

Abstract
Activation of cannabinoid receptors inhibits voltage-gated Ca2+ channels and activates K+ channels, reminiscent of other G-protein-coupled signaling pathways that produce presynaptic inhibition. We tested cannabinoid receptor agonists for effects on excitatory neurotransmission between cultured rat hippocampal neurons. Reducing the extracellular Mg2+ concentration to 0.1 mm elicited repetitive, transient increases in intracellular Ca2+ concentration ([Ca2+]i spikes) that resulted from bursts of action potentials, as measured by combined whole-cell current clamp and indo-1-based microfluorimetry. Pharmacological characterization indicated that the [Ca2+]i spikes required glutamatergic synaptic transmission. Cannabinoid receptor ligands inhibited stereoselectively the frequency of [Ca2+]i spiking in the rank order of potency: CP 54,939 > CP 55,940 > Win 55,212-2 > anandamide, with EC50 values of 0.36, 1.2, 2.7, and 71 nm, respectively. CP 55,940 was potent, but not efficacious, and reversed the inhibition produced by Win 55,212-2, indicating that it is a partial agonist. Inhibition of [Ca2+]i spiking by Win 55,212-2 was prevented by treatment of cultures with active, but not heat-treated, pertussis toxin. Win 55,212-2 (100 nm) inhibited stereoselectively CNQX-sensitive excitatory postsynaptic currents (EPSCs) elicited by presynaptic stimulation with an extracellular electrode, but did not affect the presynaptic action potential or currents elicited by direct application of kainate. Consistent with a presynaptic site of action, Win 55,212-2 increased both the number of response failures and the coefficient of variation of the evoked EPSCs. In contrast, cannabimimetics did not affect bicuculline-sensitive inhibitory postsynaptic currents. Thus, activation of cannabinoid receptors inhibits the presynaptic release of glutamate via an inhibitory G-protein.
Keywords: cannabinoid, glutamate, presynaptic inhibition, G-protein, intracellular calcium, indo-1, patch clamp, hippocampal culture
In addition to its widespread recreational use, Δ-9-tetrahydrocannabinol, the principal psychoactive ingredient in marijuana, has significant therapeutic potential (Abood and Martin, 1992). Cannabinoids have been reported to be clinically effective analgesics and useful in the treatment of glaucoma, bronchial asthma, diarrhea, muscle spasticity, and convulsions (Howlett, 1995). However, established clinical use of cannabinoids has been limited to their use as antiemetics and appetite stimulants in cancer and AIDS patients (Weinroth et al., 1995). Potential clinical applications have provided a rationale for synthesis of water-soluble cannabinoid analogs (e.g., CP 55,940 and CP 54,939) (Johnson and Melvin, 1986) and aminoalkylindoles (e.g., Win 55,212-2) (Bell et al., 1991; D’Ambra et al., 1992) that have proven to be highly selective and potent activators of cannabinoid receptors in vitro and in vivo (Devane et al., 1988; Compton et al., 1992; Compton et al., 1993).
The cannabinoid receptor is a member of the seven helix transmembrane, G-protein-linked receptor superfamily (Matsuda et al., 1990). Both mRNA for this receptor, and binding sites for radiolabeled cannabinoids, are distributed widely throughout the CNS, including cerebral cortex, hippocampus, basal ganglia, and cerebellum (Herkenham et al., 1990; Jansen et al., 1992; Mailleux and Vanderhaeghen, 1992;Thomas et al., 1992; Matsuda et al., 1993). Identification of an endogenous ligand, anandamide (Devane et al., 1992; Deutsch and Chin, 1993; Felder et al., 1993; Dimarzo et al., 1994), that activates cannabinoid receptors suggests that anandamide, in conjunction with the cannabinoid receptor, forms a neuromodulatory system (Mechoulam et al., 1994).
Activation of cannabinoid receptors inhibits adenylate cyclase via an inhibitory G-protein (Howlett et al., 1986; Bidaut-Russell et al., 1990; Childers et al., 1992; Compton et al., 1993). Similar to other receptors that interact with inhibitory G-proteins, cannabinoid receptors also couple to ion channels (Martin et al., 1994). Cannabimimetics inhibit N- and Q-type voltage-gated Ca2+ channels via pertussis toxin (PTX)-sensitive G-proteins (Caulfield and Brown, 1992; Mackie and Hille, 1992; Mackie et al., 1995; Pan et al., 1996). Cannabinoids enhance activation of A-type K+ currents in cultured hippocampal neurons via PTX-sensitive G-proteins (Deadwyler et al., 1993), and an inwardly rectifying K+ channel inXenopus oocytes co-injected with GIRK1 and cannabinoid receptor mRNA (Henry and Chavkin, 1995). Cannabinoids also inhibit long-term potentiation of the Schaeffer collateral commissural fiber–CA1 synapse (Nowicky et al., 1987; Collins et al., 1994), suggesting that the cannabinoid system may modulate glutamatergic synaptic transmission. Thus, cannabinoid receptor signal transduction is reminiscent of several well-characterized neurotransmitter receptor systems, including those of adenosine and the opioids, that couple to inhibitory G-proteins to produce presynaptic inhibition of synaptic transmission in the hippocampus (Thompson et al., 1993).
In this report, we test directly the hypothesis that activation of cannabinoid receptors inhibits glutamatergic synaptic transmission. Our data are consistent with this hypothesis in that synaptic activity blocked completely by glutamate receptor antagonists also was inhibited by cannabinoid receptor agonists. Additional pharmacological characterization of this effect determined that both endogenous and synthetic cannabinoid receptor agonists activate potently and stereoselectively a presynaptic receptor that inhibits the release of glutamate via an inhibitory G-protein. Modulation of synaptic glutamate release may account for the psychoactive, analgesic, and anticonvulsant effects of the cannabinoids.
MATERIALS AND METHODS
Materials. Materials were obtained from the following companies: indo-1 AM and indo-1 pentapotassium salt, Molecular Probes, Eugene, OR; PTX, List Biological Laboratories, Campbell, CA; anandamide, (−)-bicuculline methchloride, kainic acid, NMDA, (±)−2-amino-5-phosphonopentanoic acid, CGS19755, and CNQX, RBI, Natick, MA; Win 55,212-2 and Win 55,212-3, Sterling-Winthrop, Rensselaer, NY, or RBI; CP 54,939 (desacetyllevonantradol) and CP 55,940 (levorotatory enantiomer), Pfizer, Groton, CT; media and sera, GIBCO, Grand Island, NY; and all other reagents, Sigma, St. Louis, MO.
Cell culture. Rat hippocampal neurons were grown in primary culture as described previously (Wang et al., 1994) with minor modifications. Fetuses were removed on embryonic day 17 from maternal rats anesthetized with CO2 and killed by decapitation. Hippocampi were dissected and placed in Ca2+ and Mg2+-free HEPES-buffered Hank’s salt solution (HHSS), pH 7.45. HHSS was composed of the following (in mm): HEPES 20, NaCl 137, CaCl2 1.3, MgSO4 0.4, MgCl2 0.5, KCl 5.0, KH2PO4 0.4, Na2HPO4 0.6, NaHCO3 3.0, and glucose 5.6. Cells were dissociated by trituration through a 5 ml pipette and a flame-narrowed Pasteur pipette. Cells were pelleted and resuspended in DMEM without glutamine, supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 U/ml and 100 μg/ml, respectively). Dissociated cells then were plated at a density of 50,000 cells/well onto 25-mm-round 1 cover glasses that had been coated with poly-d-lysine (0.1 mg/ml), and washed with H2O. Neurons were grown in a humidified atmosphere of 10% CO2 and 90% air (pH = 7.4) at 37°C, and fed every 7 days by exchange of 30% of the media with DMEM supplemented with 10% horse serum and penicillin/streptomycin. Cells used in these experiments were cultured without mitotic inhibitors for a minimum of 2 weeks.
[Ca2+]i measurement.[Ca2+]i was determined using a previously described dual-emission microfluorimeter (Werth and Thayer, 1994) to monitor indo-1 (Grynkiewicz et al., 1985). Cells were loaded with 2 μm indo-1 acetoxymethyl ester for 45 min at 37°C in HHSS containing 0.5% bovine serum albumin. Loaded cells were placed in a flow-through chamber (Thayer et al., 1988), and experiments were performed at room temperature. The chamber was mounted on an inverted microscope, and cells were superfused with HHSS containing 10 μm glycine at a rate of 1–2 ml/min for 15 min before starting an experiment. Superfusion solutions were selected with a multiport valve coupled to several reservoirs.
For excitation of indo-1, light from a 75 W Xe arc lamp was passed through a 350/10 nm band-pass filter (Omega Optical, Brattleboro, VT). Excitation light was reflected from a dichroic mirror (380 nm), through a 70 × phase-contrast oil immersion objective (Leitz, numerical aperture 1.15). Emitted light was reflected sequentially from dichroic mirrors (440 and 516 nm), through band-pass filters (405/20 and 495/20 nm, respectively), to photomultiplier tubes operating in photon-counting mode (Thorn EMI, Fairfield, NJ). Cells were illuminated with transmitted light (580 nm long pass) and visualized with a video camera placed after the second emission dichroic. Recordings were defined spatially with a rectangular diaphragm. The 5 V photomultiplier output was integrated by passing the signal through an eight-pole Bessel filter at 2.5 Hz. This signal then was input into two channels of an analog-to-digital converter (Indec Systems, Sunnyvale, CA) sampling at 1 Hz.
After completion of each experiment, cells were wiped from the microscope field using a cotton-tipped applicator, and background light levels were determined (typically <5% of cell counts). Autofluorescence from cells that had not been loaded with dye was not detectable. Records were corrected later for background, and the ratios recalculated. Indo-1 was calibrated by converting the ratios to [Ca2+]i by the equation [Ca2+]i =Kdβ(R − Rmin)/(Rmax − R), in which R is the 405/495 nm fluorescence ratio. TheKd used for indo-1 was 250 nm, and β was the ratio of the emitted fluorescence at 495 nm in the absence and presence of calcium.Rmin, Rmax, and β were determined in ionomycin-permeabilized cells in calcium-free (1 mm EGTA) and 5 mmCa2+ buffers. The system was recalibrated after any adjustments. Values of Rmin,Rmax, and β ranged from 0.35–0.38, 4.23–4.34, and 3.0–3.95, respectively.
