

Abstract
Neurodegeneration may occur secondary to glutamate-triggered Ca2+ influx through any of three routes: NMDA channels, voltage-sensitive Ca2+ channels (VSCC), and Ca2+-permeable AMPA/kainate channels (Ca-A/K). This study aims to examine Ca2+ ion dynamics in the generation of excitotoxic injury by correlating the relative amounts of45Ca2+ that flow into cortical neurons through each of these routes over a 10 min epoch (“10 min Ca2+loads;” a measure of influx rate), with resultant levels of intracellular free Ca2+ ([Ca2+]i) and subsequent injury. Neurons possessing Ca-A/K make up a small subset (∼13%) of cortical neurons in culture, which can be identified by a histochemical stain based on kainate-stimulated Co2+ uptake (Co2+(+) neurons) and which are unusually vulnerable to AMPA/kainate receptor-mediated injury. Initial studies using brief kainate exposures (to selectively destroy Co2+(+) neurons) along with kainate-triggered 45Ca2+ influx measurements suggested that kainate causes rapid Ca2+influx into Co2+(+) neurons (comparable to that caused by NMDA). Influx through both Ca-A/K and NMDA channels increased proportionately with extracellular Ca2+, suggesting that these channels have high Ca2+ permeability.
When cultures were subjected to exposures that gave similar 10 min Ca2+ loads through different routes, comparable levels of injury were observed, suggesting that net intracellular Ca2+ accumulation is a critical determinant of injury. However, the relationship between [Ca2+]i and influx was less direct: although exposures that gave the lowest or highest 10 min Ca2+ loads showed correspondingly lower or higher mean [Ca2+]i responses, there appears to be a wide range of exposures over which individual neuronal differences and sequestration/buffering mechanisms obscure [Ca2+]i as a reflection of influx rate.
Keywords: calcium, neurotoxicity, excitotoxicity, glutamate, Fura-2, neuron
Glutamate-triggered Ca2+ influx, under pathological conditions, may set into motion processes that culminate in neurodegeneration. In cortical culture, brief periods of activation of NMDA receptors, which gate channels with high Ca2+permeability (MacDermott et al., 1986), trigger extensive neuronal injury. In contrast, prolonged periods of activation of generally poorly Ca2+-permeable AMPA/kainate channels are necessary to induce comparable widespread injury (Choi, 1992). Such slow AMPA/kainate receptor-mediated injury likely reflects neuronal depolarization and secondary Ca2+ influx through voltage-sensitive Ca2+ channels (VSCC) (Murphy and Miller 1989; Weiss et al., 1990).
However, recent studies have demonstrated a third important route of glutamate-triggered Ca2+ entry; certain subpopulations of central neurons possess AMPA/kainate receptors gating channels with direct Ca2+ permeability (Iino et al., 1990; Pruss et al., 1991; Brorson et al., 1992). The demonstration that these neurons are unusually sensitive to Ca2+-dependent AMPA/kainate receptor-mediated injury suggests that the ability of Ca2+ions to gain entry through this direct route somehow underlies their vulnerability (Brorson et al., 1994; Turetsky et al., 1994; Weiss, 1994a; Yin et al., 1994a,b).
The sequence of events linking Ca2+ entry to neurodegeneration is only beginning to be explored. A full understanding of these mechanisms, however, requires an understanding of Ca2+ dynamics as it enters the cell through any of several routes and reaches the target sites at which it mediates its injurious effects. Although Ca2+ imaging studies have generally found poor relationships between agonist exposures, intracellular free Ca2+ concentrations ([Ca2+]i), and degeneration (until terminal stages of injury when a sharp rise in [Ca2+]iindicates imminent death) (Michaels and Rothman, 1990; Dubinsky and Rothman, 1991; Randall and Thayer, 1992; Dubinsky, 1993), the recent finding that NMDA and kainate induced similar [Ca2+]i rises in spinal neurons while NMDA caused much more damage suggested that the route through which Ca2+ enters the cell may determine its propensity to cause injury (Tymianski et al., 1993). In contrast, the finding that brief NMDA exposure triggered high 45Ca2+ influx rates (compared with kainate) that paralleled its potent ability to trigger injury suggested the possibility that total Ca2+load is of primary importance (Hartley et al., 1993).
The present work, which extends a recent brief report examining the rate of Ca2+ influx into neurons with Ca2+-permeable AMPA/Kainate channels (Ca-A/K) (Lu et al., 1995), seeks to correlate Ca2+ entry through each of the three primary routes with [Ca2+]i homeostasis and subsequent neuronal injury.
MATERIALS AND METHODS
Tissue culture. Dissociated mixed neocortical cell suspensions were prepared from fetal Swiss–Webster mice (gestational age 14–16 d) and plated on a previously established confluent layer of cortical astrocytes (see below) in 24-well tissue culture plates at 1–2 × 105 cells/cm2, generally as described previously (Rose et al., 1993). The initial plating medium was Eagle’s Minimum Essential Medium (MEM; Earle’s salts, supplied glutamine-free) supplemented with 10% heat-inactivated horse serum, 10% heat-inactivated fetal bovine serum, glutamine (total, 2 mm), and glucose (total, 21 mm). Cultures were kept in a 37°C/5% CO2 incubator. After 4–6 d in vitro, non-neuronal cell division was halted by exposure to 10−5m cytosine arabinoside for 1–3 d. The cells were then shifted into a maintenance medium identical to the plating medium but lacking fetal serum. Subsequent media replacement occurred twice a week. Cultures were studied after 13–14 d in vitro.
Glial cultures were prepared using the same protocol as for mixed cell cultures, except that cortices were removed from early postnatal mice (1–3 d postnatal) and plated directly on Falcon Primaria culture plates in media supplemented with epidermal growth factor (final concentration 10 ng/ml).
Neurotoxicity experiments. Toxic exposures to NMDA or kainate were performed in room air at 22–25°C, in a humidified atmosphere, in a HEPES-buffered control salt solution (HSS) with the following composition (in mm): 130 Na+, 5.4 K+, 0.8 Mg2+, 1.8 Ca2+ (except as indicated), 130.6 Cl−, 20 HEPES, pH 7.4, at 25°C, and 15 glucose. Ten micromolars MK-801 were added to all kainate exposures, and 10 μm glycine was added to all NMDA exposures. Brief (10 min and 1 hr kainate “pretreatments”) toxic exposures were terminated by replacing the exposure solution with MEM + glucose and returning the cultures to the CO2 incubator.
Evaluation of neuronal cell loss. Overall neuronal injury was assessed 12–15 hr after start of exposure (for continuous prolonged exposures) or 18–24 hr after start of exposure (for 10 min to 1 hr exposures) by examination of cultures with phase-contrast microscopy at 100–400×; previous experience has suggested that the excitatory amino acid-induced injury of cultured neurons and glia can be reliably estimated in this manner. In some experiments, this examination was verified by subsequent bright-field examination of trypan blue staining (0.4% for 5 min), which labels debris and nonviable cells.
