

Abstract
Abnormalities of mitochondrial energy metabolism may play a role in normal aging and certain neurodegenerative disorders. In this regard, complex I of the electron transport chain has received substantial attention, especially in Parkinson’s disease. The conventional method for studying complex I has been quantitation of enzyme activity in homogenized tissue samples. To enhance the anatomic precision with which complex I can be examined, we developed an autoradiographic assay for the rotenone site of this enzyme. [3H]dihydrorotenone ([3H]DHR) binding is saturable (KD = 15–55 nm) and specific, and Hill slopes of 1 suggest a single population of binding sites. Nicotinamide adenine dinucleotide (NADH) enhances binding 4- to 80-fold in different brain regions (EC50 = 20–40 μm) by increasing the density of recognition sites (Bmax). Nicotinamide adenine dinucleotide phosphate also increases binding, but NAD+ does not. In skeletal muscle, heart, and kidney, binding was less affected by NADH. [3H]DHR binding is inhibited by rotenone (IC50 = 8–20 nm), meperidine (IC50 = 34–57 μm), amobarbitol (IC50 = 375–425 μm), and MPP+(IC50 = 4–5 mm), consistent with the potencies of these compounds in inhibiting complex I activity. Binding is heterogeneously distributed in brain with the density in gray matter structures varying more than 10-fold. Lesion studies suggest that a substantial portion of binding is associated with nerve terminals. [3H]DHR autoradiography is the first quantitative method to examine complex I with a high degree of anatomic precision. This technique may help to clarify the potential role of complex I dysfunction in normal aging and disease.
Keywords: mitochondria, electron transport chain, complex I, [3H]dihydrorotenone, rotenone, MPP+, amobarbitol, autoradiography
Energy-consuming reactions are fueled by the hydrolysis of phosphate bonds, principally in the form of ATP. The primary mechanism by which this high-energy intermediate is produced can be oxidative or glycolytic, depending on the specific tissue studied. In neurons, ATP is produced primarily during oxidative phosphorylation, although a small quantity is derived from glycolysis (Erecinska and Silver, 1989).
Oxidative phosphorylation requires the coordinated activity of five enzymes of the inner mitochondrial membrane: complexes I, II, III, IV, and V (Hatefi, 1985). The proximal four enzymes, collectively known as the electron transport chain (ETC), convert the potential energy in reduced adenine nucleotides [nicotinamide adenine dinucleotide (NADH) and FADH2] into a form capable of supporting ATP synthase activity. Reducing equivalents (electrons) enter the ETC at two sites: complex I (NADH:ubiquinone oxidoreductase) or complex II (succinate:ubiquinone oxidoreductase) and undergo sequential oxidation-reduction (redox) reactions. Complex I and II reduce ubiquinone (coenzyme Q) to ubiquinol (Cooper and Clark, 1994). Complex III (ubiquinone:cytochrome c oxidoreductase) couples the oxidation of ubiquinol to cytochrome c reduction (Rieske, 1976). Complex IV (cytochrome c:oxygen oxidoreductase) catalyzes the oxidation of cytochrome c, ultimately reducing molecular oxygen and producing water (Caughey et al., 1976; Capaldi, 1990). Potential energy released during redox cycling drives the extrusion of protons across the inner mitochondrial membrane at three sites: complex I, III, and IV. This electrochemical gradient supports the synthesis of ATP by complex V (Caughey et al., 1976; Capaldi et al., 1994).
Interest in the role of the mitochondrion and especially complex I in neurological disease has surged since the description of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced parkinsonism (Davis et al., 1979). A metabolite of MPTP, 1-methyl-4-phenylpyridinium (MPP+), is selectively toxic to dopaminergic neurons by inhibiting complex I (Nicklas et al., 1985). Moreover, a defect in complex I has been reported in muscle (Bindoff et al., 1989), platelets (Shoffner et al., 1991), and brain (Schapira et al., 1990a) in patients with Parkinson’s disease. The defect in brain is reportedly restricted to the substantia nigra (Schapira et al., 1990b); however, the distribution of complex I in brain has not been described.
Conventional assays of complex I are performed in tissue homogenates or mitochondrial preparations. Anatomical resolution is limited by dissection techniques and the quantity of tissue needed for the assay. Moreover, complex I assays are labile and highly dependent on tissue preparation protocols (Trijbels et al., 1993). Using an analog of rotenone, the insecticide that defines the catalytic activity of complex I by its inhibition (Cooper and Clark, 1994), we have developed a stable, reproducible, and quantitative method to examine complex I with a high degree of anatomic precision (Greenamyre et al., 1992). We now detail the pharmacological specificity, regional distribution, and the effect of complex I substrate on [3H]dihydrorotenone ([3H]DHR) binding.
MATERIALS AND METHODS
Chemicals. All chemicals were purchased from Sigma (St. Louis, MO) except MPP+, which was obtained from Research Biochemicals (Natick, MA).
Tissue. All animal-use procedures were performed in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University Committee on Animal Resources. Male Sprague–Dawley rats (200–225 gm) were killed by decapitation, and the brains were rapidly removed, coated with embedding matrix (Lipshaw, Detroit, MI), and frozen under powdered dry ice. Twenty micrometer horizontal sections containing striatum, hippocampus, and cerebellum were used in pharmacological experiments. For distribution studies, coronal sections at 11 levels were examined, 4 anterior (6.70, 5.20, 1.70, and 0.20 mm) and 7 posterior to bregma (0.80, 2.30, 3.60, 5.80, 6.80, 10.80, and 13.30 mm) according to the atlas of Paxinos and Watson (1986). Sections were cut on a cryostat and thaw-mounted onto poly-l-lysine-coated slides. Samples of kidney, heart, and striated muscle were prepared in an identical manner. Slide-mounted tissue sections were stored at −70°C until the time of assay.
[3H]DHR autoradiography.[3H]DHR binding was performed as described previously (Greenamyre et al., 1992), with minor modification. All experiments were performed at room temperature in 50 mm Tris-HCl containing 1% BSA, pH 7.6. [3H]DHR (55 Ci/mmol, custom synthesized by Amersham Life Science, Arlington Heights, IL) was included at a final concentration of 5 nm except in saturation studies, in which the concentration ranged from 1 to 100 nm. Nonspecific binding was defined by 100 μm rotenone [dissolved in dimethylsulfoxide (DMSO)]. The final concentration of DMSO never exceeded 1% and did not alter binding. A 2 hr incubation was used except during association studies when the incubation ranged from 0.5 to 6 hr. A three-step rinse followed the incubation. Slides were placed for 60 min in Tris-HCl/bovine serum albumin buffer, twice for 5 min in 25 mm Tris-HCl, pH 7.6, and once for 15 sec in distilled H2O. In dissociation studies, the initial rinse varied from 0.5 to 30 hr in length. After the rinses, sections were rapidly dried under a stream of warm air.