Electrophysiology. Whole-cell recordings were obtained from cultured neurons using pipettes (3–5 MΩ resistance) pulled from borosilicate glass (Narashige, Greenvale, NY) on a Sutter Instruments (Novato, CA) P-87 micropipette puller. For recording EPSCs, kainate-gated currents, and action potentials, pipettes were filled with a solution containing (in mm): k-gluconate 130, KCl 10, NaCl 10, BAPTA 10, HEPES 10, glucose 10, MgATP 5, Na2GTP 0.3, pH 7.2 with KOH, 300 mOsm/kg. Whole-cell recordings were established in an extracellular solution containing (in mm): NaCl 127, KCl 5, CaCl2 10, MgCl2 0.9, glucose 5, and HEPES 10, pH 7.4 with NaOH. For experiments measuring EPSCs and kainate-evoked currents, after the gigaohm seal was formed, the external solution was changed to one containing (in mm): NaCl 140, KCl 5, CaCl23, MgCl2 6, glucose 5, HEPES 10, pH 7.4 with NaOH, 310 mOsm/kg with sucrose. High divalent ion concentrations were used to suppress polysynaptic responses. Polysynaptic responses were suppressed further by using sparse cultures and selecting pairs of cells that were alone in the microscope field (400 ×). The high extracellular Mg2+ concentration ([Mg2+]o) together with the absence of glycine served to isolate the non-NMDA component of the synaptic response. In some EPSC experiments, the external solution contained 10 μm bicuculline methchloride; results were similar with or without bicuculline in the bath. Action potentials were recorded in an extracellular solution that contained (in mm): NaCl 143, KCl 5, CaCl2 1.3, MgCl2 0.9, glucose 5, HEPES 10, pH 7.4 with NaOH, 310 mOsm/kg with sucrose. IPSCs were recorded as for EPSCs, except that the extracellular solution contained 50 μm AP-5 and 10 μm CNQX, and the pipettes were filled with a solution containing (in mm): KCl 140, BAPTA 10, HEPES 10, glucose 10, MgATP 5, Na2GTP 0.3, QX314 1, pH 7.2 with KOH, 300 mOsm/kg. For NMDA-gated currents, the pipette solution contained (in mm) CsMeSO3 125, CsCl 15, CaCl23, BAPTA 11, HEPES 20, MgATP 5, Na2GTP 0.3, pH 7.2 with CsOH, 300 mOsm/kg, and the external recording solution contained (in mm): KCl 5, NaCl 137, CaCl2 1.3, HEPES 20, glucose 5, and (in μm): glycine 10, strychnine 2, bicuculline methchloride 10, CNQX 10, and TTX 0.3, pH 7.4 with NaOH, 310 mOsm/kg with sucrose. Combined electrophysiology and microfluorimetry experiments were conducted in extracellular solution similar to that used for synaptic transmission recordings except that CaCl2 was 1.3 mm, MgCl2 was either 0.9 or 0.1 mm, and 10 μm glycine was added. Na2GTP was omitted from the pipette solution, and 200 μm indo-1 pentapotassium salt, the cell-impermeant form of the fluorescent calcium chelator, was substituted for BAPTA. Indo-1 fluorescence was measured as described above in [Ca2+]i measurementexcept that background light levels were collected in the cell-attached configuration. Solutions were applied by a gravity-fed superfusion system; exchange of solutions was complete within 10 sec. Drugs were applied until an apparent steady-state inhibition was achieved.
Whole-cell currents were recorded using an Axopatch 200A patch-clamp amplifier and the BASIC-FASTLAB interface system (Indec systems). For combined electrophysiology and microfluorimetry, membrane potential recordings were filtered at 25 Hz (four-pole Bessel low-pass filter) and sampled every 8 msec (slow sweeps, see Fig. 2) or were filtered at 200 Hz and sampled every 1 msec (fast sweeps, see Fig. 2). Synaptic transmission and current-clamp recordings were filtered at 2 kHz and sampled every 100 μsec. EPSCs were elicited every 10 sec, and IPSCs were elicited every 15 sec by a 0.1 msec pulse delivered by a concentric-bipolar, extracellular, stimulating electrode positioned near the cell body of a nearby neuron. The postsynaptic cell was voltage clamped at −70 mV. Action potentials were evoked every 5 sec by current injection through the patch pipette or every 15 sec by a concentric-bipolar, extracellular, stimulating electrode positioned near the cell body and recorded in whole-cell current-clamp mode. Kainate and NMDA-gated currents were recorded from cells held at −70 mV and elicited by a 15 sec bath application of agonist (100 μm) applied every 5 min. These currents were filtered at 20 Hz and sampled every 10 msec. Displayed currents were not corrected for leak.
Fig. 2.

Low [Mg2+]o-induced [Ca2+]i spiking is driven by bursts of action potentials. [Ca2+]i (upper trace) and membrane potential (Vm,lower trace) recorded from a hippocampal neuron in whole-cell current clamp. Initially, the neuron was bathed in a solution containing 0.9 mm[Mg2+]o; thearrow indicates the onset of exposure to 0.1 mm[Mg2+]o. Action potentials are truncated in this record by low sampling rate (125 Hz) and filter setting (25 Hz). Data collection was interrupted briefly after ∼1 min and again after ∼3 min, as indicated bygaps in the traces. Inset, [Ca2+]i (two upper traces) and Vm (two lower traces) recorded from the same neuron with a higher sampling rate (1 kHz) and filter setting (200 Hz) during a burst in either 0.9 or 0.1 mm[Mg2+]o. The onset of the bursts have been aligned to illustrate that the burst in low [Mg2+]o lasts longer, contains more action potentials, and triggers a much larger [Ca2+]i transient. All traces are representative of four experiments.
Data analysis. The frequency of [Ca2+]i spiking was calculated from data collected during a 10 min window starting 5 min after changing the bath to 0.1 mm[Mg2+]o for control (Fcontrol) and from a 5 min window starting 5 min after drug application for drug-treated (F). Percent inhibition was calculated by the formula ((Fcontrol − F)/Fcontrol) × 100. The concentration–response curve was fit by a logistic equation of the form % inhibition = ((IMax − IMin)/(1 + (X/EC50)b)) + IMin, where X is the agonist concentration, IMin andIMax are the % inhibition calculated forX = 0, and for an “infinite” concentration, respectively, and b is a slope factor that determines the steepness of the curve (De Lean et al., 1978). EC50 values were calculated by a nonlinear, least-squares curve fitting algorithm using Fig.P software (Biosoft).
Whole-cell currents were analyzed as follows. The mean current measured during the 5 msec (EPSC and IPSC) or 5 sec (kainate-gated current and NMDA-gated current) before the stimulus artifact was designated as holding current and the maximum inward current measured at any point between the upstroke and decay of the current was designated as peak current for a given response. Percent inhibition was calculated by the formula ((Imax − I)/Imax) × 100, in whichI for a given treatment was determined by subtracting the average holding current from the average peak current of several sweeps obtained after inhibition had reached apparent equilibrium during that treatment, and Imax was determined by applying the same procedure to several sweeps obtained just before drug application.
Synaptic responses were analyzed for changes in the number of failures and changes in the coefficient of variation (CV). The failure of synaptic transmission was assessed by inspecting visually individual sweeps for significant deflection from the baseline noise during 20 msec after the stimulus artifact (Ulrich and Huguenard, 1995). CV analysis was used to determine synaptic site of action of cannabinoid agonists (Faber and Korn, 1991). CV was calculated from 10 to 30 consecutive sweeps. CV = (varsignal)0.5/M, where M is the mean EPSC amplitude and thevarsignal is the variance of the recorded EPSC amplitudes corrected for the variance of the background noise using the relation varsignal = varEPSC − varnoise. Background noise was estimated by subtracting two periods of averaged recording of baseline before the stimulus artifact from each other (Sayer et al., 1989). Changes in the ratio of the CVs squared (r), r = CV2control/CV2drug were compared with the modification factor (π), π = Mdrug/Mcontrol(Ulrich and Huguenard, 1995).
Changes in the action potential were assessed by measuring the resting membrane potential, action potential threshold, action potential duration, and action potential amplitude. Resting membrane potential was determined by calculating the mean membrane potential during the 5 msec preceding current injection. Action potential threshold was defined as the inflection point of the derivative of the membrane potential during current injection. Amplitude was measured from threshold to the peak, and the action potential duration was measured at half peak amplitude.
Data are presented as mean ± SEM. Statistical comparisons were made by Student’s t test or ANOVA with Bonferroni post-test.
RESULTS
Reduced [Mg2+]o induces a stable pattern of glutamatergic synaptic activity
Reducing [Mg2+]oelicits a repetitive pattern of excitatory electrical activity and [Ca2+]i spiking in synaptically connected central neurons in primary culture (Kudo and Ogura, 1986; Abele et al., 1990; Rose et al., 1990; Robinson et al., 1993). In rat hippocampal cultures, reducing [Mg2+]o to 0.1 mm elicited repetitive [Ca2+]i spikes (Fig.1A). Spike frequency and amplitude decreased over the first 5 min to stabilize at a mean frequency of 0.15 ± 0.08 Hz (mean ± SD, n = 215) and amplitude of 395 ± 32 nm (n = 26). This frequency is in good agreement with the frequency of [Ca2+]i spikes observed in response to treatment of cultured cortical (Robinson et al., 1993) and hippocampal (Kudo and Ogura, 1986; Ogura et al., 1987) neurons with Mg2+-free buffers. [Ca2+]i spiking was dependent on glutamatergic synaptic activity, as indicated by complete block of spiking by the non-NMDA receptor antagonist CNQX (Fig.1B). Superfusion of 10 μm CNQX onto cells spiking in the presence of 0.1 mm[Mg2+]o blocked spiking activity completely and reversibly in each of five cells tested. The spiking activity also had a component mediated by NMDA receptors, as indicated by inhibition of [Ca2+]i spiking by the NMDA receptor antagonist CGS19755 (Fig. 1C). Superfusion of 10 μm CGS19755 reduced [Ca2+]i spiking frequency by 61 ± 19% and decreased amplitude by 74 ± 11% (n = 5). TTX (1 μm) completely blocked all [Ca2+]i spiking activity induced by 0.1 mm[Mg2+]o (Fig.1D, n = 5). This pharmacological characterization of 0.1 mm[Mg2+]o-induced [Ca2+]i spiking reveals that activation of glutamatergic synapses is required to produce the spikes. The sensitivity of the 0.1 mm[Mg2+]o-induced spiking to all drugs tested in this study was independent of the initial frequency of spiking. Recording changes in [Ca2+]i spike frequency provides a relatively straightforward method to study the effects of drugs on glutamatergic synaptic activity (Abele et al., 1990; Rose et al., 1990; Robinson et al., 1993).
