

Abstract
Basic fibroblast growth factor (bFGF) is a potent neurotrophic factor that regulates cell proliferation and differentiation during neuronal development. Here we report that fetal hippocampal neurons chronically treated with bFGF displayed larger [Ca2+]i increases than nontreated neurons in response to high K+-induced depolarization. This [Ca2+]i response was abolished by nicardipine and was little affected by treatments that depleted intracellular Ca2+ stores, thus reflecting the activities of L-type voltage-dependent Ca2+ channels. Whole-cell recordings also demonstrated increased high-voltage-activated Ca2+ currents in bFGF-treated neurons, whereas low-voltage-activated Ca2+currents remained unchanged. bFGF-stimulated increase in Ca2+ response was not observed in neurons treated with cycloheximide or actinomycin D, indicating that protein and RNA synthesis were required for this effect. Visualization using a fluorescent dihydropyridine analog revealed that bFGF-treated neurons expressed increased amounts of L-type Ca2+ channels on the cell body. In addition, bFGF-treated neurons acquired distinctive morphology of neurites that was characterized by markedly increased neuritic branching. The branching points in neurites were associated with clusters of L-type Ca2+ channels and resultant “Ca2+ hotspots” that showed large [Ca2+]i increases in response to membrane depolarization. Concurrent application of nicardipine completely blocked the bFGF-stimulated increase in neuritic branching. Therefore, bFGF enhances the expression of functional L-type Ca2+channels on the cell body and neurites of fetal hippocampal neurons, which may play an important role in the regulation of their differentiation and the establishment of their neurite morphology.
Keywords: basic fibroblast growth factor, hippocampal neurons, neuronal development, calcium, voltage-dependent Ca2+channel, expression, channel distribution, neurite branching
Neuronal development proceeds through several steps of sequential processes, including proliferation of neural precursor cells, their migration, and their differentiation into mature neurons with specialized morphological and functional characteristics. These processes are under strong influences of various cellular interactions that are mediated by cell-surface molecules and diffusible trophic factors.
Basic fibroblast growth factor (bFGF) is a single-chain polypeptide composed of 146 amino acids and is well known as a potent mitogen for various cell types. High levels of bFGF and its receptors are found during nervous system development (Gonzalez et al., 1990; Wanaka et al., 1991), and bFGF stimulates proliferation of neuronal progenitor cells (Gensburger et al., 1987; Vescovi et al., 1993; DeHamer et al., 1994). In addition to its mitogenic effect, bFGF acts as a neurotrophic factor based on its ability to support the survival of neurons of various brain regions (Morrison et al., 1986; Walicke et al., 1986;Ferrari et al., 1989; Matsuda et al., 1990) and to promote neuronal differentiation (Ray et al., 1993; Vicario-Abejon et al., 1995), including the establishment of neurite morphology (Hatten et al., 1988;Aoyagi et al., 1994); however, much is unknown concerning the precise mechanisms by which bFGF exerts these multiple and potent effects on neuronal cells.
Changes in the expression patterns of ion channels are likely to be an important feature of neuronal development. Developmental changes in neuronal electrical excitability, which is determined by ion channel expression, are also considered to be regulated by many kinds of environmental factors such as direct interactions with other cell types (Wu and Barish, 1994) and neurotrophic factors (Dourado and Dryer, 1994). Voltage-dependent Ca2+ channels (VDCCs), the major pathways for Ca2+ influx across the plasma membrane, are the first of the voltage-activated ion channels to be detected during development of various neurons (Spitzer, 1979), and they undergo progressive changes in their expression or localization (Yaari et al., 1987; McCobb et al., 1989; Gottmann et al., 1991; Thompson and Wong, 1991; Gruol et al., 1992; Desarmenien et al., 1993).
Ca2+ is an important intracellular messenger known to control many aspects of neuronal functions such as enzyme activity, gene expression, synaptic function, and neurotransmitter release. Ca2+ influx during developmental stages can influence neuronal differentiation processes (Spitzer, 1994) including neurite outgrowth (Cohan et al., 1987; Mattson and Kater, 1987) and cell survival (Collins and Lile, 1989; Ghosh et al., 1994). Therefore, changes in Ca2+ channel expression could profoundly affect elaboration of neuronal phenotypes in the developing nervous system.
Because of the particular importance of Ca2+ in neuronal development, we were interested in the possible roles of bFGF in the regulation of Ca2+ channel expression. Here we provide evidence that bFGF enhances the expression of L-type VDCCs in cultured fetal hippocampal neurons, which is closely associated with its morphogenic effect on the neurites of these neurons.
MATERIALS AND METHODS
Culture medium. Modified Eagle’s medium is composed of 8.73 mg/ml Eagle’s minimal essential medium powder (Nissui Pharmaceuticals, Tokyo, Japan) supplemented with 8.2 mg/mld-glucose, 0.29 mg/ml l-glutamine, 1.7 mg/ml NaHCO3, and 94 μg/ml sodium pyruvate. Where indicated, 10% (v/v) heat-inactivated fetal bovine serum (FBS) (Sanko-Junyaku, Tokyo, Japan) was added. Human transferrin (100 μg/ml) (Sigma, St. Louis, MO), bovine insulin (5 μg/ml, Becton Dickinson, Bedford, MA), progesterone (20 nm, Sigma), and putrescine (100 μm, Sigma) were added to the medium for serum-free culture. We found that in this serum-free condition nicardipine showed cytotoxicity, decreasing the number of surviving neurons when added chronically to the culture medium. Therefore, in the experiments presented in Figure 8 we used B-27 supplement (Life Technologies, Gaithersburg, MD), which was found to eliminate the cytotoxicity of nicardipine. The effects of bFGF on Ca2+ responses and neurite morphogenesis were similarly observed in the presence of either supplement.
Fig. 8.

L-type VDCC activity is involved in bFGF-stimulated neuritic branching. A, Effect of nicardipine (5 μm) on bFGF-stimulated increase in neuritic branching. bFGF and nicardipine were applied at 2 DIV.n = 12 ∼ 20 for each treatment. **p < 0.01 versus control (Duncan’s multiple-range test). B–D, Representative examples of hippocampal neurons at 4 DIV that received no treatment (B), 10 ng/ml bFGF (C), and 10 ng/ml bFGF + 5 μm nicardipine (D) from 2 DIV. Note the marked increase in fine branches along the neurites of the bFGF-treated neuron (C). Nicardipine prevented formation of these branches. Scale bar, 25 μm.
Cell culture. The culture of dissociated hippocampal neurons was prepared according to the methods described previously (Okuda et al., 1994), with some modifications. Plastic 35 mm dishes (Becton Dickinson) and 15-mm-round glass coverslips (Matsunami, Osaka, Japan) were treated with 0.1% polyethyleneimine (Sigma) dissolved in 0.15 m borate buffer, pH 8.4. The coverslips were cleaned and sterilized in 70% ethanol before they were coated. They were then rinsed twice with sterile distilled water. Whole brains were isolated from embryonic day 18 Wistar rats (SLC, Shizuoka, Japan), and the hippocampi were further dissected out. The tissue was cut into pieces with knives and incubated with 0.25% trypsin (1:250, Difco, Detroit, MI) and 0.01% deoxyribonuclease I (Sigma) at 37°C for 15 min. The incubation was terminated by the addition of heat-inactivated horse serum (Cell Culture Lab, Cleveland, OH). The tissue fragments were centrifuged at 1200 rpm for 5 min. The pellets were suspended again in modified Eagle’s medium containing 10% FBS, and single cells were dissociated by gently passing the suspension through a plastic tip. The cell suspension was passed through two sheets of nylon net (25 μm mesh) to remove cell lumps and was diluted with the medium containing 10% FBS. The suspension was plated on 15-mm-round glass coverslips to obtain a final density of 2.5 × 104cells/cm2 for the experiments on the [Ca2+]i measurement and the binding of (−)-(4,4-difluoro-7-styryl-4-bora-3a,4a-diaza)-3-(s-indacene)propionic acid-labeled dihydropyridine (STBodipy-DHP, Molecular Probes, Eugene, OR), and 2.5 × 103 cells/cm2 for the experiments on the neurite morphology. For whole-cell recording experiments, the suspension was plated on 35 mm plastic dishes to obtain a final density of 1 × 104cells/cm2. They were cultured at 37°C in a humidified 5% CO2/95% air atmosphere. To prevent proliferation of glial cells, the FBS-containing medium was changed to the serum-free medium within 12 hr (0.5 d) after plating the cells. bFGF was added to the cultures at 0.5 or 2 d in vitro (DIV). When added at 0.5 DIV, bFGF was supplemented again at 2 DIV. When the effects of actinomycin D (Sigma), cycloheximide, nicardipine or ω-conotoxin GVIA (ω-CTx, Alomone Labs, Jerusalem, Israel) on the action of bFGF were examined, each drug was added to cultures 30 min before the application of bFGF.