The modulatory effects of NADH, NAD+, and nicotinamide adenine dinucleotide phosphate (NADPH) on [3H]DHR binding were examined over a concentration range of 0.1–1000 μm. In competition experiments, the complex I inhibitors rotenone (0.5–100 nm), meperidine (0.5–1000 μm), amobarbitol (5–5000 μm), and MPP+ (0.2–50 mm) were included in the incubation.
Sections were mounted in x-ray cassettes with tritium standards (Amersham Microscales) and apposed to Hyperfilm (Amersham) for 1–4 weeks. Films were developed in Kodak D-19, stopped in 10% acetic acid, and fixed in Kodak Fixer.
Entorhinal cortex lesions. Male Sprague–Dawley rats (270–300 gm) received stereotaxic lesions of the left medial entorhinal cortex under Chloropent anesthesia (chloral hydrate, 42.5 mg/ml + pentobarbital sodium, 8.86 mg/ml + magnesium sulfate, 21.2 mg/ml; 0.3–0.4 ml/100 gm of body weight, i.p.) using a slight modification of the procedure described by Levisohn and Isacson (1991). Rats were placed in a Kopf stereotaxic apparatus with the incisor bar set at −3.4 mm. A 0.06 m solution of NMDA, pH 7.4, was prepared and drawn into a 5 μl Hamilton syringe. After exposure of the skull, the needle was placed −8.3 mm posterior to bregma and angled 10° outward with the needle tip at the midline. The needle was then moved 3.25 mm laterally, and a small hole was drilled in the skull to the dura. The needle was lowered 6 mm from the dura and 0.75 μl was infused over 3 min; then the needle was raised 1 mm for a second infusion of 0.75 μl. The syringe was then withdrawn and repositioned −8.8 mm posterior, 3.65 mm lateral, and 5 mm deep for the final infusion of 0.75 μl. The craniotomy was packed with gel foam and scalp wounds were clipped.
Data analysis. [3H]DHR binding in autoradiographic images was quantified on an MCID image analysis system (Imaging Research, St. Catharine’s, Ontario, Canada) by relating optical density to calibrated tritium standards. For saturation and competition experiments, data from the striatum (str), dentate gyrus (dg), and molecular layer of the cerebellum (cmol) were analyzed with Ligand, an iterative nonlinear curve-fitting program (Munson and Rodbard, 1980). To determine whether Hill slopes differed significantly from unity, one-sample t tests were performed.
RESULTS
Characterization of [3H]DHR binding
In preliminary experiments, high levels of nonspecific binding prevented accurate determination of specific [3H]DHR binding. As noted by Horgan et al. (1968), albumin reduced nonspecific binding to ∼10–20% of total [3H]DHR binding under routine assay conditions. Therefore, albumin (1%) was included in all experiments; more concentrated solutions did not further reduce nonspecific binding (data not shown). In addition, preliminary experiments showed that [3H]DHR binding reached equilibrium within 2 hr and remained stable for at least 6 hr (data not shown). Thus, a 2 hr incubation was used routinely. Other experiments showed that extensively prewashing tissue sections in buffer for 30–60 min to remove endogenous NADH did not affect binding (data not shown).
As shown previously (Greenamyre et al., 1992), [3H]DHR binding was saturable with an affinity in the low nanomolar range (Fig. 1).KD values were 15.4 ± 2.7 nm in cmol, 26 ± 5.1 nm in dg, and 54.7 ± 24.0 nm in str. Hill slopes ranged from 0.95 to 0.98, suggesting that [3H]DHR interacted with a single population of sites. Consistent with this, a Scatchard plot of the data was linear (Fig. 1). As seen with other ligand-binding systems, nonspecific binding increased linearly with ligand concentration. In the presence of 1% albumin at a [3H]DHR concentration of 5 nm, 80–90% of total binding was specific.
Fig. 1.

Top, Saturation isotherm of [3H]DHR binding in the cmol in the absence of NADH. Bottom, Scatchard transformation of the data. This experiment was performed four times with similar results.
Effects of adenine nucleotides
NADH enhanced [3H]DHR binding in a concentration-dependent manner, with a Hill slope that did not differ significantly from 1 (Fig. 2). The EC50 was 20–40 μm (Table1). As shown in Figure 3, the enhancement of binding by NADH was attributable primarily to an increase in the number of [3H]DHR binding sites (Bmax). NADH (200 μm) caused a mean sixfold increase inBmax in str, an eightfold increase in dg, and a ninefold increase in cmol (Table 2). NADPH also enhanced the binding of [3H]DHR, but NAD+ did not (Table 1, Fig. 2). In addition to increasing the Bmax, NADH also caused a moderate decrease in [3H]DHR affinity (Table2).
Fig. 2.

Effects of NAD+, NADH, and NADPH on [3H]DHR binding in str. The concentration of adenine nucleotides ranged from 1 to 1000 μm. Values represent mean ± SEM (n = 4). Inset, Hill plot of NADH enhancement. The Hill slope did not differ significantly from 1.
Table 1.
Regional EC50 values (μm) for NADH and NADPH enhancement of [3H]DHR binding
| Nucleotide | Striatum | Dentate gyrus | Cerebellum |
|---|---|---|---|
| NADH | 29.2 ± 1.5 | 20.7 ± 2.4 | 37.1 ± 7.3 |
| NADPH | 36.9 ± 5.7 | 37.6 ± 2.4 | 52.6 ± 30.2 |
Values represent mean ± SEM; n = 4. [3H]DHR concentration was 5 nm.
Fig. 3.

NADH enhances binding by increasing the number of binding sites. Top, Saturation isotherm of [3H]DHR binding in the str showingspecific binding in the absence and presence of NADH.Bottom, Scatchard transformation of binding data in the absence and presence of NADH. This experiment was performed four times with similar results.
Table 2.
Regional [3H]DHR binding parameters in the absence and presence of 200 μm NADH
| Striatum | Dentate gyrus | Cerebellum | |
|---|---|---|---|
| Basal (no NADH) | |||
| KD | 54.7 ± 24.0 | 26.0 ± 5.1 | 15.4 ± 2.7 |
| Bmax | 190 ± 70 | 260 ± 60 | 170 ± 40 |
| nH | 0.98 ± 0.02 | 0.97 ± 0.02 | 0.95 ± 0.03 |
| Enhanced (200 μm NADH) | |||
| KD | 86.0 ± 29.7* | 95.9 ± 22.1 | 88.7 ± 16.9* |
| Bmax | 1150 ± 300* | 2110 ± 550* | 1490 ± 280* |
| nH | 1.00 ± 0.01 | 0.99 ± 0.01 | 1.00 ± 0.01 |
Values represent mean ± SEM; n = 5. KD, nm; Bmax, fmol/mg protein. *p < 0.05 compared with basal conditions by paired t test.