Fig. 1.

Pharmacological characterization of 0.1 mm[Mg2+]o-induced [Ca2+]i spiking in cultured rat hippocampal neurons. A, Stable [Ca2+]i spiking was induced by superfusion with 0.1 mm[Mg2+]o during the time indicated by the horizontal bar (n = 26). Note that the frequency of [Ca2+]i spiking was high initially, then decreased to become stable after 5 min of exposure to 0.1 mm[Mg2+]o. [Ca2+]i spiking required low Mg2+ buffer, because spiking disappeared when [Mg2+]o was restored to 0.9 mm. In separate experiments, CNQX (10 μm, n = 5) (B) and TTX (1 μm, n = 5) (D) blocked low [Mg2+]o-induced [Ca2+]i spiking completely, whereas CGS19755 (10 μm,n = 5) (C) inhibited, but did not completely block, spiking. Treatments were applied during the times indicated by the horizontal bars. Scale bars apply to all traces. Low [Mg2+]o was applied throughout the recordings shown inB–D.
Low [Mg2+]o (0.1 mm)-induced [Ca2+]i spiking is driven by bursts of action potentials
To characterize the nature of the electrical activity that underlie [Ca2+]i spikes, we conducted combined whole-cell current-clamp and microfluorimetric recordings. In Figure 2, membrane potential (lower trace) was recorded simultaneously with indo-1-based [Ca2+]i measurements (upper trace). Virtually all neurons in these hippocampal cultures had some level of spontaneous activity. Typically, as shown in the initial 1 min of the recording in Figure 2, we observed either occasional, single action potentials that failed to produce a detectable increase in [Ca2+]i or a spontaneous burst of action potentials that produced a small transient [Ca2+]i increase as shown in Figure 2 immediately preceding the reduction in [Mg2+]o. After reducing [Mg2+]o to 0.1 mm, periodic bursts of action potentials were generated coincident with large [Ca2+]i spikes. In Figure2 inset, a recording of a spontaneous burst and a burst recorded in 0.1 mm[Mg2+]o are superimposed. Similar high time-resolution recordings from four cells revealed that exposure to 0.1 mm[Mg2+]o increased burst duration dramatically (3.4 ± 0.6 sec) and the number of action potentials per burst (30 ± 6) relative to spontaneous bursts in physiological [Mg2+]o(1.0 ± 0.2 sec and 11 ± 2 action potentials). The increased burst intensity in low [Mg2+]oalso produced a threefold increase in [Ca2+]i spike amplitude. A similar pattern of bursting electrical activity has been observed in hippocampal and cortical cultures after complete removal of [Mg2+]o (Abele et al., 1990; Rose et al., 1990; Robinson et al., 1993). We found that reducing [Mg2+]o to 0.1 mm rather than omitting it altogether decreased the frequency slightly and enhanced the stability and periodicity of bursts, rendering the method more suitable for assessing effects of drugs on glutamatergic synaptic activity (Abele et al., 1990).
Cannabinoid receptor agonists inhibit stereoselectively [Ca2+]i spiking
We evaluated the effects of cannabimimetics on 0.1 mm[Mg2+]o-induced [Ca2+]i spiking. Our initial test for a potential role of cannabinoid receptors in modulating glutamatergic synaptic activity was to take advantage of the stereoisomers of Win 55,212, which differ in affinity for cannabinoid receptors by well over 1000-fold (D’Ambra et al., 1992; Jansen et al., 1992; Kuster et al., 1993). As shown in Figure 3, superfusion with 10 nm Win 55,212-3, the inactive isomer, was without effect, although subsequent application of the active enantiomer, Win 55,212-2, completely blocked low [Mg2+]o-induced [Ca2+]i spiking. Overall, 10 nm Win 55,212-2 inhibited spiking frequency by 86 ± 4%, whereas 10 nm Win 55,212-3 did not affect [Ca2+]i spiking in any of the seven cells tested.
Fig. 3.

Stereospecific inhibition of 0.1 mm[Mg2+]o-induced [Ca2+]i spiking by cannabinoid receptor agonists. Win 55,212-3 and Win 55,212-2 (10 nm) were applied sequentially, as indicated byhorizontal bars. The cell was exposed to 0.1 mm[Mg2+]o throughout the recording. Win 55,212-3, a biologically inactive stereoisomer of the potent cannabinoid receptor agonist Win 55,212-2, did not affect low [Mg2+]o-induced [Ca2+]i spiking, but Win 55,212-2 blocked [Ca2+]ispiking completely. The trace shown is representative of seven experiments.
Synthetic cannabinoid receptor agonists are potent inhibitors of [Ca2+]i spiking frequency
We tested two classes of synthetic cannabinoid receptor agonists for their ability to inhibit, in a concentration-dependent manner, low [Mg2+]o-induced excitatory activity. CP 54,939 and CP55,940 are chemical derivatives of cannabinol (Johnson and Melvin, 1986). Win 55,212-2 is an aminoalkylindole, a group of compounds with potent cannabimimetic actions (Bell et al., 1991; D’Ambra et al., 1992). As shown in Figure4, these compounds all potently reduced the frequency of 0.1 mm[Mg2+]o-induced [Ca2+]i spiking. Increasing the concentration of these cannabimimetic drugs produced a graded reduction in the frequency of the spikes. Spike amplitude also decreased with increasing concentrations of the drugs, although this effect was more variable and only became apparent at concentrations that produced at least 50% inhibition of spike frequency. EC50 values were calculated by fitting the concentration response data plotted in Figure 4D with a logistic equation as described in Materials and Methods. EC50 values for CP 54,939, CP 55,940, and Win 55,212-2 were 0.36 ± 0.05, 1.2 ± 0.7, and 2.7 ± 0.3 nm, respectively, in reasonable agreement with previously published potency and rank order, which varies somewhat between preparations (see Discussion). The slope factors (b) for CP 54,939, CP 55,940, and Win 55,212-2 were 0.89 ± 0.09, 0.98 ± 0.46, and 1.6 ± 0.26, respectively. Because known drug concentrations were perfused continually in these experiments, b can be interpreted as the Hill coefficient nH (De Lean et al., 1978). Thus, for CP 54,939 and CP 55,940, a single class of noncooperative binding sites is suggested, because bapproximates 1. Interestingly, the b value for Win 55,212-2, an aminoalkylindole, appears greater than the values calculated for the cannabinoid analogs CP 54,939 and CP 55,940. A more complete structure activity relationship is required to determine the significance of this observation to the interaction of these drugs with the cannabinoid receptor.
Fig. 4.

Inhibition of low [Mg2+]o-induced [Ca2+]i spiking by the aminoalkylindole Win 55,212-2 and the synthetic cannabinoids CP54,939 and CP55,940. A–C, Representative traces illustrate concentration-dependent inhibition of 0.1 mm[Mg2+]o-induced [Ca2+]i spiking by Win 55,212-2, CP54,939, and CP55,940. Drug application is indicated by thebars, and 0.1 mm[Mg2+]o was superfused throughout the recordings. Scale bars apply to all traces.D, Concentration–response curves of Win 55,212-2, CP54,939, and CP55,940 inhibition of 0.1 mm[Mg2+]o-induced [Ca2+]i spike frequency. CP54,939 (solid circles) was the most potent of the tested compounds, EC50 0.36 ± 0.05 nm. Win 55,212-2 (open circles) also was a potent inhibitor of low Mg2+-induced [Ca2+]i spiking, EC50 2.7 ± 0.3 nm. CP55,940 (solid squares) maximally inhibited spiking frequency by only 44 ± 10%, although it was very potent, EC50 1.2 ± 0.7 nm. Data points represent at least three experiments and are expressed as mean ± SEM. Curves were fit by a logistic equation of the form % inhibition = ((IMax − IMin)/(1 + (X/EC50)b)) + IMin, where X is the drug concentration, IMin andIMax are the % inhibition calculated forX = 0 and for an “infinite” concentration, respectively, and b is a slope factor that determines the steepness of the curve. EC50 values were calculated by a nonlinear, least-squares curve fitting algorithm using Fig.P software (Biosoft) and are expressed as mean ± SEM.
CP55,940 is a partial agonist
Because CP 55,940, even at 1000 nm, a concentration over 800 times the EC50, produced maximal reduction in [Ca2+]i spiking frequency of only 44 ± 10%, we explored the possibility that this compound was a partial agonist in this system. This experiment is shown in Figure5. We elicited [Ca2+]i spiking in a hippocampal neuron by reducing the [Mg2+]o to 0.1 mm, which was maintained throughout the recording. Spiking then was blocked completely by application of the full agonist Win 55,212-2 (100 nm). In the continued presence of Win 55,212-2, application of 100 nm CP 55,940 restored [Ca2+]i spiking activity to 48 ± 9% of control, indicating that CP 55,940 antagonized the activity of Win 55,212-2. Thus, CP 55,940 is a partial agonist. Partial agonist properties for cannabinoid receptor-mediated inhibition of Ca2+ currents have been described previously for anandamide in NG108-15 cells (Mackie et al., 1993) and for CP 55,940 in sympathetic neurons injected with rat brain cRNA encoding the cannabinoid receptor (Pan et al., 1996).
Fig. 5.
