

Abstract
An intrachromosomal duplication containing the PMP22gene is associated with the human hereditary peripheral neuropathy Charcot-Marie-Tooth disease type 1A, and PMP22 overexpression as a consequence of increased PMP22 gene dosage has been suggested as causative event in this frequent disorder of peripheral nerves. We have generated transgenic mice that carry additional copies of the pmp22 gene to prove that increased PMP22 gene dosage is sufficient to cause PNS myelin deficiencies. Mice carrying approximately 16 and 30 copies of the pmp22 gene display a severe congenital hypomyelinating neuropathy as characterized by an almost complete lack of myelin and marked slowing of nerve conductions. Affected nerves contain an increased number of nonmyelinating Schwann cells, which do not form onion bulbs but align in association with axons. The mutant Schwann cells are characterized by a premyelination-like state as indicated by the expression of embryonic Schwann cell markers. Furthermore, continued Schwann cell proliferation is observed into adulthood. We hypothesize that Schwann cells are impaired in their differentiation into the myelinating phenotype, leading to a disorder comparable to severe cases of hereditary motor and sensory neuropathies. Our findings, combined with the analysis of heterozygous and homozygous PMP22-deficient mice, indicate that aberrant pmp22 gene copy numbers cause various forms of myelination defects.
Keywords: PMP22, myelin, Schwann cell, dysmyelination, Charcot-Marie-Tooth disease, gene dosage, transgenic mouse
Mutations affecting the myelin proteins PMP22, Cx32, and P0 have been linked to the peripheral neuropathy Charcot-Marie-Tooth disease (CMT) (for review, see Suter and Snipes, 1995a). The most common form (70–80%) of familial and spontaneous CMT (CMT1A) is associated with a 1.5 megabase intrachromosomal duplication on chromosome 17p11.2, which contains the PMP22 gene (Lupski et al., 1991; Raeymaekers et al., 1991; Matsunami et al., 1992; Patel et al., 1992; Timmerman et al., 1992; Valentijn et al., 1992a). Less commonly, nonconservative missense point mutations within the putative transmembrane regions of PMP22 have also been found in CMT1A (Valentijn et al., 1992b; Roa et al., 1993a,b) and the congenital hypomyelinating peripheral neuropathy Dejerine-Sottas syndrome (DSS) (Roa et al., 1993c). The morphological abnormalities of these human disorders are comparable to those found in the natural mouse mutantsTrembler (Tr) and Trembler-J(Tr-J), both of which are also mutated in thepmp22 gene (Suter et al., 1992a,b; Suter et al., 1993). Deletions of the same chromosomal segment, which is duplicated in CMT1A, are linked to hereditary neuropathy with liability to pressure palsies (HNPP) (Chance et al., 1993), and the causative role of PMP22 in HNPP has been demonstrated by the generation of PMP22-deficient mice that exhibit a phenotype comparable to the human disease (Adlkofer et al., 1995).
PMP22 belongs to a family of hydrophobic proteins with four potential membrane-associated domains (Spreyer et al., 1991; Welcher et al., 1991). Structurally, PMP22 resembles proteolipid protein (PLP), the major myelin protein of the CNS, and an intriguing parallel exists in the genetics of PMP22 and PLP in that in both cases, altering gene dosage and point mutations lead to severe myelin deficiencies (Suter and Patel, 1994; Nave and Boespflug-Tanguy, 1996). Evidence derived from the analysis of PMP22-deficient mice and observations in human CMT1A patients have established that PMP22 is crucial for the correct development of peripheral nerves, the maintenance of axons, and the determination of myelin thickness and stability (Adlkofer et al., 1995). The exact function of PMP22 in the PNS, however, remains elusive (Suter and Snipes, 1995b).
In vitro experiments have suggested that PMP22, in addition to its role as a component of compact PNS myelin (Snipes et al., 1992;Haney et al., 1996), may be involved in the regulation of the cell cycle and apoptosis (Manfioletti et al., 1990; Fabbretti et al., 1995;Zoidl et al., 1995). A broad role of PMP22 is supported further by its widespread expression throughout embryonic mouse development (Baechner et al., 1995), its gene regulation by a complex regulatory system including a myelinating Schwann cell-specific and a ubiquitously expressed promoter (Suter et al., 1994), and the finding of an extended PMP22 gene family with high expression in various tissues outside of the PNS (Marvin et al., 1995; Taylor et al., 1995; Taylor and Suter, 1996).
In this report, we describe the phenotype caused by highly increasedpmp22 gene dosage in transgenic mice, which leads to a severe peripheral neuropathy reminiscent of DSS.
MATERIALS AND METHODS
Isolation of a cosmid containing the pmp22gene. Molecular biology techniques were performed according to standard protocols. A 129Sv mouse genomic pTCF cosmid library was screened using a 2 kb BamHI-EcoRI fragment of the 5′-flanking region of the pmp22 gene (5′-probe; see Fig. 1). For the characterization of clones and transgenic animals by Southern blot analysis, the same 5′-probe and an additional 3′-probe [1.1-kb-long SfiI-SacII fragment (Adlkofer et al., 1995)] was used. For the determination of the transgene copy number, an N-CAM probe (Cremer et al., 1993) was used as internal standard. Signal intensity values were determined using the PhosphorImager device (Molecular Dynamics, Kemsing, UK).
Fig. 1.

Generation of PMP22-transgenic mice.A, Structure of the genomic PMP22 cosmid clone pTCF-6.1. Identified exons are indicated by filled boxes and numbered below; cosmid-derived sequences are represented by open boxes. The DNA probes used for identification and characterization of the clone (3′- and 5′-probes) are indicated bythick lines. The 43-kb-long NruI fragment of pTCF-6.1 was purified on a 0.6% agarose gel, isolated by electroelution, and microinjected into fertilized oocytes.S, SalI; K,KpnI; B, BamHI;N, NruI. B, Southern blot analysis of BamHI-digested genomic DNA of PMP22-transgenic and wild-type founder mice using the 5′-probe (lanes 2 and 3 are wild type, andlanes 1, 4, and 5 represent the transgenic lines TgN247, TgN248, and TgN249, respectively).
Generation of transgenic mice. The 43 kb insert of cosmid pTCF-6.1 was released by NruI restriction endonuclease digest and separated on a 0.6% agarose gel. DNA was isolated using the Biotrap electroelutor (Schleicher & Schuell, Dassel, Germany). DNA was desalted and equilibrated in the injection buffer (5 mmTris-HCl, pH 7.5/5 mm NaCl/0.1 mm EDTA), using a Centricon 30 concentrator (Amicon, Beverly MA), and was sterile-filtered with a prewashed Ultrafree-MC filter unit (0.22 μm, Millipore, Bedford MA). The DNA was diluted to 2 ng/μl and microinjected into the pronucleus by standard procedures. Zygotes were recovered from the oviduct of superovulated B6C3F1 females mated with males of the same hybrid. CD2F1 hybrids were used as surrogates.
RNA isolation and Northern blotting. Tissue samples for total RNA isolation were dissected and immediately snap-frozen in liquid nitrogen until further processing. RNA was isolated using the TRIzol reagent (Life Technologies, Gaithersburg, MD) according to the manufacturer’s recommendations. RNA was resuspended in DEPC-treated water, and the amount of RNA was determined by measurement of OD(260/280); 10 μg of RNA was separated on a 1.25% denaturing agarose gel. RNA was blotted onto a nylon membrane (Hybond-N+, Amersham, Buckinghamshire, UK) by capillary blotting and immobilized at 80°C for 2 hr. After prehybridization of the membrane, PMP22 and P0 mRNA were detected by hybridization with the respective radioactively labeled cDNA probe. For quantitative analysis, band intensity data were collected using the PhosphorImager device and analyzed with the National Institutes of Health Image program (public domain program, written by Wayne Rasband at National Institutes of Health and available from the Internet by anonymous FTP fromzippy.nimh.nih.gov).
