFERENCES
- 1.Abramson SN, Li Y, Culver P, Taylor P. An analog of lophotoxin reacts covalently with Tyr190 in the α-subunit of the nicotinic acetylcholine receptor. J Biol Chem. 1989;264:12666–12672. [PubMed] [Google Scholar]
- 2.Bertrand D, Bertrand S, Ballivet M. Pharmacological properties of the homomeric α7 receptor. Neurosci Lett. 1992;146:87–90. doi: 10.1016/0304-3940(92)90179-b. [DOI] [PubMed] [Google Scholar]
- 3.Blount P, Merlie J. Molecular basis of the two nonequivalent ligand binding sites of the muscle nicotinic acetylcholine receptor. Neuron. 1989;3:349–357. doi: 10.1016/0896-6273(89)90259-6. [DOI] [PubMed] [Google Scholar]
- 4.Boulter J, Connolly J, Deneris E, Goldman D, Heinemann S, Patrick J. Functional expression of two neuronal nicotinic acetylcholine receptors from cDNA clones identifies a gene family. Proc Natl Acad Sci USA. 1987;84:7763–7767. doi: 10.1073/pnas.84.21.7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen JB, Blanton MP, Chiara DC, Sharp SD, White BH. Structural organization of functional domains of the nicotinic acetylcholine receptor. J Cell Biochem [Suppl] 1992;16E:217. [Google Scholar]
- 6.Cohen BN, Figl A, Quick MW, Labarca C, Davidson N, Lester HA. Regions of β2 and β4 responsible for differences between the steady state dose-response relationships of the α3β2 and α3β4 neuronal nicotinic receptors. J Gen Physiol. 1995;105:745–764. doi: 10.1085/jgp.105.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corringer PJ, Galzi JL, Eisele JL, Bertrand S, Changeux JP, Bertrand D. Identification of a new component of the agonist binding site of the nicotinic alpha 7 homooligomeric receptor. J Biol Chem. 1995;270:11749–11752. doi: 10.1074/jbc.270.20.11749. [DOI] [PubMed] [Google Scholar]
- 8.Craig DA. The Cheng-Prusoff relationship: something lost in the translation. Trends Pharmacol Sci. 1993;14:89–91. doi: 10.1016/0165-6147(93)90070-z. [DOI] [PubMed] [Google Scholar]
- 9.Czajkowski C, Karlin A. Agonist binding site of Torpedo electric tissue nicotinic acetylcholine receptor: a negatively charged region of the δ subunit within 0.9 nm of the α subunit binding site disulfide. J Biol Chem. 1991;266:22603–22612. [PubMed] [Google Scholar]
- 10.Czajkowski C, Kaufmann C, Karlin A. Negatively charged amino acid residues in the nicotinic receptor γ subunit that contribute to the binding of acetylcholine. Proc Natl Acad Sci USA. 1993;90:6285–6289. doi: 10.1073/pnas.90.13.6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dennis M, Giraudat J, Kotzyba-Hibert F, Goeldner M, Hirth C, Chang J-Y, Lazure C, Chretien M, Changeux J-P. Amino acids of the Torpedo marmorata acetylcholine receptor α subunit labelled by photoaffinity ligand for acetylcholine binding site. Biochemistry. 1988;27:2346–2357. doi: 10.1021/bi00407a016. [DOI] [PubMed] [Google Scholar]
- 12.Dewan JC, Grant GA, Sacchettini JC. Crystal structure of κ-bungarotoxin at 2.3-Å resolution. Biochemistry. 1994;33:13147–13154. doi: 10.1021/bi00248a026. [DOI] [PubMed] [Google Scholar]
- 13.Duvoisin RM, Deneris ES, Boulter J, Patrick J, Heinemann S. The functional diversity of the neuronal nicotinic acetylcholine receptors is increased by a novel subunit: β4. Neuron. 1989;3:487–496. doi: 10.1016/0896-6273(89)90207-9. [DOI] [PubMed] [Google Scholar]
- 14.Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S. α9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell. 1994;79:705–715. doi: 10.1016/0092-8674(94)90555-x. [DOI] [PubMed] [Google Scholar]
- 15.Figl A, Cohen BN, Quick MW, Davidson N, Lester HA. Regions of β4-β2 subunit chimeras that contribute to the agonist selectivity of neuronal nicotinic receptors. FEBS Lett. 1992;308:245–248. doi: 10.1016/0014-5793(92)81284-s. [DOI] [PubMed] [Google Scholar]
- 16.Fiordalisi JJ, Al-Rabiee R, Chiappinelli VA, Grant GA. Site-directed mutagenesis of κ-bungarotoxin: implications for neuronal receptor specificity. Biochemistry. 1994;33:3872–3877. doi: 10.1021/bi00179a011. [DOI] [PubMed] [Google Scholar]
- 17.Fu DX, Sine SM. Competitive antagonists bridge the α-γ subunit interface of the acetylcholine receptor through quaternary ammonium-aromatic interactions. J Biol Chem. 1994;269:26152–26157. [PubMed] [Google Scholar]
- 18.Galzi J, Revah F, Black D, Goeldner M, Hirth C, Changeux J-P. Identification of a novel amino acid α-tyrosine 93 within the cholinergic ligands-binding sites of the acetylcholine receptor by photoaffinity labeling. J Biol Chem. 1990;265:10430–10437. [PubMed] [Google Scholar]
- 19.Halvorsen SW, Berg DK. Affinity labeling of neuronal acetylcholine receptor subunits with an α-neurotoxin that blocks receptor function. J Neurosci. 1987;7:2547–2555. [PMC free article] [PubMed] [Google Scholar]
