

Abstract
Nicotinic acetylcholine receptor (nAChR) genes are regulated by muscle electrical activity. E-box sequences found in their promoters are necessary for this regulation. However, many muscle genes contain E-boxes, yet are not regulated by muscle depolarization. This suggests that other elements are necessary, perhaps working in conjunction with E-boxes, to confer depolarization-dependent control onto promoter activity. We have used direct DNA injection into muscle as an in vivo assay to identify and characterize these additional elements. Mutagenesis and expression assays identified multiple elements within the first 81 base pairs (bp) of the nAChR δ-subunit promoter that contribute to its regulation by muscle electrical activity. Within this 81 bp sequence, two regions of DNA were identified that were capable of conferring activity-dependent regulation onto a heterologous promoter. The stronger of these two putative enhancers was characterized further. It is a 47 bp sequence that contains an E-box along with sequences similar to the SV40 core enhancer and an SP1 site. Site-directed mutagenesis identified residues within each of these sequences that were necessary for enhancer activity. Furthermore, methylation interference DNA footprinting assays showed increased nuclear protein binding to sequences within both these enhancers after muscle denervation, and this pattern of binding was very similar to that observed with nuclear protein isolated from myotube extracts. These latter results suggest that similar mechanisms may mediate increased nAChR expression during muscle development and after muscle denervation.
Keywords: gene expression, electrical activity, promoter, muscle, transcription, denervation
The neuromuscular junction (NMJ) provides a relatively simple system to study the effects that presynaptic neuronal activity have on postsynaptic gene expression. Nerve-induced muscle electrical activity alters the distribution, composition, and expression of muscle nicotinic acetylcholine receptors (nAChR) (Hall and Sanes, 1993). Specifically, muscle electrical activity suppresses extrajunctional expression of the embryonic-type (α2βγδ) nAChRs (Goldman et al., 1988). Characterization of the mechanisms mediating this activity-dependent expression will not only further our understanding of synapse formation at the NMJ, but also may identify mechanisms that contribute to synaptic plasticity in the adult nervous system.
The control of nAChR expression by muscle activity is largely at the transcriptional level. Both in vivo and in vitrogene transfer experiments have shown that nAChR α-, γ-, and δ-subunit gene promoters can confer activity-dependent expression onto a reporter gene (Merlie and Kornhauser, 1989; Chahine et al., 1992; Simon et al., 1992; Bessereau et al., 1994; Tang et al., 1994;Gilmour et al., 1995). Within these promoters, E-box sequences (CANNTG) have been identified that are necessary for activity-dependent expression (Bessereau et al., 1994; Tang et al., 1994; Su et al., 1995).
These results suggested that trans-acting myogenic factors may mediate activity-dependent expression via binding to theircis-acting E-box elements. Consistent with this possibility is the observation that the myogenic factors also are regulated by muscle depolarization (Eftimie et al., 1991; Chahine et al., 1993) and can regulate nAChR promoter activity (Gilmour et al., 1991; Prody and Merlie, 1991; Berberich et al., 1993; Bessereau et al., 1993). Therefore, muscle activity first may modulate the expression of certain myogenic factors that subsequently bind to and regulate expression from nAChR promoters. However, this does not explain why other muscle-specific, E-box-containing genes are not also regulated by muscle activity. The simplest explanation may be that additional factors bind other nearby DNA elements and, in conjunction with myogenic factor binding to E-boxes, confer activity-dependent expression on the nAChR genes. Identification of these additional sequences will not only confirm this hypothesis, but also provide the necessary probes for identifying the proteins that bind these sequences.
To this end, we assayed the activity of wild-type and mutant rat δ-promoter expression constructs in innervated and denervated muscle tissue. These assays identified multiple elements located throughout the first 81 bp of the δ-promoter that contribute to its regulation by muscle activity. However, no single mutation abolished this regulation completely unless nucleotides (nt) −5 to −81 were deleted. Within this piece of DNA, we identified two regions that appeared to function as activity-dependent enhancers. The stronger of the two enhancers contains an E-box that was shown previously to be necessary for activity-dependent control of the mouse δ-promoter (Tang et al., 1994). This enhancer also contains sequences similar to the SV40 core enhancer (Khoury and Gruss, 1983) and an SP1 site. The importance of each of these sequences in activity-dependent regulation was confirmed by site-directed mutagenesis.
MATERIALS AND METHODS
Plasmids. For the enhancer experiments, a minimal enkephalin promoter construct (MEK pXP2) was made by placing a 135 bp fragment of the enkephalin promoter (Comb et al., 1986), extending from nt −72 to +60, relative to the transcriptional start site, in theSmaI and filled in BglII sites of the pXP2 vector (Nordeen, 1988). Oligonucleotides containing δ-promoter sequences (Chahine et al., 1992) as indicated in Figure 5 (δ −81/−32, δ −47/−1) were synthesized with a BamHI restriction enzyme site included on their 5′ ends and a BglII restriction enzyme site included on their 3′ ends. These oligos then were placed in the BamHI site of the MEK pXP2 construct. These oligos also were subcloned into BSSK(+) for sequence determination.
Fig. 5.

Two enhancers from the δ −102 bp promoter can confer activity-dependent regulation onto the MEK promoter. Diagram at the top of the figure shows the 102 bp δ-promoter region with putative regulatory elements indicated by boxes andrectangles. Below this are diagrams of the various constructs containing fragments of the δ-promoter subcloned in front of an MEK promoter driving Luc expression. These constructs were injected into innervated and denervated EDL muscles along with CMVCAT for normalization. One week later, muscles were harvested and Luc and CAT activities were measured. Results are shown with SD on the right side of the figure along with the fold differences. Expression levels are reported relative to the MEK Luc construct. Numbers in parenthesis indicate the number of animals injected. p-values (shown in brackets) were calculated using a one-tailed t test.
For the mutation and deletion studies on the δ-promoter, we used the pXP δ550/4 construct described previously (Chahine et al., 1992), containing sequences −550 to +11 relative to the transcriptional start site. Scanner–linker mutations (slm) were generated by creating a series of 5′ and 3′ deletions on this construct using Exonuclease III. These deleted DNAs were sequenced to define their 5′ and 3′ ends. Deletions that had 3′ ends exactly 17 bp upstream of an appropriate 5′ deletion were ligated with a 17 bp linker inserted in between. One exception is δ −550 slm −44/−29, which has only a 16 bp deletion between ends with a 17 bp linker inserted. The sequence of the linker is CAGATCTCGAGCTCCAC. This linker contains three unique restriction sites: BglII, XhoI, and SacI. The slm δ −550 slm −21/−5 also was assayed in a δ-promoter construct whose 5′ end was at position −119. This construct was generated by PCR amplification using an oligonucleotide with a 5′ end at −119 and another oligonucleotide complementary to plasmid sequences flanking the 3′ end of the δ-promoter insert. The PCR fragment then was cut withHindIII and cloned into theSmaI–HindIII sites of BSSK(+) for sequencing and subsequently cut with BamHI–HindIII and subcloned into the BamHI–HindIII sites of the pXP1 vector.