Overall neuronal cell injury was also quantitatively assessed by the measurement of lactate dehydrogenase (LDH), released by damaged or destroyed cells, in the extracellular fluid (Koh and Choi, 1987). A small amount of LDH is always present in the media of cultures carried through the exposure protocol but without addition of excitatory amino acids. This background amount, determined on sister cultures within each experiment, was subtracted from values obtained in treated cultures. Specific efflux of LDH induced by glutamate exposure (after background subtraction) has been shown to be linearly proportional to the number of neurons damaged or destroyed, and no specific LDH efflux occurred when pure astrocyte cultures were similarly exposed to glutamate (Koh and Choi, 1987). In each experiment, LDH values were scaled to the mean value obtained by prolonged (>12 hr) maximal NMDA exposures (50 μm in 10 mm Ca2+ or 300 μm in 1.8 mm Ca2+) that control experiments have demonstrated to reliably kill >90% of all neurons.
To assess damage to the Co2+(+) population, cultures were Co2+-loaded immediately before fixing and developing the Co2+ stain. Specific destruction of Co2+(+) neurons was assessed as the difference between the mean number of intact cells in several sham-washed cultures and the number in several sister cultures exposed to excitotoxic agonists, expressed as a percentage of the former.
45Ca2+ influx studies. Cultures were washed with HSS (25°C) containing the indicated concentration of extracellular Ca2+ ([Ca2+]e) and then incubated in the presence of agonist, in HSS spiked with45CaCl2 in proportion to its Ca2+concentration (50 nm45CaCl2 for 0.4 mm Ca2+ HSS; 225 nm45Ca2+ for 1.8 mm Ca2+HSS; and 1250 nm45Ca2+ for 10 mm Ca2+ HSS) for 10 min. Kainate exposures were all in the presence of MK-801 (10 μm). Exposure solution was then washed out (5 washes), the cells were lysed by addition of 0.2% SDS solution, and the cell lysate was counted. Counts in matched blanks (sister cultures identically exposed, in the same [Ca2+]e, except for the absence of agonist) were subtracted separately from counts in corresponding agonist exposures to yield the 45Ca2+ influx specifically induced by agonist application. Raw counts after background influx subtraction were then normalized to the number of counts observed in sister cultures exposed to 50 μm NMDA in 10 mm [Ca2+]e (typically, ∼12,000–16,000 cpm, = 100%) so that data from several repetitions of the experiments, carried out on cultures from different platings containing different absolute numbers of neurons, could be combined.
Co2+ labeling. Co2+ labeling was performed as described (Pruss et al., 1991; Turetsky et al., 1994) with minor modifications. Cultures were Co2+-loaded by exposure to kainate (100 μm) with Co2+ (2.5 mm) in uptake buffer (139 mm sucrose, 57.5 mm NaCl, 5 mm KCl, 2 mmMgCl2, 1 mm CaCl2, 12 mm glucose, 10 mm HEPES, pH 7.6) for 15 min. Cultures were then washed in uptake buffer with 3 mm EDTA to remove extracellular Co2+ and incubated in 0.05% (NH4)2S for 5 min to precipitate intracellular Co2+, followed by washing in uptake buffer (3×) and fixation (4% paraformaldehyde, 30 min). For silver enhancement, cultures were washed three times in development buffer (292 mm sucrose, 15.5 mm hydroquinone, 42 mm citric acid) and incubated in 0.1% AgNO3 in development buffer at 50–55°C. This solution was changed at 15 min intervals while enhancement was monitored periodically by microscopic observation. When enhancement was complete (usually after 30–50 min), the reaction was terminated by washing three times in warm development buffer.
[Ca2+]i measurements. Cultures were loaded with Fura-2 by incubating them in the dark (25°C, room air) for 30 min in HSS containing 5 μm Fura-2 acetoxymethyl ester (Fura-2 AM; Molecular Probes, Eugene, OR), 0.2% pluronic acid, and 0.4% dimethyl sulfoxide (DMSO). After loading, the cultures were washed (5 ×) with HSS and kept in the dark for 30 more min. Cultures were then mounted in a microscope-stage adaptor and viewed with an inverted microscope (Nikon Diaphot equipped with Xenon epifluorescence optics). Cells were alternately illuminated with 340 and 380 nm light from a Xenon source, and fluorescence-imaged (at 510 nm) by a Hamamatsu intensified CCD camera. Perimeters of neuronal somata were outlined, and data were gathered on an 80486-based computer using Fluor software from Universal Imaging (West Chester, PA). [Ca2+]i was determined by the equation [Ca2+]i = Kd · (Fmin/Fmax){(R − Rmin)}/{(Rmax − R)}, using Kd = 224 nm. The system was recalibrated after any adjustments to the apparatus.
Chemicals and reagents. NMDA and kainate were from Sigma (St. Louis, MO), and MK-801 was from Research Biochemicals (Natick, MA). Tissue culture media and serum were from Life Technologies (Grand Island, NY)
RESULTS
NMDA channels and Ca2+-permeable AMPA/kainate channels permit rapid 45Ca2+ influx
To compare rates of NMDA- and kainate-triggered Ca2+accumulation in cortical neurons, 45Ca2+ influx over a 10 min epoch (“10 min Ca2+ load”) was measured during different agonist exposures (see Materials and Methods). Although the values obtained literally reflect relative amounts of Ca2+ influx, as influx is carried out for fixed 10 min periods, they provide measures of relative influx rates (quantity of influx per fixed unit of time) that can be compared between different excitotoxin exposures. In each experiment,45Ca2+ accumulation was normalized to the maximal influx seen in sister cultures exposed to 50 μmNMDA in the presence of 10 mm Ca2+ media (“control” influx), after subtraction of background influx. Previous studies have demonstrated that similar agonist exposures to pure astrocyte cultures cause little influx (Hartley et al., 1993), indicating that measured influx is neuronal.
As reported previously (Hartley et al., 1993), kainate triggered much less 45Ca2+ influx than NMDA (Fig.1). Also, when the [Ca2+]e was varied during these exposures, 45Ca2+ influx increased almost proportionately in the case of NMDA exposures, whereas with kainate exposures, increasing [Ca2+]eevoked less-than-proportional increases in influx (Fig. 1).
Fig. 1.

Kainate- and NMDA-triggered45Ca2+ influx depends on the [Ca2+]e. 45Ca2+influx was measured in sister cultures during 10 min exposures to NMDA or kainate in the presence of the indicated [Ca2+]e, as described (see Materials and Methods). Values represent summation of seven experiments, 18–30 cultures each condition. *, Difference from kainate-induced influx in the same [Ca2+]e; #, difference from influx with the same agonist in 1.8 mm[Ca2+]e (p < 0.01 by two-tailed t test).
Kainate triggers Ca2+ accumulation through VSCC as well as through Ca-A/K. Neurons possessing Ca-A/K can be identified by a histochemical stain based on kainate-stimulated Co2+ uptake (Co2+(+) cells); the specificity of the stain is indicated by the failure of NMDA or high K+ to substitute for kainate in triggering Co2+ uptake (Pruss et al., 1991; Turetsky et al., 1994). In cortex, Co2+(+) neurons constitute a distinct subset (∼13%) (Turetsky et al., 1994; our unpublished observations) that is unusually vulnerable to kainate toxicity.