Pharmacology of [3H]DHR binding
The effects of four complex I inhibitors on [3H]DHR binding were examined in competition studies. The natural compound, rotenone, inhibited binding with an IC50 of 8–20 nm and a Hill coefficient that was not significantly different from 1 (Fig.4, Table 3). Meperidine had an IC50 of 34–57 μm and a Hill coefficient of 1. Amobarbitol, a less potent rotenone site blocker, inhibited [3H]DHR binding with an IC50 of ∼400 μm (Table3). Amobarbitol did not compete for 100% of the [3H]DHR binding sites (Fig. 4). MPP+ inhibits complex I activity with an IC50 in the low millimolar range (Ramsay et al., 1987); in our assay, it inhibited [3H]DHR binding with an IC50 of 4–5 mm. Unlike [3H]DHR and rotenone, amobarbitol and MPP+ both had Hill coefficients significantly >1 (Table 3). There was an excellent correlation between IC50 values for [3H]DHR binding versus IC50 values for complex I enzyme activity (from the literature), for inhibitors ranging 100,000-fold in potency (Fig. 5;R2 = 0.99; p < 0.0001). Although NADH markedly enhanced its binding, the IC50 and Hill coefficient of MPP+ did not differ in the absence or presence of NADH (data not shown), and in either case, MPP+was able to compete fully for [3H]DHR binding (Fig. 6). Double-reciprocal plots of [3H]DHR saturation experiments performed in the presence of 0, 1, and 5 mmMPP+ indicated that the interaction of MPP+ with the [3H]DHR binding site was competitive (Fig. 7). In the dg, theKD and Bmaxwere 17.8 ± 4.0 nm and 1.16 ± 0.08 pmol/mg tissue in the absence of MPP+, 25.1 ± 5.7 nm and 1.07 ± 0.06 pmol/mg tissue in the presence of 1 mm MPP+, and 133.1 ± 74.9 nm and 1.79 ± 0.48 pmol/mg tissue in the presence of 5 mmMPP+ (n = 4).
Fig. 4.

Competition for striatal [3H]DHR binding sites by rotenone, meperidine, amobarbitol, and MPP+. Values represent mean ± SEM (n = 4).
Table 3.
IC50 values and Hill slopes of complex I inhibitors competing for [3H]DHR binding
| Inhibitor | Striatum | Dentate gyrus | Cerebellum |
|---|---|---|---|
| Rotenone | |||
| IC50(nm) | 19.9 ± 5.0 | 11.2 ± 2.0 | 8.2 ± 1.3 |
| nH | 1.07 ± 0.04 | 1.06 ± 0.03 | 1.10 ± 0.08 |
| Meperidine | |||
| IC50 (μm) | 34.4 ± 1.3 | 56.7 ± 2.2 | 50.9 ± 6.9 |
| nH | 1.03 ± 0.05 | 1.12 ± 0.11 | 1.14 ± 0.09 |
| Amobarbitol | |||
| IC50 (μm) | 427 ± 36 | 374 ± 25 | 426 ± 33 |
| nH | 3.08 ± 0.36* | 2.83 ± 0.23** | 2.04 ± 0.11** |
| MPP+ | |||
| IC50 (mm) | 3.96 ± 0.49 | 4.73 ± 0.56 | 4.56 ± 0.24 |
| nH | 1.65 ± 0.31* | 1.66 ± 0.12* | 1.82 ± 0.12** |
Values represent mean ± SEM; n = 4. [3H]DHR concentration was 5 nm, NADH concentration was 200 μm. *p < 0.05, **p < 0.005, compared with 1 by one-samplet test.
Fig. 5.

Correlation between IC50values for [3H]DHR binding obtained in the current study and IC50 values for complex I enzyme activity obtained from the literature.R2 = 0.999; p < 0.0001. Superscript numbers refer to references from which IC50 values were obtained:1Ueno et al., 1994; 2Earley et al., 1987; 3Filser and Werner, 1988;4Andreani et al., 1994;5Ramsay et al., 1991.
Fig. 6.

[3H]DHR binding is completely displaced by MPP+ under both basal (no added NADH) and enhanced (200 μm NADH) conditions. The IC50 and Hill coefficient were not changed by manipulating NADH concentrations.
Fig. 7.

Inhibition of [3H]DHR binding by MPP+ is competitive. [3H]DHR saturation studies were performed in the presence of 0, 1, and 5 mmMPP+. The double-reciprocal plot demonstrates that increasing concentrations of MPP+ decreases the apparent affinity of [3H]DHR binding without changing the number of binding sites.
Regional distribution of [3H]DHR binding
Basal
Of the tissues assayed, kidney and heart had, by far, the highest levels of [3H]DHR binding (Fig.8, Table 4). Without supplemental NADH, binding in the renal medulla (369.8 ± 17.1 fmol/mg tissue) was more than 10-fold higher than in the external plexiform layer (epl) of the olfactory bulb (34.8 ± 17.1 fmol/mg tissue), the brain region with greatest binding. [3H]DHR binding in the heart (315.9 ± 17.1 fmol/mg tissue) was nearly as dense as in the renal medulla.
Fig. 8.
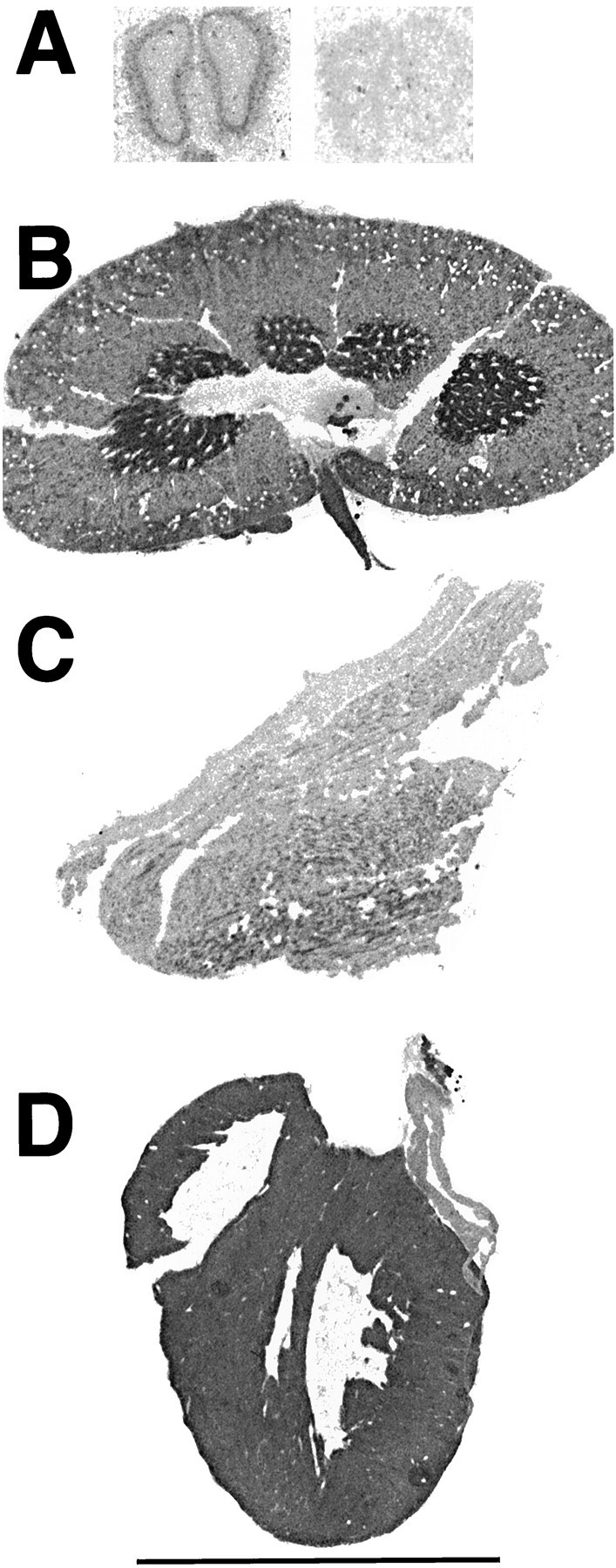
Basal [3H]DHR binding (in the absence of added NADH) in olfactory bulb (A), kidney (B), skeletal muscle (C), and heart (D). In A, the image on the rightrepresents nonspecific binding. All images were captured, processed, and printed identically. Scale bar, 1 cm.