Internal deletions of the δ-promoter (−550/+11) were created by digestion of the 3′ end of δ −550/+11 with Exonuclease III. After sequence analysis to define the 3′ ends, deletions were subcloned into a pXP vector just upstream of a small δ-promoter fragment harboring the δ-subunit gene transcription start site. All mutations and deletions generated also were subcloned into BSSK(+) for sequence analysis.
The mouse wild-type and E1 E-box mutant (mutation 5) δ-promoter expression constructs (Simon and Burden, 1993) were kindly provided by Dr. S. J. Burden. Because these promoters contained restriction enzyme sequences also found in the multiple cloning site of the plasmid, they had to be subcloned into the pXP2 expression vector in two stages. First we isolated a KpnI–BamHI 3′ promoter fragment and subcloned it into the KpnI–BglII sites of pXP2. Second, we isolated the remaining 5′ δ-promoter fragment using HindIII–KpnI and subcloned it into the HindIII–KpnI sites of the pXP2 vector containing the mouse 3′ δ-promoter fragment.
Point mutations within the −47 to −1 region, used in the enhancer assays, were created by PCR using oligonucleotides complementary to sequences within the −47 to −1 region, containing single or multiple point mutations, and an oligonucleotide complementary to plasmid sequences. Amplified PCR products containing point mutations within the −47 to −1 region then were subcloned into BSSK(+) for sequence determination and subsequently subcloned upstream of the MEK in the MEK pXP2 construct.
The CMVCAT expression vector harbors the cytomegalovirus (CMV) promoter (Boshart et al., 1985) driving chloramphenicol acetyltransferase (CAT) expression. The CMVβgal expression vector harbors the CMV promoter driving β-galactosidase expression (β-gal). The SV40nlβ-gal expression vector harbors an SV40 promoter driving expression of β-galactosidase containing a nuclear localization sequence (SVnlβ-gal). RSV Luc (De Wet et al., 1987) and MCKpXP contain the Rous sarcoma virus (RSV) and muscle creatine kinase (MCK) (Jaynes et al., 1986) promoters, respectively, driving luciferase (Luc) expression.
Muscle denervation and DNA injections. Rats, ∼1 month old, were anesthetized with ether, and the lower left hindlimb was denervated by removing a 3 mm section of sciatic nerve just below the hip. This procedure ensures that regeneration does not occur during the course of our experiments. Four to five days after denervation, DNA solutions were injected into innervated and denervated extensor digitorum longus (EDL) muscles of anesthetized rats.
DNA, for injection into muscle, was purified twice on CsCl gradients. Before injection, 150–200 μg of CMVCAT along with 150–200 μg of one of the pXP expression constructs were mixed and precipitated two times with 150 mm NaCl and two volumes of ethanol. The final precipitate was rinsed two times with 70% ethanol and dried briefly. This DNA was resuspended at 10 mg/ml in 150 mm NaCl, and ∼10 μl was injected into each of four different locations along the length of the EDL muscle.
Procedures for injecting DNA into skeletal muscle were based on those described previously by Wolff (1991). We used a 100 μl Hamilton syringe and a 0.5-inch-long, 27 gauge needle that was fitted with a Williams collar exposing 2–3 mm of the end of the needle. DNA was injected slowly into the EDL muscle through the skin.
Muscle extracts and reporter gene assays. One week after injection, the EDL muscle was dissected out of the leg, rinsed in ice-cold PBS, minced with a razor blade, and placed into 2 ml of extraction buffer containing (in mm): potassium phosphate 100, pH 7.8, EDTA 1, PMSF 1. Samples were homogenized immediately with a polytron (setting 4) for 20 sec and placed on ice. After homogenization, samples were placed in an ice-water bath and sonicated twice for 10 sec using a Cole–Parmer ultrasonic homogenizer set at 70%. Ice-cold extraction buffer (200 μl) containing 10% Triton X-100 then was added, and the samples were mixed by vortexing. Insoluble material was removed by centrifugation in a Ti50 rotor (Beckman, Fullerton, CA) at 45,000 rpm for 15 min at 4°C. The supernatant was transferred to a new tube, and 100 μl was used to assay for Luc activity. Another 50–100 μl of the extract was used to assay for CAT activity. Luc and CAT assays were performed as described previously (Neumann et al., 1987; Brasier et al., 1989). CMVCAT activity was used to normalize for differences between samples that might result from different amounts of DNA injected into the muscle or variability in the uptake of DNA. CMVCAT expression did not appear to be regulated by muscle activity.
Muscle sectioning and staining. One week after injection of β-gal constructs into innervated and denervated EDL muscle, the animals were perfused with 4% paraformaldehyde in PBS. EDL muscles were dissected out and incubated overnight at 4°C in a 30% sucrose/4% paraformaldehyde PBS solution. The tissue then was embedded in OCT and frozen on dry ice. Samples were stored at −80°C until sectioning. Cryostat sections (30 μm) were taken, placed on polylysine-coated slides, and allowed to air dry. Staining for β-gal was performed by adding 50 μl of staining solution (PBS containing 5 mmK3Fe(CN)6, 5 mmK4Fe(CN)6, 2 mm MgCl2, and 1 mg/ml X-gal) to sections. Coverslips were added, and edges were sealed with rubber cement. Slides then were placed in a sealed humidified container at 37°C overnight. After β-gal staining, rubber cement was removed, and sections were soaked in PBS for 10 min to remove coverslips. Sections then were stained with eosin (0.5%) and mounted with permount.
Cell culture. The mouse muscle cell line C2C12 was cultured as described previously (Evans et al., 1987).