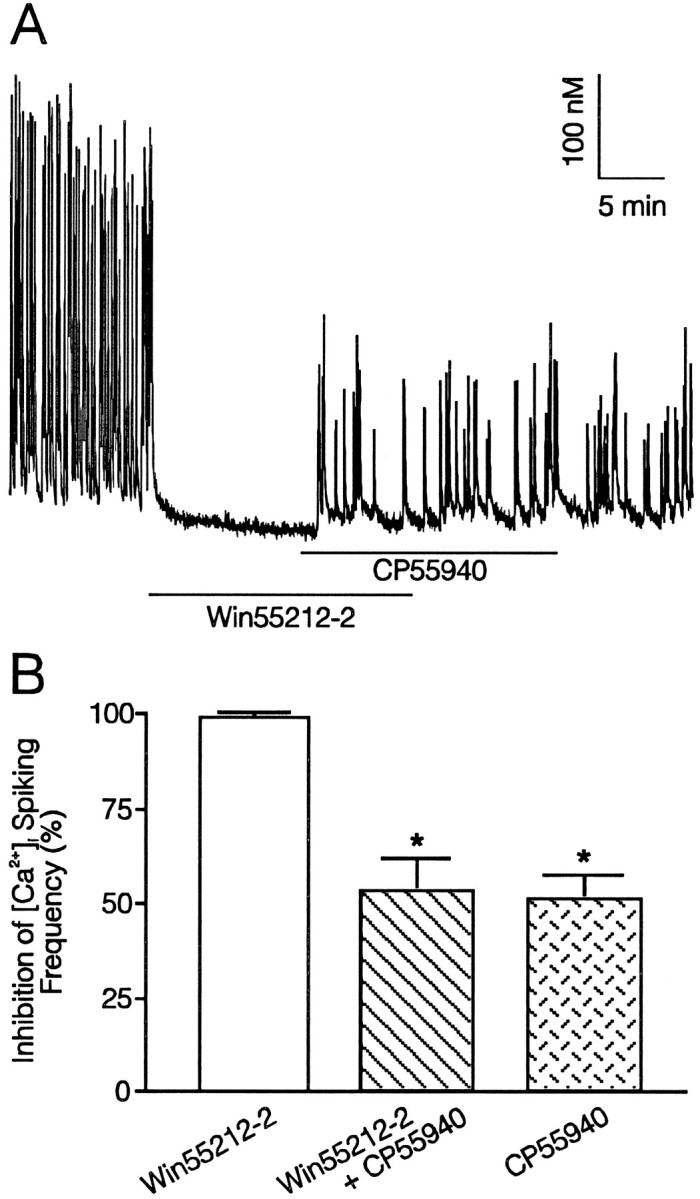
CP55,940 is a partial agonist. A, The synthetic cannabinoid CP55,940 (100 nm) partially reverses 100 nm Win 55,212-2 inhibition of low [Mg2+]o-induced [Ca2+]i spiking. Application of Win 55,212-2 and CP55,940 is indicated by thebars, and 0.1 mm[Mg2+]o buffer was superfused throughout the recording. B, Histogram summarizing reduction in [Ca2+]i spike frequency produced by Win 55,212-2 and CP55,940 (n = 8). Error bars represent SEM. Asterisks indicate significantly different from Win 55,212-2 application alone;p < 0.01 (ANOVA with Bonferroni post-test).
Anandamide inhibits [Ca2+]i spiking
Anandamide is synthesized and metabolized in brain (Devane et al., 1992; Deutsch and Chin, 1993; Dimarzo et al., 1994) and binds to cannabinoid receptors, inhibiting both adenylate cyclase and voltage-gated Ca2+ channels (Felder et al., 1993). Thus, anandamide represents an endogenous cannabinoid receptor ligand. In the present study, anandamide reduced the frequency of 0.1 mm[Mg2+]o-induced [Ca2+]i spiking with an EC50 of 71 ± 39 nm (Fig.6), in reasonable agreement with potency and affinity values reported for anandamide, which range from 20 nm, the IC50 for Ca2+ channel inhibition (Mackie et al., 1993), to 543 nm, the Ki for displacement of [3H]-CP 55,940 from cloned cannabinoid receptors expressed in murine Ltk (L)-cells (Felder et al., 1993). Anandamide (3 μm) inhibited [Ca2+]i spiking by 84 ± 8% (n = 3), similar to the maximal block observed with Win 55,212-2 and CP 54,939. However, the concentration–response curve was very broad, spanning a four-log change in concentration, and the slope factor, b, was 0.59 ± 0.19, considerably less than 1. The shallow slope of the dose–response curve may have resulted from nonspecific inhibition of glutamatergic synaptic transmission at high concentrations. A significant decrease in anandamide concentration owing to metabolism was unlikely, because the bath was superfused continually with drug; the 200 μl chamber was perfused at a rate of 1–2 ml/min, resulting in a complete bath exchange every 10 sec. Because partial agonist properties of anandamide have been reported previously (Mackie et al., 1993; Fride et al., 1995), we conducted experiments similar to those described above using CP 55,940 (Fig. 5) to test directly whether anandamide was a partial agonist in inhibiting [Ca2+]i spiking in the present study. Coapplication of 100 nm, 300 nm, 1 μm, and even 3 μm (n = 3) anandamide together with 100 nm Win 55,212-2 failed to reverse inhibition of 0.1 mm[Mg2+]o-induced [Ca2+]i spiking produced by Win 55,212-2 alone (data not shown). Thus, we were unable to detect partial agonist properties of anandamide in inhibiting [Ca2+]i spiking in the present study.
Fig. 6.

Anandamide inhibits glutamatergic synaptic transmission. A, B, Representative traces illustrate anandamide inhibition of 0.1 mm[Mg2+]o-induced [Ca2+]i spiking. Anandamide (10 nm) (A) or (1000 nm) (B) was superfused onto the cells at the times indicated by the horizontal bars. [Mg2+]o (0.1 mm) was superfused throughout the recording.C, Concentration–response curve of inhibition of low [Mg2+]o-induced [Ca2+]i spike frequency by anandamide. Data points represent at least three experiments and are expressed as mean ± SEM. EC50 = 71 ± 39 nm. The curve was fit by a logistic equation of the form % inhibition = ((IMax − IMin)/(1 + (X/EC50)b)) + IMin, where X is the drug concentration, IMin andIMax are the % inhibition calculated forX = 0 and for an “infinite” concentration, respectively, and b is a slope factor that determines the steepness of the curve. EC50 values were calculated by a nonlinear, least-squares curve fitting algorithm using Fig.P software (Biosoft) and are expressed as mean ± SEM.
Cannabinoid receptor agonists inhibit [Ca2+]i spiking via an inhibitory G-protein
Cannabinoid receptor-mediated inhibition of adenylyl cyclase (Howlett et al., 1986), activation of K+ channels (Deadwyler et al., 1993; Henry and Chavkin, 1995), inhibition of Ca2+ channels (Caulfield and Brown, 1992; Mackie and Hille, 1992; Mackie et al., 1993; Mackie et al., 1995; Pan et al., 1996), and, potentially, activation of phospholipase A2 (Audette et al., 1991) are mediated by coupling to inhibitory G-proteins. We investigated whether inhibition of [Ca2+]i spiking by cannabimimetics described here also was mediated by an inhibitory G-protein by treating the cultures with PTX. Cultures treated with either 500 ng/ml PTX for 24 hr, 1 μg/ml cholera toxin (CTX) for 4 hr, or 500 ng/ml heat-inactivated (15 min, 95°C) PTX for 24 hr were compared with untreated control cultures. As shown in Figure7A, Win 55,212-2 (100 nm) failed to inhibit 0.1 mm[Mg2+]o-induced [Ca2+]i spiking in cultures pretreated with PTX (n = 9). In contrast, heat-inactivated PTX was without effect, as indicated by a full inhibition of 0.1 mm[Mg2+]o-induced [Ca2+]i spiking by 100 nm Win 55,212-2 (n = 6). Treating the cultures with CTX did not affect the inhibition of [Ca2+]i spiking produced by Win 55,212-2 (n = 6). These results are summarized in Figure 7C, in which Win 55,212-2 induced inhibition of 0.1 mm[Mg2+]o-induced [Ca2+]i spiking was left intact after treatment with CTX or heat-inactivated PTX, but prevented by prior treatment with PTX (p < 0.001).
Fig. 7.

Treatment with PTX prevents inhibition of glutamatergic synaptic transmission by Win 55,212-2. A, PTX pretreatment (500 ng/ml, 24–28 hr) prevented 100 nm Win 55,212-2 inhibition of 0.1 mm[Mg2+]o-induced [Ca2+]i spiking.B, Heat-inactivated PTX (15 min, 95°C) pretreatment (500 ng/ml, 24–28 hr) did not affect 100 nm Win 55,212-2 inhibition of low [Mg2+]o-induced [Ca2+]i spiking.C, Histogram summarizing the effects of pretreating cultured hippocampal neurons with PTX, CTX (1 μg/ml, 4 hr), and heat-inactivated PTX on 100 nm Win 55,212-2 inhibition of low [Mg2+]o-induced [Ca2+]i spiking (n = 9 for control and PTX treated, n = 6 for CTX treated and heat-inactivated PTX treated). Error bars represent SEM. Asterisks indicate significantly different from control; p < 0.001 (ANOVA with Bonferroni post-test).
Cannabinoid receptor agonists inhibit the presynaptic release of glutamate
Data presented thus far indicate clearly that cannabinoid receptor agonists interfere with an excitatory pattern of electrical activity that requires activation of glutamatergic synapses. To test whether the cannabimimetics were acting directly on glutamatergic synaptic transmission, and further resolve whether the effects of these drugs were pre- or postsynaptic, we recorded excitatory postsynaptic currents (EPSCs) using the whole-cell configuration of the patch clamp. The presynaptic cell was stimulated with an extracellular bipolar concentric electrode, and EPSCs were recorded in extracellular solution that contained no glycine and 6 mm[Mg2+]o to reduce the probability of polysynaptic responses. As shown in Figure8, presynaptic stimulation for 100 μsec elicited reproducible EPSCs. Experiments in which EPSCs exhibited a multicomponent waveform or were not coupled strictly in one-to-one fashion with the stimulus were not included in the data set. The 14 experiments that met these criteria had a mean latency of 4.1 ± 0.7 msec, a mean rise time (t1/2) of 1.5 ± 0.1 msec, a mean amplitude of 463 ± 97 pA, and an average recovery time constant (τ) of 4.6 ± 0.4 msec. In nine experiments, superfusion of 10 μm CNQX inhibited reversibly synaptic transmission by 93 ± 2% (Fig. 8A, B); the noncompetitive NMDA receptor antagonist CGS19755 (10 μm) had no effect (3 ± 11% inhibition,n = 4), indicating that under these conditions, the EPSCs were mediated entirely by glutamate activation of non-NMDA receptors. As shown in Figure 8, A and B, superfusion of 100 nm Win 55,212-3, the inactive enantiomer, did not affect EPSC amplitude (0.3 ± 0.8%;n = 6) significantly. In contrast, the active compound Win 55,212-2 (100 nm) inhibited EPSC amplitude by 86 ± 4% (p < 0.05; n = 10, paired t test). We also tested the partial agonist CP 55,940 (1 μm) for effects on EPSCs and found, similar to [Ca2+]i spiking experiments, that CP 55,940 showed modest efficacy producing maximal inhibition of 60 ± 6% (n = 5). Furthermore, the inhibition produced by 100 nm Win 55,212-2 (92 ± 3%) was partially reversed by 100 nm CP 55,940 (71 ± 4%, n = 4, p < 0.01, pairedt test).
Fig. 8.