Ca2+ imaging. Changes in [Ca2+]i at somatic regions were detected by a standard microfluorometrical technique using fura-2, according to the methods described previously (Tanaka et al., 1993). Briefly, the cultured cells at 4 DIV (unless indicated otherwise) were incubated in a medium containing 10 μm fura-2 acetoxymethyl ester at 37°C for 60 min. The cells then were rinsed with HEPES-buffered balanced salt solution (BSS) of the following composition (in mm): 130 NaCl, 5 KCl, 1.8 CaCl2, 10 d-glucose, and 20 HEPES, pH 7.4 with NaOH. The coverslip with the fura-2-loaded cells was mounted on the stage of an inverted microscope (Nikon, Tokyo, Japan). The cultures then were perfused continuously with BSS at a rate of 3.5 ml/min (chamber volume 0.3 ml) throughout the experiments. An image analysis system FC-200 (Mitsubishi Kasei, Tokyo, Japan) was used to monitor the ratio of the intensity of fura-2 fluorescence excited at 340 nm and 360 nm. For calibration of free Ca2+ concentrations, 10 μm fura-2 was suspended in standard solutions of 50 mm PIPES-KOH buffer, pH 6.8, containing EGTA and CaCl2 at calculated ratios (Harafuji and Ogawa, 1980), and illuminated by 340 nm and 360 nm beams alternately in vitro (Kudo and Ogura, 1986). Intracellular Ca2+ responses were quantified by peak amplitudes. Baseline Ca2+ levels were subtracted from the peak amplitude values in individual cells.
To test voltage-dependent Ca2+ responses, the cells were depolarized with 50 mm KCl solution (KCl was substituted for equimolar NaCl in BSS) for 15 sec, which was applied from a rapid perfusion system consisting of an array of 280 μm inner diameter capillary tubes positioned within 200 μm from the cells under study. The solution was fed by gravity (∼300 μl/min). Other drugs applied acutely were also perfused through the same capillary tubes. For experiments involving VDCC blockers, 50 mm K+solution was applied repeatedly at 10–12 min intervals, and each blocker was applied 3–6 min before the second application of 50 mm K+.
We noted that the amplitude of Ca2+ responses was consistent among neurons from the same sister cultures, but it varied considerably among those from different sister cultures. Therefore, in all sets of experiments, the effects of drug treatments were evaluated by comparison of responses of neurons obtained from the same sister cultures.
For the experiments on [Ca2+]i responses in neurites, the cultured cells were incubated in a medium containing 10 μm fluo-3 acetoxymethyl ester and 0.05% Pluronic F127 at 37°C for 60 min. Other procedures followed those in the experiments with fura-2 described above, except for the use of devices with a confocal laser microscope (see below). All experiments were performed at room temperature (23–27°C).
Whole-cell recording. Whole-cell voltage-clamp recordings were made from the hippocampal neurons with neurites at 4 DIV, unless indicated otherwise. A culture dish was settled on the stage of an inverted microscope (Olympus, Tokyo, Japan) and was perfused at 3.5 ml/min with an extracellular solution of the following composition (in mm): 110 choline chloride, 30 tetraethylammonium chloride (Sigma), 10 HEPES, 10 d-glucose, 1 MgCl2, 10 CaCl2, and 0.05% bovine serum albumin (Sigma), pH adjusted to 7.3 with tetraethylammonium hydroxide (Sigma). The recording patch pipette (3–5 MΩ) was filled with the intracellular solution of the following composition (in mm): 120 CsCl, 20 tetraethylammonium chloride, 10 HEPES, 10 EGTA, 2 MgCl2, and 3 Mg-ATP, pH adjusted to 7.3 with CsOH. An array of capillary tubes was positioned within 300 μm from the cell under study, and the extracellular solution containing 1 μm tetrodotoxin (Sigma) and 5 mm 4-aminopyridine was perfused continuously (∼300 μl/min). Membrane currents were recorded under voltage-clamp conditions using a patch-clamp amplifier CEZ-2300 (Nihon Kohden, Tokyo, Japan). The liquid junction potential between external and internal solutions was measured with a 3 m KCl-agar electrode to calibrate the actual membrane potential. Capacity transients were subtracted on-line by analog compensation in the amplifier. Series resistance compensation was not used routinely. Cell membrane capacitance was calculated after entering the whole-cell configuration, from the integral of the capacity transient elicited by a 10 mV hyperpolarizing pulse from −60 mV. Peak amplitude of Ca2+current was divided by the cell membrane capacitance value to obtain Ca2+ current density. Cells were held at −60 mV, and the membrane potential was stepped every 15 sec through a 1.75 sec prepulse to −100 mV, followed by a 250 msec depolarizing test pulse to a potential between −60 mV and +60 mV. Recorded currents were filtered at 5 kHz, monitored on an oscilloscope and a pen recorder, and stored at 20 kHz on a digital audiotape recorder RD-125T (TEAC, Tokyo, Japan) for later off-line analysis using an analysis program (QP-120J, Nihon Kohden) on a personal computer. All experiments were performed at room temperature (20–25°C).
STBodipy-DHP binding. Binding of STBodipy-DHP to hippocampal neurons was examined according to the methods described previously (Knaus et al., 1992). The cultured cells on coverslips were incubated for 5 min in the dark at room temperature with 10 nmSTBodipy-DHP in BSS. After incubation, the cells were rapidly washed twice for 30 sec with ice-cold BSS containing 1% bovine serum albumin, and they were rinsed additionally with ice-cold BSS. The coverslip then was immediately mounted on the stage of a confocal laser microscope for observation of the fluorescence.
Confocal microscopy. Confocal imaging was carried out with a laser scanning confocal system MRC-600 (BioRad, Hercules, CA) equipped with an inverted microscope (Nikon), an argon ion laser, and a host computer system integrated with an optical disk for image storage. All image generation and processing operations were performed by software provided with the confocal system. For the measurements of STBodipy-DHP and fluo-3 fluorescence, the cells were illuminated with the excitation wavelengths of 514 nm and 488 nm, and the fluorescence images were obtained through 550 nm and 515 nm long-pass filters, respectively. The cells were viewed first with a 20 × 0.5 NA objective using a phase-contrast condenser to search a field to be tested, and then with a 60 × 1.4 NA oil immersion objective to obtain fluorescence images. The images of STBodipy-DHP fluorescence were composed of whole horizontal sections of hippocampal neurons at 1 μm intervals (∼20 images), and the changes in the intensity of fluo-3 fluorescence caused by the 50 mm K+ challenge were taken from look-through projection, with the spatial filter aperture fully opened. To make quantitative measurements, the same neutral density filter and level of photomultiplier output were used for all images in the same set of experiments. In the experiments using STBodipy-DHP, all images were taken within 10 min after incubation. Under these experimental conditions, the fluorescence of each dye was stable, and photobleaching could be reduced to a minimum. To quantify the intensity of STBodipy-DHP fluorescence on the cell body, the pixel intensity values (0–275) obtained by maximum pixel mode were averaged for each cell within the image of the somatic region. The vertical position of the maximal value at each pixel apparently followed that of plasma membrane, indicating that STBodipy-DHP fluorescence was associated primarily with plasma membrane, not internal membranes. Changes in the intensity of fluo-3 fluorescence were also expressed by maximum pixel values between 0 and 275.
Evaluation of neurite branching. Evaluation of neurite branching was performed according to the methods described previously (Aoyagi et al., 1994), with some modifications. Briefly, we selected neurons with neurites longer than their soma diameters at 2 DIV, which were free from contact with other cells, and recorded their locations in the culture dish using an ACAS 470 work station (Meridian, Okemos, MI). These cells were photographed and then drugs were added to the cultures. The same cells were again photographed 48 hr later (at 4 DIV). The number of branch points along the longest neurite was counted by tracing the photographs on a digitizing tablet. If the selected neurons died within 48 hr, data from these cells were discarded. Neurons with many neuritic spine-like protrusions were frequently encountered in bFGF-treated cultures. Neurite–neurite branches are differentiated from neurite–spine branches according to the length of their collaterals (Yu et al., 1994; Papa et al., 1995). We defined the branches with collateral >20 μm as neurite–neurite branches.
Statistics. Data are presented as mean ± SEM. Statistical significance was evaluated with either Student’st test or one-way ANOVA followed by Duncan’s multiple-range test.
Drugs and chemicals. The bFGF used in the present study is CS23 (a generous gift from Takeda Chemical Industries, Osaka, Japan), a modified human bFGF in which serine is substituted for cysteine at amino acid residues 70 and 88 to prevent conformational changes and increase stability. The biological activity of CS23 on brain neurons is virtually the same as that of wild-type human bFGF and bovine bFGF (Abe et al., 1990). Nicardipine was a generous gift from Yamanouchi Pharmaceutical (Tsukuba, Japan). Thapsigargin was purchased from Research Biochemicals (Natick, MA). All other chemicals were purchased from Wako Chemicals (Osaka, Japan), unless specified otherwise.