RNase protections. RNase protections were performed on RNA isolated from transiently transfected C2C12 myotubes by the GTC/CsCl method as described previously (Chirgwin et al., 1979). To assay for transcription originating at the MEK start site, an antisense probe was generated from an 865 bp HindIII–EcoRI fragment of the MEK pXP2 subcloned into BSSK(+). This fragment contained the 135 bp MEK promoter, including its transcriptional start site, together with 730 bp from the 5′ end of the Luc cDNA. To normalize for variability in transfection efficiency between samples, RNase protections also were performed on the same RNA sample using an antisense CAT probe. The CAT probe was generated using a 270 bpBglII–EcoRI fragment from the 5′ end of the CAT gene cloned into the BamHI–EcoRI sites of BSSK(+). RNase protections were performed using RNase T2 as described previously (Saccomanno et al., 1992).
Dimethylsulfate (DMS) DNA footprinting. DNA probes were made from a δ-promoter clone containing 102 bp upstream of the transcriptional start site. One end of the probe was32P-labeled by Klenow fill-in reaction, isolated on a polyacrylamide gel, eluted, and purified by phenol–chloroform–isoamyl alcohol (25:24:1) extraction and ethanol precipitation. C2C12 myotube nuclear extracts were prepared by the method of Dignam et al. (1983). Innervated and denervated muscle nuclear extracts were prepared by the method of Hahn and Covault (1990). DNA–protein binding reactions and DMS footprinting were performed by incubating either C2C12 myotube (200 μg), innervated (160 μg) or denervated (160 μg) nuclear extracts in 125 μl of binding buffer [25 mm HEPES, pH 7.9, 100 mm NaCl, 10% glycerol, 1 mg/ml BSA, 0.5 mm DTT, 0.2 mm PMSF, 50 μg/ml poly(dI-dC)] with 20,000 cpm of labeled probe (∼0.1–0.2 ng) for 20 min at room temperature. After the binding reaction, samples were treated with 75 μl of DMS diluted 1:200 in DMS reaction buffer (50 mm sodium cacodylate, pH 8.0, 1 mm EDTA) for 2 min at 25°C. DMS reaction was terminated with DMS stop solution (1.5 mm sodium acetate, pH 7.0, 1 m β-mercaptoethanol, 250 μg/ml yeast tRNA). Samples then were phenol–chloroform extracted and precipitated with ethanol. Strand cleavage at methylated guanine residues then was carried out as outlined in the chemical DNA sequencing technique of Maxam and Gilbert (1980).
RESULTS
In vivo expression of DNA injected into innervated and denervated muscle
Before characterizing nAChR promoter activity in vivo, we determined the number of muscle fibers and the number of nuclei within a single fiber expressing the injected DNA. For these experiments, we used two different β-gal expression vectors. CMVβ-gal was used to assay for the number and type of cells expressing the injected DNA, and SV40nlβ-gal was used to determine the distribution of nuclei expressing injected DNA within an individual muscle fiber. Figure 1A shows a β-gal-stained muscle cross-section that was injected previously with the CMVβ-gal construct. This analysis showed that expression of the injected DNA is localized to muscle fibers in the vicinity of the injection site. However, the level of expression is quite variable among the expressing fibers. This likely reflects the amount of DNA taken up by the fiber and the location of expressing nuclei in relationship to where the cross-section was taken. Quantitation of the number of fibers expressing this construct indicate that ∼50–60 EDL fibers were routinely able to incorporate this DNA into their nuclei and express it.
Fig. 1.

β-Galactosidase reporter genes identify muscle fibers and nuclei expressing injected DNA. A, Cross-section (30 μm thick) of EDL muscle injected with 150 μg of CMVβ-gal.B, Longitudinal section (30 μm) of EDL muscle injected with 150 μg of SV40nlβ-gal. Sections were stained for β-galactosidase activity (blue) and subsequently stained with eosin. Photographs were taken with a Zeiss Axiophot microscope using the 20× objective for pictures of longitudinal sections and the 10× objective for pictures of cross-sections.
We also determined whether there were any differences in DNA uptake and expression between innervated and denervated muscle fibers. No significant difference in the number of fibers expressing β-gal was observed, although denervated muscle did show slightly reduced β-gal activity compared with innervated muscle (data not shown). This latter observation is consistent with that reported by Gunderson et al. (1993)and appears to reflect a change in β-gal stability at muscle denervation.
To determine whether the injected DNA diffuses throughout the muscle fiber or remains localized to the site of injection, we used an SV40nlβ-gal expression construct. Longitudinal sections were cut from these muscles and stained for β-gal activity (Fig. 1B). Because the β-gal product is nuclear-localized, it allowed us to ascertain the distribution of this DNA in the injected muscle fiber. Expression appears to be localized to a small region of the muscle fiber (0.27 ± 0.12 mm), presumably representing the site of injection.
We investigated next whether this system could be used to study the effects of muscle electrical activity on nAChR promoter expression. First, we assayed for the effects muscle denervation had on promoters not expected to be induced by muscle denervation. Two promoters were chosen for this analysis: (1) the MCK promoter, which is not regulated by muscle electrical activity (Chahine et al., 1992), and (2) the Rous sarcoma virus promoter (RSV) promoter, which is induced by a calcium/calmodulin-dependent protein kinase (Kapiloff et al., 1991) and, therefore, may be downregulated after denervation. Activities of these promoters after direct injection of MCKpXP or RSV Luc into innervated and denervated muscle are shown in Figure2A. Consistent with our expectations, we find that muscle denervation had little effect on the MCK promoter and downregulated the RSV promoter.
Fig. 2.