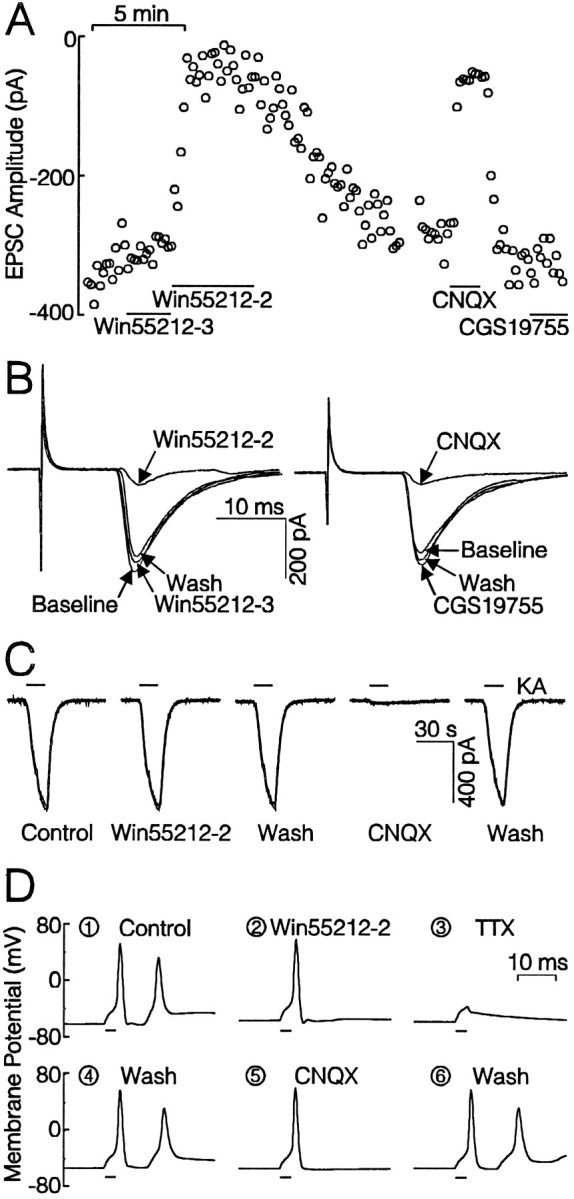
Cannabinoid receptor activation inhibits glutamatergic neurotransmission. A, Plot of EPSC amplitude versus time showing that Win 55,212-3 (100 nm) had no effect, but Win 55,212-2 (100 nm) elicited substantial and reversible inhibition of EPSC amplitude. CNQX (10 μm) blocked EPSCs completely, whereas CGS19755 (10 μm) had no effect. Drugs were applied as indicated by the horizontal bars. Data collection was stopped briefly during the break in the plot at ∼15 min.B, EPSCs recorded during the experiment in A. Traces are time-superimposed averages of three EPSCs recorded during the indicated treatment. The large spike at theleft of the records is stimulus artifact. Scale bars apply to both sets of currents. C, Win 55,212-2 (100 nm) did not inhibit whole-cell currents evoked by application of 100 μm kainate. CNQX (10 μm) blocked kainate currents completely. The horizontal bars indicate kainate application.D, Win 55,212-2 (100 nm) did not affect the action potential waveform elicited by current injection. In normal recording solution, current injection evoked an action potential followed by a secondary EPSP (D1). Win 55,212-2 blocked the EPSP (D2). TTX (0.3 μm) abolished both action potential and secondary EPSP (D3). CNQX (10 μm) also blocked secondary EPSP (D5) reversibly (D6). The bars below each trace indicate when current was injected.
Win 55,212-2 (100 nm) did not significantly affect inward currents elicited by exogenously applied kainate (100 μm), as shown in Figure 8C. When Win 55,212-2 was present during the second application of kainate, the second response was 99 ± 4% (n = 5) of the first. CNQX blocked completely the kainate-induced currents (n = 4). Thus, 100 nm Win 55,212-2 did not affect directly AMPA- or kainate-activated ion channels. Although the recording conditions used for evoked synaptic currents eliminate an NMDA receptor-mediated component, the 0.1 mm[Mg2+]o induced [Ca2+]i spiking activity was inhibited significantly by NMDA receptor antagonists. Thus, we tested NMDA-activated currents for sensitivity to cannabimimetics. In control experiments, the second current evoked by 100 μm NMDA was 91 ± 5% of the first (n = 7). Win 55,212-2 (100 nm) did not affect the second response significantly (79 ± 4%), relative to the rundown seen in control (n = 7).
Because Δ-9-tetrahydrocannabinol at micromolar concentrations will inhibit voltage-gated Na+ channels (Turkanis et al., 1991), we explored the possibility that Win 55,212-2 might inhibit the presynaptic action potential evoked by field stimulation. Action potentials were evoked in six cells held in whole cell current clamp by direct current injection. Win 55,212-2 (100 nm) had no effect (paired t test) on resting membrane potential (−60 ± 1 vs −59 ± 1 mV), action potential threshold (−36 ± 3 vs −34 ± 3 mV), action potential duration (1.2 ± 0.2 vs 1.1 ± 0.1 msec), or action potential amplitude (87 ± 4 vs 88 ± 4 mV). In four cells, current injection elicited a rapid action potential followed by a secondary depolarization that we determined was an excitatory postsynaptic potential (EPSP) (Fig. 8D1). Presumably, the action potential was exciting a local circuit, possibly an autapse, that fed back onto the current-clamped cell. Win 55,212-2 (100 nm) had no effect on the evoked action potential, although it blocked the EPSP completely (Fig. 8D2). TTX blocked both the action potential and EPSP (Fig. 8D3). After washout of TTX (Fig. 8D4), 10 μmCNQX was found to block the EPSP completely (Fig. 8D5). Thus, in the same cell, we found that Win 55,212-2 had no effect on the action potential waveform at a concentration that blocked the EPSP completely. Similar results were obtained in cells stimulated by extracellular electrode (n = 3). We hypothesize that Win 55,212-2 activates presynaptic cannabinoid receptors that subsequently inhibit the release of glutamate into the synaptic cleft.
In experiments such as that represented in Figure 8A, fluctuation in the amplitude of the EPSCs reflects probabilistic release of transmitter. We analyzed synaptic transmission experiments for changes in the CV of the evoked EPSCs as described by Faber and Korn (1991). This analysis assumes CV2 to be inversely proportional to quantal content and independent of the size of a quantum. Thus, the ratio of the squared CVs (r) before and after drug treatment (r = CV2control/CV2drug) should remain at unity for a purely postsynaptic effect that would alter the size of the quanta, not the probability, of their release. A presynaptic site of action for the cannabinoids predicts that these drugs should produce a reduction in r at least as much as a reduction in the amplitude of the EPSCs. In Figure 9A,r values calculated for Win 55,212-2 (solid circles), CP 55,940 (solid triangles), and CNQX (open squares) are plotted versus the modification factor (π). π is the fraction of EPSC amplitude that remains in the presence of drug (π = EPSCdrug/EPSCcontrol). The mean r and π for 100 nm Win 55,212-2 were 0.08 ± 0.03 and 0.18 ± 0.04, respectively (n = 10). In nine of the experiments, 10 μm CNQX also was applied (open squares), presumably acting postsynaptically. The meanr and π for 10 μm CNQX were 0.4 ± 0.1 and 0.09 ± 0.02, respectively (n = 9). Whereas all of the points from cannabimimetic treatment (solid symbols) were on or below the diagonal (slope = 1), those in the presence of CNQX (open symbols) all were above the diagonal. Finding that for the cannabimimetics, mean r was smaller than the mean π is consistent with a presynaptic site of action, because the reduction in EPSC amplitude can be accounted for entirely by a reduction in quantal content. As shown in Figure 9B, the appropriately scaled EPSC in the presence of Win 55,212-2 could be superimposed on the control. A reduction in the scale, but not the shape, of the EPSC waveform after cannabinoids suggests that the same synaptic boutons were activated under both conditions (Ulrich and Huguenard, 1995). Win 55,212-2 (100 nm) also increased the number of synaptic failures from 4 ± 4% in control to 40 ± 11% (Fig. 9C). CP 55,940 (1 μm) increased response failures from 3 ± 2% to 25 ± 11%.
Fig. 9.

Cannabinoid agonists act presynaptically to inhibit neurotransmission. A, Plot of the ratio of the CVs squared (r = CV2control/CV2drug) versus the ratio of the EPSC amplitude (π = Mdrug/Mcontrol) shows that all the points generated in the presence of 100 nm Win 55,212-2 (solid circles) and 1 μm CP55,940 (solid triangles) lay on or below the diagonal (r = π), whereas points generated in the presence of 10 μm CNQX (open squares) lay above the diagonal. B, After scaling to the peak of control, the average EPSC of five sweeps in the presence of 100 nm Win 55,212-2 was superimposed on that of control. The current surge before the EPSC is stimulus artifact.C, Summary of successful synaptic responses during control and after application of 100 nm Win 55,212-2. In the presence of 100 nm Win 55,212-2, successful synaptic responses were reduced from 96 ± 4% (control) to 60 ± 11%,n = 10, *p < 0.01 (paired ttest).
Thus, four lines of evidence point to a presynaptic site of action for the reduction in glutamatergic neurotransmission produced by the cannabinoids. Cannabinoids did not affect the action potential that serves as the stimulus for evoked EPSCs, nor did the drugs affect the direct activation of glutamate-gated ion channels. Cannabinoids did increase both the coefficient of variation and the number of response failures for evoked EPSCs.
Cannabinoid receptor activation does not affect GABA-mediated inhibitory neurotransmission
In primary rat hippocampal cultures, the [Ca2+]i spiking activity results from complex electrical activity that includes an inhibitory tone that also might be susceptible to modulation by cannabimimetics. To test whether cannabimimetics affect inhibitory synaptic transmission, we recorded IPSCs using the whole-cell configuration of the patch clamp. The presynaptic cell was stimulated with an extracellular bipolar concentric electrode, and IPSCs were recorded with 140 mm Cl− in the patch pipette producing large inward currents (Wilcox and Dichter, 1994). As shown in Figure 10, IPSCs were blocked completely by 10 μm bicuculline, confirming that they were mediated by GABA-activated ion channels. The IPSCs were not affected by 100 nm Win 55,212-2 (4 ± 6%,n = 10). Furthermore, CP 55,940 (1 μm) had no effect on evoked IPSCs (n = 3). Thus, cannabinoid receptor agonists had no effect on GABAergic synaptic transmission in these rat hippocampal cultures.
Fig. 10.