RESULTS
bFGF enhances developmental increases in high K+-induced Ca2+ response
Hippocampal neurons obtained from 18-d-old rat embryos displayed development of depolarization-induced intracellular Ca2+responses. At 0.5 DIV, application of 50 mm KCl for 15 sec caused a slow and slight increase in [Ca2+]iin 17 of 51 cells tested (Fig. 1A). Although the cells are not morphologically differentiated at this developmental stage, most of them are probably neurons because the presence of glial cells in fresh cultures is minimal (Banker and Cowan, 1977). Hippocampal neurons extended neurites as the culture period proceeded, and both the amplitude of the high K+-induced Ca2+ transients and the percentage of cells responding to high K+ gradually increased, which indicates that they had undergone neuronal differentiation (Fig.1A,B, Table 1). In the bFGF-treated cultures, neuritic complexity was increased (see below), and the developmental increase in the amplitude of Ca2+ transients was enhanced (Fig.1A,B). When 10 ng/ml bFGF was added to the culture medium at 0.5 DIV, the amplitude of Ca2+transients measured at 2 DIV was increased significantly, and this increased Ca2+ response in bFGF-treated cultures was maintained to 4 DIV (Fig. 1B, solid circles). Moreover, bFGF was also effective when added to the medium from 2 DIV (Fig. 1B, solid squares), and the amplitude of the Ca2+ transients caused by high K+ was significantly increased even when measured 24 hr later (at 3 DIV). On the other hand, the developmental increase in the percentage of cells responding to high K+depolarization was not affected by the chronic treatment with bFGF (Table 1). The effects of varying concentrations of bFGF were also examined on the development of intracellular Ca2+responses. As shown in Figure 1C, bFGF increased the amplitude of high K+-induced Ca2+ transients in a concentration-dependent manner, within a range of 1–30 ng/ml.
Fig. 1.

Chronic treatment with bFGF increases depolarization-induced Ca2+ responses in cultured hippocampal neurons. A, Representative recordings of Ca2+ transients evoked by 15 sec application of 50 mm K+ to hippocampal neurons at 0.5 DIV (left) and 4 DIV (right). Enhancement of Ca2+ response was observed in bFGF-treated (10 ng/ml) neurons (top) compared with control neurons (bottom). B, Time-dependent increase in the amplitude of depolarization-induced Ca2+ responses in control neurons (open circles) and bFGF-treated neurons. bFGF increased Ca2+ responses when added to the medium either from 0.5 DIV (left arrow, solid circles) or from 2 DIV (right arrow,solid squares). Plots are averaged data from neurons that showed detectable [Ca2+]i increases. Percentages of responding cells are shown in Table 1.n = 17 ∼ 48 for each time point.Asterisks indicate significant differences from control; *p < 0.05, **p < 0.01 (Student’s t test). C,Concentration-dependent effect of bFGF on Ca2+ responses at 4 DIV. bFGF was applied from 0.5 DIV at different concentrations (1–30 ng/ml; n = 57–66). *p < 0.05, **p < 0.01 versus control (C) by Duncan’s multiple-range test.D, Identification of the pathway for Ca2+influx. Before the second challenge with 50 mmK+, 20 μm Cd2+, 5 μm nicardipine, or 300 nm ω-CTx was applied (6 min for Cd2+, 3 min for others) to neurons at 4 DIV. The peak amplitudes of Ca2+ transients evoked by the second challenge with 50 mm K+ (hatched bars) were compared with those evoked by the first one (open bars) in both control (C) and bFGF-treated (F) neurons. Values are averages of data from 9 ∼ 29 cells. Asterisks indicate significant differences from the control group; **p < 0.01 (Duncan’s multiple range test).
Table 1.
Development of sensitivity to 50 mmK+
| Age in culture (DIV) | Control | 10 ng/ml bFGF |
|---|---|---|
| 0.5 | 17/51 (33.3%) | |
| 2 | 20 /27 (74.1%) | 23 /31 (74.2%) |
| 3 | 34 /39 (87.2%) | 42 /51 (82.4%) |
| 4 | 48 /50 (98.0%) | 36 /36 (100%) |
Values shown are the numbers of cells that showed detectable [Ca2+]i rise in response to 50 mmK+ versus the numbers of cells examined. Percentages of responding cells are given in parentheses. bFGF was added to cultures at 0.5 DIV.
To confirm that the observed Ca2+ responses to high K+ depolarization were mediated by Ca2+ influx through VDCCs, we tested the effect of Cd2+, a broad-spectrum VDCC blocker. Ca2+ transients were elicited repeatedly in the same neurons by the application of high K+ solution at 12 min intervals, and 20 μmCd2+ was applied 6 min before the second challenge with high K+. In both control and bFGF-treated neurons, Cd2+ markedly reduced the amplitudes of Ca2+transients caused by the second challenge with high K+, compared with those caused by the first challenge (Fig.1D). Ca2+ response to later high K+ challenge after washout of Cd2+ showed a tendency to recover (data not shown). To clarify further the subclasses of VDCCs responsible for the high K+-induced Ca2+ transients, we examined the effects of the selective VDCC blockers nicardipine (an L-type VDCC blocker) and ω-CTx (an N-type VDCC blocker). In both control and bFGF-treated neurons, 10 μm nicardipine almost completely abolished the Ca2+ transients caused by high K+; the amplitude of [Ca2+]i rise in the presence of nicardipine fell to ∼5% of that elicited by the first high K+ challenge (Fig. 1D). On the other hand, ω-CTx had no significant effect on the high K+-induced Ca2+ response, although a small decrease (∼13%) in the amplitude was noted in bFGF-treated neurons. These results show that depolarization-induced Ca2+responses in our hippocampal cultures are initiated by the opening of L-type VDCCs.
Ca2+-induced Ca2+ release does not contribute to the bFGF-enhanced Ca2+ responses
Ca2+-induced Ca2+ release from intracellular Ca2+ stores is thought to be a mechanism that amplifies Ca2+ signals in various cell types and can be triggered by Ca2+ influx via VDCCs (Lipscombe et al., 1988). Therefore, the enhanced Ca2+ responses in bFGF-treated neurons might be attributable to an increased amplification of Ca2+ signals by Ca2+-induced Ca2+ release. To test this possibility, we performed two pharmacological approaches.
First, caffeine was administered to empty the intracellular Ca2+ stores (Tsien and Tsien, 1990; Miller, 1991) before the challenge with high K+ solution. Application of 50 mm caffeine for 15 sec caused small elevations of [Ca2+]i in both control (18 of 49 neurons tested) and bFGF-treated (7 of 29 neurons tested) cultures. No significant differences were observed between control and bFGF-treated neurons in the amplitude of Ca2+ transients elicited by caffeine (Fig. 2A, open columns) and in the percentage of responding cells. Subsequent challenge with high K+ solution caused much larger Ca2+ transients in both control and bFGF-treated neurons compared with that induced by caffeine. As shown in Figure2A (hatched columns), the amplitude of the Ca2+ transients caused by high K+ was significantly larger in bFGF-treated neurons compared with that in control neurons, even after the releasable Ca2+ was depleted from intracellular stores by caffeine.
Fig. 2.
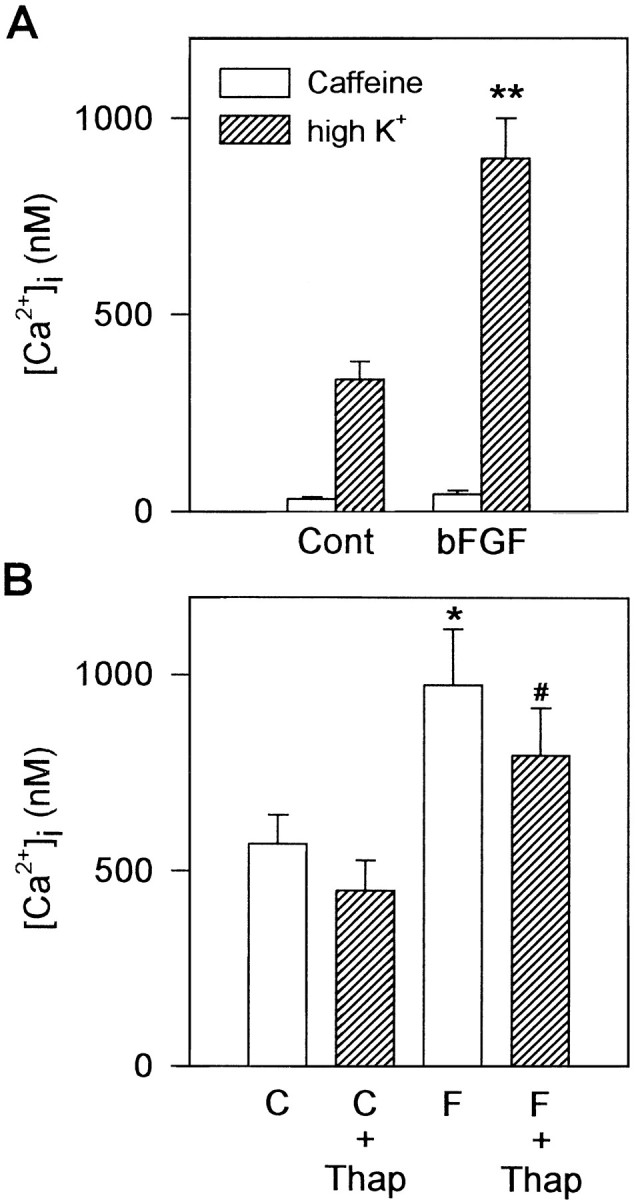
Intracellular Ca2+ stores are not major contributors to depolarization-induced Ca2+responses. A, Caffeine (50 mm) was administered for 15 sec, before the challenge with 50 mmK+. Caffeine-induced Ca2+ transients (open bars) were small in both control and bFGF-treated neurons. Even after the application of caffeine, Ca2+responses to high K+ depolarization (hatched bars) were significantly larger in bFGF-treated neurons compared with those in control neurons. B, Thapsigargin (Thap; 1 μm) was administered for 3 min, before the high K+ challenge. Although the amplitude of Ca2+ transients was reduced slightly by thapsigargin pretreatment (hatched bars) in both control (C) and bFGF-treated (F) neurons compared with groups not treated with thapsigargin (open bars), bFGF-treated neurons still showed larger Ca2+ responses compared with controls. Significance of difference was determined using Duncan’s multiple-range test; *p < 0.05, **p < 0.01 versus control group; #p < 0.05 versus thapsigargin-treated control group.