Table 4.
Regional distribution of basal and NADH-enhanced [3H]DHR binding
| Region | Basal | Enhanced | % Enhancement |
|---|---|---|---|
| Olfactory bulb | |||
| Glomerular layer | 8.17 ± 1.77 (24) | 266.1 ± 19.2 (59) | 3157 |
| External plexiform layer | 34.8 ± 7.12 (100) | 452.9 ± 26.4 (100) | 1201 |
| Internal granular layer | 13.4 ± 1.74 (39) | 157.6 ± 11.3 (39) | 1073 |
| Basal ganglia | |||
| Striatum | 9.16 ± 1.18 (26) | 132.8 ± 9.43 (29) | 1350 |
| Globus pallidus | 3.35 ± 0.28 (10) | 38.10 ± 3.70 (8) | 1037 |
| Substantia nigra | 4.08 ± 1.58 (12) | 131.0 ± 13.0 (29) | 3111 |
| Cerebral cortex | |||
| Parietal, outer | 17.9 ± 2.85 (52) | 227.7 ± 9.65 (50) | 1170 |
| Parietal, inner | 10.6 ± 0.88 (31) | 88.11 ± 7.93 (20) | 730 |
| Septal region | |||
| Lateral septal nucleus | 21.5 ± 3.33 (62) | 189.6 ± 11.0 (42) | 783 |
| Diagonal band | 8.25 ± 4.52 (24) | 129.1 ± 10.8 (29) | 1465 |
| Nucleus accumbens | ND | 219.8 ± 5.85 (49) | — |
| Thalamus | |||
| Reticular | 6.88 ± 3.22 (20) | 71.80 ± 8.68 (16) | 944 |
| Ventroposteromedial | 13.0 ± 10.0 (37) | 130.0 ± 17.6 (29) | 900 |
| Laterodorsal | 4.76 ± 1.65 (14) | 175.8 ± 23.0 (39) | 3592 |
| Habenula | 6.48 ± 1.91 (19) | 110.0 ± 15.7 (24) | 1597 |
| Medial geniculate | 2.22 ± 1.19 (6) | 115.1 ± 19.5 (25) | 5086 |
| Hippocampus | |||
| CA1 | |||
| Stratum moleculare | 24.2 ± 3.95 (70) | 218.0 ± 23.7 (48) | 799 |
| Stratum radiatum | 14.1 ± 2.44 (41) | 229.0 ± 28.8 (51) | 1519 |
| Stratum pyramidale | 13.2 ± 3.24 (38) | 63.4 ± 12.9 (14) | 381 |
| Stratum oriens | 12.5 ± 3.10 (36) | 217.9 ± 16.7 (48) | 1636 |
| CA2 | 16.7 ± 3.91 (48) | 220.7 ± 18.7 (49) | 1218 |
| CA3 | 12.9 ± 3.73 (37) | 244.1 ± 26.2 (54) | 1795 |
| Dentate gyrus | 21.2 ± 4.22 (61) | 327.1 ± 29.8 (72) | 1446 |
| Forebrain, other | |||
| Hypothalamus | 7.76 ± 3.31 (22) | 157.6 ± 9.24 (35) | 1932 |
| Amygdala, basolateral | 13.1 ± 3.87 (38) | 127.2 ± 17.8 (28) | 869 |
| Brainstem | |||
| Superior colliculus | |||
| Superficial gray layer | 10.0 ± 3.60 (29) | 199.6 ± 24.0 (44) | 1904 |
| Optic nerve layer | 2.38 ± 1.69 (7) | 82.56 ± 15.9 (18) | 3369 |
| Periaqueductal gray | ND | 120.7 ± 23.0 (27) | — |
| Interpeduncular nucleus | 10.1 ± 4.56 (29) | 245.9 ± 10.2 (54) | 2334 |
| Oculomotor nucleus | ND | 244.5 ± 15.8 (54) | — |
| Facial nucleus | 6.82 ± 1.08 (20) | 216.3 ± 28.5 (48) | 3072 |
| Hypoglossal nucleus | ND | 236.8 ± 18.2 (52) | — |
| Cerebellum | |||
| Molecular layer | 3.61 ± 0.08 (10) | 283.4 ± 35.0 (63) | 7750 |
| Granular layer | 3.52 ± 1.61 (10) | 167.8 ± 15.5 (37) | 4668 |
| Kidney | |||
| Cortex | 161.1 ± 19.9 (463) | 451.6 ± 15.5 (100) | 174 |
| Medulla | 369.8 ± 26.7 (1060) | 571.8 ± 10.5 (126) | 55 |
| Muscle | |||
| Striated | 163.6 ± 94.3 (470) | 207.9 ± 76.3 (46) | 27 |
| Cardiac | 315.9 ± 17.1 (908) | 544.3 ± 11.0 (120) | 72 |
Enhanced binding was performed in the presence of 200 μm NADH; [3H]DHR concentration was 5 nm. Binding is in units of fmol/mg tissue; values represent mean ± SEM (n = 4). ND, Not distinguishable from background (% external plexiform layer binding).