Cannabinoid receptor activation does not affect GABA-mediated inhibitory neurotransmission. Inward IPSCs were recorded from a −70 mV holding potential with 140 mminternal Cl− as described in Materials and Methods. Win 55,212-2 (100 nm) had no effect on the amplitude of IPSCs evoked by stimulation of the presynaptic cell with an extracellular electrode. IPSCs were blocked by 10 μm bicuculline. Drugs were applied at the times indicated by the horizontal bars. Inset,Representative IPSC traces during control, Win 55,212-2 treatment and bicuculline treatment were superimposed. Each trace is the average of five consecutive sweeps recorded during the indicated treatment.
DISCUSSION
In rat hippocampal cultures, cannabinoid receptor agonists inhibited both glutamatergic synaptic transmission elicited by reducing [Mg2+]o, which excites the entire network of neurons in the culture (Robinson et al., 1993), and glutamatergic EPSCs elicited by direct stimulation of the presynaptic neuron. These effects were clearly mediated by cannabinoid receptors, as indicated by pharmacological criteria: (1) The agonists were potent; subnanomolar concentrations inhibited neurotransmission. (2) The inhibition was stereoselective. (3) The compounds varied in efficacy. (4) The rank order of potency was consistent with previously reported values at cannabinoid receptors. (5) The receptors coupled to inhibitory G-proteins, as indicated by blockade of the cannabinoid effects by pretreatment with PTX.
We concluded that activation of cannabinoid receptors inhibited the release of glutamate presynaptically. Win 55,212-2 decreased the amplitude of EPSCs evoked by stimulation of the presynaptic neuron, but did not affect currents elicited by the exogenous application of kainate or NMDA. This effect was not a result of preventing the stimulus from reaching the nerve terminal, because Win 55,212-2 did not affect the evoked action potential. Indeed, in cells that participated in local networks, the drug left the action potential intact but completely blocked the EPSP. Furthermore, cannabimimetics did not affect IPSCs elicited in the same manner. Win 55,212-2 increased both the CV of the synaptic response and the number of failures, providing additional evidence of a presynaptic site of action. CV analysis requires certain assumptions. Of primary concern is that the same synaptic boutons are activated in the presence of drugs. This appears to be the case for the experiments described here, because the EPSC waveform changed in scale but not shape. Cannabinoid activation of K+ channels (Deadwyler et al., 1993; Henry and Chavkin, 1995) may produce a presynaptic hyperpolarization that contributes to the inhibition of EPSC amplitude. However, a postsynaptic shunting of the synaptic currents is unlikely to account for the reduction in amplitude, because even a 10-fold change in membrane conductance would be predicted to only moderately influence the EPSC (Spruston et al., 1993). N- and P-type Ca2+ channels control the release of glutamate in the hippocampus (Luebke et al., 1993; Wheeler et al., 1994; Piser et al., 1995; Scholz and Miller, 1995), and activation of cannabinoid receptors inhibits these channels (Mackie et al., 1993; Mackie et al., 1995; Pan et al., 1996), suggesting that cannabimimetics may inhibit excitatory neurotransmission by modulating the influx of Ca2+ into the nerve terminal.
Other functional and anatomical evidence suggests a presynaptic locus for cannabinoid receptors. Cannabinoids inhibit electrically evoked, cholinergically mediated contraction of isolated vas deferens and ileum (Roth, 1978; Nye et al., 1985; Pertwee et al., 1992; Compton et al., 1993). Cannabinoids inhibit depolarization-evoked synaptosomal Ca2+ influx (Harris and Stokes, 1982; Okada et al., 1992), and bind with high affinity to synaptosomal membranes (Devane et al., 1988). Lesions of striatal projections to substantia nigra eliminated cannabinoid receptor binding in substantia nigra (Herkenham et al., 1991; Glass et al., 1993), and Win 55,212-2 attenuated GABAergic activity in the substantia nigra evoked by stimulation of the striatum (Miller and Walker, 1995), suggesting that cannabinoids might modulate the release of GABA. Cannabimimetics were without effect on IPSCs evoked in hippocampal cultures. Taken together, these results indicate that cannabinoid receptors are distributed widely and suggest that in many cases, they are localized presynaptically to modulate the release of several different neurotransmitters.
Cannabinoid mediated changes in glutamatergic neurotransmission were consistent with other cannabinoid mediated responses. The stereoisomers of Win 55,212-2 showed a marked difference in potency, and the three synthetic compounds tested inhibited excitatory synaptic activity in the rank order CP 54,939 (EC50 = 0.36 nm) > CP 55,940 (EC50 = 1.2 nm) > Win 55,212-2 (EC50 = 2.7 nm). We found the potency of these compounds in reasonable agreement with their binding to brain membranes (Devane et al., 1988; Kuster et al., 1993), inhibition of electrically stimulated contraction of peripheral smooth muscle (Pertwee et al., 1992; Compton et al., 1993), inhibition of adenylyl cyclase in the case of CP 54,939 (Howlett et al., 1988), and inhibition of Ca2+ currents (Mackie and Hille, 1992). The drugs were 10-fold more potent in this assay than for binding to brain slices (Herkenham et al., 1990; Jansen et al., 1992;Thomas et al., 1992) and 100 times more potent in this assay than for inhibition of adenylate cyclase in the case of CP 55,940 and Win 55,212-2 (Childers et al., 1992; Compton et al., 1993). The rank order of potency for these compounds has varied somewhat, depending on the particular assay system, although CP 55,940 was consistently found to be more potent than Win 55,212-2, possibly because of the interaction of the ligands with different residues on the receptor (Song and Bonner, 1995).
Anandamide inhibited glutamatergic synaptic transmission with an EC50 of 71 nm, in good agreement with the range of affinity and potency values reported for anandamide. In murine L-cells, anandamide competes with CP 55,940 for binding to cannabinoid receptors with a Kiof 543 nm and inhibits cAMP production with an IC50 of 160 nm (Felder et al., 1993). Anandamide competes with CP 55,940 for binding to P2 membranes with aKi of 101 nm (Smith et al., 1994) and binds to mouse forebrain membranes with aKD of 143 nm (Hillard et al., 1995). In N18 neuroblastoma cells, anandamide inhibits Ca2+ current with an IC50of 20 nm, but only 61% efficacy relative to the full agonist Win 55,212-2 (Mackie et al., 1993). Anandamide inhibits electrically evoked contraction of ileum and vas deferens with IC50s of 289 and 61 nm, respectively (Pertwee et al., 1995). Thus, anandamide is ∼10- to 100-fold less potent than the synthetic cannabinoids and aminoalkylindoles tested in this study. In some preparations, the apparent potency of anandamide is attenuated significantly by degradation, as indicated by its enhanced potency in the presence of phenylmethylsulphonyl fluoride, which inhibits degradation (Hillard et al., 1995; Pertwee et al., 1995). In the present study, the concentration–response curve for anandamide was broad, which may have resulted from nonspecific inhibition of synaptic activity at high concentrations of anandamide, or metabolism of the compound, which we did not attempt to prevent. However, continual superfusion of anandamide in the experiments reported here renders a significant decrease in drug concentration owing to metabolism unlikely. Consistent with this interpretation, the inhibition of glutamatergic synaptic transmission described here is among the more potent effects of anandamide reported to date.
The data presented here suggest that CP 55,940 is a partial agonist. CP 55,940 maximally inhibited Ca2+ spiking by only 44%, compared with complete inhibition by both Win 55,212-2, and CP 54,939. Similarly, Pan et al. (1996) found that CP 55,940 produced only 38% inhibition of N-type Ca2+ channels in rat sympathetic neurons injected with CB1 receptor cRNA. In contrast, in NG108-15 cells, Win 55,212-2 and CP 55,940 elicited identical inhibition of voltage-gated Ca2+ channels, and these compounds fully occluded Ca2+ channel inhibition by each other (Mackie and Hille, 1992). Mackie and Hille (1993) found that anandamide had partial agonist activity compared with Win 55,212-2 for inhibition of Ca2+ channels in N18 cells, and low doses of anandamide attenuated both inhibition of adenylate cyclase and behavioral effects evoked by Δ-9-tetrahydrocannabinol (Fride et al., 1995). We found that anandamide exhibited a broad dose–response curve, but micromolar concentrations inhibited [Ca2+]i spiking by greater than 80%, indicating that anandamide is nearly a full agonist for inhibition of glutamatergic synaptic transmission in hippocampal cultures. We confirmed these results by demonstrating that CP 55,940, but not anandamide, reversed inhibition by the full agonist Win 55,212-2. Thus, the efficacy of CP 55,940 and anandamide appears to be reversed for inhibition of Ca2+ current in N18 neuroblastoma cells versus inhibition of Ca2+current in a neuronal expression system and inhibition of excitatory synaptic transmission in cultured hippocampal neurons. These intriguing discrepancies may result from differences in receptor–effector coupling or the existence of distinct cannabinoid receptor subtypes (Munro et al., 1993) and isoforms (Shire et al., 1995).
The partial agonist characteristics of CP 55,940 may prove to be a desirable therapeutic attribute. Many drugs that interfere with glutamate neurotransmission produce psychotomimetic side effects (Piercey et al., 1988; Tricklebank et al., 1989). Better tolerated drugs appear to be less efficacious inhibitors of glutamate receptor activation, but retain neuroprotective efficacy, consistent with reduction, but not abolition, of glutamate receptor activation. Such drugs include the NMDA open-channel blocker memantine (Chen et al., 1992) and adenosine analogs, which reduce glutamate release presynaptically (Arvin et al., 1989; Scholz and Miller, 1991; Scanziani et al., 1992).
Glutamate is the predominant excitatory neurotransmitter in the brain (Orrego and Villanueva, 1993). Most neurons receive glutamatergic input (Seeburg, 1993), and many neurotransmitters produce presynaptic inhibition of glutamatergic synaptic transmission (Thompson et al., 1993). For these reasons, it is possible that presynaptic inhibition of glutamate release by the cannabinoid neuromodulatory system accounts for many of the pharmacological effects of cannabimimetics and is consistent with their potential clinical application as anticonvulsant, analgesic, and neuroprotective agents.
Footnotes
This work was supported by grants from National Institutes of Health (DA07304) and the National Science Foundation (IBN9412654). T.M.P. was supported by Training Grant T32DA07234. The synthetic cannabinoids CP 54,939 and CP 55,940 were gifts of Pfizer Inc. The aminoalkylindoles Win 55,212-2 and Win 55,212-3 were gifts from Sterling Winthrop.
Correspondence should be addressed to Stanley A. Thayer, Department of Pharmacology, University of Minnesota Medical School, 3-249 Millard Hall, 435 Delaware Street SE, Minneapolis, MN 55455.