Second, we used another compound, thapsigargin, which depletes intracellular Ca2+ stores by inhibition of the endoplasmic reticulum Ca2+-ATPase (Thastrup et al., 1990). Most of the hippocampal neurons did not respond to a 3 min perfusion of 1 μm thapsigargin; a small sustained elevation of basal [Ca2+]i was observed in a minor population (45 ± 21 nm in control, 10 of 36 neurons tested; 27 ± 4 nm in bFGF-treated, 4 of 39 neurons tested). After the treatment with thapsigargin, the amplitude of Ca2+ transients elicited by high K+ was reduced slightly in both control and bFGF-treated neurons. The degree of reduction, however, was almost the same in both groups (by ∼20%). The amplitude of depolarization-induced Ca2+ transients was still significantly larger in bFGF-treated neurons compared with control (Fig. 2B).
Because the concentrations of caffeine and thapsigargin that were used were reported to be sufficient for depleting internal Ca2+stores in a number of studies (Verkhratsky and Shmigol, 1996), we suggest that the development of the intracellular Ca2+stores was poor in hippocampal neurons under the present experimental conditions, and the contribution of the intracellular Ca2+stores to depolarization-induced Ca2+ transients was relatively small. Thus, we conclude that the enhanced Ca2+responses in bFGF-treated neurons are attributable not to an increased amplification by Ca2+-induced Ca2+ release but to an increased Ca2+ influx through VDCCs.
Involvement of protein synthesis in bFGF action
bFGF is known to activate its specific receptor tyrosine kinases and also to activate other protein kinases as a cascade of intracellular events (Jaye et al., 1992; Creuzet et al., 1995). Therefore, the observed effect of bFGF may be mediated by phosphorylation of Ca2+ channel proteins by these protein kinases, because VDCC functions can be modulated by phosphorylation (Dolphin, 1996). If so, bFGF also should be effective when applied acutely, because phosphorylation-based modulation of ion channel activities is expected to manifest quickly. Acute application of bFGF (20 ng/ml), however, did not enhance but rather slightly depressed the amplitude of Ca2+ transients caused by high K+depolarization (Fig. 3A,B). We also tested the effect of a 1 hr incubation of hippocampal neurons with 10 ng/ml bFGF just before the [Ca2+]imeasurement. bFGF again failed to enhance high K+-induced Ca2+ response (data not shown). These results suggest, although indirectly, that phosphorylation of Ca2+ channel proteins is not likely to be the mechanism of bFGF action.
Fig. 3.

Involvement of protein synthesis in bFGF action.A, B, Acute application of bFGF did not enhance the high K+-induced Ca2+ transients. Challenges with 50 mm K+ were performed four times at 8 min intervals. bFGF (20 ng/ml) was applied for 3 min before and during the third challenge. The effect was estimated as the relative amplitude of the Ca2+ transients, setting those induced by the second challenge as 100%. A shows the representative recording and B shows the mean values of Ca2+transients at second, third, and fourth challenges with 50 mm K+ (from left toright, n = 17). C, D, Effects of cycloheximide (CHX) and actinomycin D (Act D) on bFGF action. Hippocampal neurons were incubated with 0.1 μm cycloheximide or 1 nmactinomycin D during bFGF treatment from 2 to 4 DIV. The amplitude of Ca2+ transients evoked by 50 mm K+was normalized to control (Cont). Both cycloheximide and actinomycin D virtually abolished the effect of bFGF (n = 93–104). Asterisksindicate significant differences from the control group; **p < 0.01 (Duncan’s multiple-range test).
Another possible mechanism is that bFGF may increase the amount of Ca2+ channel proteins themselves on the plasma membrane. In other words, the effect of bFGF may be mediated by enhancement of new protein synthesis. To test this possibility, hippocampal neurons were incubated with the protein synthesis inhibitor cycloheximide (0.1 μm) or the RNA synthesis inhibitor actinomycin D (1 nm) during the bFGF treatment from 2 to 4 DIV. These inhibitors did not produce any cell damage at the concentrations used. As shown in Figure 3C,D, both cycloheximide and actinomycin D virtually abolished bFGF-stimulated increase in Ca2+ response. These results suggest that neosynthesis of proteins and RNAs is required for this effect of bFGF.
Whole-cell Ca2+ current is enhanced by bFGF treatment
To assess more directly whether the amount of VDCCs is increased in bFGF-treated cultures, we measured voltage-activated Ca2+ currents (ICa) using whole-cell patch-clamp recording techniques. Membrane potentials of hippocampal neurons grown in the absence and presence of bFGF (10 ng/ml) from 0.5 to 4 DIV were held at −60 mV, and whole-cellICa was evoked by step depolarization over the range of −60 to +60 mV from the membrane potential prepulsed to −100 mV. In both control and bFGF-treated neurons, low-voltage-activatedICa, which was activated at relatively low membrane potential (−50 to −40 mV), and high-voltage-activatedICa, which was activated at relatively high membrane potential (−30 to −20 mV), were observed, in good agreement with previous reports (Yaari et al., 1987; Ozawa et al., 1989). bFGF (10 ng/ml) treatment significantly increased high-voltage-activatedICa (−10 to +20 mV). Representative current traces evoked by depolarization to 0 mV were shown in Figure4A. The peakICa measured at 0 mV test potential was 693 ± 55 pA (n = 5) in control and 1109 ± 119 pA (n = 6) in bFGF-treated neurons (Fig.4B). On the other hand, low-voltage-activatedICa activated at −50 to −40 mV remained unchanged (Fig. 4B). We also evaluated the amplitude of low-voltage-activated transient ICa by subtracting the sustained current amplitude remaining at the end of the voltage step from the peak current amplitude, and again observed no significant difference between control and bFGF-treated neurons (data not shown). These results indicate that bFGF treatment specifically increased high-voltage-activated ICa.
Fig. 4.

Development of ICaduring bFGF treatment. A, Representative current traces of ICa from control neurons (middle) and from bFGF-treated neurons (bottom) at 4 DIV. ICa was evoked by voltage pulses to 0 mV from a holding potential of −100 mV (as indicated at top). B,Current–voltage relationship of ICa in control (n = 5, open circles), bFGF-treated (10 ng/ml) (n = 6, solid squares), and bFGF + cycloheximide-treated (0.1 μm) (n = 3, solid triangles) neurons at 4 DIV. Currents were evoked by step depolarization over the range of −60 to +60 mV from a holding potential of −100 mV. The peak amplitude ofICa was measured and plotted against test potential. Note that bFGF treatment significantly increased high-voltage-activated ICa, whereas low-voltage-activated ICa remained unchanged. Asterisks indicate significant differences from the control group; *p < 0.05 (Student’st test). C, Development ofICa density in the absence (open bars) and presence (hatched bars) of bFGF at indicated concentrations (in ng/ml). Cell membrane capacitance was measured, and the ICa density at 0 mV test potential was calculated. The ICa density increased with the length of culture period (0.5 to 4 DIV). bFGF significantly enhanced the developmental increase of theICa density. CHX (0.1 μm) blocked the enhancement of theICa density by bFGF treatment. Thenumber of neurons examined is given inparentheses. Asterisk indicates significant difference from the control group (C); *p < 0.05 (Duncan’s multiple-range test).
The enhancement of ICa under bFGF treatment could be the consequences of an increase in specific ion conductance or an enlargement of membrane surface. Thus, we also measured cell membrane capacitance as an indicator of the size of the neurons and calculated the ICa density at 0 mV test potential. The ICa density increased with the length of the culture period, and bFGF significantly enhanced the developmental increase in the ICa density (Fig.4C). On the other hand, no significant difference in the cell membrane capacitance was found between control and bFGF-treated neurons (at 4 DIV, 35.8 ± 2.2 pF in control, n = 5 vs 37.7 ± 3.8 pF in bFGF-treated, n = 6). bFGF affected the ICa density in a concentration-dependent manner (Fig. 4C). Moreover,ICa measurement was performed on the neurons that received 0.1 μm cycloheximide during bFGF treatment. Cycloheximide prevented enhancement of the ICadensity increase by bFGF (Fig. 4B,C), which was consistent with the results obtained from [Ca2+]i measurement experiments (Fig.3C). These results suggest that the bFGF-stimulated increase in ICa is attributable to an increase in the density of functional VDCCs on the plasma membrane.