Preparation of sciatic nerve homogenates and Western blot analysis. Sciatic nerves were removed and immediately snap-frozen in liquid nitrogen until processing. Nerves were homogenized in 200 μl of 8 m urea and cleared from debris at 10,000 × g for 5 min, and protein concentration of the supernatant was determined (Bio-Rad protein assay, Bio-Rad, Richmond, CA). Ten micrograms of protein in sample buffer were separated on a 12.5% SDS-PAGE gel, and the proteins were transferred onto a nitrocellulose membrane (Schleicher & Schuell) using a semi-dry blotting apparatus (Trans-Blot SD semi-dry transfer cell; Bio-Rad). Transfer efficiency was controlled by Ponceau-S staining of the membrane and by Coomassie brilliant blue staining of the gel. Membranes were blocked in 2% skimmed milk powder in PBS. Antibodies directed against PMP22 (Susi 4) (Pareek et al., 1993) or directed against the myelin protein P0 (P0raba; gift from Dr. M. Filbin, Hunter College, NY, NY) were both applied at a dilution of 1:1000 at 4°C for 16 hr. Secondary antibodies (horse radish peroxidase-coupled polyclonal goat anti-rabbit antisera; Sigma, St. Louis, MO) at a dilution of 1:5000 were applied for 1–2 hr at room temperature after fourfold washing of the membrane with PBS/0.2% Tween 20. After repeated washing to remove excess secondary antibodies, detection of the immunoreactive products was performed by visualization of chemiluminescence (ECL Western blot detection reagents; Amersham, Arlington Heights, IL) on x-ray films (Fuji, Stamford, CT).
Light microscopic analysis. Six micrometer frozen sections from the quadriceps muscle were stained by NADH tetrazolium reductase histochemistry according to standard protocols and analyzed and photographed on an Axiophot microscope. Ten micrometer cross-cryosections of the sciatic nerves of 8-, 21-, and 70-d-old wild-type and PMP22-transgenic mice were stained with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI) (Boehringer Mannheim, Mannheim, Germany) according to the manufacturer’s recommendations. Photographic slides were analyzed using a Power Macintosh 6100/60 computer using the National Institutes of Health Image program (see above). For semithin sections, nerves embedded as for electron microscopy were cut in 2 μm sections and stained with alkaline toluidine blue.
Immunohistology. The localization of low-affinity NGF receptor (LNGFR), N-CAM, L1, and glial fibrillary acidic protein (GFAP) was carried out by indirect immunofluorescence on fresh-frozen sections of femoral quadriceps nerves as described (Giese et al., 1992;Montag et al., 1994).
Proliferation analysis. Mice were injected with 5-bromo-2′-deoxyuridine (BrdU) intraperitoneally (50 mg/kg in PBS) at various time points (21 and 70 d after birth). The animals were killed with an overdose of Nembutal (Abbott Laboratories, North Chicago, IL) 18 hr after the administration of the nucleotide analog that allows for immunocytochemical detection of proliferating cells after incorporation into replicating DNA (Gratzner, 1982). Sciatic nerves were fixed in situ for 20 min, postfixed in 4% paraformaldehyde (PFA) in PBS, and embedded in paraffin. BrdU immunocytochemistry was carried out using a monoclonal, biotinylated antibody (1:50; Caltag, San Francisco, CA).
Tissue preservation and electron microscopy. Electron microscopy of sciatic nerves of transcardially perfused experimental and control mice (2% PFA and 2% glutaraldehyde in 0.1 mcacodylate buffer) was performed according to Adlkofer et al. (1995).
Electrophysiological analysis. The methods for electrophysiological analysis have been described (Adlkofer et al., 1995). Six PMP22-transgenic and five wild-type control animals (sibling of two litters) were examined. Latencies, amplitudes, duration of the compound muscle action potential (CMAP or M-response) (Kimura, 1989), and the F-wave elicitability, latency, and duration were analyzed in the sciatic nerve (Hartung et al., 1988; Kimura, 1989). Because CMAP amplitudes were very low in PMP22-transgenic mice, 10–20 M-responses in these animals were averaged. Nerve conduction velocities of control mice were similar to those reported earlier (Low and McLeod, 1975;Pollard and McLeod, 1980; Montag et al., 1994; Adlkofer et al., 1995). Double-sided U test (significance level 0.05) was used to assess the statistical significance of differences in the nerve conduction studies between PMP22-transgenic and control mice (Instat, GraphPad, San Diego, CA).
RESULTS
Isolation of a cosmid clone containing the mouse genomicpmp22 gene and generation of transgenic mice
On the basis of the examination of mouse genomic DNA by Southern blot analysis, we estimated that the pmp22 gene spans ∼40 kb (data not shown). Thus, we decided to screen a mouse cosmid library to isolate a full-length mouse genomic pmp22 clone. Using the 5′-flanking region of the pmp22 gene as a probe (5′-probe containing exons 1b and 2) (Fig.1A), we isolated five independent overlapping cosmid clones ranging in their insert size from 32.5 to 42.5 kb (data not shown). One of these clones (pTCF-6.1; insert size, 42.5 kb) (Fig. 1A) contained the entirepmp22 gene flanked by ∼7 kb upstream nontranscribed region and >4 kb downstream nontranscribed region. The 5′-untranslated exons 1a and 1b were mapped by Southern blot analysis, and identity was confirmed by DNA sequencing (Adlkofer et al., 1995) (data not shown). Exon 5 was localized by Southern blot analysis using a probe specific for the 3′-flanking region (Adlkofer et al., 1995) (3′-probe in Fig.1A), and the presence of the complete exon 5 was confirmed by PCR analysis (data not shown). The purified 43 kDaNruI-excised insert of pTCF-6.1, which included ∼0.2 kb of vector sequences on either end, was used for microinjection into fertilized mouse oocytes of the F1 hybrid strain B6C3F1 to generate PMP22-transgenic mice.
Copy number of pmp22 genes in PMP22-transgenic mouse lines
PMP22-transgenic mice were identified by Southern blot analysis ofBamHI-digested mouse genomic DNA using the 5′-probe described above (Fig. 1B). Quantitative analysis using an internal control standard (see Materials and Methods) and phosphor-imaging identified three transgenic founder mice (out of 17 viable progenies) with gene copy numbers of approximately 16 [TgN(pmp22Ge)248Zbz = TgN248; nomenclature according to ILAR News, 1992] and 30 [TgN(pmp22Ge)247Zbz = TgN247 and TgN(pmp22Ge)249Zbz = TgN249]. Detailed additional analysis correlated with concatemeric head-to-tail integration of the transgenes (data not shown). Stable transgenic lines were established from all three founder animals, and no significant differences were observed in the phenotype and pathology between the three lines. Thus, the following analysis shows representative data obtained using line TgN248 (unless indicated differently).
Characterization of PMP22 expression in PMP22-transgenic mice
Northern blot analysis was performed to examine the regulation of PMP22 expression in transgenic animals. In agreement with the tissue-specific expression pattern of the endogenous pmp22gene in wild-type control mice, no PMP22 mRNA was detected in kidney, stomach, thymus, spleen, and liver of transgenic animals at postnatal day 21 (data not shown). Low expression of PMP22 mRNA was found in the heart and the lung. Although no overexpression was observed in the lung of PMP22-transgenic mice (data not shown), a relative PMP22 overexpression of approximately twofold was found in the heart (for examples, see Fig. 2A, lanes 1–4), but no defects could be detected in this tissue by histological analysis (data not shown). P0 expression was not detectable, demonstrating that PMP22 expression in the heart is not attributable to innervation. As expected on the basis of the observed severe myelin deficiency in sciatic nerves of PMP22-transgenic mice (see below), PMP22 mRNA was decreased to 54 ± 32% (n = 5) if normalized to the amount of total RNA and compared with wild-type animals (for example, see lanes 5 and 6 in Fig. 2A,C). Because PMP22 and P0 show a similar regulation in myelinating Schwann cells, we used a cDNA probe specific to P0 as an internal standard for myelin-specific genes and reprobed the Northern blot membranes. On the basis of the decreased expression of the P0 mRNA (26 ± 7%; n = 6) in PMP22-transgenic animals (lanes 5 and 6 in Fig.2B,C), an approximately twofold overexpression of PMP22 mRNA was observed in the sciatic nerves of 21-d-old PMP22-transgenic animals when standardized to the levels of P0 mRNA expression. Western blot analysis using antibodies directed against PMP22 or P0 revealed a dramatic decrease in protein expression of both myelin proteins in PMP22-transgenic mice (Fig. 2D). Interestingly, this reduction was much more pronounced than at the mRNA level observed previously in Northern blot analysis.