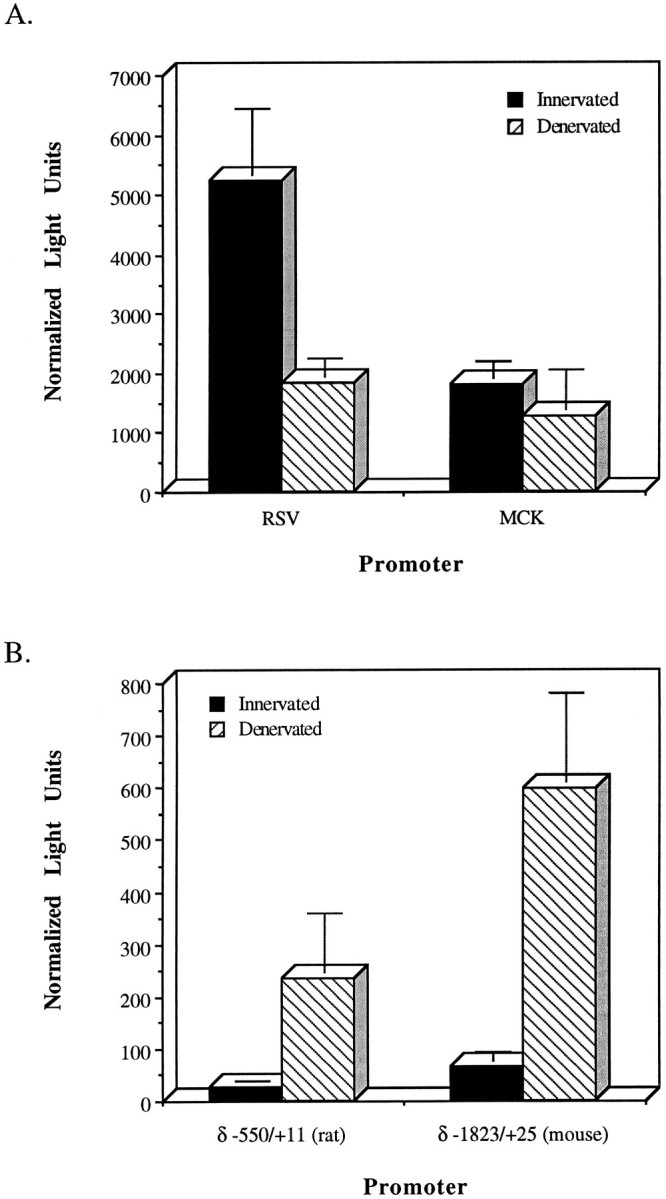
Direct injection of expression vectors containing RSV, MCK, or nAChR promoters shows specific induction of nAChR promoter activity after muscle denervation. A, Expression of the RSV Luc and MCK pXP constructs injected into innervated and denervated EDL muscle. B, Expression of the rat δ −550/+11 pXP and mouse δ −1823/+25 pXP constructs injected into innervated and denervated EDL muscle. Innervated and denervated EDL muscles were injected with the expression vectors (above) along with CMVCAT for normalization. One week later, EDL muscles were harvested and assayed for Luc and CAT activity. Luc light units were normalized to CAT activity. The bars indicate SD.
To confirm that direct DNA injection into muscle was suitable for analyzing nAChR promoter activity in response to muscle denervation, we injected expression vectors harboring two different nAChR δ-promoters. One promoter, mouse δ −1823/+25, previously had been shown to be induced by muscle denervation in transgenic animals (Simon et al., 1992) and, therefore, serves as a positive control. The other promoter, rat δ −550/+11, previously had been shown to be regulated by muscle activity in vitro (Chahine et al., 1992). Direct injection of δ −1823/+25 pXP or δ −550/+11 pXP into innervated and denervated muscle revealed that these promoters were induced eight- and sixfold, respectively, in the denervated muscle fibers (Fig.2B).
Mutational analysis of the δ-promoter identifies sequences within −81 to −5 that are necessary for electrical activity-dependent regulation
Previous studies showed that activity-dependent regulation of the rat δ-subunit gene was contained within a 102 bp region just upstream of the transcriptional start site (Chahine et al., 1992). Therefore, we mutated specific regions within the first 100 nt upstream of the start site and assayed their effect on promoter activity, in vivo, by direct injection of DNA into muscle.
To confirm that this system would reveal an effect, if the appropriate mutation were created, we used a wild-type (δ −1823 wt) and an E-box mutant (δ −1823 E1 mut 5) mouse δ-promoter. The E-box mutation within the δ-promoter previously was shown to significantly decrease activity-dependent expression in vivo (Tang et al., 1994). Injection of these DNAs into innervated and denervated muscle revealed significant loss in activity-dependent regulated expression from the mutant promoter (Fig. 3).
Fig. 3.

Mutation of the E-box sequence in the mouse δ-promoter suppresses activity-dependent regulation. Schematic representation of the mouse δ −1823/+25 promoter is shown. Putative transcription factor binding sites within 100 bp upstream of the transcription start site are shown. Expression of injected δ −1823/+25 pXP or δ −1823/+25 E1 mutation 5 pXP in innervated and denervated muscle relative to the wild-type construct, along with the fold difference, is indicated to the right of the constructs. Numbers in parenthesis indicate number of animals injected. Luc light units were normalized to co-injected CMVCAT activity. p-values (shown in brackets) were calculated using a one-tailed t test. Putative regulatory elements are indicated by rectangles along the sequence and labeled at the bottom of the figure. The E-box element in δ −1823 E1 mut 5 is black to indicate that it was mutated. The enhancer X is a previously identified enhancer with no homology to known regulatory elements (W. Walke and D. Goldman, unpublished observations) (Simon and Burden, 1993).
We confirmed the importance of this E-box in mediating activity-dependent expression of the rat δ-promoter by creating an slm that replaced the rat δ-promoter’s E-box and 3′ flanking DNA with a synthetic 17 bp sequence (see Materials and Methods). This mutation (δ −550 slm −21/−5) reduced activity-dependent expression by ∼40% (Fig. 4). A similar but more robust effect was observed when we used δ −119 as the parent for creating this mutation (compare δ −119 wt with δ −119 slm −21/−5 in Fig. 4). Surprisingly, two other slm mutations (slm −44/−29 and slm −81/−65) that altered sequences upstream of the E-box also reduced the δ-promoter’s response to muscle denervation (Fig. 4). It is interesting that mutation slm −81/−65 caused lower expression, whereas mutation slm −44/−29 resulted in higher expression in both innervated and denervated muscle. This is consistent with the disruption of a positive regulatory element in slm −81/−65 and disruption of a negative element in slm −44/−29. To ensure that specific effects were not contributed by the introduced linker, experiments were repeated with constructs in which the linker was removed. No significant differences were found between constructs containing linker and those lacking the linker. These data suggest that multiple elements contribute to activity-dependent control of δ-subunit gene expression.
Fig. 4.

Elements within nt −81 to −5 of the δ-promoter are necessary for activity-dependent regulation. The various slm and internal deletions of the δ −550/+11 promoter are represented schematically, with the numbers indicating the nt bordering regions replaced or deleted. These expression vectors were co-injected with CMVCAT into innervated and denervated EDL muscles. After 1 week, muscles were harvested and Luc and CAT activities were determined. Results are shown with SD and fold differences on the rightside of the figure. Values are reported as a percentage of δ −550 wt expression. Numbers in parenthesis indicate the number of animals injected. p-values (shown in brackets) were calculated using a one-tailed t test.