In brain, basal [3H]DHR binding (5 nm, without supplemental NADH) was heterogeneously distributed (Table 4). Binding in the epl was more than twice as high as in the majority of 30 other brain regions examined. Relatively high levels of [3H]DHR were bound in the lateral septum and hippocampal formation. Binding in most structures, including certain thalamic, basal ganglia, and brainstem nuclei, was only 20–30% of that found in the epl. Delineation of specific thalamic nuclei was difficult. A laminar pattern was present in the cerebral cortex, with denser labeling of superficial layers. In contrast, differentiation of lamina within the cerebellar cortex was difficult. Specific binding could not be detected in several regions, mostly in brainstem. There was negligible binding in white matter regions, such as the corpus callosum.
Enhanced
In the presence of 200 μm NADH (enhanced binding), [3H]DHR binding (5 nm) increased dramatically in most regions of the brain (Fig. 9, Table 4). In contrast, NADH modulation of [3H]DHR binding in non-neural tissues was much less pronounced, but did vary regionally (Fig. 9, Table 4). Only in the renal cortex did [3H]DHR binding increase substantially (174%).
Fig. 9.

Enhancement of [3H]DHR binding by 200 μm NADH in skeletal muscle (A), kidney (B), and brain (C). Note the marked enhancement in brain relative to other tissues. All images were captured, processed, and printed identically. Scale bar, 1 cm.
In certain brain structures, including the cerebellar cortex, substantia nigra, and superior colliculus, binding increased 20-fold or more. A minority of brain regions, such as the basolateral amygdala, septum, and reticular nucleus of the thalamus, had a smaller response to NADH (less than 10-fold increase). Of the areas surveyed, the smallest response to NADH, a fourfold increase, occurred in the CA1 pyramidal cell layer, and the most striking potentiation was seen in cmol. Under basal conditions, only four structures contained lesser amounts of [3H]DHR than the cmol. In the presence of 200 μm NADH, only two regions possessed more label, the epl and dg. The distribution of [3H]DHR binding sites in the presence of 200 μm NADH is shown in Figure10.
Fig. 10.
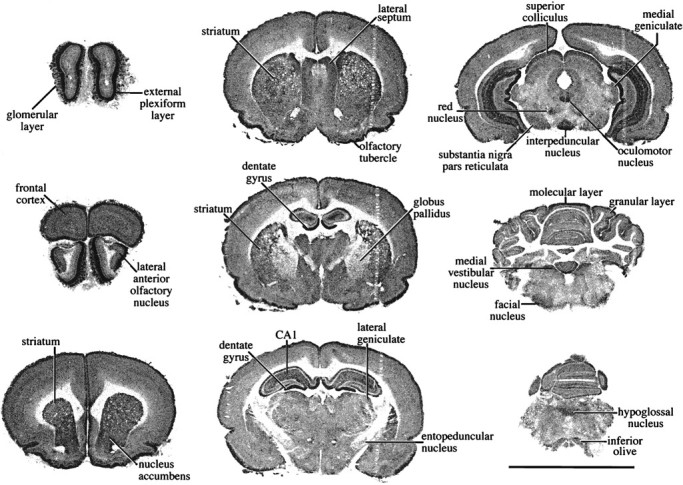
Regional distribution of brain [3H]DHR binding in the presence of 200 μm NADH. All images were captured, processed, and printed identically. Scale bar, 1 cm.
Effects of entorhinal cortex lesions
One week after injection of NMDA into the medial entorhinal cortex (n = 3), there was a modest local decrease in binding (25–30%), particularly in superficial cortical layers, and a more substantial loss of binding (51%; 24.7 ± 4.1 vs 12.2 ± 4.3 fmol/mg tissue; p < 0.05) in the middle one-third of the molecular layer of the dg (Fig. 11).
Fig. 11.

Excitotoxic lesions of entorhinal cortex result in local and distant losses of [3H]DHR binding to complex I. A, One week after the NMDA infusion, there is a moderate (30%) local loss of binding, particularly in the superficial layers of entorhinal cortex (thickarrows). In the middle one-third of the molecular layer of the dg, the region to which the lesioned entorhinal neurons project, there is a 50% decrease in binding (thin arrows). Thus, it appears that a substantial portion of complex I is associated with synaptic terminals in this region. B, Unlesioned hemisphere from the same brain.
DISCUSSION
We developed an autoradiographic binding assay to study complex I using [3H]DHR. Radioligand assays of complex I are not new. In electron transport particle preparations, [14C]-labeled complex I inhibitors rotenone (Horgan et al., 1968) and piericidin A (Gutman et al., 1970) have been used to probe this enzyme. These earlier studies were hampered by substantial nonspecific binding, which was reduced, although not satisfactorily, by albumin. The low specific activity of [14C]rotenone (2.36 mCi/mmol), more than 20,000-fold lower than that of [3H]DHR, undoubtedly contributed to the high level of nonspecific binding. As can be seen in Figure 1, nonspecific binding increases linearly with ligand concentration; however, the high specific activity of [3H]DHR facilitates working at low ligand concentrations at which the signal-to-noise ratio is excellent, and >80% of binding is specific. In contrast, to obtain a reliable signal using [14C]rotenone, much higher concentrations are required, accompanied by a linear increase in nonspecific binding. For example, in the classic study of Horgan et al. (1968), thelowest concentration of [14C]rotenone used in the binding assay was ∼125 nm (estimated from their Fig. 2), which is higher than the highest concentration of [3H]DHR used in our study. Finally, as discussed below, NADH markedly enhances specific binding without affecting nonspecific binding, further improving the signal-to-noise ratio. Thus, the high degree of specific binding obtained with [3H]DHR compared with [14C]rotenone is not surprising.
Specific [3H]DHR binding was defined as that binding that was displaceable by a saturating concentration of rotenone. Further evidence of the specificity of [3H]DHR binding was obtained by detailed competition studies using well known inhibitors of complex I. Rotenone inhibited binding with an IC50 of 8–20 nm, consistent with its potency as a complex I inhibitor (Horgan et al., 1968). The competition data yielded a Hill slope of 1, which suggests a simple competition for [3H]DHR binding sites. Meperidine inhibited binding with an IC50 of ∼50 μm, close to its IC50 for enzyme activity of ∼200 μm. Amobarbitol, one of the first complex I inhibitors described (Ernster et al., 1955), inhibits mitochondrial function at concentrations in the high micromolar-to-low millimolar range (Ernster et al., 1963). We found that amobarbitol inhibited [3H]DHR binding with an IC50 of 400 μm, but did not displace ∼30% of specifically bound [3H]DHR. Unlike rotenone, amobarbitol had Hill slopes of 2–3. MPP+ is a weak complex I inhibitor, having an IC50 of 5–10 mm in submitochondrial particles (Hoppel et al., 1987) and electron transport particles (Ramsay et al., 1987). In the [3H]DHR binding assay, MPP+ had an IC50 of 4–5 mm and a Hill slope that was significantly >1; these binding parameters were not changed by the absence or presence of NADH. Both the rank order and absolute potency of these inhibitors indicate that the pharmacology of [3H]DHR binding is consistent with the rotenone site of complex I (Fig. 5).