Fig. 2.

Analysis of the expression of PMP22 and P0. Northern blot analysis of PMP22-transgenic and wild-type mice (A–C). The same blot was hybridized first with a PMP22 cDNA probe (A) and subsequently with P0 cDNA probe (B). Ethidium bromide-stained agarose gel (C; 18S RNA) is shown as quantitation control. RNA was isolated from heart (lanes 1–4) and from sciatic nerves (lanes 5 and 6) of 21-d-old PMP22-transgenic mice (lanes 1, 3, and 5) and wild-type (lanes 2, 4, and 6) siblings (note that the exposure times of the different blots and probes were not identical). Western blot analysis of PMP22-transgenic and wild-type mice (D). Crude sciatic nerve homogenates (20 μg of protein) of PMP22-transgenic mice (lanes 1and 3; TgN248) and wild-type littermates (lanes 2 and 4) were separated on 12.5% SDS-PAGE and blotted to nitrocellulose membranes. Proteins were probed with antibodies specific for PMP22 (lanes 1 and2) or P0 (lanes 3 and4).
Behavioral characteristics of PMP22-transgenic mice
Two of the founder mice (TgN247 and TgN249) showed a distinct phenotype characterized mainly by an unsteady gait, a trait that was stably transmitted to the progeny. The founder of TgN248 was not visibly affected and yielded only 27% (n = 168) transgenic progeny in the F1 generation (which was phenotypically and pathologically not distinguishable from TgN247 and TgN249), indicative of a mosaic animal. In general, PMP22-transgenic males show poor breeding efficiency, and the litter size of PMP22-transgenic females was reduced significantly (data not shown).
First signs of behavioral abnormalities appear 2 weeks after birth as manifested by a slight shivering of the animals, which is most obvious at the head. At 4 weeks, an unsteady gait becomes apparent. Signs of muscle atrophy, in the form of distal body shape slenderness, are present at the age of 2–3 months but are variable in extent between individual animals. Progression in severity of the disease with age often culminates in pronounced paralysis of the hindlimbs. Generally, the lifespan of PMP22-transgenic mice exceeds 8 months.
Electrophysiology
Motor nerve conduction studies in all examined 10- to 12-week-old PMP22-transgenic mice showed the typical electrophysiological signs of a severe dysmyelinating polyneuropathy, i.e., increased latencies and decreased amplitudes of CMAP responses with temporal dispersion in both the sciatic and facial nerves (Fig. 3). Motor nerve conduction velocities were reduced severely in the sciatic nerve of PMP22-transgenic mice. In general, a large inter-individual variability in the severity of the electrophysiological impairment in PMP22-transgenic mice was observed, for example, with facial nerve M-response latencies ranging from 3.8 msec to as much as 14.2 msec (range of normal control mice, 0.8–1.24 msec; see Fig. 3). We could not obtain M-responses after proximal sciatic nerve stimulation in three PMP22-transgenic mice, indicating either severe dysmyelination or complete conduction failure. F-wave studies showed reproducible late compound motor responses in only two PMP22-transgenic mice, with a highly increased latency (37 and 42.5 msec as compared with a mean ± SD of 4.1 ± 0.2 msec in five control mice after proximal sciatic nerve stimulation). Threshold stimulus values were increased significantly in the facial and sciatic nerve (distal stimulation site) of PMP22-transgenic mice (data not shown), indicating possible axonal damage. Reliable measurements of the absolute refractory time of the facial nerve could not be obtained in PMP22-transgenic mice because of M-response polyphasia and signal background. We also could not record mixed afferent nerve potentials in the sciatic nerve of PMP22-transgenic mice even after averaging >100 stimuli, indicating either complete conduction failure or profound dispersion of nerve potential components leading to phase cancellation attributable to severe dysmyelination. This latter finding indicates that afferent fibers are affected similarly to motor fibers.
Fig. 3.

Nerve conduction analysis of PMP22-transgenic mice and wild-type littermates. Top, Summary of the results of the nerve conduction studies in the facial (left) and sciatic nerves (right; Mean ± SD;n = number of animals analyzed) of 10-week-old PMP22-transgenic and wild-type control mice. We were unable to elicit proximal sciatic nerve CMAP responses in two PMP22-transgenic mice. Therefore, the conduction velocity in the sciatic nerve could be determined in only three mice of this group and may therefore be slightly overestimated. Bottom, Redrawn original recordings of CMAP responses after stimulation of the facial nerve (left) and the distal sciatic nerve (right) of a 79-d-old control mouse (top row) and of a 79-d-old PMP22-transgenic sibling (bottom row). Note the different time and amplitude scales. Recordings were reproducible on repeated stimulation.
Morphological analysis by light microscopy
Two micrometer semithin cross-sections of the sciatic nerve were prepared and stained with alkaline toluidine blue. No detectable myelin was seen in 21-d-old PMP22-transgenic mice (compare Fig.4a and 4a′) or in 10- or 70-d-old animals (data not shown). Abnormal myelination in nerves of wild-type animals was never observed.
Fig. 4.
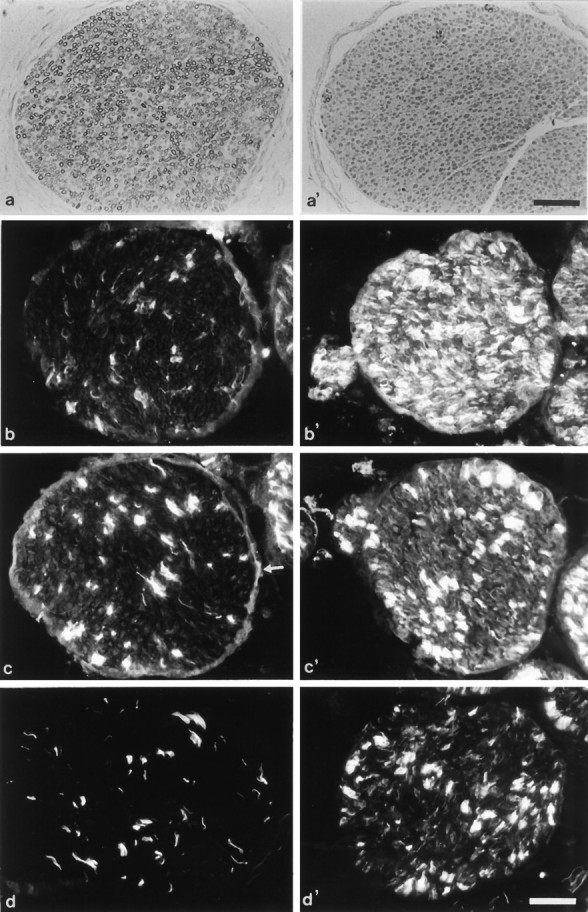
Assessment of myelination in semithin sections and immunohistological analysis. Cross-sections (2 μm) of 21-d-old wild-type (a) and PMP22-transgenic mice (a′) were stained with toluidine blue. Note the lack of detectable myelin (a′). Immunohistological localization of the cell surface molecules LNGFR (b,b′), N-CAM (c, c′), and L1 (d, d′) in femoral quadriceps nerves of adult wild-type (b–d) and mutant mice (b′–d′). Although LNGFR is prominently upregulated in mutant mice (b, b′), N-CAM is moderately (c, c′) and L1 weakly increased (d, d′). Note that the nonmyelinating axon-Schwann cell units also are labeled stronger for N-CAM and L1 in the mutants (c′, d′) than in the wild-type mice (c, d). Arrow inc marks the weakly N-CAM-immunoreactive perineurium of a wild-type nerve. Scale bars, 50 μm.