Dr. Piser’s present address: Cardiovascular Pharmacology, Pharmacia and Upjohn, 301 Henrietta Street, Kalamazoo, MI 49001.
REFERENCES
- 1.Abele AE, Scholz KP, Scholz WK, Miller RJ. Excitotoxicity induced by enhanced excitatory neurotransmission in cultured hippocampal pyramidal neurons. Neuron. 1990;4:A413–A419. doi: 10.1016/0896-6273(90)90053-i. [DOI] [PubMed] [Google Scholar]
- 2.Abood ME, Martin BR. Neurobiology of marijuana abuse. Trends Pharmacol Sci. 1992;13:201–206. doi: 10.1016/0165-6147(92)90064-d. [DOI] [PubMed] [Google Scholar]
- 3.Arvin B, Neville L, Pan J, Roberts P. 2-Cloroadenosine attenuates kainic acid-induced toxicity within the rat striatum: relationship to release of glutamate and Ca2+influx. Br J Pharmacol. 1989;98:225–235. doi: 10.1111/j.1476-5381.1989.tb16886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Audette CA, Burstein SH, Doyle SA, Hunter SA. G-protein mediation of cannabinoid-induced phospholipase activation. Pharmacol Biochem Behav. 1991;40:559–563. doi: 10.1016/0091-3057(91)90363-7. [DOI] [PubMed] [Google Scholar]
- 5.Bell MR, D’Ambra TE, Kumar V, Eissenstat MA, Herrmann JLJ, Wetzel JR, Rosi D, Philion RE, Daum SJ, Hlasta DJ, Kullnig RK, Ackerman JH, Haubrich DR, Luttinger DA, Baizman ER, Miller MS, Ward SJ. Antinociceptive (aminoalkyl)indoles. J Med Chem. 1991;34:1099–1110. doi: 10.1021/jm00107a034. [DOI] [PubMed] [Google Scholar]
- 6.Bidaut-Russell M, Devane WA, Howlett AC. Cannabinoid receptors and modulation of cyclic AMP accumulation in the rat brain. J Neurochem. 1990;55:21–26. doi: 10.1111/j.1471-4159.1990.tb08815.x. [DOI] [PubMed] [Google Scholar]
- 7.Caulfield MP, Brown DA. Cannabinoid receptor agonists inhibit Ca current in NG108-15 neuroblastoma cells via a pertussis toxin-sensitive mechanism. Br J Pharmacol. 1992;106:231–232. doi: 10.1111/j.1476-5381.1992.tb14321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen HSV, Pellegrini JW, Aggarwal SK, Lei SZ, Warach S, Jensen FE, Lipton SA. Open-channel block of N -methyl-d-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor mediated neurotoxicity. J Neurosci. 1992;12:4427–4436. doi: 10.1523/JNEUROSCI.12-11-04427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Childers SR, Fleming L, Konkoy C, Marckel D, Pacheco M, Sexton T, Ward S. Opioid and cannabinoid receptor inhibition of adenylyl cyclase in brain. Ann NY Acad Sci. 1992;654:33–51. doi: 10.1111/j.1749-6632.1992.tb25954.x. [DOI] [PubMed] [Google Scholar]
- 10.Collins DR, Pertwee RG, Davies SN. The action of synthetic cannabinoids on the induction of long-term potentiation in the rat hippocampal slice. Eur J Pharmacol. 1994;259:R7–R8. doi: 10.1016/0014-2999(94)90666-1. [DOI] [PubMed] [Google Scholar]
- 11.Compton DR, Gold L, Ward SJ, Balster R, Martin BR. Aminoalkylindole analogs: cannabimimetic activity of a class of compounds structurally distinct from (delta)9-tetrahydrocannabinol. J Pharmacol Exp Ther. 1992;263:1118–1126. [PubMed] [Google Scholar]
- 12.Compton DR, Rice KC, de Costa BR, Razdan RK, Melvin LS, Johnson MR, Martin BR. Cannabinoid structure-activity relationships: correlation of receptor binding and in vivo activities. J Pharmacol Exp Ther. 1993;265:218–226. [PubMed] [Google Scholar]
- 13.D’Ambra TE, Estep KG, Bell MR, Eissenstat MA, Josef KA, Ward SJ, Haycock DA, Baizman ER, Casiano FM, Beglin NC, Chippari SM, Grego JD, Kullnig RK, Daley GT. Conformationally restrained analogues of pravadoline: nanomolar potent, enantioselective, (aminoalkyl)indole agonists of the cannabinoid receptor. J Med Chem. 1992;35:124–135. doi: 10.1021/jm00079a016. [DOI] [PubMed] [Google Scholar]
- 14.Deadwyler SA, Hampson RE, Bennett BA, Edwards TA, Mu J, Pacheco MA, Ward SJ, Childers SR. Cannabinoids modulate potassium current in cultured hippocampal neurons. Receptors Channels. 1993;1:121–134. [PubMed] [Google Scholar]
- 15.De Lean A, Munson PJ, Rodbard D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am J Physiol. 1978;235:E97–E102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- 16.Deutsch DG, Chin SA. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem Pharmacol. 1993;46:791–796. doi: 10.1016/0006-2952(93)90486-g. [DOI] [PubMed] [Google Scholar]
- 17.Devane WA, Dysarz FA, Johnson MR, Melvin LS, Howlett AS. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- 18.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 19.Dimarzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- 20.Faber DS, Korn H. Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J. 1991;60:1268–1294. doi: 10.1016/S0006-3495(91)82162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Felder CC, Briley EM, Axelrod J, Simpson JT, Mackie K, Devane WA. Anandamide, an endogenous cannabimimetic eicosanoid, binds to the cloned human cannabinoid receptor and stimulates receptor-mediated signal transduction. Proc Natl Acad Sci USA. 1993;90:7656–7660. doi: 10.1073/pnas.90.16.7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fride E, Barg J, Levy R, Saya D, Heldman E, Mechoulam R, Vogel Z. Low doses of anandamides inhibit pharmacological effects of Δ9-tetrahydracannabinol. J Pharmacol Exp Ther. 1995;272:699–707. [PubMed] [Google Scholar]
- 23.Glass M, Faull R, Dragunow M. Loss of cannabinoid receptors in the substantia nigra in Huntington’s disease. Neuroscience. 1993;56:523–527. doi: 10.1016/0306-4522(93)90352-g. [DOI] [PubMed] [Google Scholar]
- 24.Grynkiewicz G, Peonie M, Tsien RY. A new generation of calcium indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 25.Harris RA, Stokes JA. Cannabinoids inhibit calcium uptake by brain synaptosomes. J Neurosci. 1982;2:443–447. doi: 10.1523/JNEUROSCI.02-04-00443.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henry DJ, Chavkin C. Activation of inwardly rectifying potassium channels (GIRK1) by co-expressed rat brain cannabinoid receptors in Xenopus oocytes. Neurosci Lett. 1995;186:91–94. doi: 10.1016/0304-3940(95)11289-9. [DOI] [PubMed] [Google Scholar]
- 27.Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, Rice KC. Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herkenham M, Lynn AB, de Costa BR, Richfield EK. Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res. 1991;547:267–274. doi: 10.1016/0006-8993(91)90970-7. [DOI] [PubMed] [Google Scholar]
- 29.Hillard C, Edgemond W, Campbell W. Characterization of ligand binding to the cannabinoid receptor of rat brain membranes using a novel method—application to anandamide. J Neurochem. 1995;64:677–683. doi: 10.1046/j.1471-4159.1995.64020677.x. [DOI] [PubMed] [Google Scholar]
- 30.Howlett A. Pharmacology of cannabinoid receptors. Annu Rev Pharmacol Toxicol. 1995;35:607–634. doi: 10.1146/annurev.pa.35.040195.003135. [DOI] [PubMed] [Google Scholar]
- 31.Howlett AC, Qualy JM, Khachatrian LL. Involvement of Gi in the inhibition of adenylate cyclase by cannabimimetic drugs. Mol Pharmacol. 1986;29:307–313. [PubMed] [Google Scholar]
- 32.Howlett AC, Johnson MR, Melvin LS, Milne GM. Nonclassical cannabinoids analgetics inhibit adenylate cyclase: development of a cannabinoid receptor model. Mol Pharmacol. 1988;33:297–302. [PubMed] [Google Scholar]
- 33.Jansen EM, Haycock DA, Ward SJ, Seybold VS. Distribution of cannabinoid receptors in rat brain determined with aminoalkylindoles. Brain Res. 1992;575:93–102. doi: 10.1016/0006-8993(92)90428-c. [DOI] [PubMed] [Google Scholar]
- 34.Johnson MR, Melvin LS. The discovery of nonclassical cannabinoid analgetics. In: Mechoulam R, editor. Cannabinoids as therapeutic agents. CRC; Boca Raton, FL: 1986. pp. 121–145. [Google Scholar]
- 35.Kudo Y, Ogura A. Glutamate-induced increase in intracellular Ca2+ concentration in isolated hippocampal neurons. Br J Pharmacol. 1986;89:191–198. doi: 10.1111/j.1476-5381.1986.tb11135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuster JE, Stevenson JI, Ward SJ, D’Ambra TE, Haycock DA. Aminoalkylindole binding in rat cerebellum: selective displacement by natural and synthetic cannabinoids. J Pharmacol Exp Ther. 1993;264:1352–1363. [PubMed] [Google Scholar]
- 37.Luebke JI, Dunlap K, Turner TJ. Multiple calcium channel types control glutamatergic synaptic transmission in the hippocampus. Neuron. 1993;11:895–902. doi: 10.1016/0896-6273(93)90119-c. [DOI] [PubMed] [Google Scholar]
- 38.Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci USA. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mackie K, Devane WA, Hille B. Anandamide, an endogenous cannabinoid, inhibits calcium currents as a partial agonist in N18 neuroblastoma cells. Mol Pharmacol. 1993;44:498–503. [PubMed] [Google Scholar]
- 40.Mackie K, Lai Y, Westenbroek R, Mitchell R. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J Neurosci. 1995;15:6552–6561. doi: 10.1523/JNEUROSCI.15-10-06552.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mailleux P, Vanderhaeghen J. Distribution of neuronal cannabinoid receptor in the adult rat brain: a comparative receptor binding radioautography and in situ hybridization histochemistry. Neuroscience. 1992;48:655–668. doi: 10.1016/0306-4522(92)90409-u. [DOI] [PubMed] [Google Scholar]