Based on hyperbolic saturation isotherms, Hill slopes of 1, and linear Scatchard plots, it appears that [3H]DHR binds to a single class of sites. Unlike earlier studies of [14C]rotenone binding (Horgan et al., 1968), we did not find sigmoid binding curves that would suggest multiple, interacting binding sites; however, we did not use the extremely high ligand concentrations of the earlier studies. Moreover, it is possible that [3H]DHR binds to more than one site, each having a very similar affinity. This could produce linear Scatchard plots and Hill slopes of 1. Consistent with this possibility are the steep Hill slopes for amobarbitol and MPP+, which may indicate positive cooperativity of multiple binding sites. MPP+ inhibition of complex I has been suggested to be a two-site process. According to this scheme, MPP+ initially binds to a hydrophilic site accessible to the matrix compartment, resulting in partial inhibition. Subsequent access to a hydrophobic site within the mitochondrial membrane produces complete inhibition (Singer and Ramsay, 1994). An interaction of this nature may account for the high Hill numbers obtained in the current study. In addition, although MPP+ has a steep Hill slope and [3H]DHR does not, double-reciprocal plots indicate that the interaction of these compounds at this site is competitive. The fact that amobarbitol did not compete for all [3H]DHR binding sites may reflect the presence of distinct classes of binding sites with different pharmacologies.
NADH dramatically increased the number of [3H]DHR binding sites, probably by an allosteric mechanism. The EC50 for NADH stimulation of [3H]DHR binding (20–40 μm) closely matches theKm of the enzyme for NADH (Hatefi, 1985). Complex I can be resolved into membrane-based and peripheral components (Hofhaus et al., 1991). The NADH recognition site is believed to reside in the hydrophilic arm of the enzyme, which projects into the mitochondrial matrix. The rotenone site, localized to the membrane adjacent to the ubiquinone binding site (Pilkington et al., 1993), is a substantial distance from the NADH site. After reduction by NADH, conformational changes in complex I have been demonstrated by cross-linking experiments (Belogrudov and Hatefi, 1994). In addition, NADH modifies electron spin resonance spectra of complex I, consistent with structural alterations in the region of Fe-S cluster 2 (deJong et al., 1994). Although the mechanism of NADH enhancement of binding remains to be defined, enzyme reduction likely induces conformational changes that facilitate access of [3H]DHR to the membrane-based recognition site. Because NADH binding to the “proximal” hydrophilic portion of the enzyme modulates the binding of [3H]DHR to the “distal” hydrophobic segment, the response to NADH can be used to probe the ability of the enzyme complex to undergo allosteric modification. Only reduced adenine nucleotides are capable of inducing these changes in complex I, as NAD+ minimally affected [3H]DHR binding.
Within the brain, there were marked regional differences in the degree to which NADH enhanced binding. In those structures in which full saturation studies were performed without and with NADH, the increase in Bmax varied from 600% in str to 900% in cmol. This difference in NADH response was even more striking when brain was compared with other organs. In non-neural tissue, the average enhancement was only 82% (Table 4). The reasons for these differences are not known, but are unlikely to be related to different levels of residual NADH because extensive prewashes did not alter binding. It has been suggested that there may be tissue-specific isozymes of complex I (Clay and Ragan, 1988). Regional variations in the EC50 values for NADH and NADPH, and in the affinities of rotenone and dihydrorotenone, provide further support for this hypothesis.
The binding of [3H]DHR is also enhanced by NADPH. Direct oxidation of NADPH by complex I has been reported byHatefi (1973). In those studies, the pattern of Fe-S cluster reduction suggested entry of reducing equivalents at a site distinct from NADH. In addition, adjacent to complex I in the mitochondrial membrane is NAD(P)H transhydrogenase (Hatefi and Hanstein, 1973). Conceivably, transfer of hydride ions from NADPH to endogenous NAD+ could contribute to this phenomenon.
[3H]DHR binding is heterogeneously distributed in the brain as are two other enzymes of the ETC, succinate dehydrogenase (complex II; Pandykula, 1952) and cytochrome oxidase (complex IV; Hevner et al., 1995). The density of [3H]DHR binding sites varied more than 10-fold across brain regions. As with other ETC enzymes, complex I appears to be concentrated in neurons; binding in white matter structures is negligible, even under enhanced conditions. In addition, the highest levels of binding in brain are found in the epl, dg, and cmol, which are relatively acellular regions with high densities of excitatory synapses. This finding is consistent with the work of Wong-Riley (1989)showing that cytochrome oxidase is preferentially concentrated in synaptic terminals and dendrites. A distant lesion of entorhinal cortex, which removes synaptic input to a portion of the dg, dramatically decreases binding in this region and lends support to the contention that a substantial portion of complex I (50%) is associated with synaptic terminals. Functional studies also support the notion that ETC enzymes, including complex I, are concentrated in regions of high synaptic density. For example, experimental manipulations that alter neuronal firing patterns across several synapses result in alterations of succinate dehydrogenase and cytochrome oxidase activity (Wong-Riley, 1979; Marshall et al., 1981; Porter et al., 1994; Blandini and Greenamyre, 1995). We have shown recently that [3H]DHR binding to complex I regulates regionally in a manner identical to these other enzymes (Blandini et al., 1995).
The assay described in this report may be useful for studying human neurological disorders. Selective defects in complex I have been implicated in the pathogenesis of MPTP-induced parkinsonism (Nicklas et al., 1985), idiopathic Parkinson’s disease (Schapira et al., 1990b), Leber’s hereditary optic neuropathy (Smith et al., 1994), and idiopathic dystonia (Benecke et al., 1992). Previously, there has been no technique that could provide quantitative information about complex I with a high degree of anatomic precision. [3H]DHR autoradiography has this capability, but it is not without limitations. Enzyme activity is not measured in our assay. Instead, quantitative information about the rotenone binding site of complex I is obtained. Nonetheless, because rotenone inhibition defines the activity of complex I, defects in [3H]DHR binding can be expected to reflect functional changes in the enzyme. Moreover, the modulatory effect of NADH provides an indication of the functional integrity of the complex—its ability to regulate allosterically.
In summary, we have developed and characterized an assay for complex I based on the binding of [3H]DHR to the enzyme. [3H]DHR binding is specific, saturable, and has the pharmacological profile of a ligand interacting with the rotenone site. Binding in brain is enhanced by NADH, apparently by an allosteric mechanism that reveals more binding sites. Autoradiography of [3H]DHR binding to brain sections provides a simple, reproducible method for obtaining, with excellent spatial resolution, quantitative information about complex I.