The examination of other organs and tissues did not reveal any differences between PMP22-transgenic mice and wild-type littermates, with the exception of neurogenic muscle atrophy (see below) and decreased body fat (probably attributable to feeding difficulties).
Characterization of Schwann cell antigen expression
To characterize the differentiation status of Schwann cells overexpressing PMP22, the expression of a set of proteins typically associated with Schwann cells of a pre- or nonmyelinating state was investigated on fresh-frozen sections of peripheral nerves of wild-type and mutant adult mice. These markers include the cell surface components low-affinity NGF receptor (LNGFR), N-CAM, and L1, and the cytoskeletal component GFAP (Jessen and Mirsky, 1991; Martini, 1994).
We have chosen the femoral quadriceps nerve for this analysis because it contains only a few nonmyelinating axon-Schwann cell units, which express these molecules in the wild type at high levels (Fig. 4b–d) (Giese et al., 1992). In mutant mice, a strong upregulation of LNGFR by most, if not all, Schwann cells was characteristic (Fig. 4b′). N-CAM was upregulated by the developmentally abnormal Schwann cells of the mutant but to a significantly lower degree than LNGFR (Fig. 4c′). L1 was weakly upregulated by some abnormal Schwann cells of the nerve (Fig.4d′). Interestingly, both N-CAM and L1 were also upregulated by nonmyelinating axon-Schwann cell units (Fig.4c′,d′), suggesting that PMP22 overexpression might have an additional subtle effect on this Schwann cell type, which has recently been shown to express low levels of endogenous PMP22 (Haney et al., 1996). Finally, the cytoskeletal component GFAP was confined to nonmyelinating Schwann cells in the wild type and was weakly upregulated in the mutant nerve (not shown).
Electron microscopy
In sciatic nerves of 10-, 21-, and 72-d-old wild-type mice, large caliber axons were surrounded by compact myelin (Fig.5a). All Schwann cells formed a typical basal lamina, and endoneurial collagen fibrils were abundant between the axon-Schwann cell units.
Fig. 5.
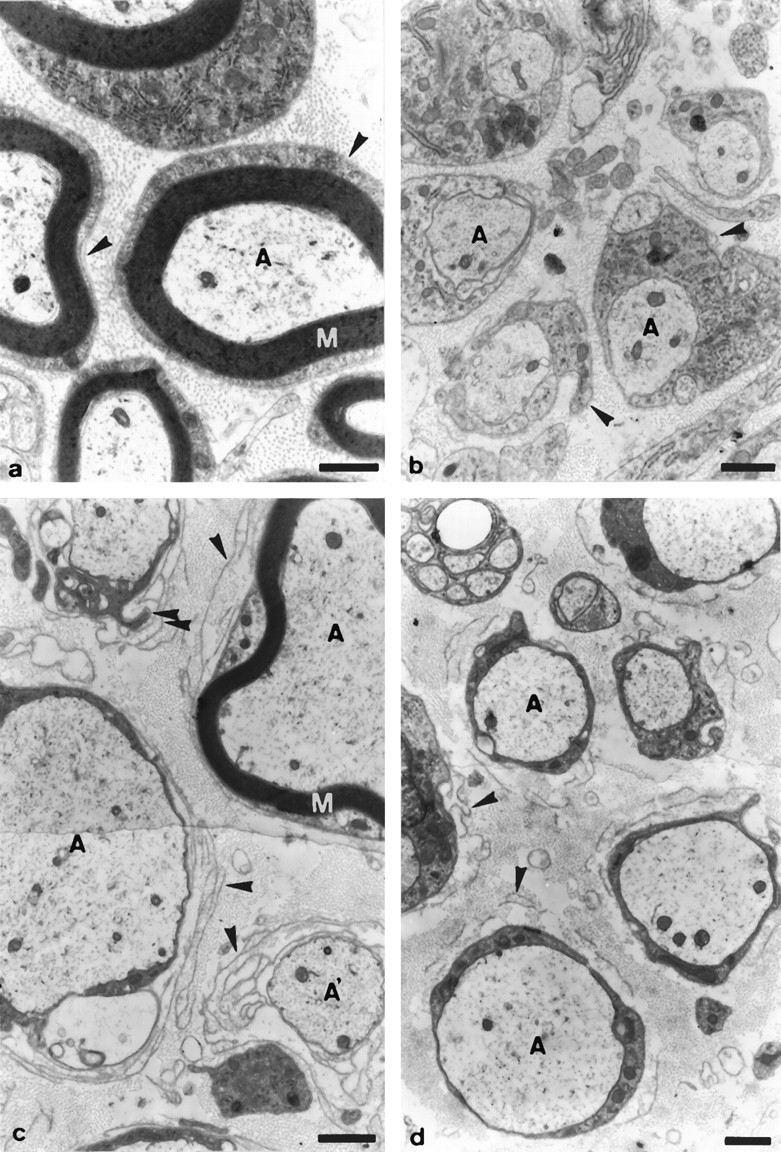
Ultrastructural examination of sciatic nerves. Electron microscopy of sciatic nerves of 10- (a, b), 21- (c), and 72-d-old (d) wild-type (a) or PMP22-transgenic mice (b–d; TgN248). In 10-d-old wild-type mice (a), larger-caliber axons (A) are associated with compact myelin (M), whereas in transgenic littermates (b), larger-caliber axons are ensheathed by Schwann cells, which do not form myelin. Arrowhead points to Schwann cell basal lamina. In 21-d-old PMP22-transgenic mice (c), most of the larger-caliber axons are associated with Schwann cells, which do not form myelin; however, ∼10% of the axons having acquired a 1:1 ratio with Schwann cells are surrounded by a thin sheath of compact myelin (M). Note the prominent pockets of Schwann cell basal laminae (arrowheads), which are occasionally filled with Schwann cell processes of high electron density (double arrowheads). A′, Axons devoid of an ensheathing Schwann cell are found that are surrounded by pockets of basal laminae, indicative of the previous presence of a Schwann cell (arrowheads). In 72-d-old PMP22-transgenic mice (d), larger-caliber axons (A) are always associated with Schwann cells devoid of myelin. Profiles of supernumerary Schwann cell basal laminae (arrowheads) are seen consistently but appear less prominent than in 21-d-old mice. Scale bars, 1 μm.
Morphology of the sciatic nerves of the corresponding mutant littermates was very different and showed evidences of a severe disruption of myelin formation (Figs. 5b–d). In 10-d-old mice, myelin sheaths were completely absent, although most of the Schwann cells had formed a basal lamina and had acquired a 1:1 ratio with their axons (Fig. 5b). In rare cases, two or more Schwann cells were associated with one central axon (data not shown). No degenerating myelin or myelin debris was detectable in 10-d-old mice, suggesting that the absence of myelin was not the result of myelin degeneration but the consequence of disrupted myelin formation from the onset. In 21-d-old animals, the situation was similar to that seen in 10-d-old mice, with three exceptions (Fig. 5c). First, the cytoplasm of most of the Schwann cells associated with large caliber axons was much more electron-dense than that of 10-d-old mutant mice or 21-d-old wild-type littermates forming myelin. Second, each axon-Schwann cell unit was surrounded by multiple “pockets” of basal laminae, which usually contained very thin collagen fibrils, indicative of Schwann cell origin (Bunge et al., 1986). Occasionally, in some of these supernumerary pockets, dark Schwann cell processes were detectable. Third, a few larger-caliber axons were surrounded by unusually thin sheaths of compacted myelin. Such profiles were always surrounded by multiple pockets of Schwann cell basal laminae. Rarely, degenerating myelin and myelin debris were detectable (data not shown). At postnatal day 72, compact myelin was no longer seen, which is in line with the occurrence of some degenerating myelin profiles in 21-d-old mice (Fig. 5d, and data not shown). Supernumerary Schwann cell basal laminae were still frequent, but often appeared disorganized or even disrupted and only very rarely contained Schwann cell processes (Fig. 5d).