Increased expression of L-type Ca2+ channels on the soma under bFGF treatment
Next we performed another set of experiments to verify increased expression of VDCCs in bFGF-treated neurons. Here the expression of L-type VDCCs was quantified by the binding of STBodipy-DHP to living hippocampal neurons. STBodipy-DHP is a useful agent for visualization of L-type VDCCs (Knaus et al., 1992). Subcellular regions expressing the DHP-receptor domain of L-type VDCCs were visualized clearly by confocal microscopy (Fig.5A,B). In control neurons, STBodipy-DHP fluorescence was distributed unevenly on the cell body. Each neuron had a “hot region” where L-type VDCCs were densely expressed (Fig. 5A). The intense labeling did not seem to be attributable to the thickness of the cells and cytoplasmic fluorescence, because maximum pixel mode was used to construct the images and no obvious difference was observed in cell thickness between the hot region and the other areas of soma. To check the specificity of STBodipy-DHP binding to L-type VDCCs, displacement experiments were carried out using the VDCC blockers nicardipine and ω-CTx. In the neurons pretreated with 10 μm nicardipine, the hot region was absent (data not shown). The averaged fluorescence intensity on the cell bodies decreased to 54% of control for the neurons pretreated with nicardipine (Fig. 5C). In contrast, 300 nmω-CTx had little effect on the binding of STBodipy-DHP (Fig.5C). Thus, STBodipy-DHP recognizes L-type VDCCs in hippocampal neurons.
Fig. 5.

Visualization of L-type VDCCs on rat hippocampal neurons by STBodipy-DHP. A, B, Pseudo-color confocal images of STBodipy-DHP fluorescence in living hippocampal neurons at 3 DIV, cultured in the absence (A) and presence (B) of 10 ng/ml bFGF for 24 hr. The number of DHP-binding sites shown as fluorescence intensity increased in bFGF-treated neurons. Scale bar, 25 μm. C, Competition of the binding of STBodipy-DHP (10 nm) with nicardipine (Nic) (10 μm) and ω-CTx(300 nm). Nicardipine markedly reduced fluorescence intensity, whereas ω-CTx had no effect. Asteriskindicates significant difference from the control group (c); **p < 0.01 (Student’s t test).D, Correlation of the magnitude of Ca2+response with the amount of L-type VDCCs. Ca2+ responses to 50 mm K+ depolarization were measured by fura-2 microfluorometry. After that, the same cultures were used for STBodipy-DHP binding experiments. The amplitudes of [Ca2+]i responses are plotted against the intensities of STBodipy-DHP fluorescence for individual neurons.Open circles, control; solid circles, bFGF-treated (10 ng/ml). Significant correlation between the two parameters was obtained (r = 0.31, p < 0.05).
After 24 hr treatment with 10 ng/ml bFGF from 2 DIV, the capacity of STBodipy-DHP binding on the cell bodies of hippocampal neurons increased significantly, compared with that of nontreated neurons (Fig.5B). The averaged fluorescence intensity on the cell bodies increased to 146% of control, whereas the characteristic pattern of the distribution of STBodipy-DHP fluorescence was retained. The hot region increased both in area and in fluorescence intensity. Furthermore, with careful observation we noted that the expression of L-type VDCCs was increased along the neurites (described below). We also examined high K+-induced Ca2+ responses in the same neurons before STBodipy-DHP binding experiments. The intensity of STBodipy-DHP fluorescence and the amplitude of [Ca2+]i responses showed a significant correlation (r = 0.31, p < 0.05; Fig.5D). Thus, the enhanced Ca2+ response by bFGF treatment was associated with the increased expression of L-type VDCCs on the cell body.
Distribution of L-type Ca2+ channels along neurites
As mentioned above, neuritic complexity was increased and the expression of L-type VDCCs seemed to increase along the neurites of bFGF-treated neurons. Therefore, we further examined the distribution of L-type VDCCs on the neurites based on the observations of STBodipy-DHP fluorescence. For this purpose, we used hippocampal neurons plated at a low density. When plated at 2500 cells/cm2, individual neurons extended neurites without any contact with other neighboring neurons. Application of 10 ng/ml bFGF to low-density culture markedly increased the number of branches of the longest neurite of hippocampal neurons (Fig.6A–C). When visualized by STBodipy-DHP fluorescence, mild expression of L-type VDCCs was observed along the neurites of bFGF-treated neurons, whereas in control neurons the expression was weak, present only on the proximal part of neurites (Fig. 6D–F). In addition, we could observe several fluorescent “hotspots,” which were assumed to represent clusters of L-type VDCCs, on the distal part of neurites of bFGF-treated neurons. These hotspots were expressed in a punctate appearance, overlapping with either the branch points or varicosities (enlargements on the neurites) along the neurites (Fig.6H,I,K,L). No such fluorescent hotspots were observed on the neurites of control neurons (Fig. 6G,J).
Fig. 6.
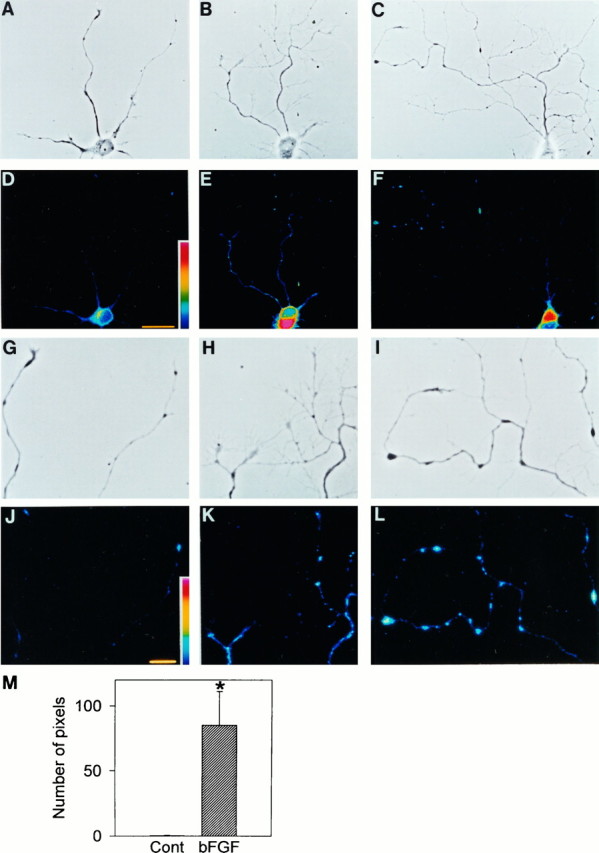
Subcellular distribution of L-type VDCCs in hippocampal neurons visualized by STBodipy-DHP. Confocal images were taken from 4 DIV hippocampal neurons cultured in the absence (D) and presence (E,F) of 10 ng/ml bFGF from 2 DIV.A–C show phase-contrast photographs of the same neurons in D–F, respectively.G–I show the neurites of neurons inA–C, respectively, at higher magnification, andJ–L represent corresponding fluorescence images. Note that several fluorescent “hotspots” are observed along the neurites in bFGF-treated neurons, which overlap with the branch points or varicosities. No such fluorescent hotspots are observed on the neurites of control neurons. Scale bars in D and inJ represent 25 μm and 10 μm, respectively.M, Quantification of L-type VDCCs on the neurites of hippocampal neurons (see Results for details). The amount of L-type VDCCs on the neurites is increased in bFGF-treated neurons (n = 5, hatched bar), compared with control neurons (n = 5, open bar). *p < 0.01 versus control (Student’st test).
To evaluate the expression of L-type VDCCs on the neurites quantitatively, we selected neurons that had complexly branching neurites in bFGF-treated cultures and size-matched neurons in control cultures. We measured the averaged intensity of STBodipy-DHP fluorescence on the cell body of control neurons and made it a criterion for evaluating the quantity of L-type VDCCs on the longest neurite. The number of pixels that had a fluorescence intensity above the criteria was remarkably increased along the longest neurite of bFGF-treated neurons (Fig. 6M). This indicates that bFGF increased the expression of L-type VDCCs on the neurites as well as on the cell body.
Furthermore, to test whether the clusters of L-type VDCCs could result in altered [Ca2+]i regulation at the branch points of neurites, we examined the intracellular Ca2+responses to high K+ depolarization at these branch points. The Ca2+-sensitive dye fluo-3 was used, and Ca2+ responses to high K+-induced depolarization were measured using a confocal laser microscope. As shown in Figure 7
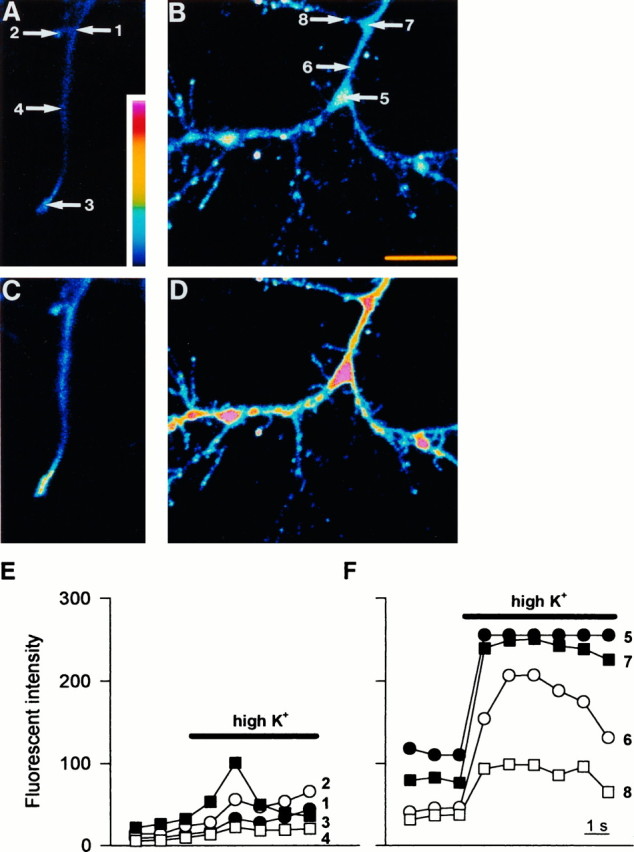
, neurites of bFGF-treated neurons showed large Ca2+ responses compared with neurites of control neurons. Moreover, within the neurites of bFGF-treated neurons, Ca2+ responses to high K+ depolarization were much larger at the branch points compared with adjacent regions not forming branches (Fig.7B,D,F). Therefore, it is suggested that L-type VDCC hotspots function as “Ca2+hotspots” at the branch points of neurites of bFGF-treated neurons.