- 20.Higuchi R. Recombinant PCR. In: Innis MA, Gelfand GH, Sninsky JJ, White TJ, editors. PCR protocols, a guide to methods and applications. Academic; San Diego: 1990. [Google Scholar]
- 21.Hussy N, Ballivet M, Bertrand D. Agonist and antagonist effects of nicotine on chick neuronal nicotinic receptors are defined by α and β subunits. J Neurophys. 1994;72:1317–1326. doi: 10.1152/jn.1994.72.3.1317. [DOI] [PubMed] [Google Scholar]
- 22.Kao PN, Dwork AJ, Kaldany R-RJ, Silver MS, Wideman J, Stein S, Karlin A. Identification of the α subunit half-cystine specifically labeled by an affinity reagent for the acetylcholine receptor binding site. J Biol Chem. 1984;259:11662–11665. [PubMed] [Google Scholar]
- 23.Leff P, Dougall IG. Further concerns over Cheng-Prusoff analysis. Trends Pharmacol Sci. 1993;14:110–112. doi: 10.1016/0165-6147(93)90080-4. [DOI] [PubMed] [Google Scholar]
- 24.Luetje CW, Patrick J. Both α- and β-subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. J Neurosci. 1991;11:837–845. doi: 10.1523/JNEUROSCI.11-03-00837.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luetje CW, Patrick J, Séguéla P. Nicotine receptors in the mammalian brain. FASEB J. 1990a;4:2753–2760. doi: 10.1096/fasebj.4.10.2197155. [DOI] [PubMed] [Google Scholar]
- 26.Luetje CW, Wada K, Rogers S, Abramson SN, Tsuji K, Heinemann S, Patrick J. Neurotoxins distinguish between different neuronal nicotinic acetylcholine receptor subunit combinations. J Neurochem. 1990b;55:632–640. doi: 10.1111/j.1471-4159.1990.tb04180.x. [DOI] [PubMed] [Google Scholar]
- 27.Luetje CW, Piattoni M, Patrick J. Mapping of ligand binding sites of neuronal nicotinic acetylcholine receptors using chimeric α subunits. Mol Pharmacol. 1993;44:657–666. [PubMed] [Google Scholar]
- 28.Middleton RE, Cohen JB. Mapping of the acetylcholine binding site of the nicotinic acetylcholine receptor: [3H]nicotine as an agonist photoaffinity label. Biochemistry. 1991;30:6987–6997. doi: 10.1021/bi00242a026. [DOI] [PubMed] [Google Scholar]
- 29.Nowak MW, Kearney PC, Sampson JR, Saks ME, Labarca CG, Silverman SK, Zhong W, Thorson J, Abelson JN, Davidson N, Schulz PG, Dougherty DA, Lester HA. Nicotinic receptor binding site probed with unnatural amino acid incorporation in intact cells. Science. 1995;268:439–442. doi: 10.1126/science.7716551. [DOI] [PubMed] [Google Scholar]
- 30.Papke RL, Boulter J, Patrick J, Heinemann S. Single-channel currents of rat neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes. Neuron. 1989;3:589–596. doi: 10.1016/0896-6273(89)90269-9. [DOI] [PubMed] [Google Scholar]
- 31.Papke RL, Duvoisin RM, Heinemann S. The amino terminal half of the nicotinic β-subunit extracellular domain regulates the kinetics of inhibition by neuronal bungarotoxin. Proc R Soc Lond [Biol] 1993;252:141–148. doi: 10.1098/rspb.1993.0058. [DOI] [PubMed] [Google Scholar]
- 32.Pedersen SE, Cohen JB. d -Tubocurarine binding sites are located at the α-γ and α-δ subunit interfaces of the nicotinic acetylcholine receptor. Proc Natl Acad Sci USA. 1990;87:2785–2789. doi: 10.1073/pnas.87.7.2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Role LW. Diversity in primary structure and function of neuronal nicotinic acetylcholine receptor channels. Curr Opin Neurobiol. 1992;2:254–262. doi: 10.1016/0959-4388(92)90112-x. [DOI] [PubMed] [Google Scholar]
- 34.Sargent PB. The diversity of neuronal nicotinic acetylcholine receptors. Annu Rev Neurosci. 1993;16:403–443. doi: 10.1146/annurev.ne.16.030193.002155. [DOI] [PubMed] [Google Scholar]
- 35.Sheridan RP, Nilakantan R, Dixon JS, Venkataraghavan R. The ensemble approach to distance geometry: application to the nicotinic pharmacophore. J Med Chem. 1986;29:899–906. doi: 10.1021/jm00156a005. [DOI] [PubMed] [Google Scholar]
- 36.Sine SM. Molecular dissection of subunit interfaces in the acetylcholine receptor: identification of residues that determine curare selectivity. Proc Natl Acad Sci USA. 1993;90:9436–9440. doi: 10.1073/pnas.90.20.9436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wheeler SV, Chad JE, Foreman R. Residues 1 to 80 of the N-terminal domain of the β subunit confer neuronal bungarotoxin sensitivity and agonist sensitivity on neuronal nicotinic receptors. FEBS Lett. 1993;332:139–142. doi: 10.1016/0014-5793(93)80500-t. [DOI] [PubMed] [Google Scholar]
- 38.Williams M, Robinson JL. Binding of the nicotinic cholinergic antagonist dihydro-β-erythroidine to rat brain tissue. J Neurosci. 1984;4:2906–2911. doi: 10.1523/JNEUROSCI.04-12-02906.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]