The finding of abnormally thin but structurally intact myelin in 21-d-old animals may be explained by somatic transgene inactivation, like methylation or chromosomal rearrangements, that allowed some Schwann cells to escape from PMP22 overexpression. Because such inactivation processes are likely to be only partially effective, the remaining PMP22 overexpression level may still lead to an unstable myelin structure that is rapidly degraded, similar to the observed pathology of CMT1A patients with comparably mild PMP22 overexpression (Gabreels-Festen et al., 1996).
In contrast to the Schwann cells associated with large caliber axons, the Schwann cells ensheathing small caliber axon groups were normally organized in mutant mice and were neither of darker appearance nor surrounded by multiple pockets of basal laminae (Fig. 5d; data not shown).
Muscle pathology in PMP22-transgenic mice
Histological analysis of the quadriceps muscle of 10-week-old mice using NADH tetrazolium reductase staining revealed an even distribution of intermingled type I (dark muscle fibers) and type II (light muscle fibers) in wild-type animals (Fig.6A). In PMP22-transgenic animals, however, extensive fiber-type grouping was visible (Fig.6B), and at more advanced stages, definite signs of neurogenic atrophy were seen, such as large areas containing mainly atrophic fibers intermingled with smaller groups of hypertrophic muscle fibers (Fig. 6C). Even at this stage, however, no muscle fiber degeneration was present, indicating the absence of any relevant myopathic component in the disease.
Fig. 6.

Histological analysis of the quadriceps muscle of PMP22-transgenic and wild-type mice. Normal chessboard-like distribution pattern of types I (slow-twitch muscle,dark) and II (fast twitch muscle, light) muscle fibers of wild-type animals (A). PMP22-transgenic animals (B, C) show prominent segregation and grouping of types I and II fibers (B) and few hypertrophic fibers (arrows) but no significant atrophy. C, Late stage of neurogenic myopathy with type grouping and large groups of atrophic fibers. Scale bar, 25 μm.
Aberrant Schwann cell proliferation in the sciatic nerve of PMP22-transgenic mice
Isolation of total RNA from the sciatic nerves of PMP22-transgenic 21-d-old animals consistently yielded double the amount of RNA compared with that of wild-type littermates, indicating a potential increase in cell number (data not shown). Ten micrometer frozen cross- and longitudinal sections of the sciatic nerves of 8-, 21-, and 70-d-old mice were used to assess cell number. 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI) staining revealed an approximately twofold increase of nuclei in 8-d-old mutant animals when compared with wild-type littermates and an approximately fourfold increase in 70-d-old mutant nerves (Fig. 7). Analysis of longitudinal sections by electron microscopy showed that most of the supernumerary cells are Schwann cells that have axonal contact and are aligned along the axon (Fig. 8C); however, these Schwann cells fail to build up myelin. These results are in agreement with the microscopic findings that there are no signs of a significant number of invading macrophages, endoneurial fibroblasts, or supernumerary Schwann cells not in contact with axons in the sciatic nerves of mutant mice (data not shown). Proliferation analysis as examined by injection of 5-bromo-2′-deoxyuridine (BrdU) and subsequent immunohistochemical analysis of sciatic nerves revealed a robust increase in the number of proliferating cells in 21-d-old PMP22-transgenic mice compared with wild-type littermates (Fig.8A,B). Similar results were obtained in 70-d-old mice (data not shown).
Fig. 7.

Cell number in cross-sections of sciatic nerves. Sciatic nerves of 10-week-old wild-type mice (A) and PMP22-transgenic siblings (B) were analyzed histologically after DAPI staining. Scale bar, 50 μm. Ten micrometer cryosections of PMP22-transgenic and wild-type mice of various ages were stained with DAPI, and the nuclei were counted (C). The bars indicate the relative surplus of nuclei in the sciatic nerves of PMP22-transgenic mice compared with wild-type animals. p values (two-tailed Student’st test) are given.
Fig. 8.

Schwann cell proliferation in the sciatic nerve of PMP22-transgenic and wild-type mice. Longitudinal sections (2 μm) of BrdU-labeled sciatic nerves of 21-d-old wild-type mice (A) and PMP22-transgenic littermates (B). Frequent incorporation of BrdU in PMP22-transgenic mice indicates proliferation of Schwann cells (arrows inB), whereas only rare signals in the wild type are detectable. Scale bar, 25 μm. Ultrathin longitudinal section of a sciatic nerve of a 10-week-old PMP22-transgenic mouse (C). A large caliber axon (black asterisks) is ensheathed by darkly appearing Schwann cell processes (arrows in C). White asterisks indicate Schwann cell perikarya with direct contact to the axon. Note that their frequency is unusually high. The two neighboring Schwann cell nuclei at the left possibly indicate that a Schwann cell mitosis takes place (arrowheads, endoneurial collagen). Scale bar, 5 μm.
DISCUSSION
We report the characterization of three transgenic mouse lines that carry additional copies of the pmp22 gene. Each line showed an identical phenotype in all parameters examined, despite differences in the copy number of transgenes. We attribute this finding to the high number of PMP22 transgenes present, which probably leads to a massive overdose of PMP22 expression in all three lines. Nevertheless, we observed only a twofold overexpression of PMP22 mRNA in the mutant animals relative to the mRNA levels of the major PNS myelin protein P0. When normalized to total RNA, both PMP22 and P0 mRNA levels were reduced, and this downregulation was quantitatively even more pronounced at the protein level, in agreement with the virtually complete absence of myelin. Interestingly, similar downregulations of P0 mRNA have also been observed in other myelin-deficient mouse strains with either null mutations in the Krox-20 gene (Topilko et al., 1994) or point mutations in pmp22 (Tr; Bascles et al., 1992). In PMP22-transgenic mice, the finding of a twofold absolute overexpression of PMP22 mRNA in heart tissue suggests that the observed absolute underexpression of PMP22 in the sciatic nerve is a tissue-specific phenomenon, probably attributable to the predominant dysmyelination phenotype.
The relatively small overexpression of PMP22 compared with the level of P0 and the number of transgenes also has been seen in other transgenic mouse mutants that carry multiple copies of myelin genes, including PLP and DM20 (Nadon et al., 1994). It is important to realize, however, that the overexpression of transgenes may vary considerably throughout development, particularly in PMP22-transgenic mice, because the observed phenotype indicates that the critical time period in which overexpression of PMP22 affects Schwann cell development is right at the start of myelination. In line with these results, PMP22-deficient mice are delayed significantly in myelination, suggesting that PMP22-deficiency also affects Schwann cell differentiation (Adlkofer et al., 1995).
Comparison of the phenotypes of PMP22-transgenic mice and hereditary peripheral neuropathies
PMP22-transgenic mice reveal that strong overexpression of PMP22 is sufficient to cause a very robust myelin deficiency with the typical clinical and electrophysiological signs of a severe dysmyelinating polyneuropathy. This dysmyelination is likely attributable to the highly increased PMP22 gene dosage, because transgenic rats that carry a lower copy number of PMP22 transgenes exhibit a more CMT1A-like demyelinating phenotype, which is characterized by classical Schwann cell onion bulb formation (Sereda et al., 1996). In contrast, PMP22-transgenic mice are characterized mainly by complete lack of myelin, Schwann cell proliferation, and the presence of redundant basal laminae. We interpret these findings such that a high pmp22 gene dosage in transgenic mice shows the consequences of one extreme of a continuum of phenotypes associated with different levels of PMP22 expression. Mice that are completely deficient in PMP22 develop prominent focal hypermyelination (tomacula) and subsequent myelin degeneration (Adlkofer et al., 1995). A similar but less pronounced pathology is observed in mice carrying only one functional pmp22 copy, which genetically and pathologically mimics HNPP patients. Three PMP22 gene copies in heterozygous CMT1A patients (Gabreels-Festen et al., 1996) at first approximation lead to quite normal myelination followed by demyelination, with a more severe phenotype in homozygous CMT1A patients (four pmp22 gene copies) (Killian and Kloepfer, 1979). Finally, the high pmp22 gene copy number in the mice reported here is characterized by the most severe defect, essentially showing nearly complete absence of myelin. Thus, the combined data support the hypothesis that altered PMP22 gene dosage is not compatible with normal Schwann cell development and homeostasis.