Footnotes
This work was supported by a Mallinckrodt Scholar Award (J.T.G.), the National Parkinson Foundation Center of Excellence at the University of Rochester, Public Health Service Grants NS33779 and AG11755, and the Rochester Area Pepper Center.
Correspondence should be addressed to Dr. J. Timothy Greenamyre, Emory University School of Medicine, Department of Neurology, Woodruff Memorial Building, Suite 6000, P.O. Drawer V, Atlanta, GA 30322.
Dr. Higgins’ current address: Ohio State University School of Medicine, Department of Neurology, 1654 Upham Drive, Columbus, OH 43210.
REFERENCES
- 1.Andreani A, Rambaldi M, Locatelli A, Leoni A, Ghelli A, Degli Espositi M. Thienylvinylindoles as inhibitors of mitochondrial NADH dehydrogenase. Pharm Acta Helv. 1994;69:15–20. doi: 10.1016/0031-6865(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 2.Belogrudov G, Hatefi Y. Catalytic sector of complex I (NADH:ubiquinone oxidoreductase): subunit stoichiometry and substrate induced conformation changes. Biochemistry. 1994;33:4571–4576. doi: 10.1021/bi00181a018. [DOI] [PubMed] [Google Scholar]
- 3.Benecke R, Strumper P, Weiss H. Electron transfer complex I defect in idiopathic dystonia. Ann Neurol. 1992;32:683–686. doi: 10.1002/ana.410320512. [DOI] [PubMed] [Google Scholar]
- 4.Bindoff LA, Birch-Martin M, Cartlidge NEF, Parker WD, Turnbull DM. Mitochondrial function in Parkinson’s disease. Lancet. 1989;1:49. doi: 10.1016/s0140-6736(89)90291-2. [DOI] [PubMed] [Google Scholar]
- 5.Blandini F, Greenamyre JT. Effects of subthalamic nucleus lesions on mitochondrial enzyme activity in the rat basal ganglia. Brain Res. 1995;669:59–66. doi: 10.1016/0006-8993(94)01233-8. [DOI] [PubMed] [Google Scholar]
- 6.Blandini F, Porter RHP, Greenamyre JT. Autoradiographic study of mitochondrial complex I and glutamate receptors in the basal ganglia of rats after unilateral subthalamic lesion. Neurosci Lett. 1995;186:99–102. doi: 10.1016/0304-3940(95)11292-5. [DOI] [PubMed] [Google Scholar]
- 7.Capaldi RA. Structure and function of cytochrome c oxidase. Annu Rev Biochem. 1990;59:569–596. doi: 10.1146/annurev.bi.59.070190.003033. [DOI] [PubMed] [Google Scholar]
- 8.Capaldi RA, Aggeler R, Turina P, Wilkens S. Coupling between catalytic sites and the proton channel in F1F0-type ATPases. Trends Biochem Sci. 1994;19:284–289. doi: 10.1016/0968-0004(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 9.Caughey WS, Wallace WJ, Volpe JA, Yoshikawa S. Cytochrome c oxidase. In: Boyer PD, editor. The enzymes. Academic; New York: 1976. pp. 299–345. [Google Scholar]
- 10.Clay VJ, Ragan CI. Evidence of the existence of tissue specific isoenzymes of mitochondrial NADH dehydrogenase. Biochem Biophys Res Commun. 1988;157:1423–1428. doi: 10.1016/s0006-291x(88)81034-9. [DOI] [PubMed] [Google Scholar]
- 11.Cooper JM, Clark JB. The structural organization of the mitochondrial respiratory chain. In: Schapira AHV, DiMauro S, editors. Mitochondrial disorders in neurology. Butterworth-Heinemann; New York: 1994. pp. 1–30. [Google Scholar]
- 12.Davis GC, Williams AC, Markey SP, Ebert MH, Caine ED, Reichert CM, Kopin IJ. Chronic parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1979;1:249–254. doi: 10.1016/0165-1781(79)90006-4. [DOI] [PubMed] [Google Scholar]
- 13.deJong AMPh AMPh, Kotylar AB, Albracht SPJ. Energy-induced structural changes in NADH:Q oxidoreductase of the mitochondrial respiratory chain. Biochim Biophys Acta. 1994;1186:163–171. doi: 10.1016/0005-2728(94)90175-9. [DOI] [PubMed] [Google Scholar]
- 14.Earley FGP, Patel SD, Ragan CI, Attardi G. Photolabelling of a mitochondrially encoded subunit of NADH dehydrogenase with [3H]dihydrorotenone. FEBS Lett. 1987;219:108–113. doi: 10.1016/0014-5793(87)81200-0. [DOI] [PubMed] [Google Scholar]
- 15.Erecinska M, Silver IA. ATP and brain function. J Cereb Blood Flow Metab. 1989;9:2–19. doi: 10.1038/jcbfm.1989.2. [DOI] [PubMed] [Google Scholar]
- 16.Ernster L, Jalling O, Low H, Lindberg O. Alternate pathways of mitochondrial DPNH oxidation studies with amytal. Exp Cell Res (Suppl) 1955;3:124–132. [PubMed] [Google Scholar]
- 17.Ernster L, Dallner G, Azzone GF. Differential effects of rotenone and amytal on mitochondrial electron and energy transfer. J Biol Chem. 1963;238:1124–1131. [Google Scholar]
- 18.Filser M, Werner S. Pethidine analogues, a novel class of potent inhibitors of mitochondrial NADH:ubiquinone reductase. Biochem Pharmacol. 1988;37:2551–2558. doi: 10.1016/0006-2952(88)90245-6. [DOI] [PubMed] [Google Scholar]
- 19.Greenamyre JT, Higgins DS, Eller RV. Quantitative autoradiography of dihydrorotenone binding to complex I of the electron transport chain. J Neurochem. 1992;59:746–749. doi: 10.1111/j.1471-4159.1992.tb09431.x. [DOI] [PubMed] [Google Scholar]
- 20.Gutman M, Singer TP, Casida JE. Studies on the respiratory chain-linked reduced nicotinamide adenine dinucleotide dehydrogenase: XVII. Reaction sites of piericidin A and rotenone. J Biol Chem. 1970;245:1992–1997. [PubMed] [Google Scholar]
- 21.Hatefi Y. Oxidation of reduced triphosphopyridine nucleotide by submitochondrial particles from beef heart. Biochem Biophys Res Commun. 1973;50:978–984. doi: 10.1016/0006-291x(73)91502-7. [DOI] [PubMed] [Google Scholar]
- 22.Hatefi Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu Rev Biochem. 1985;54:1015–1069. doi: 10.1146/annurev.bi.54.070185.005055. [DOI] [PubMed] [Google Scholar]
- 23.Hatefi Y, Hanstein WG. Interactions of reduced and oxidized triphosphopyridine nucleotides with the electron-transport system of bovine heart mitochondria. Biochemistry. 1973;12:3515–3522. doi: 10.1021/bi00742a026. [DOI] [PubMed] [Google Scholar]