To estimate the relative rate of kainate-triggered45Ca2+ influx through Ca-A/K, we made use of the observation that the Co2+(+) neuronal population is substantially (∼90%) destroyed 24 hr after a brief (1 hr) exposure to 100 μm kainate (kainate pretreatment) (Turetsky et al., 1994; Lu et al., 1995); these exposures cause little damage to the background, Co2+(−) neurons. Thus, we estimated kainate-triggered Ca2+ influx into Co2+(+) neurons as the difference (Δ influx) between influx in cultures in which the Co2+(+) population had been largely destroyed (by such a kainate pretreatment the day before) and influx obtained in sister cultures preexposed to sham wash alone.
Consistent with our previous report (Lu et al., 1995), such pretreatments substantially decreased kainate-triggered45Ca2+ influx (Table 1). Identical pretreatments had little or no effect on NMDA or high K+ (100 mm)-triggered45Ca2+ influx (Table 1). Thus, the pretreatments do not cause a generalized loss of neuronal channel function, and the decrements in kainate-triggered influx most likely reflect specific loss of Co2+(+) neurons. Although the estimated kainate-triggered Ca2+ influx into Co2+(+) neurons is substantially less than NMDA-triggered influx with either 1.8 or 10 mm[Ca2+]e (Table 1), Co2+(+) neurons make up only ∼13% of neurons in cortical culture, whereas NMDA channels are expressed on virtually all neurons. Thus, normalizing influx to the small size of the Co2+(+) population (Δ influx/0.13) provides estimated “per-neuron” influx rates that are comparable with NMDA-triggered influx rates in the presence of the same [Ca2+]e concentrations (Table 1). In addition, like NMDA receptor-mediated influx, estimated kainate-triggered influx into Co2+(+) neurons increases markedly with increasing [Ca2+]e, suggesting that Ca-A/K are not saturated at these [Ca2+]e levels.
Table 1.
The decrement (Δ influx) in kainate-triggered45Ca2+ influx after kainate pretreatment provides an estimate of the rate of kainate-triggered influx into Co2+ (+) neurons
| Agonist | [Ca2+]emm | Sham wash cultures (% max. influx) | Pretreated cultures (% max. influx) | Δ influx | Δ influx/0.13 |
|---|---|---|---|---|---|
| Kainate (100 μm) | 1.8 | 8.78 ± 0.66 | 4.88 ± 1.32* | 3.9 ± 1.98 | 15–45% |
| 10 | 24.61 ± 1.72# | 9.39 ± 0.81*# | 15.2 ± 2.53# | 97–136% | |
| NMDA (20 μm) | 1.0 | 9.49 ± 0.85# | |||
| 1.8 | 17.92 ± 2.42 | 14.87 ± 1.46 | |||
| High K+ (100 mm) | 1.8 | 8.34 ± 1.1 | 8.77 ± 1.11 |
45Ca2+ influx was measured in sister cultures exposed for 10 min to either kainate, NMDA, or high K+ in the presence of the indicated [Ca2+]e in cultures either preexposed to sham wash or pretreated with kainate [100 μm × 1 hr; an exposure that triggers selective degeneration of >90% of the Co2+(+) neurons; see Materials and Methods]. Influx is scaled to maximal NMDA (50 μm, 10 mm[Ca2+]e)-induced45Ca2+ influx (=100) after subtraction of background counts. Note that kainate pretreatment causes a substantial decrement in kainate-triggered influx while causing little decrement in NMDA- or high K+-triggered influx, and that normalizing this decrement in influx for the small size of the Co2+(+) population (∼13% of cortical neurons) provides an estimated rate of kainate-triggered influx into Co2+(+) neurons comparable to that at which NMDA triggers influx into most neurons. Values represent summations of 3–10 experiments (12–40 cultures each condition). *, Influx rate different from sham-washed cultures; #, influx rate different from 1.8 mm[Ca2+]eexposures to same agonist (p < 0.01 by two-tailedt test).
Ca2+ influx is a primary determinant of injury
Subsequent experiments examined the relationship between Ca2+ influx and subsequent neurodegeneration. As discussed above, brief (several-minute) intense periods of NMDA receptor activation are sufficient to trigger widespread neurodegeneration (in 5 min exposure to cortical cultures, the LD50 for NMDA is ∼120 μm) (Koh and Choi, 1988), which parallels a rapid induction of 45Ca2+ accumulation (Hartley et al., 1993). In contrast, with lower exposures that induce lower rates of Ca2+ influx (Fig. 1) (Hartley et al., 1993), widespread injury still results after more prolonged durations of exposure (the LD50 for NMDA in 20–24 hr exposure is ∼16 μm) (Koh and Choi, 1988). With such a low NMDA exposure (20 μm, 1.8 mm[Ca2+]e), we found injury to increase with increasing duration of exposure, further suggesting that net Ca2+ accumulation may be a critical determinant of injury (Fig. 2A).
Fig. 2.
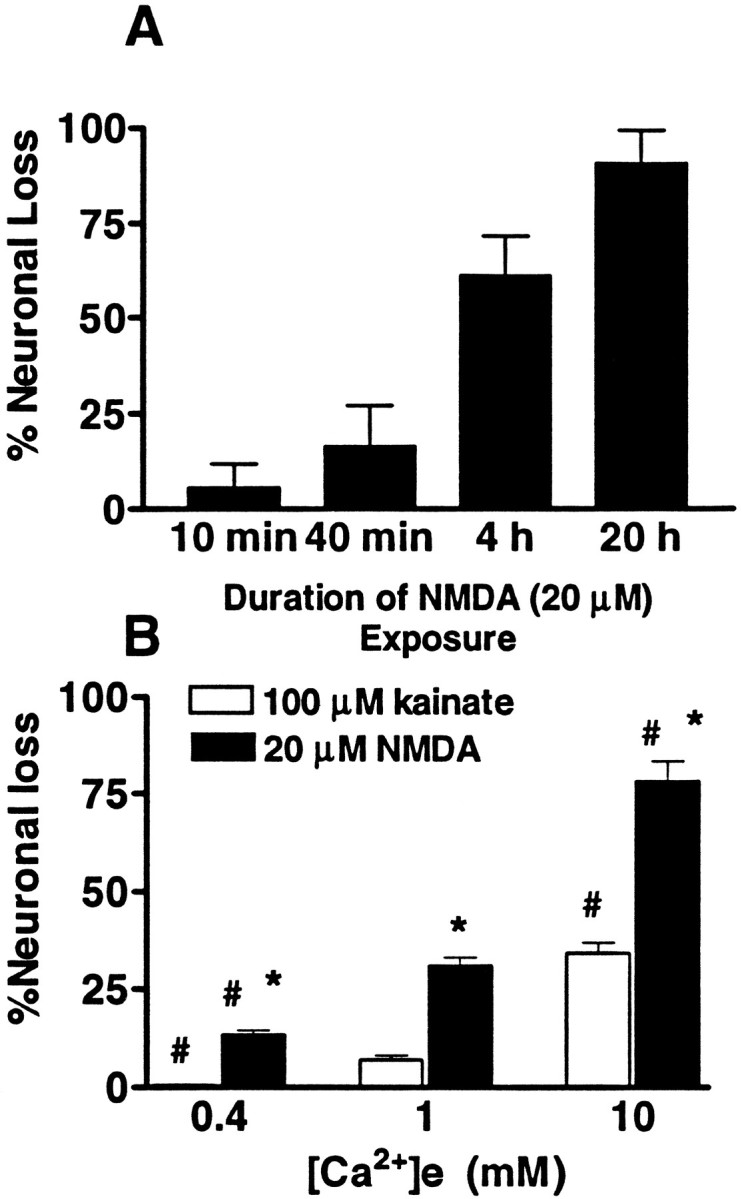
Time and [Ca2+]edependence of excitotoxic injury. A, Cultures were exposed continuously to NMDA (20 μm, 1.8 mm [Ca2+]e) in room air (25°C). Exposures were terminated after the indicated interval by addition of MK-801 (10 μm), and injury was assessed as described (qualitatively by morphological examination and quantitatively by measurement of LDH release) 20 hr after exposure onset. Values represent summation of four experiments, 16 cultures each condition. B, Cultures were exposed continuously to NMDA or to kainate in room air (25°C) in the presence of the indicated [Ca2+]e, and injury was assessed, as above, after 12–15 hr. Values represent summation of seven experiments, 22–27 cultures each condition. *, Difference from kainate-induced cell loss in the same [Ca2+]e; #, difference from cell loss with the same agonist in 1.0 mm[Ca2+]e (p < 0.01 by two-tailed t test).