Based on these data, one would predict that a deletion spanning nt −81 to −5 would cause complete loss of activity-dependent expression. Indeed, Figure 4 shows that such a deletion does render the δ-promoter completely insensitive to muscle activity. However, if the deletion spans nt −52 to −5, partial activity-dependent regulation is retained.
Two regions within the δ-subunit 102 bp promoter can confer electrical activity-dependent regulation onto a heterologous promoter
Expression vector δ −550 Δ−81/−5 (Fig. 4) showed that elements mediating activity-dependent control of the δ-promoter resided within a region of DNA spanning nt −81 to −5. However, slm targeting specific subregions of this DNA were not able to abrogate this regulation completely (Fig. 4). This suggested to us that there may be redundant subdomains of the −81 to −5 region that possessed elements both necessary and sufficient for conferring activity-dependent regulation onto the δ-promoter. To identify these putative activity-dependent regulatory subregions, we turned to enhancer assays. For these studies, we synthesized different oligonucleotides that spanned different regions of the 102 bp δ-promoter (oligo −106/−72; oligo −81/−32; oligo −47/−1; and oligo −25/+10). These oligonucleotides were subcloned upstream of the MEK promoter in the pXP expression vector. These constructs first were transfected into C2C12 and NIH3T3 cells to evaluate whether they had enhancer activity. Only oligos −81/−32 and −47/−1 were able to confer enhanced expression on the MEK promoter. Oligo −81/−32 was shown to act as a general enhancer, whereas oligo −47/−1 acted as a muscle-specific enhancer (W. Walke and D. Goldman, unpublished observations). Therefore, we focused on these two oligos in the following experiments.
δ-Enhancer–MEK expression vectors were injected into innervated and denervated muscle and their activity assayed 1 week later (Fig.5). The MEK promoter alone did not show a statistically significant response to muscle activity (Fig. 5). However, addition of either δ-enhancer (oligo −81/−32 or oligo −47/−1) to the MEK promoter increased its activity and conferred onto it depolarization-dependent regulation (Fig. 5). δ −47/−1 MEK showed the most robust regulation on muscle denervation, with expression in denervated muscle being approximately six- to sevenfold higher than in innervated muscle. This change in δ −47/−1 MEK promoter activity on muscle denervation is similar to that observed for the δ −550 promoter (Figs. 2, 4). In addition, oligo −47/−1 confers activity-dependent expression onto the MEK promoter in either orientation, albeit with only a 2.6-fold induction when it is in the 3′ to 5′ direction (Fig. 5). Neither the E-box nor the SVCE sequences on their own were able to increase MEK promoter activity or confer activity-dependent regulation onto the MEK promoter (data not shown).
To confirm that the δ-promoter enhancer sequences were conferring increased activity on the MEK promoter, we performed RNase protection assays on transiently transfected C2C12 myotubes with probes that allowed us to distinguish between promotion starting within the MEK promoter versus promotion starting within the δ-subunit fragments. Figure 6 shows the results of these experiments. Using our 865 nt MEK probe, a protected band at 800 bp would indicate transcription originating at the MEK start site, whereas protection of a larger 865 bp band would indicate promotion originating upstream of the MEK promoter. Cotransfection with MCKCAT and subsequent RNase protection for CAT RNA allowed us to normalize for transfection efficiency. Densitometric analysis showed that promoter activity originating at the MEK start site is increased ∼nine- and 15-fold by fragments δ −81/−32 and δ −47/−1, respectively, located upstream of the MEK promoter, and that promoter activity originating from within the δ-promoter fragment is minimal by comparison.
Fig. 6.

The δ-subunit gene enhancers increase promotion from the transcriptional start site of the enkephalin gene. C2C12 myotubes were transfected with MEK pXP2, δ −47/−1 MEK pXP2, or δ −81/−32 MEK pXP2 along with MCKCAT. Cells were harvested 48 hr after transfection, and RNA was isolated for mapping the transcriptional start site by RNase protection assays. The diagram at thetop illustrates the organization of the transfected constructs containing the δ-subunit gene enhancer 5′ of the MEK promoter, which flanks the transcriptional start site of the enkephalin gene (indicated by the arrow). The arrow above the δ-enhancer is meant to indicate potential transcriptional start sites. The thick line below the top diagram indicates the region of DNA represented in the antisense RNA probe. Illustratedbelow the probe are possible RNase-resistant products that are expected to be generated depending on whether transcription begins at the enkephalin or δ-gene start sites. The bottom halfof the figure shows results from an RNase protection assay. RNA isolated from C2C12 cells transfected with the MEK pXP2, δ −47/−1 MEK pXP2, or δ −81/−32 MEK pXP2 constructs, and cotransfected with MCKCAT DNA, was probed with the 865 nt probe and a 270 bp CAT probe. Protection of a product at 800 nt indicates promoter activity originating from the MEK start site, which is increased by the presence of the δ-sequences. Protection of the 270 nt probe was used to assay MCKCAT expression to account for transfection variations.
An E-box within the −47 to −1 region is necessary but requires adjacent sequences to confer electrical activity-dependent regulation onto a minimal promoter
The above experiments indicated that activity-dependent regulation of δ-promoter activity could be mediated by a combinatorial action of two regions of DNA, one containing enhancer X (nt −81 to −32) and the other containing nt −47 to −1. This latter region was a more robust activity-dependent enhancer than the enhancer X region (Fig. 5) and, therefore, was examined in greater detail. This sequence contains an E-box, an SP1-like sequence, and a sequence similar to the SV40 core enhancer (Khoury and Gruss, 1983). To determine whether residues within each of these putative elements are necessary for mediating activity-dependent control of promoter expression, we used site-directed mutagenesis to change residues in each of these putative elements. We chose to mutate specific residues that are likely to interact with trans-acting regulatory proteins as revealed by DNA methylation interference assays (see next section; Fig. 8). Figure 7 shows the sequence of the mutations made in δ −47/−1, which were placed upstream of the MEK promoter. These constructs were injected into innervated and denervated muscle, and the results of their expression, along with the wild-type enhancer, is indicated to the right. These data indicate that mutation in any of these elements abrogates δ −47/−1 enhancer activity, in addition to causing a loss of activity-dependent regulation.
Fig. 8.