With respect to human peripheral neuropathies, the morphological phenotype of PMP22-transgenic mice is almost identical to a specific subtype of hereditary motor and sensory neuropathies type III (autosomal recessive HMSN III), which is characterized by amyelination and basal lamina onion bulb formation (Gabreel-Festen et al., 1992;Nara et al., 1995). Recent findings in transgenic mice carrying null mutations of the Krox-20 (Schneider-Maunoury et al., 1993; Topilko et al., 1994) and SCIP genes (Bermingham et al., 1995) exhibit a phenotype similar to HMSN III, which may indicate that specific mutations in transcription factors that are involved in the determination of the myelinating Schwann cell phenotype are also potential candidate disease genes. Thus, it is tempting to speculate that mutations in these transcription factors may lead to a dysregulation of PMP22gene expression and in turn to a closely related hypomyelinating phenotype as observed in PMP22-transgenic animals.
Consequences of PMP22 overexpression and comparison to PMP22 point mutations
The phenotype of PMP22-transgenic animals is similar but not identical to that of Tr mice, which carry a point mutation in the pmp22 gene (Henry et al., 1983; Henry and Sidman, 1988). Severe hypomyelination and increased Schwann cell number are the main common morphological features, and prominent pockets of redundant basal laminae surrounding hypomyelinated axons have been described in both mouse mutants (Low, 1977). The etiology of excessive basal laminae is not clear but differs in Tr compared with PMP22-transgenic mice. During the development of Tr nerves, supernumerary Schwann cells move away from the axons and form classical onion bulbs as the result of demyelination, Schwann cell proliferation, and remyelination. Subsequently, these redundant Schwann cells are believed to extend sprouts that later degenerate or retract, leaving basal laminae behind as a fingerprint of this process (Low, 1977). In contrast, classical cellular onion bulb formation was not observed in PMP22-transgenic mice, and the supernumerary Schwann cells were found aligned along the axons. Thus, the detected excessive basal laminae are likely to be the direct result of proliferation, migration, and possible transient extensions of Schwann cell processes associated with axons. Alternatively, the observed redundant basal laminae might be remnants of Schwann cells that have undergone cell death, possibly attributable to PMP22 overexpression.
Schwann cell development and proliferation in PNS myelination mutants
Increased Schwann cell number and aberrant Schwann cell proliferation into adulthood are common features in PNS hypomyelinating mutants; however, this phenomenon seems to be caused by different mechanisms. In longitudinal sections of sciatic nerves of Krox-20 null mutants, the nuclei of redundant nerve-resident cells appear mainly spherical and are distributed randomly (Topilko et al., 1994). In contrast, Schwann cell nuclei in PMP22-transgenic mice retain their characteristically elongated shape, in agreement with the finding that supernumerary Schwann cells retain axonal contact. Immunohistological analysis of PMP22-transgenic nerves reveals a strong upregulation of LNGFR, a moderate increase of N-CAM, and minor upregulation of L1 and GFAP. This characteristic expression profile of Schwann cell development markers in combination with the positions of the supernumerary Schwann cells along single axons indicates that the differentiation state of mutant Schwann cells in PMP22-transgenic mice closely resembles the characteristics of normal Schwann cells just before myelination (Jessen and Mirsky, 1991). It is an attractive hypothesis that the mutant Schwann cells begin to differentiate toward the myelinating phenotype and concomitantly start to overexpress PMP22 massively (Suter et al., 1994). This pulse of strong PMP22 overexpression may not allow further differentiation, thereby explaining the dysmyelination phenotype, or possibly may even cause cell death, as suggested by complementary in vitro studies (Fabbretti et al., 1995). If the latter speculation is correct, the observed aberrant proliferation of Schwann cells into adulthood may provide a mechanism to replace the continuously dying cells.
It should be stressed, however, that the mutant Schwann cells in PMP22-transgenic mice have established a 1:1 relationship with large caliber axons and laid down basal laminae. Furthermore, no morphological abnormalities of nonmyelinating Schwann cells ensheathing small caliber axons were observed. This is in contrast to a mouse model with increased Schwann cell death, which is caused by the expression of the bacterial diphtheria toxin A chain under the control of the P0 gene promoter (Messing et al., 1992). Those mutants show a pronounced secondary response of nonmyelinating Schwann cells, including proliferation and retraction of processes, suggesting a dynamic equilibrium between myelinating and nonmyelinating Schwann cells.
In summary, although the elucidation of the mechanistic basis of the observed Schwann cell differentiation defects in mice with strongly increased pmp22 gene dosage requires additional in vivo and in vitro studies to clarify the function of PMP22 in normal and mutant organisms, these animals demonstrate that impaired Schwann cell differentiation is a likely disease mechanism in hereditary peripheral neuropathies.
Footnotes
This work was supported by a grant from the Swiss National Science Foundation to U.S. We thank C. Zgraggen and S. Kasper for excellent technical assistance, and Dr. V. Taylor for critically reading this manuscript.
Correspondence should be addressed to Ueli Suter, Institute of Cell Biology, Swiss Federal Institute of Technology, ETH-Hoenggerberg, CH-8093 Zurich, Switzerland.
Dr. Dembic’s present address: Institute for Immunology and Rheumatology, Fr. Quamsgt. 1, N-0172 Oslo, Norway.
REFERENCES
- 1.Adlkofer K, Martini R, Aguzzi A, Zielasek J, Toyka KV, Suter U. Hypermyelination and demyelinating peripheral neuropathy in PMP22-deficient mice. Nature Genet. 1995;11:274–280. doi: 10.1038/ng1195-274. [DOI] [PubMed] [Google Scholar]
- 2.Baechner D, Liehr T, Hameister H, Altenberger H, Grehl H, Suter U, Rautenstrauss B. Widespread expression of the peripheral myelin protein-22 gene (PMP22) in the neural and non-neural tissues during murine development. J Neurosci Res. 1995;42:735–741. doi: 10.1002/jnr.490420602. [DOI] [PubMed] [Google Scholar]
- 3.Bascles L, Bonnet J, Garbay B. Expression of the PMP22 gene in trembler mutant mice: comparison with the other myelin protein genes. Dev Neurosci. 1992;14:336–341. doi: 10.1159/000111680. [DOI] [PubMed] [Google Scholar]
- 4.Bermingham JRJ, O’Connell S, Arroyo E, Powell F, Kalla K, McEvilly R, Scherer SS, Rosenfeld MG. Mutation of the POU domain transcription factor TST-1/OCT6/SCIP in mice produces neuronal and myelinating Schwann cell defects. Soc Neurosci Abstr. 1995;21:5. [Google Scholar]