As prolonged periods of AMPA/kainate receptor activation are necessary to induce widespread injury to cultured cortical neurons, (Choi, 1992), subsequent experiments (paralleling the 45Ca2+influx experiments described above) examined the [Ca2+]e dependence of neurotoxicity caused by prolonged (12–15 hr) kainate or NMDA exposures under conditions identical to those used in the influx experiments (HSS, 25°C). Under these conditions, although the maximal injury produced by intense kainate exposures (100 μm, 10 mm[Ca2+]e) was considerably less than that produced by a relatively low (20 μm) NMDA exposure in the same [Ca2+]e, both NMDA and kainate induced neurotoxicity, which increased with increasing [Ca2+]e concentrations (Fig.2B).
To directly examine the hypothesis that net accumulation of Ca2+, rather than the route of Ca2+ entry, is the primary determinant of the extent of excitotoxic injury, we next compared injury caused by exposures estimated to give similar initial influx rates (estimated as 10 min Ca2+ loads) through different routes. We first compared the injury induced by Ca2+ influx through NMDA channels with that induced by influx through VSCC. To do this, cultures were kainate-pretreated (to destroy Co2+(+) neurons, so that influx through Ca-A/K would not be a factor) and exposed for 12–14 hr to either kainate (100 μm, 10 mm [Ca2+]e) or to NMDA (20 μm, 1 mm[Ca2+]e), exposure levels that gave similar 10 min Ca2+ loads (Table 1). Supporting the idea that net Ca2+ accumulation is an important determinant of injury under these conditions, these exposures caused comparable submaximal degrees of neuronal injury (as assessed by measurement of the release of the cytosolic enzyme LDH into the media) (Fig.3A).
Fig. 3.
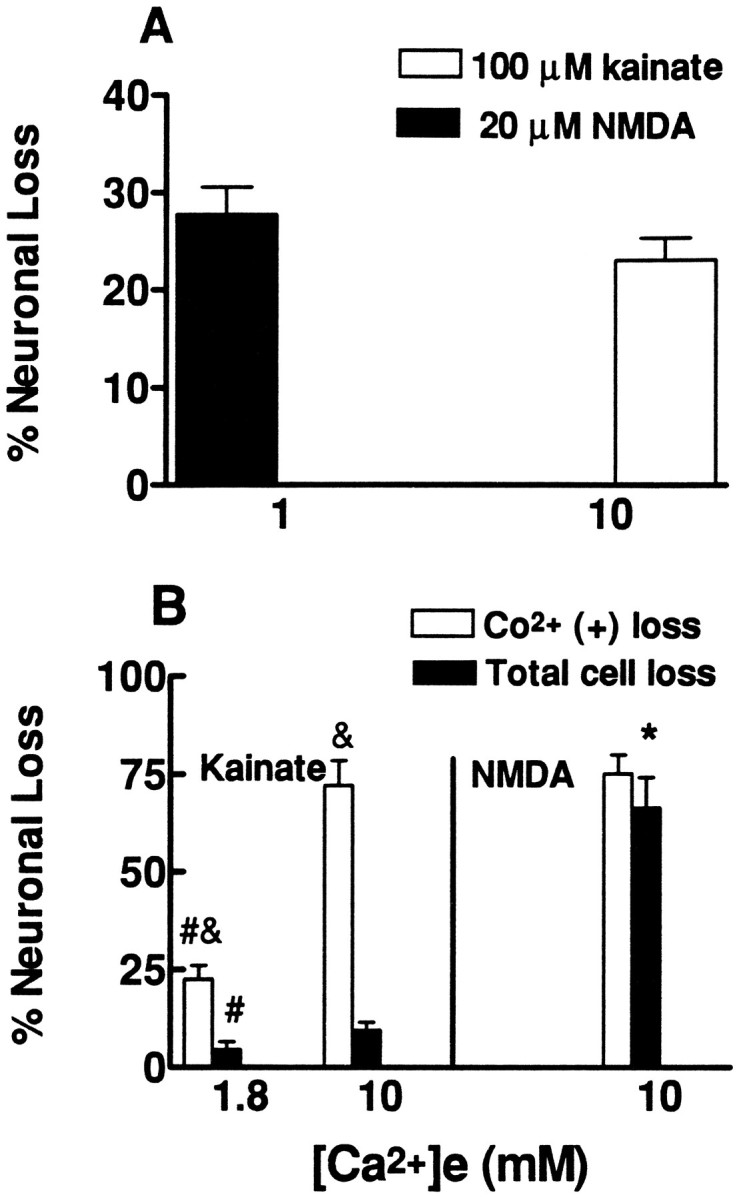
Kainate and NMDA exposures that cause similar 10 min Ca2+ loads induce similar levels of neurodegeneration.A, Cultures were pretreated to selectively kill the Co2+(+) neurons (see Materials and Methods). The next day they were exposed continuously to NMDA (20 μm) or kainate (100 μm) in the presence of the indicated [Ca2+]e, and injury was assessed, as described (qualitatively by morphological examination and quantitatively by measurement of LDH release), after 12–14 hr. Note that 45Ca2+ influx experiments found these exposures to trigger similar initial rates of Ca2+ influx through VSCC and NMDA channels (see Table 1). Values represent summation of seven experiments, 22–27 cultures each condition.B, Cultures were exposed to NMDA (50 μm) or kainate (100 μm) for 10 min in the presence of the indicated [Ca2+]e. Assessment of injury to the Co2+(+) population (by direct counts of labeled cells) as well as to the overall neuronal population (qualitatively by morphological examination and quantitatively by measurement of LDH release) was made after 20–24 hr, as described. Note that the NMDA exposure and the kainate exposure in 10 mm[Ca2+]e were estimated to trigger similar rates of influx through NMDA channels and through Ca-A/K into Co2+(+) neurons (see Table 1). Seven experiments, 25–27 cultures each condition, were compiled for assessment of Co2+(+) neuronal loss, and three experiments, 12 cultures each condition, for assessment of total neuronal loss. *, Difference from kainate-induced cell loss to same cell type in the same [Ca2+]e; #, difference from same type of cell loss in 10 mm [Ca2+]e; &, difference from total cell loss after same exposure (p < 0.01 by two-tailed ttest).