Fig. 7.
Ca2+ increases at neurites in response to high K+ depolarization. A, B, Pseudo-color images of fluo-3 fluorescence in the neurite of a control neuron (A) and a bFGF-treated neuron (B) under resting conditions; C, D, corresponding images during exposure to 50 mmK+. The neurite of the control neuron showed a modest increase in Ca2+, whereas that of the bFGF-treated neuron showed a robust increase. Note that Ca2+ responses are much larger at the branch points of neurite (5 and7) than at adjacent regions of neurite (6 and 8). Scale bar, 10 μm.E, F, Time course of the changes in the intensity of fluo-3 fluorescence after exposure to 50 mmK+. Numbers correspond to the regions shown in A and B. At the branch points of bFGF-treated neurite (5 and 7), Ca2+ responses are larger and faster compared with adjacent regions (6 and 8).
Possible involvement of L-type Ca2+ channel expression in bFGF-induced neurite branching
Because several studies suggest a close relationship between Ca2+ influx and the establishment of neurite morphology (Neely and Nicolls, 1995), we inferred that bFGF-promoted neurite branching was relevant to the increased expression of L-type VDCCs along the neurites. This possibility was examined by testing the effects of VDCC blockers on neurite morphogenesis. As shown in Figure8A, nicardipine (5 μm) alone did not affect the branch formation of the longest neurite of hippocampal neurons, but when added together with bFGF, completely blocked the branching-promoting effect of bFGF (10 ng/ml). Moreover, the density of spine-like protrusions along the neurites was increased in bFGF-treated neurons. Nicardipine also eliminated these bFGF-induced sprouting of spines along the neurites (Fig. 8B–D).
DISCUSSION
We have demonstrated in the present study that fetal hippocampal neurons chronically exposed to bFGF acquire increased responsiveness mediated by the opening of L-type VDCCs. This was observed as an increased [Ca2+]i response to high K+-induced membrane depolarization. The selective L-type VDCC blocker nicardipine virtually abolished the [Ca2+]i response (Fig. 1D). Moreover, the results of thapsigargin and caffeine experiments (Fig. 2) suggest that the contribution of Ca2+-induced Ca2+ release to this response was minimal. Therefore, the depolarization-induced Ca2+ response in our hippocampal primary cultures well represents the activities of L-type VDCCs, which allowed us to estimate the amount of functional Ca2+channels using [Ca2+]i measurement experiments.
The most well established separation among various VDCC subclasses is that between the low-voltage-activated and the high-voltage-activated channels (Tsien et al., 1988; Hess, 1990; Swandulla et al., 1991), L-type VDCC being one of the major high-voltage-activated channels present in hippocampal neurons (Ozawa et al., 1989). Indeed, whole-cell voltage-clamp recordings demonstrated that high-voltage-activated Ca2+ currents were significantly larger in bFGF-treated neurons than in nontreated neurons (Fig. 4). A specific enhancement in high-voltage-activated Ca2+ conductance was demonstrated because cell membrane capacitance, which reflected cell size and membrane surface area, was not affected by bFGF treatment. In contrast, low-voltage-activated Ca2+ currents were not altered by bFGF treatment, which suggests distinct regulation of these two major Ca2+ channel types.
The increased Ca2+ channel responses could be a consequence of covalent modification of Ca2+ channel proteins themselves by protein phosphorylation. Voltage-dependent Ca2+ currents can be facilitated by phosphorylation (Dolphin, 1996). Furthermore, bFGF mediates its effects through specific high-affinity receptors that contain a tyrosine kinase activity and initiates a cascade of intracellular events that leads to the activation of other protein kinases (Jaye et al., 1992; Creuzet et al., 1995). Indeed, Ca2+ currents in several cell types, including retinal glial cells (Puro and Mano, 1991) and subpopulations of ventromedial hypothalamic neurons (Koike et al., 1993), are enhanced by acute treatment with bFGF, which may be mediated by channel protein phosphorylation; however, this does not seem to be the case with hippocampal primary cultures in the present study. Rather, it is concluded for the following reasons that the increased Ca2+responses are attributable to the increased expression of functional L-type Ca2+ channel proteins on the plasma membrane. First, the effects of bFGF required chronic exposure of hippocampal neurons: acute treatment with bFGF had no effect on high K+-induced Ca2+ response. Second, cycloheximide and actinomycin D completely blocked the bFGF-stimulated increase in high K+-induced Ca2+ response and high-voltage-activated Ca2+ currents, indicating that new protein synthesis is required for these effects. Third, increased Ca2+ responses were accompanied by the increased capacity of STBodipy-DHP binding on bFGF-treated neurons.
VDCCs are composed of distinct classes of interacting subunits: α1, α2/δ, β, and γ (Dunlap et al., 1995). Of these, the α1 subunit plays a major role in the sense that it forms a channel pore, can function as a Ca2+channel by itself, and represents basic characteristics of Ca2+ channel subclasses. α1C and α1D subunits represent brain L-type Ca2+channels, both of which possess a DHP-binding domain. STBodipy-DHP is a fluorescent DHP analog that recognizes this DHP-binding domain (Knaus et al., 1992). Therefore, the increased capacity of STBodipy-DHP binding reflects an increase in the amount of α1C and/or α1D subunit proteins on the cell membrane. This increase can be accomplished by several mechanisms. That is, the increase in the amount of channel proteins may be exerted directly by increased expression of genes encoding α1C and/or α1Dsubunit proteins. Alternatively, changes in the expression of the other subunit proteins may affect the amount of functional VDCCs expressed on the plasma membrane. For example, a recent report showed that β2a subunit co-transfected with α1C subunit in human embryonic kidney cells markedly increased the membrane localization of α1C subunit proteins (Chien et al., 1995). Degradation rate can also determine the amount of channel proteins; the increased expression of nicotinic receptors in muscle fibers is caused not only by increased protein synthesis (Devreotes and Fambrough, 1975) but also by a decrease in the rate of receptor protein degradation (Shyng et al., 1991). These alternatives should be addressed in future studies by measuring the amount of Ca2+channel subunit mRNAs. It should be noted that the activation of FGF receptors can lead to genomic alterations through the induction of immediate early genes (Altin et al., 1991; Ferhat et al., 1993). Some of the proteins encoded by these genes act as transcription factors regulating the expression of other genes involved in cellular growth and differentiation (Sheng and Greenberg, 1990). There is also evidence that immediate early genes c-fos and c-jun can upregulate membrane Ca2+ conductance in a rat pheochromocytoma cell line (Cavalie et al., 1994).
The initial fura-2 Ca2+ imaging experiments were concerned with somatic Ca2+ responses. The results of whole-cell recording experiments also provided information mainly about somatic Ca2+ channels, and may not reflect the properties of dendritic Ca2+ channels caused by space-clamp problems. On the other hand, confocal image analysis of STBodipy-DHP binding revealed specialized distribution of L-type VDCCs on the cell bodies as well as on neurites of hippocampal neurons. Each neuron had a hot region on the cell body. Such distribution is similar to that observed in hippocampal neurons in vivo. Clustering of L-type VDCCs is usually observed in the somatic region from which the major dendrite originates (Westenbroek et al., 1990). bFGF enhanced the expression of L-type VDCCs without disturbing this distribution pattern within the cell body (Fig. 5B). Furthermore, bFGF treatment induced characteristic distribution of L-type VDCCs at the branching points of neurites (Fig. 6). These branch points showed large [Ca2+]i increases in response to high K+-induced membrane depolarization, as revealed by fluo-3 confocal imaging (Fig. 7). These findings are particularly intriguing because one of the most prominent effects of bFGF on fetal hippocampal neurons is an increase in the number of axonal branches (Miyagawa et al., 1993; Aoyagi et al., 1994). Consistent with previous studies, we observed a marked increase in the number of branching points along the neurites of bFGF-treated neurons (Figs. 6, 8).
The mechanisms by which neuritic branching is formed are not yet clear, but several lines of evidence suggest that Ca2+ plays an important role (Yu et al., 1994; Neely and Nicolls, 1995). Manivannan and Terakawa (1994) reported that depolarizing current injection into varicosities induced rapid sprouting of filopodia in a Ca2+-dependent manner. We propose here that bFGF promotes the formation of clusters of L-type VDCCs along the neurites, and these clusters serve as Ca2+ hotspots. Evidence has suggested that the cluster of Ca2+ channels can mediate [Ca2+]i rise enough to activate many kinds of Ca2+-dependent biochemical processes in that region (Silver et al., 1990). Therefore, Ca2+ increase at the hotspots could cause cytoskeletal rearrangement (Yu et al., 1994; Neely and Nicolls, 1995), which leads to the formation of neuritic branching (Aoyagi et al., 1995). Indeed, we have found in the present study that nicardipine completely blocks the increase in neuritic branching induced by bFGF (Fig. 8), indicating that a bFGF-induced increase in L-type VDCC activities and an increase in neuritic branching are closely associated events.