- 42.Martin BR, Welch SP, Abood M. Progress toward understanding the cannabinoid receptor and its second messenger systems. Adv Pharmacol. 1994;25:341–397. doi: 10.1016/s1054-3589(08)60437-8. [DOI] [PubMed] [Google Scholar]
- 43.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 44.Matsuda LA, Bonner TL, Lolait SJ. Localization of cannabinoid receptor mRNA in rat brain. J Comp Neurol. 1993;327:535–550. doi: 10.1002/cne.903270406. [DOI] [PubMed] [Google Scholar]
- 45.Mechoulam R, Hanus L, Martin BR. Search for endogenous ligands of the cannabinoid receptor. Biochem Pharmacol. 1994;48:1537–1544. doi: 10.1016/0006-2952(94)90197-x. [DOI] [PubMed] [Google Scholar]
- 46.Miller AS, Walker JM. Effects of a cannabinoid on spontaneous and evoked neuronal activity in the substantia nigra pars reticulata. Eur J Pharmacol. 1995;279:179–185. doi: 10.1016/0014-2999(95)00151-a. [DOI] [PubMed] [Google Scholar]
- 47.Munro S, Thomas K, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 48.Nowicky AV, Teyler TJ, Vardaris RM. The modulation of long-term potentiation by delta-9-tetrahydrocannabinol in the rat hippocampus. Brain Res Bull. 1987;19:663–672. doi: 10.1016/0361-9230(87)90052-9. [DOI] [PubMed] [Google Scholar]
- 49.Nye JS, Seltzman HH, Pitt CG, Snyder SH. High-affinity cannabinoid binding sites in brain membranes labeled with [3H]-5′-trimethylammonium delta 8-tetrahydrocannabinol. J Pharmacol Exp Ther. 1985;234:784–791. [PubMed] [Google Scholar]
- 50.Ogura A, Lijima T, Amano T, Kudo Y. Optical monitoring of excitatory synaptic activity between cultured hippocampal neurons by a multi-site Ca2+ fluorometry. Neurosci Lett. 1987;78:69–74. doi: 10.1016/0304-3940(87)90563-5. [DOI] [PubMed] [Google Scholar]
- 51.Okada M, Urae A, Mine K, Shoyama Y, Iwasaki K, Fujiwara M. The facilitating and suppressing effects of Δ9-tetrahydrocannabinol on the rise in intrasynaptosomal Ca2+ concentration in rats. Neurosci Lett. 1992;140:55–58. doi: 10.1016/0304-3940(92)90680-6. [DOI] [PubMed] [Google Scholar]
- 52.Orrego F, Villanueva S. The chemical nature of the main central excitatory transmitter: a critical appraisal based upon release studies and synaptic vesicle localization. Neuroscience. 1993;56:539–555. doi: 10.1016/0306-4522(93)90355-j. [DOI] [PubMed] [Google Scholar]
- 53.Pan X, Ikeda SR, Lewis DL. The rat brain cannabinoid receptor modulates N-type Ca2+ channels in a neuronal expression system. Mol Pharmacol. 1996;49:707–714. [PubMed] [Google Scholar]
- 54.Pertwee RG, Stevenson LA, Elrick DB, Mechoulam R, Corbett AD. Inhibitory effects of certain enantiomeric cannabinoids in the mouse vas deferens and the myenteric plexus preparation of guinea-pig small intestine. Br J Pharmacol. 1992;105:980–984. doi: 10.1111/j.1476-5381.1992.tb09088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pertwee RG, Fernando SR, Griffin G, Abadji V, Makriyannis A. Effect of phenylmethylsulphonyl fluoride on the potency of anandamide as an inhibitor of electrically evoked contractions in two isolated tissue preparations. Eur J Pharmacol. 1995;272:73–78. doi: 10.1016/0014-2999(94)00618-h. [DOI] [PubMed] [Google Scholar]
- 56.Piercey M, Hoffmann W, Kaczkofsky P. Functional evidence for PCP-like effects of the anti-stroke candidate MK-801. Psychopharmacology. 1988;96:561–562. doi: 10.1007/BF02180042. [DOI] [PubMed] [Google Scholar]
- 57.Piser TM, Lampe RA, Keith RA, Thayer SA. Complete and reversible block by ω-grammotoxin SIA of glutamatergic synaptic transmission between cultured rat hippocampal neurons. Neurosci Lett. 1995;201:1–4. doi: 10.1016/0304-3940(95)12169-2. [DOI] [PubMed] [Google Scholar]
- 58.Robinson HPC, Kawahara M, Jimbo Y, Torimitsu K, Kuroda Y, Kawana A. Periodic synchronized bursting and intracellular calcium transients elicited by low magnesium in cultured cortical neurons. J Neurophysiol. 1993;70:1606–1616. doi: 10.1152/jn.1993.70.4.1606. [DOI] [PubMed] [Google Scholar]
- 59.Rose K, Christine C, Choi D. Magnesium removal induces paroxysmal neuronal firing and NMDA receptor-mediated neuronal degeneration in cortical cultures. Neurosci Lett. 1990;115:313–317. doi: 10.1016/0304-3940(90)90474-n. [DOI] [PubMed] [Google Scholar]
- 60.Roth S. Stereospecific presynaptic inhibitory effect of delta-9-tetrahydrocannabinol on cholinergic transmission in the myenteric plexus of the guinea pig. Can J Physiol Pharmacol. 1978;56:968–975. doi: 10.1139/y78-154. [DOI] [PubMed] [Google Scholar]
- 61.Sayer RJ, Redman SJ, Andersen P. Amplitude fluctuations in small EPSPs recorded from CA1 pyramidal cells in the guinea pig hippocampal slice. J Neurosci. 1989;9:840–850. doi: 10.1523/JNEUROSCI.09-03-00840.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scanziani M, Capogna M, Gahwiler BH, Thompson SM. Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron. 1992;9:919–927. doi: 10.1016/0896-6273(92)90244-8. [DOI] [PubMed] [Google Scholar]
- 63.Scholz KP, Miller RJ. Analysis of adenosine actions on Ca2+ currents and synaptic transmission in cultured rat hippocampal pyramidal neurons. J Physiol (Lond) 1991;435:373–393. doi: 10.1113/jphysiol.1991.sp018515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scholz KP, Miller RJ. Developmental changes in presynaptic calcium channels coupled to glutamate release in cultured rat hippocampal neurons. J Neurosci. 1995;15:4612–4617. doi: 10.1523/JNEUROSCI.15-06-04612.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seeburg PH. The Trends Neurosci/TIPS lecture—the molecular biology of mammalian glutamate receptor channels. Trends Neurosci. 1993;16:359–365. doi: 10.1016/0166-2236(93)90093-2. [DOI] [PubMed] [Google Scholar]
- 66.Shire D, Carillon C, Kaghad M, Calandra B, Rinaldicarmona M. An amino-terminal variant of the central cannabinoid receptor resulting from alternative splicing. J Biol Chem. 1995;270:3726–3731. doi: 10.1074/jbc.270.8.3726. [DOI] [PubMed] [Google Scholar]
- 67.Smith PB, Compton DR, Welch SP, Razdan RK, Mechoulam R, Martin BR. The pharmacological activity of anandamide, a putative endogenous cannabinoid, in mice. J Pharmacol Exp Ther. 1994;270:219–227. [PubMed] [Google Scholar]
- 68.Song ZH, Bonner TI. A lysine residue of the cannabinoid receptor is critical for receptor recognition by several agonists but not WIN55212-2. Soc Neurosci Abstr. 1995;21:2060. [PubMed] [Google Scholar]
- 69.Spruston N, Jaffe DB, Williams SH, Johnston D. Voltage- and space-clamp errors associated with the measurement of electrotonically remote synaptic events. J Neurophysiol. 1993;70:781–802. doi: 10.1152/jn.1993.70.2.781. [DOI] [PubMed] [Google Scholar]
- 70.Thayer SA, Sturek M, Miller RJ. Measurement of neuronal Ca2+ transients using simultaneous microfluorimetry and electrophysiology. Pflügers Arch. 1988;412:216–223. doi: 10.1007/BF00583753. [DOI] [PubMed] [Google Scholar]
- 71.Thomas BF, Wei X, Martin BR. Characterization and autoradiographic localization of the cannabinoid binding site in rat brain using [3H]11-OH-Δ9-THC-DMH. J Pharmacol Exp Ther. 1992;263:1383–1390. [PubMed] [Google Scholar]
- 72.Thompson SM, Capogna M, Scanziani M. Presynaptic inhibition in the hippocampus. Trends Neurosci. 1993;16:222–227. doi: 10.1016/0166-2236(93)90160-n. [DOI] [PubMed] [Google Scholar]
- 73.Tricklebank M, Singh L, Oles R, Preston C, Iversen S. The behavioral effects of MK-801: a comparison with antagonists acting non-competitively and competitively at the NMDA receptor. Eur J Pharmacol. 1989;167:127–135. doi: 10.1016/0014-2999(89)90754-1. [DOI] [PubMed] [Google Scholar]
- 74.Turkanis SA, Partlow LM, Karler R. Delta-9-tetrahydrocannabinol depresses inward sodium current in mouse neuroblastoma cells. Neuropharmacology. 1991;30:73–77. doi: 10.1016/0028-3908(91)90045-d. [DOI] [PubMed] [Google Scholar]
- 75.Ulrich D, Huguenard JR. Purinergic inhibition of GABA and glutamate release in the thalamus: implications for thalamic network activity. Neuron. 1995;15:909–918. doi: 10.1016/0896-6273(95)90181-7. [DOI] [PubMed] [Google Scholar]
- 76.Wang G, Randall R, Thayer S. Glutamate-induced intracellular acidification of cultured hippocampal neurons demonstrates altered energy metabolism resulting from Ca2+ loads. J Neurophysiol. 1994;72:2563–2569. doi: 10.1152/jn.1994.72.6.2563. [DOI] [PubMed] [Google Scholar]
- 77.Weinroth SE, Parenti DM, Simon GL. Wasting syndrome in AIDS: pathophysiologic mechanisms and therapeutic approaches. Infect Agent Dis. 1995;4:76–94. [PubMed] [Google Scholar]
- 78.Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci. 1994;14:348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wheeler DB, Randall A, Tsien RW. Role of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- 80.Wilcox KS, Dichter MA. Paired pulse depression in cultured hippocampal neurons is due to a presynaptic mechanism independent of GABAB autoreceptor activation. J Neurosci. 1994;14:1775–1788. doi: 10.1523/JNEUROSCI.14-03-01775.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]