Next, to compare injury resulting from influx through NMDA channels with that resulting from influx through Ca-A/K, normal (not pretreated) cultures were exposed for 10 min to kainate (100 μm, 10 mm [Ca2+]e) or to NMDA (50 μm, 10 mm [Ca2+]e). This kainate exposure was estimated to cause a similar rate of Ca2+ entry through Ca-A/K into Co2+(+) neurons as the NMDA exposure was estimated to induce into the overall neuronal population (Table 1). The observation that the kainate exposure gave a comparable level of injury to the Co2+(+) population as the NMDA exposure did to both the Co2+(+) population and the overall neuronal population (Fig. 3B) further supports the hypothesis that a high rate of intracellular Ca2+accumulation underlies the high vulnerability of Co2+(+) neurons to AMPA/kainate receptor-mediated injury. For comparison, when sister cultures were identically exposed to kainate (100 μm), but in the presence of only 1.8 mm[Ca2+]e [an exposure that caused less Ca2+ influx into Co2+(+) neurons (Table 1)], resultant damage to the Co2+(+) population was considerably less (Fig. 3B).
[Ca2+]i: nonlinear relationship to Ca2+ influx rate
Fura-2 fluorescent imaging techniques were used to examine the relationship between agonist-triggered influx through different routes and resultant [Ca2+]i levels in neuronal somata. As previous studies have demonstrated that [Ca2+]i can be maintained at near normal levels after termination of a brief but lethal excitotoxic exposure, until an abrupt rise signals impending death (Randall and Thayer, 1992;Dubinsky, 1993; Tymianski et al., 1993), we desired to image [Ca2+]i during continuous prolonged agonist exposures, but before death ensued. In control toxicity experiments, little (<3%) neuronal cell death (as assessed by trypan blue staining) was observed immediately after 2 hr exposures to NMDA (20 μm, 10 mm [Ca2+]e) or to kainate (100 μm, 10 mm[Ca2+]e) at 25°C. However, after 3 hr exposures to these same conditions, a minority of neurons (18–22%) stained with trypan blue dye, indicating intervening loss of viability; with lower [Ca2+]e, less death occurred (Table 2). Thus, [Ca2+]ilevels were recorded in cortical cultures during 3 hr exposures to NMDA (20 μm) or to kainate (100 μm) with varying [Ca2+]e levels (Fig. 4; Table2). On average, these exposures caused [Ca2+]i to rise rapidly and then to fall off somewhat over time. Greater [Ca2+]e values were associated with greater mean [Ca2+]ivalues that began after onset of the exposures. These differences in [Ca2+]i values generally persisted throughout the duration of the exposures, suggesting that initial differences in influx rates were largely maintained.
Table 2.
[Ca2+]i levels (mean ± SD) in cortical neurons during 2 hr kainate or NMDA exposures
| Drugs | [Ca2+]emm | Live cells | Dead cells | ||
|---|---|---|---|---|---|
| [Ca2+]i nm | Log [Ca2+]i | [Ca2+]inm | Log [Ca2+]i | ||
| Kainate (100 μm) | 1.8 | 376 ± 190 | 2.53 ± 0.21 | 470 ± 248 | 2.62 ± 0.24 |
| n = 171 | n = 9 | ||||
| 10 | 616 ± 259 | 2.75 ± 0.19* | 961 ± 294 | 2.96 ± 0.17 | |
| n = 199 | # | n = 34 | |||
| NMDA (20 μm) | 0.4 | 226 ± 116 | 2.31 ± 0.17 | ||
| n = 89 | & | ||||
| 1 | 428 ± 173 | 2.59 ± 0.22 | |||
| n = 131 | |||||
| 1.8 | 590 ± 285 | 2.73 ± 0.19 | 698 ± 281 | 2.81 ± 0.18 | |
| n = 246 | & | n = 25 | |||
| 10 | 752 ± 457 | 2.79 ± 0.27 | 782 ± 301 | 2.86 ± 0.17 | |
| n = 160 | & | n = 44 | |||
Cortical cultures were exposed to kainate or NMDA in HSS (25°C), as indicated, in the presence of the indicated [Ca2+]e, and neuronal viability was examined at the end of the 3 hr exposure by staining with trypan blue dye. Values in each category represent the mean ± SD of [Ca2+]i response of all neurons, after first averaging [Ca2+]i responses in each neuron over the first 2 hr of the exposure. All statistical comparisons were carried out on mean log [Ca2+]i values so as not to disproportionately weigh high-end [Ca2+]i values and to make statistical demonstration of differences more rigorous. *, Difference from kainate exposures in 1.8 mm [Ca2+]e; #, difference from dead cells after same exposure; &, difference from NMDA in 1 mm [Ca2+]e (for all indicated comparisons, p < 10−3 by ANOVA with Bonferroni post hoc test).
Fig. 4.

Mean neuronal [Ca2+]i levels during prolonged NMDA or kainate exposures depend on [Ca2+]e. Mean [Ca2+]i levels are plotted during 3 hr exposures to NMDA (top) or kainate (bottom), in HSS (25°C) with the indicated [Ca2+]e. Neuronal viability was examined at the end of the exposure by staining with trypan blue dye. *, Plot of [Ca2+]i values in neurons that were dead at the end of the exposure. For all other plots, only neurons that remained viable throughout the exposure were included. Tracings represent mean values from 34 (dead cell tracing) to 246 neurons from five or more experiments each condition. Baseline recordings (not to scale on these graphs) were stable for at least 5 min before addition of agonist.
In each condition, [Ca2+]i levels were averaged over the first 2 hr after onset of exposure (Table 2). Despite the near linear increase in 10 min Ca2+ loads through NMDA channels with increasing [Ca2+]e, only modest (much less than linear) increases were observed in corresponding mean [Ca2+]i levels. Also, with each [Ca2+]e, the distribution of [Ca2+]i values in individual neurons overlapped substantially with that seen at different [Ca2+]e levels (Fig. 5). With NMDA exposures in 10 mm [Ca2+]e, 22% of neurons were nonviable at the end of the 3 hr exposure (as indicated by staining with trypan blue dye). However, no significant difference was seen in [Ca2+]i values (averaged over the first 2 hr of exposure) between these neurons and those that remained viable throughout the 3 hr exposure (Table 2).
Fig. 5.

The distribution of [Ca2+]i values in cortical neurons during NMDA exposures depends on [Ca2+]e. Histograms show the distribution of log [Ca2+]ivalues (nm) in individual neurons during prolonged exposures to NMDA (20 μm) in the indicated [Ca2+]e. Values in each neuron were averaged over at least seven readings spanning the first 2 hr of the exposure. Log transformations of [Ca2+]i values are displayed to convey relative rather than absolute differences and so as not to give disproportionate weight to extreme high-end values.