Regulation of neuronal Ca2+ channels is important because entry of Ca2+ through VDCCs is a major mechanism by which changes in membrane potential can influence cellular processes. Enhanced Ca2+ influx resulting from an increased density of Ca2+ channels could modulate Ca2+-dependent factors leading to changes in neuronal excitability. Experimental evidence supports this view that Ca2+ is an important intracellular regulator of neuronal excitability in the hippocampus (Kennedy, 1989). In addition to being a determinant of the electrophysiological characteristics of many cells, voltage-activated Ca2+ influx can trigger cellular events such as the activation of enzymes, which in some cases leads to genomic responses (Ghosh and Greenberg, 1995). Indeed, vigorous elevation of Ca2+ can influence differentiation and growth of neuronal cells (Spitzer, 1994).
bFGF has been implicated in differentiation and survival of neuronal cells of various brain regions (Ray et al., 1993; Baird, 1994;Vicario-Abejon et al., 1995). From the point of view that Ca2+ is a critical signal in neuronal development, we propose that the enhanced expression of L-type VDCCs should play a particularly important role in cell growth and differentiation regulated by bFGF.
Footnotes
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science and Culture of Japan.
Correspondence should be addressed to Hiroshi Katsuki, Department of Chemical Pharmacology, Faculty of Pharmaceutical Sciences, The University of Tokyo, Bunkyo-ku, Tokyo 113, Japan.
REFERENCES
- 1.Abe K, Takayanagi M, Saito H. Effects of recombinant human basic fibroblast growth factor and its modified protein CS23 on survival of primary cultured neurons from various regions of fetal rat brain. Jpn J Pharmacol. 1990;53:221–227. doi: 10.1254/jjp.53.221. [DOI] [PubMed] [Google Scholar]
- 2.Altin JG, Kujubu DA, Raffioni S, Eveleth DD, Herschman HR, Bradshaw RA. Differential induction of primary-response (TIS) genes in PC12 pheochromocytoma cells and the unresponsive variant PC12nnr5. J Biol Chem. 1991;266:5401–5406. [PubMed] [Google Scholar]
- 3.Aoyagi A, Nishikawa K, Saito H, Abe K. Characterization of basic fibroblast growth factor-mediated acceleration of axonal branching in cultured rat hippocampal neurons. Brain Res. 1994;661:117–126. doi: 10.1016/0006-8993(94)91188-6. [DOI] [PubMed] [Google Scholar]
- 4.Aoyagi A, Saito H, Abe K. Differential effects of microtubule inhibitors on axonal branching and elongation of cultured rat hippocampal neurons. Jpn J Pharmacol. 1995;68:223–226. doi: 10.1254/jjp.68.223. [DOI] [PubMed] [Google Scholar]
- 5.Baird A. Fibroblast growth factors: activities and significance of non-neurotrophin neurotrophic growth factors. Curr Opin Neurobiol. 1994;4:78–86. doi: 10.1016/0959-4388(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 6.Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–425. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- 7.Cavalie A, Berninger B, Haas CA, Garcia DE, Lindholm D, Lux HD. Constitutive upregulation of calcium channel currents in rat phaeochromocytoma cells: role of c-fos and c-jun. J Physiol (Lond) 1994;479:11–27. doi: 10.1113/jphysiol.1994.sp020274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chien AJ, Zhao X, Shirokov RE, Puri TS, Chang CF, Sun D, Rios E, Hosey MM. Roles of a membrane-localized β subunit in the formation and targeting of functional L-type Ca2+ channels. J Biol Chem. 1995;270:30036–30044. doi: 10.1074/jbc.270.50.30036. [DOI] [PubMed] [Google Scholar]
- 9.Cohan CS, Conner JA, Kater SB. Electrically and chemically mediated increases in intracellular calcium in neuronal growth cones. J Neurosci. 1987;7:3588–3599. doi: 10.1523/JNEUROSCI.07-11-03588.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins F, Lile JD. The role of dihydropyridine-sensitive voltage-gated calcium channels in potassium-mediated neuronal survival. Brain Res. 1989;502:99–108. doi: 10.1016/0006-8993(89)90465-4. [DOI] [PubMed] [Google Scholar]
- 11.Creuzet C, Loeb J, Barbin G. Fibroblast growth factors stimulate protein tyrosine phosphorylation and mitogen-activated protein kinase activity in primary cultures of hippocampal neurons. J Neurochem. 1995;64:1541–1547. doi: 10.1046/j.1471-4159.1995.64041541.x. [DOI] [PubMed] [Google Scholar]
- 12.DeHamer MK, Buevara JL, Hannon K, Olwin BB, Calof AL. Genesis of olfactory receptor neurons in vitro: regulation of progenitor cell divisions by fibroblast growth factors. Neuron. 1994;13:1083–1097. doi: 10.1016/0896-6273(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 13.Desarmenien MG, Clendening B, Spitzer NC. In vivo development of voltage-dependent ionic currents in embryonic Xenopus spinal neurons. J Neurosci. 1993;13:2575–2581. doi: 10.1523/JNEUROSCI.13-06-02575.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Devreotes PN, Fambrough DM. Acetylcholine receptor turnover in membranes of developing muscle fibers. J Cell Biol. 1975;65:335–358. doi: 10.1083/jcb.65.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dolphin AC. Facilitation of Ca2+ current in excitable cells. Trends Neurosci. 1996;19:35–43. doi: 10.1016/0166-2236(96)81865-0. [DOI] [PubMed] [Google Scholar]
- 16.Dourado MM, Dryer SE. Regulation of A-currents by cell–cell interactions and neurotrophic factors in developing chick parasympathetic neurones. J Physiol (Lond) 1994;474:367–377. doi: 10.1113/jphysiol.1994.sp020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 18.Ferhat L, Khrestchatisky M, Roisin M-P, Barbin G. Basic fibroblast growth factor-induced increase in zif/268 and c-fos mRNA levels is Ca2+ dependent in primary cultures of hippocampal neurons. J Neurochem. 1993;61:1105–1112. doi: 10.1111/j.1471-4159.1993.tb03626.x. [DOI] [PubMed] [Google Scholar]
- 19.Ferrari G, Minozzi M-C, Toffano G, Leon A, Skaper SD. Basic fibroblast growth factor promotes the survival and development of mesencephalic neurons in culture. Dev Biol. 1989;133:140–147. doi: 10.1016/0012-1606(89)90305-9. [DOI] [PubMed] [Google Scholar]
- 20.Gensburger C, Labourdette G, Sensenbrenner M. Brain basic fibroblast growth factor stimulates the proliferation of rat neuronal precursor cells in vitro. FEBS Lett. 1987;217:1–5. doi: 10.1016/0014-5793(87)81230-9. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez AM, Buscaglia M, Ong M, Baird A. Distribution of basic fibroblast growth factor in the 18-day rat fetus: localization in the basement membranes of diverse tissues. J Cell Biol. 1990;110:753–765. doi: 10.1083/jcb.110.3.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gottmann K, Rohrer H, Lux HD. Distribution of Ca2+ and Na+ conductances during neuronal differentiation of chick DRG precursor cells. J Neurosci. 1991;11:3371–3378. doi: 10.1523/JNEUROSCI.11-11-03371.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gruol DL, Deal CR, Yool AJ. Developmental changes in calcium conductances contribute to the physiological maturation of cerebellar Purkinje neurons in culture. J Neurosci. 1992;12:2838–2848. doi: 10.1523/JNEUROSCI.12-07-02838.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harafuji H, Ogawa Y. Re-examination of the apparent binding constant of ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid with calcium around neutral pH. J Biochem. 1980;87:1305–1312. doi: 10.1093/oxfordjournals.jbchem.a132868. [DOI] [PubMed] [Google Scholar]
- 27.Hatten ME, Lynch M, Rydel RE, Sanchez J, Joseph-Silverstein J, Moscatelli D, Rifkin DB. In vitro neurite extension by granule neurons is dependent upon astroglial-derived fibroblast growth factor. Dev Biol. 1988;125:280–289. doi: 10.1016/0012-1606(88)90211-4. [DOI] [PubMed] [Google Scholar]
- 28.Hess P. Calcium channels in vertebrate cells. Annu Rev Neurosci. 1990;13:337–356. doi: 10.1146/annurev.ne.13.030190.002005. [DOI] [PubMed] [Google Scholar]
- 29.Jaye M, Schlessinger J, Dionne CA. Fibroblast growth factor receptor tyrosine kinases: molecular analysis and signal transduction. Biochim Biophys Acta. 1992;1135:185–199. doi: 10.1016/0167-4889(92)90136-y. [DOI] [PubMed] [Google Scholar]
- 30.Kennedy MB. Regulation of neuronal function by calcium. Trends Neurosci. 1989;12:417–420. doi: 10.1016/0166-2236(89)90089-1. [DOI] [PubMed] [Google Scholar]