DNA methylation interference footprinting shows nuclear factors from denervated muscle, and C2 myotubes bind to similar regions of the δ −102 bp promoter. Results from DMS footprinting analysis of the 102 bp δ-promoter region (noncoding strand) using nuclear extracts from C2C12 myotubes (C2), innervated (Inn), and denervated (Den) muscle are shown. Control lanes without nuclear extract are labeled con. Regions containing guanine residues that were protected (open circles) or hypermethylated (solid circles) are indicated with lines and are labeled along the side.AP, Putative AP2 element; X, enhancer X region;SV, SV40 core enhancer-like sequence; Sp, putative Sp1 element; and E, E-box sequence.
Fig. 7.

Site-directed mutagenesis identifies sequences in the 47 bp enhancer that are necessary to confer activity-dependent regulation onto a heterologous promoter. Diagrammed are the wild-type and various mutant constructs used in these studies. Topdiagram shows putative regulatory regions of the δ −47 bp enhancer. Below this are shown the DNA sequences of the wild-type and various mutations. Mutations are shown in boldand lowercase type. δ-Enhancer MEK Luc constructs were co-injected with CMVCAT into innervated and denervated EDL muscles and assayed for Luc and CAT activity 1 week after injection. Results are shown with SD on the right side of the figure along with the fold differences. Values are represented as a percentage of wild-type expression in denervated muscle. Results were normalized to CMVCAT expression. Numbers in parentheses indicate number of animals injected. p-values (shown inbrackets) were calculated using a one-tailed ttest.
Myotubes and denervated muscle contain nuclear factors that interact with multiple regions of the rat δ −102 bp promoter
Based on the above mutagenesis and enhancer studies, we predicted that nuclear factors would bind to the 102 bp region of the δ-subunit promoter in an activity-dependent manner. In addition, we were interested in determining whether the binding of nuclear factors from denervated muscle resembled the pattern of binding observed with that obtained from cultured C2C12 muscle myotubes. Therefore, we performed methylation interference DNA footprinting assays using nuclear extracts from C2C12 myotubes and innervated–denervated muscle (Fig.8). Innervated muscle nuclear extract had little effect on the methylation pattern. However, denervated muscle extract revealed multiple interactions along the whole length of the δ −102 bp promoter region. Increased or decreased methylation was observed in sequences corresponding to the E-box, Sp1, SVCE, enhancer X, and AP2 regions (Fig. 8). Most interesting is the similarity between binding of C2C12 myotube nuclear extract and denervated muscle extract. This latter result may indicate that similar proteins participate in activating δ-gene expression in myotubes and denervated muscle.
DISCUSSION
Direct injection of plasmid DNA into skeletal muscle was used as an in vivo expression assay to identify elements of the nAChR δ-subunit gene promoter that participate in activity-dependent control of δ-gene expression. Where comparisons could be made, our results using DNA injection into muscle were comparable to those using transgenic animals. However, one significant difference is the level of gene induction on muscle denervation. The mouse δ-promoter is induced ∼27-fold in transgenic animals (Tang et al., 1994), whereas direct injection revealed only a ninefold induction (Fig. 3). One possible explanation for this difference is damage to the innervated muscle fibers during the injection procedure resulting in muscle fiber regeneration and, therefore, increased transgene expression in the regenerated myotubes. Other possible explanations may be that, unlike transgene expression in transgenic animals, direct injection does not result in transgene integration or a eukaryotic pattern of DNA methylation. Nonetheless, it is clear that direct injection provides an alternative to transgenic animals for in vivo expression assays.
We previously had identified a 102 bp region of the rat nAChR δ-subunit gene’s 5′ flanking DNA that contains all the necessary elements to mediate activity-dependent control of gene expression (Chahine et al., 1992). In the present study, scanner–linker mutagenesis showed that multiple regions between −81 and −5 of the rat δ-promoter were important for activity-dependent expression (Fig.4). However, no single mutation completely abolished this regulated expression, unless the region between −81 and −5 was deleted. This suggested that multiple and perhaps redundant elements participated in activity-dependent control of δ-gene expression. We reasoned that these elements would be revealed most easily by studying them in isolation from other δ-promoter sequences.
Therefore, we devised a scheme to identify these various elements using an enhancer assay. Two δ-promoter sequences, −81 to −32 and −47 to −1, were found to possess activity-dependent enhancer activity (Fig.5). Although these two enhancers share a stretch of DNA corresponding to the SVCE, this piece of DNA on its own had no enhancer activity (data not shown).
The relatively small increase in promoter activity associated with enhancer X sequences −81 through −32 suggests that this enhancer may play a more minor role in mediating activity-dependent regulation of the δ-gene compared with enhancer sequences −47 to −1. Therefore, we chose to characterize further the 47 bp enhancer.
Comparison of the mouse and rat δ-promoter sequences shows two nt differences within the 47 bp enhancer. Both differences occur in the rat δ-promoters Sp1-like region. Positions −27 and −30 are G residues in the rat and A residues in the mouse. Although this is a purine–purine substitution, it may indicate that the region labeled as Sp1 in the rat sequence is not an Sp1 binding site. Additional studies are needed to test this. Because site-directed mutagenesis targeted the C residue at position −26 as necessary for mediating activity-dependent expression (Fig. 7), we interpret this to mean that the DNA sequence immediately flanking the E-box, which is conserved between rat and mouse, is necessary for this type of regulation.
It is interesting that point mutations in the 47 bp enhancer abrogate MEK promoter regulation by muscle activity (Fig. 7), yet when these same regions of the δ-gene’s 5′ flanking DNA are disrupted by slm (Fig. 4), they only partially block activity-dependent regulation. This partial block is consistent with our finding that two regions of the δ-promoter contribute to activity-dependent regulation, and one cannot eliminate completely this regulation until both regions are deleted (δ −550 Δ −81/−5) (Fig. 4).
This contextual effect may be illustrated further by the observation that point mutations in the SVCE, SP1-like sequence, or E-box all dramatically reduce MEK promoter activity in innervated and denervated muscle (Fig. 7), yet mutations in these same regions of DNA sometimes have little effect on δ-promoter activity in innervated muscle (δ-550 slm −21/−5; Fig. 4) or increase δ-promoter activity in innervated and denervated muscle (δ −550 slm −44/−29; Fig. 4). Although we favor context as an explanation for the differences observed between the δ- and MEK promoters containing mutations within the 47 bp enhancer, we cannot rule out that these differences reflect the different types of mutations created in these two expression constructs (therefore, scanner–linker vs point mutations).