In contrast to the case with NMDA, interpretation of kainate exposure data is complicated by the disparity in estimated Ca2+influx rates between the majority of neurons, into which kainate triggers a relatively slow influx, and Co2+(+) neurons, which are predicted to face much greater kainate-triggered Ca2+ influx rates. With an intense kainate exposure (100 μm, 10 mm [Ca2+]e), 15% of neurons were nonviable after 3 hr. However, unlike the case with NMDA exposures, these neurons had significantly greater [Ca2+]i levels during the first 2 hr of the exposure than surviving neurons (Fig. 4, Table 2). The observation of control experiments that such 3 hr kainate exposures (in 10 mm [Ca2+]e) induce a near complete loss of Co2+(+) neurons and the appearance of near equal numbers of trypan blue-stained neurons [123 ± 7 Co2+(+) neurons, <2 trypan blue-stained neurons/200× microscope field in untreated cultures vs <2 Co2+(+) cells, 115 ± 10 trypan blue-stained neurons after exposure, mean ± SEM, three experiments each with more than 15 fields, 2000 labeled cells counted each condition] provides indirect evidence that these neurons were Co2+(+).
To more directly examine [Ca2+]i elevations in Co2+(+) neurons during these maximal (100 μm, 10 mm [Ca2+]e) kainate exposures, pseudocolor images were obtained 3 min after onset of the exposure, followed by Co2+ staining (longer exposures interfered with staining). Note that the Co2+(+) neurons displayed unusually high [Ca2+]ivalues. For comparison, similar images were obtained after low-level (10 μm, 1.8 mm[Ca2+]e) kainate exposures, which we previously found to trigger little [Ca2+]iresponse in most neurons while preferentially elevating [Ca2+]i levels in Co2+(+) neurons (Lu et al., 1995) (Fig. 6). Quantification of results from several experiments confirmed the unusually high [Ca2+]i responses of Co2+(+) neurons during intense (100 μm, 10 mm[Ca2+]e) kainate exposures (Fig.7); mean log [Ca2+]i values (nm) in Co2+(+) neurons (3.09 ± 0.025; mean ± SEM) were significantly greater than those in Co2+(−) neurons (2.85 ± 0.013; p < 10−6; two-tailed t test).
Fig. 6.

Kainate-triggered [Ca2+]i elevations in Co2+(+) and Co2+(−) neurons: pseudocolor images. Cortical cultures were exposed to 10 μm kainate in 1.8 mm[Ca2+]e (A) or to 100 μm kainate in 10 mm[Ca2+]e (C); pseudocolor images illustrated were obtained at the end of the 3 min exposure. Immediately after imaging, cultures were Co2+-labeled, and the illustrated field was photographed (B, D). Note that Co2+(+) neurons have distinctly greater [Ca2+]i responses than Co2+(−) neurons with both of these exposures.
Fig. 7.

Co2+-positive neurons have unusually large [Ca2+]i responses during intense kainate exposures. A, [Ca2+]ilevels are plotted in individual cortical neurons before and for 3 min after addition of kainate (100 μm, 10 mm[Ca2+]e). Of the 14 neurons shown, four (indicated) were Co2+(+). B, Distribution of log [Ca2+]i values (nm) in cortical neurons (compiled from 6 experiments) 3 min after addition of kainate, as above. Values in each neuron were averages of seven to nine readings from 30 sec to 3 min after kainate addition. As in Figure 5, above, log transformations of [Ca2+]i values are displayed to convey distributions of relative rather than absolute differences and so as not to give disproportionate weight to extreme high-end values. Mean log [Ca2+]ivalues (nm) in Co2+(+) neurons (3.09 ± 0.025; mean ± SEM) were significantly greater than those in Co2+(−) neurons (2.85 ± 0.013; p < 10−6, two-tailed t test).
DISCUSSION
Correlation between Ca2+ influx and cell death
A primary aim of the present study has been to attempt to correlate the amount of Ca2+ influx through each of three routes (NMDA channels, VSCC, and Ca-A/K) with the extent of resultant neuronal cell death. Thus, present 45Ca2+influx data suggest, in agreement with our previous report (Lu et al., 1995), that the rate at which kainate triggers Ca2+ entry through Ca-A/K into Co2+(+) neurons is comparable (when normalized for the small size of the Co2+(+) population) with the high rates at which NMDA triggers Ca2+ influx, whereas VSCC permit much lower influx rates. These influx measurements reflect the density of channel expression, the single-channel Ca2+ permeability, and channel desensitization rates. Although the latter two measures are best assessed with electrophysiological techniques, present observations that NMDA-triggered influx and kainate-triggered influx through Ca-A/K appear to increase proportionately with increasing [Ca2+]e are in line with electrophysiological studies (MacDermott et al., 1986; Koh et al., 1995; Zhang et al., 1995) showing that Ca-A/K, like NMDA channels, are highly Ca2+-permeable.
To examine the role of Ca2+ entry in neurodegeneration, we first evaluated the relationship between Ca2+ influx through each of these three routes and subsequent degeneration. Consistent with previous studies showing NMDA neurotoxicity (Choi, 1992) and AMPA/kainate receptor-mediated injury to Co2+(+) neurons (Brorson et al., 1994; Turetsky et al., 1994) to be Ca2+-dependent, we found that neuronal injury triggered by Ca2+ entry through each of the three routes was increased by exposures that caused more Ca2+ entry through that route.
To gain insight into the relative importance of agonist-triggered Ca2+ load versus entry route as determinants of Ca2+-mediated injury, neuronal loss was assessed after exposures that were estimated to trigger similar initial rates of Ca2+ influx through different routes. Thus, with prolonged exposures to NMDA and kainate (calibrated to give similar 10 min Ca2+ loads through NMDA channels and through VSCC), similar overall neuronal injury resulted, whereas with brief exposures to these agonists (estimated to cause similar Ca2+ entry through NMDA channels and through Ca-A/K), NMDA triggered a degree of damage to the overall neuronal population that was comparable to the damage kainate induced in the Co2+(+) population. Although it is likely that initial influx rates do not remain constant throughout the course of the prolonged exposures, the strong concordance between estimated influx rates through differing routes and resultant injury after both short and long exposures provides support for the hypothesis that net Ca2+ accumulation is a critical determinant of injury, regardless of the route through which it gains entry to the neuron.
[Ca2+]i: relationship to influx rate, route, and death
Previous studies have generally found only weak correlations between [Ca2+]i levels during excitotoxic or ischemic exposures and subsequent degeneration (Michaels and Rothman, 1990; Dubinsky and Rothman, 1991; Tymianski et al., 1993). The observation that [Ca2+]i levels can transiently normalize after a lethal excitotoxic exposure (Randall and Thayer, 1992; Dubinsky, 1993) indicates that powerful homeostatic mechanisms can obscure a lethal Ca2+ load and raises the possibility that sequestered Ca2+, invisible to imaging, may actively contribute to the neuronal injury. Present studies extend these past studies by permitting correlations between [Ca2+]i levels in neuronal somata and estimated Ca2+ influx rates. Of note, imaging whole-cell body [Ca2+]i levels over time, using a high-affinity dye such as Fura-2, could underestimate the highest peak [Ca2+]i responses, especially those that are very transient or that occur in specific cellular domains such as the postsynaptic regions of dendrites.