- 31.Knaus H-G, Moshammer T, Friedrich K, Kang HC, Haugland RP, Glossmann H. In vivo labeling of L-type Ca2+ channels by fluorescent dihydropyridines: evidence for a functional, extracellular heparin-binding site. Proc Natl Acad Sci USA. 1992;89:3586–3590. doi: 10.1073/pnas.89.8.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koike H, Saito H, Matsuki N. Effect of fibroblast growth factors on calcium currents in acutely isolated neuronal cells from rat ventromedial hypothalamus. Neurosci Lett. 1993;150:57–60. doi: 10.1016/0304-3940(93)90107-v. [DOI] [PubMed] [Google Scholar]
- 33.Kudo Y, Ogura A. Glutamate-induced increase in intracellular Ca2+ concentration in isolated hippocampal neurones. Br J Pharmacol. 1986;89:191–198. doi: 10.1111/j.1476-5381.1986.tb11135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lipscombe D, Madison DV, Poenie M, Reuter H, Tsien RW, Tsien RY. Imaging of cytosolic Ca2+ transients arising from Ca2+ stores and Ca2+ channels in sympathetic neurons. Neuron. 1988;1:355–365. doi: 10.1016/0896-6273(88)90185-7. [DOI] [PubMed] [Google Scholar]
- 35.Manivannan S, Terakawa S. Rapid sprouting of filopodia in nerve terminals of chromaffin cells, PC12 cells, and dorsal root neurons induced by electrical stimulation. J Neurosci. 1994;14:5917–5928. doi: 10.1523/JNEUROSCI.14-10-05917.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuda S, Saito H, Nishiyama N. Effect of basic fibroblast growth factor on neurons cultured from various regions of postnatal rat brain. Brain Res. 1990;520:310–316. doi: 10.1016/0006-8993(90)91720-2. [DOI] [PubMed] [Google Scholar]
- 37.Mattson MP, Kater SB. Calcium regulation of neurite elongation and growth cone motility. J Neurosci. 1987;7:4034–4043. doi: 10.1523/JNEUROSCI.07-12-04034.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCobb DP, Best PM, Beam KG. Development alters the expression of calcium currents in chick limb motoneurons. Neuron. 1989;2:1633–1643. doi: 10.1016/0896-6273(89)90052-4. [DOI] [PubMed] [Google Scholar]
- 39.Miller RJ. The control of neuronal Ca2+ homeostasis. Prog Neurobiol. 1991;37:255–285. doi: 10.1016/0301-0082(91)90028-y. [DOI] [PubMed] [Google Scholar]
- 40.Miyagawa T, Saito H, Nishiyama N. Branching enhancement by basic fibroblast growth factor in cut neurite of hippocampal neurons. Neurosci Lett. 1993;153:29–31. doi: 10.1016/0304-3940(93)90069-w. [DOI] [PubMed] [Google Scholar]
- 41.Morrison RS, Sharma A, de Vellis J, Bradshaw RA. Basic fibroblast growth factor supports the survival of cerebral cortical neurons in primary culture. Proc Natl Acad Sci USA. 1986;83:7537–7541. doi: 10.1073/pnas.83.19.7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neely MD, Nicholls JG. Electrical activity, growth cone motility and the cytoskeleton. J Exp Biol. 1995;198:1433–1446. doi: 10.1242/jeb.198.7.1433. [DOI] [PubMed] [Google Scholar]
- 43.Okuda S, Saito H, Katsuki H. Arachidonic acid: toxic and trophic effects on cultured hippocampal neurons. Neuroscience. 1994;63:691–699. doi: 10.1016/0306-4522(94)90515-0. [DOI] [PubMed] [Google Scholar]
- 44.Ozawa S, Tsuzuki K, Iino M, Ogura A, Kudo Y. Three types of voltage-dependent calcium current in cultured rat hippocampal neurons. Brain Res. 1989;495:329–336. doi: 10.1016/0006-8993(89)90225-4. [DOI] [PubMed] [Google Scholar]
- 45.Papa M, Bundman MC, Greenberger V, Segal M. Morphological analysis of dendritic spine development in primary cultures of hippocampal neurons. J Neurosci. 1995;15:1–11. doi: 10.1523/JNEUROSCI.15-01-00001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puro DG, Mano T. Modulation of calcium channels in human retinal glial cells by basic fibroblast growth factor: a possible role in retinal pathobiology. J Neurosci. 1991;11:1873–1880. doi: 10.1523/JNEUROSCI.11-06-01873.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ray J, Peterson DA, Schinstine M, Gage FH. Proliferation, differentiation, and long-term culture of primary hippocampal neurons. Proc Natl Acad Sci USA. 1993;90:3602–3606. doi: 10.1073/pnas.90.8.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheng M, Greenberg ME. The regulation and function of c-fos and other immediate early genes in the nervous system. Neuron. 1990;4:477–485. doi: 10.1016/0896-6273(90)90106-p. [DOI] [PubMed] [Google Scholar]
- 49.Shyng S-L, Xu R, Salpeter MM. Cyclic AMP stabilizes the degradation of original junctional acetylcholine receptors in denervated muscle. Neuron. 1991;6:469–475. doi: 10.1016/0896-6273(91)90254-w. [DOI] [PubMed] [Google Scholar]
- 50.Silver RA, Lamb AG, Bolsover SR. Calcium hotspots caused by L-channel clustering promote morphological changes in neuronal growth cones. Nature. 1990;343:751–754. doi: 10.1038/343751a0. [DOI] [PubMed] [Google Scholar]
- 51.Spitzer NC. Ion channels in development. Annu Rev Neurosci. 1979;2:363–397. doi: 10.1146/annurev.ne.02.030179.002051. [DOI] [PubMed] [Google Scholar]
- 52.Spitzer NC. Spontaneous Ca2+ spikes and waves in embryonic neurons: signaling systems for differentiation. Trends Neurosci. 1994;17:115–118. doi: 10.1016/0166-2236(94)90120-1. [DOI] [PubMed] [Google Scholar]
- 53.Swandulla D, Carbone E, Lux HD. Do calcium channel classifications account for neuronal calcium channel diversity? Trends Neurosci. 1991;14:46–51. doi: 10.1016/0166-2236(91)90018-p. [DOI] [PubMed] [Google Scholar]
- 54.Tanaka T, Saito H, Matsuki N. Endogenous nitric oxide inhibits NMDA- and kainate-responses by a negative feedback system in rat hippocampal neurons. Brain Res. 1993;631:72–76. doi: 10.1016/0006-8993(93)91188-x. [DOI] [PubMed] [Google Scholar]
- 55.Thastrup O, Curren PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thompson SM, Wong RKS. Development of calcium current subtypes in isolated rat hippocampal pyramidal cells. J Physiol (Lond) 1991;439:671–689. doi: 10.1113/jphysiol.1991.sp018687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsien RW, Tsien RY. Calcium channels, stores, and oscillations. Annu Rev Cell Biol. 1990;6:715–760. doi: 10.1146/annurev.cb.06.110190.003435. [DOI] [PubMed] [Google Scholar]
- 58.Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- 59.Verkhratsky A, Shmigol A. Calcium-induced calcium release in neurones. Cell Calcium. 1996;19:1–14. doi: 10.1016/s0143-4160(96)90009-3. [DOI] [PubMed] [Google Scholar]
- 60.Vescovi AL, Reynolds BA, Fraser DD, Weiss S. bFGF regulates the proliferative fate of unipotent (neuronal) and bipotent (neuronal/astroglial) EGF-generated CNS progenitor cells. Neuron. 1993;11:951–966. doi: 10.1016/0896-6273(93)90124-a. [DOI] [PubMed] [Google Scholar]
- 61.Vicario-Abejon C, Johe KK, Hazel TG, Collazo D, McKay RDG. Functions of basic fibroblast growth factor and neurotrophins in the differentiation of hippocampal neurons. Neuron. 1995;15:105–114. doi: 10.1016/0896-6273(95)90068-3. [DOI] [PubMed] [Google Scholar]
- 62.Walicke P, Cowan WM, Ueno N, Baird A, Guillemin R. Fibroblast growth factor promotes survival of dissociated hippocampal neurons and enhances neurite extension. Proc Natl Acad Sci USA. 1986;83:3012–3016. doi: 10.1073/pnas.83.9.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wanaka A, Milbrandt J, Johnson EM., Jr Expression of FGF receptor gene in rat development. Development. 1991;111:455–468. doi: 10.1242/dev.111.2.455. [DOI] [PubMed] [Google Scholar]
- 64.Westenbroek RE, Ahlijanian MK, Catterall WA. Clustering of L-type Ca2+ channels at the base of major dendrites in hippocampal pyramidal neurons. Nature. 1990;347:281–284. doi: 10.1038/347281a0. [DOI] [PubMed] [Google Scholar]
- 65.Wu R-L, Barish ME. Astroglial modulation of transient potassium current development in cultured mouse hippocampal neurons. J Neurosci. 1994;14:1677–1687. doi: 10.1523/JNEUROSCI.14-03-01677.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yaari Y, Hamon B, Lux HD. Development of two types of calcium channels in cultured mammalian hippocampal neurons. Science. 1987;235:680–682. doi: 10.1126/science.2433765. [DOI] [PubMed] [Google Scholar]
- 67.Yu W, Ahmad FJ, Baas PW. Microtubule fragmentation and partitioning in the axon during collateral branch formation. J Neurosci. 1994;14:5872–5884. doi: 10.1523/JNEUROSCI.14-10-05872.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]