- 5.Bunge RP, Bunge MB, Eldridge CF. Linkage between axonal ensheathment and basal lamina production by Schwann cells. Annu Rev Neurosci. 1986;9:305–328. doi: 10.1146/annurev.ne.09.030186.001513. [DOI] [PubMed] [Google Scholar]
- 6.Chance PF, Alderson MK, Leppig KA, Lensch MW, Matsunami N, Smith B, Swanson PD, Odelberg SJ, Disteche CM, Bird TD. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143–151. doi: 10.1016/0092-8674(93)90058-x. [DOI] [PubMed] [Google Scholar]
- 7.Cremer H, Lange R, Christoph A, Plomann M, Vopper G, Roes J, Brown R, Baldwin S, Kraemer P, Scheff S, Barthels D, Rajewski K, Wille W. Inactivation of the N-CAM gene in mice results in size reduction of the olfactory bulb and deficits in spatial learning. Nature. 1993;367:455–459. doi: 10.1038/367455a0. [DOI] [PubMed] [Google Scholar]
- 8.Fabbretti E, Edomi P, Brancolini C, Schneider C. Apoptotic phenotype induced by overexpression of wild-type gas3/PMP22: its relation to the demyelinating peripheral neuropathy CMT1A. Genes Dev. 1995;9:1846–1856. doi: 10.1101/gad.9.15.1846. [DOI] [PubMed] [Google Scholar]
- 9.Gabreel-Festen AAWM, Joosten EMG, Gabreels FJM, Jennekens FGI, Janssen-van Kempen TW. Early morphological features in dominantly inherited demyelinating motor and sensory neuropathy (HMSN type I). J Neurol Sci. 1992;107:145–154. doi: 10.1016/0022-510x(92)90282-p. [DOI] [PubMed] [Google Scholar]
- 10.Gabreels-Festen AAWM, Bolhuis PA, Hoogendijk JE, Valentijn LJ, Eshuis EJHM, Gabreels FJM (1995) Charcot-Marie-Tooth disease type 1A: morphological phenotype of the 17p duplication versus PMP22 point mutations. Acta Neuropathol, in press. [DOI] [PubMed]
- 11.Giese KP, Martini R, Lemke G, Soriano P, Schachner M. Mouse P0 gene disruption leads to abnormal expression of recognition molecules and degeneration of myelin and axons. Cell. 1992;71:565–576. doi: 10.1016/0092-8674(92)90591-y. [DOI] [PubMed] [Google Scholar]
- 12.Gratzner HG. Monoclonal antibody to 5-bromo- and 5-iododeoxyuridine: a new reagent for the detection of DNA replication. Science. 1982;218:474–475. doi: 10.1126/science.7123245. [DOI] [PubMed] [Google Scholar]
- 13.Haney C, Snipes GJ, Shooter EM, Suter U, Garcia C, Griffin JW, Trapp BD. Ultrastructural distribution of PMP22 in Charcot-Marie-Tooth disease type 1A. J Neuropathol Exp Neurol. 1996;55:290–299. doi: 10.1097/00005072-199603000-00004. [DOI] [PubMed] [Google Scholar]
- 14.Hartung HP, Schäfer B, Heininger K, Stoll G, Toyka KV. The role of macrophages and eicosanoids in the pathogenesis of experimental allergic neuritis: serial clinical, electrophysiological, biochemical and morphological observations. Brain. 1988;111:1039–1059. doi: 10.1093/brain/111.5.1039. [DOI] [PubMed] [Google Scholar]
- 15.Henry EW, Sidman RL. Long lives for homozygous trembler mutant mice despite virtual absence of peripheral myelin. Science. 1988;241:344–346. doi: 10.1126/science.3388045. [DOI] [PubMed] [Google Scholar]
- 16.Henry EW, Cowen JS, Sidman RL. Comparison of trembler and trembler-J phenotypes: varying severity of peripheral hypomyelination. J Neuropathol Exp Neurol. 1983;42:688–706. doi: 10.1097/00005072-198311000-00008. [DOI] [PubMed] [Google Scholar]
- 17.Jessen KR, Mirsky R. Schwann cell precursors and their development. Glia. 1991;4:185–194. doi: 10.1002/glia.440040210. [DOI] [PubMed] [Google Scholar]
- 18.Killian JM, Kloepfer HW. Homozygous expression of a dominant gene for Charcot-Marie-Tooth neuropathy. Ann Neurol. 1979;5:515–522. doi: 10.1002/ana.410050604. [DOI] [PubMed] [Google Scholar]
- 19.Kimura J. F.A. Davis; Philadelphia: 1989. Electrodiagnosis in diseases of nerve and muscle: principles and practice. . [Google Scholar]
- 20.Low PA. The evolution of “onion bulbs” in the hereditary hypertrophic neuropathy of the Trembler mouse. Neuropathol Appl Neurobiol. 1977;3:81–92. [Google Scholar]
- 21.Low PA, McLeod JG. Hereditary demyelinating neuropathy in the Trembler mouse. J Neurol Sci. 1975;26:565–574. doi: 10.1016/0022-510x(75)90057-x. [DOI] [PubMed] [Google Scholar]
- 22.Lupski JR, de Oca Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, Saucedo-Cardenas O, Barker DF, Killian JM, Garcia CA, Chakravarti A, Patel PI. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- 23.Manfioletti G, Ruaro ME, Del Sal G, Philipson L, Schneider C. A growth arrest-specific (gas) gene codes for a membrane protein. Mol Cell Biol. 1990;10:2924–2930. doi: 10.1128/mcb.10.6.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martini R. Expression and functional roles of neural cell surface molecules and extracellular matrix components during development and regeneration of peripheral nerves. J Neurocytol. 1994;23:1–28. doi: 10.1007/BF01189813. [DOI] [PubMed] [Google Scholar]
- 25.Marvin KW, Wataru F, Jetten AM. Identification and characterization of a novel squamous cell-associated gene related to PMP22. J Biol Chem. 1995;270:28910–28916. doi: 10.1074/jbc.270.48.28910. [DOI] [PubMed] [Google Scholar]
- 26.Matsunami N, Smith B, Ballard L, Lensch MW, Robertson M, Albertsen H, Hanemann CO, Muller HW, Bird TD, White R, Chance PF. Peripheral myelin protein-22 gene maps in the duplication in chromosome 17p11.2 associated with Charcot-Marie-Tooth 1A. Nature Genet. 1992;1:176–179. doi: 10.1038/ng0692-176. [DOI] [PubMed] [Google Scholar]
- 27.Messing A, Behringer RR, Hammang JP, Palmiter RD, Brinster RL, Lemke G. P0 promoter directs expression of reporter and toxin genes to Schwann cells of transgenic mice. Neuron. 1992;8:507–520. doi: 10.1016/0896-6273(92)90279-m. [DOI] [PubMed] [Google Scholar]
- 28.Montag D, Giese KP, Bartsch U, Martini R, Lang Y, Blüthmann H, Karthigasan J, Kirschner DA, Wintergerst ES, Nave K-A, Zielasek J, Toyka KV, Lipp H-P, Schachner M. Mice deficient for the myelin-associated glycoprotein show subtle abnormalities in myelin. Neuron. 1994;13:229–246. doi: 10.1016/0896-6273(94)90472-3. [DOI] [PubMed] [Google Scholar]
- 29.Nadon NL, Arnheiter H, Hudson LD. A combination of PLP and DM20 transgene promotes partial myelination in the jimpy mouse. J Neurosci Res. 1994;63:822–833. doi: 10.1046/j.1471-4159.1994.63030822.x. [DOI] [PubMed] [Google Scholar]
- 30.Nara T, Akashi M, Nonaka I, Nakanishi Y, Hamano S-I, Ochiai Y, Tsuzura S. Muscle and intramuscular nerve pathology in congenital hypomyelination neuropathy. J Neurol Sci. 1995;129:170–174. doi: 10.1016/0022-510x(94)00272-p. [DOI] [PubMed] [Google Scholar]
- 31.Nave K-A, Boespflug-Tanguy O. X-linked developmental defects of myelination: from mouse mutants to human genetic disease. Neuroscientist. 1996;2:33–43. [Google Scholar]
- 32.Pareek S, Suter U, Snipes GJ, Welcher AA, Shooter EM, Murphy RA. Detection and processing of peripheral myelin protein PMP22 in cultured Schwann cells. J Biol Chem. 1993;268:10372–10379. [PubMed] [Google Scholar]