Three broad conclusions were drawn from experiments correlating 10 min Ca2+ loads through a single route (NMDA channels) with [Ca2+]i levels. First, with each exposure, individual neurons display a broad range of [Ca2+]i levels. This heterogeneity of [Ca2+]i levels could reflect both differences in agonist-triggered influx rates between neurons (perhaps because of differing levels of channel expression) and differences in homeostatic responses (comprising release from intracellular stores as well as buffering/extrusion). Second, markedly increasing the Ca2+influx rate into the neurons (by increasing [Ca2+]e between 1 and 10 mm) caused relatively little change in the distribution of [Ca2+]i levels, which overlap substantially at each exposure level. Finally, despite the close relationship between influx rate and death, [Ca2+]i levels during intense NMDA exposures (20 μm, 10 mm[Ca2+]e) did not predict the neurons that were dead at the end of the 3 hr exposure.
With kainate exposures, interpretation of [Ca2+]i response data is complicated by the presence of Co2+(+) neurons, into which kainate appears to trigger a particularly high rate of Ca2+ entry. In our previous report (Lu et al., 1995), we found Co2+(+) neurons to display significantly greater [Ca2+]irises than other neurons during very low kainate exposures (10 μm, 1.8 mm [Ca2+]e) that caused little [Ca2+]i rise in most neurons, but relatively little difference in [Ca2+]i levels between Co2+(+) and other neurons when the kainate concentration was raised to 100 μm (still with 1.8 mm[Ca2+]e). In the present study, therefore, we were initially surprised to find a significant separation in [Ca2+]i levels between Co2+(+) neurons and other neurons during the most intense (100 μm, 10 mm [Ca2+]e) kainate exposures. However, this finding may be consistent with the great disparity in estimated influx rates between Co2+(+) and (−) neurons at these exposures; increasing [Ca2+]e from 1.8 to 10 mm is estimated to result in only a modest increase in influx into Co2+(−) neurons, whereas influx into Co2+(+) neurons is estimated to increase several-fold and be greater than the influx caused by submaximal (20 μm) NMDA exposures with 10 mm [Ca2+]e.
With both kainate and NMDA exposures, despite the great variation in [Ca2+]i levels between neurons, when [Ca2+]i levels are averaged over all neurons (to obscure individual neuronal differences) a direct, although far less than linear, relationship between mean [Ca2+]i and influx rate (estimated as 10 min Ca2+ load) is observed. Based on present data, we suggest that mean [Ca2+]i levels largely reflect influx rate, but that powerful buffering/homeostatic mechanisms limit rises in [Ca2+]i until very high rates of influx (as may be observed in Co2+(+) neurons exposed to kainate in 10 mm [Ca2+]e) overwhelm these mechanisms. However, because injury does correlate strongly with estimated initial rate of Ca2+ influx, present data support the idea that sequestered Ca2+, not reflected in the [Ca2+]i measure, may play a critical role in the development of this injury. For instance, sequestration of Ca2+ ions by mitochondria (Werth and Thayer, 1994; White and Reynolds, 1995) could trigger excess production of injurious reactive oxygen species, as has been demonstrated with intense NMDA exposures (Lafon-Cazal et al., 1993; Dugan et al., 1995;Reynolds and Hastings, 1995).
Conclusion
This study is the first to attempt to correlate relative rates of excitatory amino acid-induced Ca2+ influx with resultant [Ca2+]i levels and subsequent neuronal degeneration.
An essential feature of this study is that the primary conclusions regarding correlations between Ca2+ influx, [Ca2+]i, and cell death are based on mean measures across large numbers of neurons. Such averaging is valuable in that it reveals important underlying relationships that may not be evident at the single neuron level, yet it obscures differences between individual neurons that might make them substantially more or less vulnerable than other neurons to Ca2+-mediated injury.
In addition to acute excitotoxic conditions such as stroke, present results may be relevant to excitotoxic neurodegeneration as occurs in more chronic degenerative conditions, including Alzheimer’s disease and amyotrophic lateral sclerosis. Supporting such a connection, recent studies by us and others have found that certain neurons prone to degeneration in these diseases, including basal forebrain cholinergic neurons and spinal motor neurons, are highly vulnerable to AMPA/kainate receptor-mediated injury (Hugon et al., 1989; Page et al., 1991;Rothstein et al., 1993; Weiss et al., 1994b; Carriedo et al., 1995,1996), likely reflecting possession of Ca-A/K (Yin et al., 1994a;Carriedo et al., 1995, 1996). Thus, high rates of glutamate-triggered Ca2+ influx through these channels might be one factor predisposing these neurons to degenerate under pathological conditions. However, it is clear that not all neurons possessing Ca-A/K degenerate in disease. For instance, several studies have indicated that cortical and hippocampal GABAergic neurons possess Ca-A/K (Bochet et al., 1994;Jonas et al., 1994; Yin et al., 1994b), yet there is little evidence of their preferential loss in either acute conditions, such as ischemia, or chronic conditions, such as Alzheimer’s disease. Thus, additional environmental as well as intrinsic neuronal factors most likely come into play in determining a neuron’s vulnerability in disease.
Footnotes
This work was supported by National Institutes of Health Grant NS 30884 (J.H.W.) and by grants from the Alzheimer’s Disease and Related Disorders Association (J.H.W.) and the PEW Scholars Program in the Biomedical Sciences (J.H.W.).
Correspondence should be addressed to Dr. John H. Weiss, Department of Neurology, University of California, Irvine, Irvine, CA 92717-4290.