We have formulated a model, based on the above data, to describe δ-subunit gene expression in response to muscle innervation and denervation (Fig. 9). Our methylation interference assays define five regions of the δ-promoter that appear to bind more protein in the denervated state than the innervated state (Fig. 8). These regions correspond to the E-box, SP1-like sequence, SVCE, enhancer X region, and an AP2 element. All these elements, except the AP2 element, were characterized by mutagenesis and expression assays (Figs. 4, 5, 7). The AP2 element was shown previously to be necessary for high δ-promoter activity in noninnervated myotubes (Chahine et al., 1992). Each one of these elements shows increased protein binding on muscle denervation (Fig. 8), suggesting that they contribute to high-level expression of the δ-promoter in this tissue. Therefore, our model shows protein binding to each of these elements in the denervated state (Fig. 9B). We have represented E-box binding by a myogenic heterodimer, consistent with other E-box–binding proteins (Lassar et al., 1991). The other cis-acting elements may be bound by monomers or oligomers, and all or some of these proteins may be interacting with each other.
Fig. 9.

Model for δ-promoter expression in innervated versus denervated muscle. A, Activity of the δ-promoter is low in innervated muscle because, in part, of the binding of a putative repressor in the SVCE region, which blocks activation from a putative activator binding to the enhancer X region. In addition, the absence of transcription factor binding to the E-box region and AP2 element contributes to low promoter activity in innervated muscle.B, Expression in denervated muscle is high because of loss of putative repressor binding at the SVCE site and the increased binding of activating factors to the AP2 and SVCE/E-box region.
We also have indicated that proteins binding to the E-box also interact with residues 5′ of the E-box, and comprising a portion of the SP1-like sequence, which are conserved between rat and mouse promoters. However, we cannot rule out the possibility that a separate protein (perhaps SP1) or protein complex binds to the SP1-like site.
The low-level expression of the δ-gene in innervated muscle correlates with a lack of detectable protein binding in methylation interference assays (Fig. 8). However, because this assay uses a relatively small linear piece of DNA for binding proteins in vitro, it is possible that protein–DNA interactions normally occurring in vivo are not observed. In addition, low-affinity binding may not be detected in this assay. Consistent with this is our mutagenesis and expression assays, which indicate that the enhancer X region can function as an activator (δ−550 slm −81/−65) and that the SVCE region can function as a repressor (δ −550 slm −44/−29) in innervated muscle (Fig. 4). In contrast, E-box mutations had little effect on expression in innervated muscle (Fig. 4).
These results suggest that the level of activation via enhancer X (represented by activator protein binding; Fig. 9A), repression via the SVCE (represented by repressor protein binding; Fig.9A), and the decreased binding of proteins to the E-box–SP1 and AP2 regions (represented by a lack of protein binding; Fig.9A) play an important role in determining the level of expression of this gene in innervated muscle. We have no information on whether the proteins that bind these regions in the innervated state are the same as those that bind these sequences in the denervated state. The changes in enhancer X and SVCE binding protein illustrated in Figure 9, A and B, are simply meant to indicate either different proteins, protein modification or protein conformation attributable to protein–protein interactions that would differ in the innervated and denervated states.
Our enhancer assays and mutagenesis studies (Fig. 7) indicate that the SVCE, SP1-like, and E-box sequences function as a single unit to confer activity-dependent regulation onto a heterologous promoter. Mutation of any one of these regions abrogates completely activity-dependent control of promoter activity. These elements do not function independently as activity-dependent enhancers and underscore the importance of context in determining functional activity. This explains why the same E-boxes, which are necessary for activity-dependent regulation of the nAChR genes, don’t necessarily confer activity-dependent regulation onto other muscle specific genes.
Before this study, the only cis-acting element identified to participate in activity-dependent control of nAChR gene expression had been E-boxes of the α-, γ-, and δ-subunit gene promoters (Bessereau et al., 1994; Tang et al., 1994; Su et al., 1995). The identification of additional cis-acting sequences necessary for activity-dependent regulation reported here should facilitate the identification of their binding proteins, which ultimately will allow us to understand how these proteins participate in this activation process.
Footnotes
This work was supported by grants from the National Institute of Neurological Diseases and Stroke (2 RO1 NS2153-08), the Muscular Dystrophy Association, and the Lucille P. Markey Charitable Trust. W.W. was supported by a National Research Service Award predoctoral fellowship from the National Institute of Mental Health (5 F31 MH10328-02). We thank Dr. S. J. Burden for providing the mouse δ-promoter clones, Dr. A. F. Seasholtz for providing the enkephalin promoter, and N. Goburdhun for technical assistance.
Correspondence should be addressed to Daniel Goldman, University of Michigan Mental Health Research Institute, 205 Zina Pitcher Place, Ann Arbor, MI 48109.