- 24.Hevner RF, Liu S, Wong-Riley MTT. A metabolic map of cytochrome oxidase in the rat brain: histochemical, densitometric and biochemical studies. Neuroscience. 1995;65:313–342. doi: 10.1016/0306-4522(94)00514-6. [DOI] [PubMed] [Google Scholar]
- 25.Hofhaus G, Weiss H, Leonard K. Electron microscopic analysis of the peripheral and membrane parts of mitochondrial NADH dehydrogenase (complex I). J Mol Biol. 1991;221:1027–1043. doi: 10.1016/0022-2836(91)80190-6. [DOI] [PubMed] [Google Scholar]
- 26.Hoppel CL, Grinblatt D, Kwok HC, Arora PK, Singh MP, Sayre LM. Inhibition of mitochondrial respiration by analogues of 4-phenylpyridine and 1-methyl-4-phenyl pyridinium cation (MPP+) the neurotoxic metabolite of MPTP. Biochem Biophys Res Commun. 1987;148:684–693. doi: 10.1016/0006-291x(87)90931-4. [DOI] [PubMed] [Google Scholar]
- 27.Horgan DJ, Singer TP, Casida JE. Studies on the respiratory chain-linked reduced nicotinamide adenine dinucleotide dehydrogenase: XIII. Binding sites of rotenone, piericidin A and amytal in the respiratory chain. J Biol Chem. 1968;243:834–843. [PubMed] [Google Scholar]
- 28.Levisohn LF, Isacson O. Excitotoxic lesions of the rat entorhinal cortex. Effects of selective neuronal damage on acquisition and retention of a non-spatial reference memory task. Brain Res. 1991;564:230–244. doi: 10.1016/0006-8993(91)91458-d. [DOI] [PubMed] [Google Scholar]
- 29.Marshall JF, Critchfield JW, Kozlowski MR. Altered succinate dehydrogenase activity of basal ganglia following damage to mesotelencephalic dopaminergic projection. Brain Res. 1981;212:367–377. doi: 10.1016/0006-8993(81)90469-8. [DOI] [PubMed] [Google Scholar]
- 30.Munson PJ, Rodbard D. Ligand: a versatile computerized approach for characterization of ligand-binding systems. Anal Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- 31.Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Life Sci. 1985;36:2503–2508. doi: 10.1016/0024-3205(85)90146-8. [DOI] [PubMed] [Google Scholar]
- 32.Pandykula HA. The localization of succinic dehydrogenase in tissue sections of the rat. Am J Anat. 1952;91:107–145. doi: 10.1002/aja.1000910104. [DOI] [PubMed] [Google Scholar]
- 33.Paxinos G, Watson C. Academic; Sydney: 1986. The rat brain in stereotaxic coordinates. . [DOI] [PubMed] [Google Scholar]
- 34.Pilkington SJ, Arizmendi JM, Fearnley IM, Runswick MJ, Skehel JM, Walker JE. Structural organization of complex I from bovine mitochondria. Biochem Soc Trans. 1993;21:26–31. doi: 10.1042/bst0210026. [DOI] [PubMed] [Google Scholar]
- 35.Porter RHP, Greene JG, Higgins DS, Greenamyre JT. Polysynaptic regulation of glutamate receptors and mitochondrial enzyme activities in the basal ganglia of rats with unilateral dopamine depletion. J Neurosci. 1994;14:7192–7199. doi: 10.1523/JNEUROSCI.14-11-07192.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramsay RR, McKeown KA, Johnson EA, Booth RG, Singer TP. Inhibition of NADH oxidation by pyridine derivatives. Biochem Biophys Res Commun. 1987;146:53–60. doi: 10.1016/0006-291x(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 37.Ramsay RR, Krueger MJ, Youngster SK, Gluck MR, Casida JE, Singer TP. Interaction of 1-methyl-4-phenylpyridinium ion (MPP+) and its analogs with the rotenone/piericidin binding site of NADH dehydrogenase. J Neurochem. 1991;56:1184–1190. doi: 10.1111/j.1471-4159.1991.tb11409.x. [DOI] [PubMed] [Google Scholar]
- 38.Rieske JS. Composition, structure and function of complex III of the respiratory chain. Biochim Biophys Acta. 1976;456:195–247. doi: 10.1016/0304-4173(76)90012-4. [DOI] [PubMed] [Google Scholar]
- 39.Schapira AHV, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem. 1990a;54:823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- 40.Schapira AHV, Mann VM, Cooper JM, Dexter D, Daniel SE, Jenner P, Clark JB, Marsden CD. Anatomic and disease specificity of NADHCoQ1 reductase (complex I) deficiency in Parkinson’s disease. J Neurochem. 1990b;55:2142–2145. doi: 10.1111/j.1471-4159.1990.tb05809.x. [DOI] [PubMed] [Google Scholar]
- 41.Shoffner JM, Watts RL, Juncos JL, Torrini A, Wallace DC. Mitochondrial oxidative phosphorylation defects in Parkinson’s disease. Ann Neurol. 1991;30:332–339. doi: 10.1002/ana.410300304. [DOI] [PubMed] [Google Scholar]
- 42.Singer TP, Ramsay RR. The reaction sites of rotenone and ubiquinone with mitochondrial NADH dehydrogenase. Biochim Biophys Acta. 1994;1187:198–202. doi: 10.1016/0005-2728(94)90110-4. [DOI] [PubMed] [Google Scholar]
- 43.Smith PR, Cooper JM, Govan GG, Harding AE, Schapira AHV. Platelet mitochondrial function in Leber’s hereditary optic neuropathy. J Neurol Sci. 1994;122:80–83. doi: 10.1016/0022-510x(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 44.Trijbels JMF, Scholte HR, Ruitenbeek W, Sengers RCA, Janssen AJM, Busch HFM. Problems with the biochemical diagnosis in mitochondrial (encephalo-) myopathies. Eur J Pediatr. 1993;152:178–184. doi: 10.1007/BF01956139. [DOI] [PubMed] [Google Scholar]
- 45.Ueno H, Miyoshi H, Ebisui K, Iwamura H. Comparison of the inhibitory action of natural rotenone and its stereoisomers with various NADH-ubiquinone reductases. Eur J Biochem. 1994;225:411–417. doi: 10.1111/j.1432-1033.1994.00411.x. [DOI] [PubMed] [Google Scholar]
- 46.Wong-Riley M. Changes in the visual system of the monocularly sutured or enucleated cats demonstrable with cytochrome oxidase histochemistry. Brain Res. 1979;171:11–28. doi: 10.1016/0006-8993(79)90728-5. [DOI] [PubMed] [Google Scholar]
- 47.Wong-Riley MTT. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989;12:94–101. doi: 10.1016/0166-2236(89)90165-3. [DOI] [PubMed] [Google Scholar]