REFERENCES
- 1.Bochet P, Audinat E, Lambolez B, Crepel F, Rossier J, Iino M, Tsuzuki K, Ozawa S. Subunit composition at the single-cell level explains functional properties of a glutamate-gated channel. Neuron. 1994;12:383–388. doi: 10.1016/0896-6273(94)90279-8. [DOI] [PubMed] [Google Scholar]
- 2.Brorson JR, Bleakman D, Chard PS, Miller RJ. Calcium directly permeates kainate/alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors in cultured cerebellar Purkinje neurons. Mol Pharmacol. 1992;41:603–608. [PubMed] [Google Scholar]
- 3.Brorson JR, Manzolillo PA, Miller RJ. Ca2+entry via AMPA/KA receptors and excitotoxicity in cultured cerebellar Purkinje cells. J Neurosci. 1994;14:187–197. doi: 10.1523/JNEUROSCI.14-01-00187.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carriedo SG, Yin HZ, Lamberta R, Weiss JH. In vitro kainate injury to large, SMI-32 spinal neurons is Ca2+dependent. NeuroReport. 1995;6:945–948. doi: 10.1097/00001756-199504190-00030. [DOI] [PubMed] [Google Scholar]
- 5.Carriedo SG, Yin HZ, Weiss JH. Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro . J Neurosci. 1996;16:4069–4079. doi: 10.1523/JNEUROSCI.16-13-04069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 7.Dubinsky JM. Intracellular calcium levels during the period of delayed excitotoxicity. J Neurosci. 1993;13:623–631. doi: 10.1523/JNEUROSCI.13-02-00623.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dubinsky JM, Rothman SM. Intracellular calcium concentrations during “chemical hypoxia” and excitotoxic neuronal injury. J Neurosci. 1991;11:2545–2551. doi: 10.1523/JNEUROSCI.11-08-02545.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dugan LL, Sensi SL, Canzoniero LMT, Handran SD, Rothman SM, Lin T-S, Goldberg MP, Choi DW. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N -methyl-d-aspartate. J Neurosci. 1995;15:6377–6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW. Glutamate receptor-induced45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci. 1993;13:1993–2000. doi: 10.1523/JNEUROSCI.13-05-01993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hugon J, Vallat JM, Spencer PS, Leboutet MJ, Barthe D. Kainic acid induces early and delayed degenerative neuronal changes in rat spinal cord. Neurosci Lett. 1989;104:258–262. doi: 10.1016/0304-3940(89)90585-5. [DOI] [PubMed] [Google Scholar]
- 12.Iino M, Ozawa S, Tsuzuki K. Permeation of calcium through excitatory amino acid receptor channels in cultured rat hippocampal neurones. J Physiol (Lond) 1990;424:151–165. doi: 10.1113/jphysiol.1990.sp018060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jonas P, Racca C, Sakmann B, Seeburg PH, Monyer H. Differences in Ca2+ permeability of AMPA-type glutamate receptor channels in neocortical neurons caused by differential GluR-B subunit expression. Neuron. 1994;12:1281–1289. doi: 10.1016/0896-6273(94)90444-8. [DOI] [PubMed] [Google Scholar]
- 14.Koh D-S, Geiger JRP, Jonas P, Sakmann B. Ca2+-permeable AMPA and NMDA receptor channels in basket cells of rat hippocampal dentate gyrus. J Physiol (Lond) 1995;485:383–402. doi: 10.1113/jphysiol.1995.sp020737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koh J, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 16.Koh J, Choi DW. Vulnerability of cultured cortical neurons to damage by excitotoxins: differential susceptibility of neurons containing NADPH-diaphorase. J Neurosci. 1988;8:2153–2163. doi: 10.1523/JNEUROSCI.08-06-02153.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 18.Lu YM, Yin HZ, Weiss JH. Ca2+ permeable AMPA/kainate channels permit rapid injurious Ca2+ entry. NeuroReport. 1995;6:1089–1092. doi: 10.1097/00001756-199505300-00004. [DOI] [PubMed] [Google Scholar]
- 19.MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- 20.Michaels RL, Rothman SM. Glutamate neurotoxicity in vitro: antagonist pharmacology and intracellular calcium concentrations. J Neurosci. 1990;10:283–292. doi: 10.1523/JNEUROSCI.10-01-00283.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy SN, Miller RJ. Regulation of Ca2+influx into striatal neurons by kainic acid. J Pharmacol Exp Ther. 1989;249:184–193. [PubMed] [Google Scholar]
- 22.Page KJ, Everitt BJ, Robbins TW, Marston HM, Wilkinson LS. Dissociable effects on spatial maze and passive avoidance acquisition and retention following AMPA- and ibotenic acid-induced excitotoxic lesions of the basal forebrain in rats: differential dependence on cholinergic neuronal loss. Neuroscience. 1991;43:457–472. doi: 10.1016/0306-4522(91)90308-b. [DOI] [PubMed] [Google Scholar]
- 23.Pruss RM, Akeson RL, Racke MM, Wilburn JL. Agonist-activated cobalt uptake identifies divalent cation-permeable kainate receptors on neurons and glia. Neuron. 1991;7:509–518. doi: 10.1016/0896-6273(91)90302-g. [DOI] [PubMed] [Google Scholar]
- 24.Randall RD, Thayer SA. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci. 1992;12:1882–1895. doi: 10.1523/JNEUROSCI.12-05-01882.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rose K, Goldberg MP, Choi DW. Cytotoxicity in murine cortical cell culture. In: Tyson CA, Frazier JM, editors. In vitro biological methods. Academic; San Diego: 1993. pp. 46–60. [Google Scholar]
- 27.Rothstein JD, Jin L, Dykes-Hoberg M, Kuncl RW. Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc Natl Acad Sci USA. 1993;90:6591–6595. doi: 10.1073/pnas.90.14.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turetsky DM, Canzoniero LMT, Sensi SL, Weiss JH, Goldberg MP, Choi DW. Cortical neurons exhibiting kainate-activated Co2+ uptake are selectively vulnerable to AMPA/kainate receptor-mediated toxicity. Neurobiol Dis. 1994;1:101–110. doi: 10.1006/nbdi.1994.0013. [DOI] [PubMed] [Google Scholar]
- 29.Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weiss JH, Hartley DM, Koh J, Choi DW. The calcium channel blocker nifedipine attenuates slow excitatory amino acid neurotoxicity. Science. 1990;247:1474–1477. doi: 10.1126/science.247.4949.1474. [DOI] [PubMed] [Google Scholar]
- 31.Weiss JH, Turetsky D, Wilke G, Choi DW. AMPA/kainate receptor-mediated damage to NADPH-diaphorase containing neurons is Ca2+ dependent. Neurosci Lett. 1994a;167:93–96. doi: 10.1016/0304-3940(94)91035-9. [DOI] [PubMed] [Google Scholar]
- 32.Weiss JH, Yin H, Choi DW. Basal forebrain cholinergic neurons are selectively vulnerable to AMPA/kainate receptor-mediated neurotoxicity. Neuroscience. 1994b;60:659–664. doi: 10.1016/0306-4522(94)90494-4. [DOI] [PubMed] [Google Scholar]
- 33.Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci. 1994;14:348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White RJ, Reynolds IJ. Mitochondria and Na+/Ca2+ exchange buffer glutamate-induced calcium loads in cultured cortical neurons. J Neurosci. 1995;15:1318–1328. doi: 10.1523/JNEUROSCI.15-02-01318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin H, Lindsay AD, Weiss JH. Kainate injury to cultured basal forebrain cholinergic neurons in Ca2+dependent. NeuroReport. 1994a;5:1477–1480. doi: 10.1097/00001756-199407000-00017. [DOI] [PubMed] [Google Scholar]
- 36.Yin H, Turetsky D, Choi DW, Weiss JH. Cortical neurons with Ca2+ permeable AMPA/kainate channels display distinct receptor immunoreactivity and are GABAergic. Neurobiol Dis. 1994b;1:43–49. doi: 10.1006/nbdi.1994.0006. [DOI] [PubMed] [Google Scholar]
- 37.Zhang D, Sucher NJ, Lipton SA. Co-expression of AMPA/kainate receptor-operated channels with high and low Ca2+ permeability in single rat retinal ganglion cells. Neuroscience. 1995;67:177–188. doi: 10.1016/0306-4522(94)00627-h. [DOI] [PubMed] [Google Scholar]