REFERENCES
- 1.Berberich C, Durr I, Koenen M, Witzemann V. Two adjacent E box elements and a M-CAT box are involved in the muscle-specific regulation of the rat acetylcholine receptor β-subunit gene. Eur J Biochem. 1993;216:395–404. doi: 10.1111/j.1432-1033.1993.tb18157.x. [DOI] [PubMed] [Google Scholar]
- 2.Bessereau JL, Mendelzon D, LePoupon C, Fiszman M, Changeux JP, Piette J. Muscle-specific expression of the acetylcholine receptor α-subunit gene requires both positive and negative interactions between myogenic factors, Sp1 and GBF factors. EMBO J. 1993;12:443–449. doi: 10.1002/j.1460-2075.1993.tb05676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bessereau JL, Stratford-Perricaudet LD, Piette J, Poupon CL, Changeux JP. In vivo and in vitro analysis of electrical activity-dependent expression of muscle acetylcholine receptor genes using adenovirus. Proc Natl Acad Sci USA. 1994;91:1304–1308. doi: 10.1073/pnas.91.4.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boshart M, Weber F, Jahn G, Dorsch-Hasler K, Fleckenstein B, Schaffner W. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell. 1985;41:521–530. doi: 10.1016/s0092-8674(85)80025-8. [DOI] [PubMed] [Google Scholar]
- 5.Brasier AR, Tate JE, Habener JF. Optimized use of the firefly assay as a reporter gene in mammalian cell lines. Biotechniques. 1989;7:1116–1122. [PubMed] [Google Scholar]
- 6.Chahine KG, Walke W, Goldman D. A 102 base pair sequence of the nicotinic acetylcholine receptor δ-subunit gene confers regulation by muscle electrical activity. Development. 1992;115:213–219. doi: 10.1242/dev.115.1.213. [DOI] [PubMed] [Google Scholar]
- 7.Chahine KG, Baracchini E, Goldman D. Coupling muscle electrical activity to gene expression via a cAMP-dependent second messenger system. J Biol Chem. 1993;268:2893–2898. [PubMed] [Google Scholar]
- 8.Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 9.Comb M, Birnberg NC, Seasholtz A, Herbert E, Goodman HM. A cyclic AMP- and phorbol ester-inducible DNA element. Nature. 1986;323:353–356. doi: 10.1038/323353a0. [DOI] [PubMed] [Google Scholar]
- 10.de Wet JR, Wood KV, DeLuca M, Helinski DR, Subramani S. Firefly luciferase gene: structure and expression in mammalian cells. Mol Cell Biol. 1987;7:725–737. doi: 10.1128/mcb.7.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eftimie R, Brenner HR, Buonanno A. Myogenin and MyoD join a family of skeletal muscle genes regulated by electrical activity. Proc Natl Acad Sci USA. 1991;88:1349–1353. doi: 10.1073/pnas.88.4.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans S, Goldman D, Heinemann S, Patrick J. Muscle acetylcholine receptor biosynthesis. J Biol Chem. 1987;262:4911–4916. [PMC free article] [PubMed] [Google Scholar]
- 14.Gilmour BP, Fanger GR, Newton C, Evans SM, Gardner PD. Multiple binding sites for myogenic regulatory factors are required for expression of the actylcholine receptor g-subunit gene. J Biol Chem. 1991;266:19871–19874. [PubMed] [Google Scholar]
- 15.Gilmour BP, Goldman D, Chahine KG, Gardner PD. Electrical activity suppresses nicotinic acetylcholine receptor γ-subunit promoter activity. Dev Biol. 1995;168:416–428. doi: 10.1006/dbio.1995.1091. [DOI] [PubMed] [Google Scholar]
- 16.Goldman D, Brenner HR, Heinemann S. Acetylcholine receptor α- , β- , γ- , and δ-subunit mRNA levels are regulated by muscle activity. Neuron. 1988;1:329–333. doi: 10.1016/0896-6273(88)90081-5. [DOI] [PubMed] [Google Scholar]
- 17.Gunderson K, Sanes JR, Merlie JP. Neural regulation of muscle acetylcholine receptor ε- and α-subunit gene promoters in transgenic mice. J Cell Biol. 1993;123:1535–1544. doi: 10.1083/jcb.123.6.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall Z, Sanes JR. Synaptic structure and development: the neuromuscular junction. Cell. 1993;72:99–121. doi: 10.1016/s0092-8674(05)80031-5. [DOI] [PubMed] [Google Scholar]
- 19.Hahn C-G, Covault J. Isolation of transcriptionally active nuclei from striated muscle using percoll density gradients. Anal Biochem. 1990;190:193–197. doi: 10.1016/0003-2697(90)90180-h. [DOI] [PubMed] [Google Scholar]
- 20.Jaynes JB, Chamberlain JS, Buskin JN, Johnson JE, Hauschka SD. Transcriptional regulation of the muscle creatine kinase gene and regulated expression in transfected mouse myoblasts. Mol Cell Biol. 1986;6:2855–2864. doi: 10.1128/mcb.6.8.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kapiloff MS, Mathis JM, Nelson CA, Lin CR, Rosenfeld MG. Calcium/calmodulin-dependent protein kinase mediates a pathway for transcriptional regulation. Proc Natl Acad Sci USA. 1991;88:3710–3714. doi: 10.1073/pnas.88.9.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khoury G, Gruss P. Enhancer elements (minireview). Cell. 1983;33:313–314. doi: 10.1016/0092-8674(83)90410-5. [DOI] [PubMed] [Google Scholar]
- 23.Lassar AB, Davis RL, Wright WE, Kadesch T, Murre C, Voronova A, Baltimore D, Weintraub H. Functional activity of myogenic HLH proteins requires hetero-oligomerization with E12/E47-like proteins in vivo. Cell. 1991;66:305–315. doi: 10.1016/0092-8674(91)90620-e. [DOI] [PubMed] [Google Scholar]
- 24.Maxam AM, Gilbert W. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 1980;65:499–559. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 25.Merlie JP, Kornhauser JM. Neural regulation of gene expression by an acetylcholine receptor promoter in muscle of transgenic mice. Neuron. 1989;2:1295–1300. doi: 10.1016/0896-6273(89)90067-6. [DOI] [PubMed] [Google Scholar]
- 26.Neumann JR, Morency CA, Russian KO. A novel rapid assay for chloramphenicol acetyltransferase gene expression. Biotechniques. 1987;5:444–447. [Google Scholar]
- 27.Nordeen SK. Luciferase reporter gene vectors for analysis of promoters and enhancers. Biotechniques. 1988;6:454–457. [PubMed] [Google Scholar]
- 28.Prody CA, Merlie JP. A developmental and tissue-specific enhancer in the mouse skeletal muscle acetylcholine receptor α-subunit gene regulated by myogenic factors. J Biol Chem. 1991;266:22588–22596. [PubMed] [Google Scholar]
- 29.Saccomanno CF, Bordonaro M, Chen JS, Nordstrom JL. A faster ribonuclease protection assay. Biotechniques. 1992;13:846–850. [PubMed] [Google Scholar]
- 30.Simon AM, Burden SJ. An E-box mediates activation and repression of the acetylcholine receptor δ-subunit gene during myogenesis. Mol Cell Biol. 1993;13:5133–5140. doi: 10.1128/mcb.13.9.5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simon AM, Hoppe P, Burden SJ. Spatial restriction of AChR gene expression to subsynaptic nuclei. Development. 1992;114:545–553. doi: 10.1242/dev.114.3.545. [DOI] [PubMed] [Google Scholar]
- 32.Su C-T, Huang C-F, Schmidt J. The depolarization response element in acetylcholine receptor genes is a dual-function E-box. FEBS Lett. 1995;366:131–136. doi: 10.1016/0014-5793(95)00496-v. [DOI] [PubMed] [Google Scholar]
- 33.Tang J, Jo SA, Burden SJ. Separate pathways for synapse-specific and electrical activity-dependent gene expression in skeletal muscle. Development. 1994;120:1799–1804. doi: 10.1242/dev.120.7.1799. [DOI] [PubMed] [Google Scholar]
- 34.Wolff JA, Williams P, Acsadi G, Jiao S, Jani A, Chong W. Conditions affecting direct gene transfer into rodent muscle in vivo. Biotechniques. 1991;11:474–485. [PubMed] [Google Scholar]