- 33.Patel PI, Roa BB, Welcher AA, Schoener-Scott R, Trask BJ, Pentao L, Snipes GJ, Garcia CA, Francke U, Shooter EM, Lupski JR, Suter U. The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth disease type 1A. Nature Genet. 1992;1:159–165. doi: 10.1038/ng0692-159. [DOI] [PubMed] [Google Scholar]
- 34.Pollard JD, McLeod JG. Nerve grafts in the Trembler mouse. J Neurol Sci. 1980;46:373–383. doi: 10.1016/0022-510x(80)90062-3. [DOI] [PubMed] [Google Scholar]
- 35.Raeymaekers P, Timmerman V, Nelis E, De Jonghe P, Hoogendijk JE, Baas F, Barker DF, Martin JJ, De Visser M, Bolhuis PA, Van Broeckhoven C. Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). The HMSN Collaborative Research Group. Neuromuscul Disord. 1991;1:93–97. doi: 10.1016/0960-8966(91)90055-w. [DOI] [PubMed] [Google Scholar]
- 36.Roa BB, Garcia CA, Pentao L, Killian JM, Trask BJ, Suter U, Snipes GJ, Shooter EM, Patel PI, Lupski JR. Evidence for a recessive PMP22 point mutation in Charcot-Marie-Tooth disease type 1A. Nature Genet. 1993a;5:189–194. doi: 10.1038/ng1093-189. [DOI] [PubMed] [Google Scholar]
- 37.Roa BB, Garcia CA, Suter U, Kulpa DA, Wise CA, Mueller J, Welcher AA, Snipes GJ, Shooter EM, Patel PI, Lupski JR. Charcot-Marie-Tooth disease type 1A: association with a spontaneous point mutation in the PMP22 gene. N Engl J Med. 1993b;329:96–101. doi: 10.1056/NEJM199307083290205. [DOI] [PubMed] [Google Scholar]
- 38.Roa BB, Dyck PJ, Marks HG, Chance PF, Lupski JR. Dejerine-Sottas syndrome associated with point mutation in the peripheral myelin protein 22 (PMP22) gene. Nature Genet. 1993c;5:269–272. doi: 10.1038/ng1193-269. [DOI] [PubMed] [Google Scholar]
- 39.Sereda M, Griffiths I, Pühlhofer A, Stewart H, Rossner MJ, Zimmermann F, Magyar JP, Schneider A, Hund E, Meinck H-M, Suter U, Nave KA. A rat transgenic model for Charcot-Marie-Tooth disease. Neuron. 1996;16:1049–1060. doi: 10.1016/s0896-6273(00)80128-2. [DOI] [PubMed] [Google Scholar]
- 40.Schneider-Maunoury S, Topilko P, Seitanidou T, Levi G, Cohen-Tannoudji M, Pournin S, Babinet C, Charnay P. Disruption of Krox-20 results in alteration of rhombomeres 3 and 5 in the developing hindbrain. Cell. 1993;75:1199–1214. doi: 10.1016/0092-8674(93)90329-o. [DOI] [PubMed] [Google Scholar]
- 41.Snipes GJ, Suter U, Welcher AA, Shooter EM. Characterization of a novel peripheral nervous system myelin protein (PMP22/SR13). J Cell Biol. 1992;117:225–238. doi: 10.1083/jcb.117.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spreyer P, Kuhn G, Hanemann CO, Gillen C, Schaal H, Kuhn R, Lemke G, Muller HW. Axon-regulated expression of a Schwann cell transcript that is homologous to a “growth arrest-specific” gene. EMBO J. 1991;10:3661–3668. doi: 10.1002/j.1460-2075.1991.tb04933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suter U, Patel PI. Molecular basis of inherited peripheral neuropathies. Hum Mutat. 1994;3:95–102. doi: 10.1002/humu.1380030203. [DOI] [PubMed] [Google Scholar]
- 44.Suter U, Snipes GJ. Biology and genetics of hereditary motor and sensory neuropathies. Annu Rev Neurosci. 1995a;18:45–75. doi: 10.1146/annurev.ne.18.030195.000401. [DOI] [PubMed] [Google Scholar]
- 45.Suter U, Snipes GJ. Peripheral myelin protein 22: facts and hypotheses. J Neurosci Res. 1995b;40:145–151. doi: 10.1002/jnr.490400202. [DOI] [PubMed] [Google Scholar]
- 46.Suter U, Welcher AA, Ozcelik T, Snipes GJ, Kosaras B, Francke U, Billings GS, Sidman RL, Shooter EM. Trembler mouse carries a point mutation in a myelin gene. Nature. 1992a;356:241–244. doi: 10.1038/356241a0. [DOI] [PubMed] [Google Scholar]
- 47.Suter U, Moskow JJ, Welcher AA, Snipes GJ, Kosaras B, Sidman RL, Buchberg AM, Shooter EM. A leucine-to-proline mutation in the putative first transmembrane domain of the 22 kDa peripheral myelin protein in the trembler-J mouse. Proc Natl Acad Sci USA. 1992b;89:4382–4386. doi: 10.1073/pnas.89.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suter U, Welcher AA, Snipes GJ. Progress in the molecular understanding of hereditary peripheral neuropathies reveals new insights into the biology of the peripheral nervous system. Trends Neurosci. 1993;16:50–56. doi: 10.1016/0166-2236(93)90015-e. [DOI] [PubMed] [Google Scholar]
- 49.Suter U, Snipes GJ, Schoener-Scott R, Welcher AA, Pareek S, Lupski JR, Murphy RA, Shooter EM, Patel PI. Regulation of tissue-specific expression of alternative peripheral meyelin protein-22 (PMP22) gene transcripts by two promoters. J Biol Chem. 1994;269:25795–25808. [PubMed] [Google Scholar]
- 50.Taylor V, Suter U (1996) Epithelial membrane protein-2 and epithelial membrane protein-3: two novel members of the peripheral myelin protein 22 gene family. Gene, in press. [DOI] [PubMed]
- 51.Taylor V, Welcher AA, Amgen Est Programm. Suter U. Epithelial membrane protein-1, peripheral myelin protein 22 and lens membrane protein 20 define a novel gene family. J Biol Chem. 1995;270:28824–28833. doi: 10.1074/jbc.270.48.28824. [DOI] [PubMed] [Google Scholar]
- 52.Timmerman V, Nelis E, Van Hul W, Nieuwenhuijsen BW, Chen KL, Wang S, Ben Othman K, Cullen B, Leach RJ, Hanemann CO, De Jonghe P, Raeymaekers P, van Ommen G-JB, Martin J-J, Muller HW, Vance JM, Fischbeck KH, Van Broeckhoven C. The peripheral myelin protein gene PMP-22 is contained within the Charcot-Marie-Tooth disease type 1A duplication. Nature Genet. 1992;1:171–175. doi: 10.1038/ng0692-171. [DOI] [PubMed] [Google Scholar]
- 53.Topilko P, Schneider-Maunoury S, Levi G, Baron-Van Evercooren A, Chennoufi ABY, Seitanidou T, Babinet C, Charnay P. Krox-20 controls myelination in the peripheral nervous system. Nature. 1994;371:796–799. doi: 10.1038/371796a0. [DOI] [PubMed] [Google Scholar]
- 54.Valentijn LJ, Bolhuis PA, Zorn I, Hoogendijk JE, van den Bosch N, Hensels GW, Stanton J, VP, Housman DE, Fischbeck KH, Ross DA, Nicholson GA, Meershoek EJ, Dauwerse HG, van Ommen G-JB, Bass F. The peripheral myelin gene PMP-22/GAS-3 is duplicated in Charcot-Marie-Tooth disease type 1A. Nature Genet. 1992a;1:166–170. doi: 10.1038/ng0692-166. [DOI] [PubMed] [Google Scholar]
- 55.Valentijn LJ, Baas F, Wolterman RA, Hoogendijk JE, Bosch NHA, Zorn I, Gabreels-Festen AAWM, de Visser M, Bolhuis PA. Identical point mutations of PMP-22 in Trembler-J mouse and Charcot-Marie-Tooth disease type 1a. Nature Genet. 1992b;2:288–291. doi: 10.1038/ng1292-288. [DOI] [PubMed] [Google Scholar]
- 56.Welcher AA, Suter U, De Leon M, Snipes GJ, Shooter EM. A myelin protein is encoded by the homologue of a growth arrest-specific gene. Proc Natl Acad Sci USA. 1991;88:7195–7199. doi: 10.1073/pnas.88.16.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zoidl G, Blass-Kampmann S, D’Urso D, Schmalenbach C, Müller HW. Retroviral-mediated gene transfer of the peripheral myelin protein PMP22 in Schwann cells: modulation of cell growth. EMBO J. 1995;14:1122–1128. doi: 10.1002/j.1460-2075.1995.tb07095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]