

Abstract
Environmental and pharmacological stimulation of cortical acetylcholine (ACh) efflux was determined in rats sustaining partial deafferentation of cortical cholinergic inputs. Rats were bilaterally infused with the selective cholinotoxin 192 IgG-saporin (0.005 μg/0.5 μl/site) into the frontoparietal cortex. In the first experiment, animals were pretrained to associate the onset of darkness with presentation of a palatable fruit cereal reward. The ability of this stimulus to enhance frontoparietal ACh efflux alone, and with the benzodiazepine receptor (BZR) weak inverse agonist ZK 93,426 (1.0 or 5.0 mg/kg, i.p.), was determined in lesioned and sham-lesioned rats. Intracortical infusions of 192 IgG-saporin reduced basal cortical ACh efflux by 47% of sham-lesioned values, consistent with reductions in the density of AChE-positive fibers. In spite of this deafferentation, ZK 93,426 produced a transient potentiation of the cortical ACh efflux induced by the darkness/cereal stimulus similar to that observed in control animals. In the second experiment, the ability of the more efficacious BZR partial inverse agonist FG 7142 (8.0 mg/kg, i.p.) to enhance basal cortical ACh efflux was compared in lesioned and sham-lesioned rats. Again, lesioned rats exhibited an increase comparable to control animals after FG 7142. This drug-induced stimulation of cortical ACh efflux was comparably and completely blocked in both groups by co-perfusion with tetrodotoxin (1.0 μm). These results suggest similarities in the modulation of cortical ACh efflux in intact and partially deafferented rats and indicate the potential of BZR inverse agonists for restoring transmission in animals with partial loss of cortical cholinergic inputs.
Keywords: acetylcholine, cortex, lesion, microdialysis, 192 IgG-saporin, benzodazepine receptor, inverse agonist, deafferentation
Loss of cholinergic inputs to telencephalic areas is associated with normal human aging and dementia (Bowen et al., 1976;Perry et al., 1977, 1992; Whitehouse et al., 1982). Decreases in markers of the activity of cortical acetylcholine (ACh) selectively correlate with the severity of the dementia (Sims et al., 1983; Palmer et al., 1987a,b; DeKosky et al., 1992). The effects of lesions of the cholinergic system in animals supported the hypothesis that loss of cholinergic inputs to the cortex impairs cognition (Robbins et al., 1989; Muir et al., 1992, 1994; Voytko et al., 1994; McGaughy et al., 1996). Therefore, efforts to develop pharmacological therapies for the cognitive disorders associated with cholinergic hypofunction have focused on cholinomimetic mechanisms.
Traditional approaches to the stimulation of muscarinic neurotransmission produced only limited beneficial cognitive effects (Traub and Friedman, 1992; Sahakian et al., 1993; Soares and Gershon, 1995). These approaches assume that cholinergic transmission in telencephalic areas acts largely by tonically stimulating postsynaptic receptors, a supposition which predicts that a loss of presynaptic neurons could be functionally compensated for by muscarinic agonists or cholinesterase inhibitors. The complex cognitive functions mediated by cholinergic neurons, however, are more likely the result of phasic, stimulus-bound changes in the activity of presynaptic neurons (Sillito and Murphy, 1987). Consequently, postsynaptic receptor stimulationindependent of presynaptic activity is not expected to yield beneficial cognitive effects (Drachman, 1978). An alternative pharmacological strategy to treating the consequences of cholinergic cell loss is to focus on the augmentation of residual phasic presynaptic functions (Sarter et al., 1990; Sarter and Bruno, 1994,1996a,b).
The neurobiological foundations of the GABAergic regulation of basal forebrain cholinergic neurons have been reviewed elsewhere (Sarter et al., 1990; Decker and McGaugh, 1991; Bruno and Miller, 1994; Sarter and Bruno, 1994; Sarter et al., 1994). Because benzodiazepine receptor (BZR) ligands allosterically modulate GABAergic transmission, weak or selective BZR inverse agonists (Sarter et al., 1990; Miller, 1994), by virtue of their ability to negatively modulate GABA, have been hypothesized to augment the activity of basal forebrain cholinergic neurons, thereby alleviating the cognitive impairments resulting from partial loss of cholinergic neurons (Sarter et al., 1988; Sarter and Steckler, 1989; Holley et al., 1993; Sarter et al., 1994).
Previous studies in intact animals assessing cortical ACh effluxin vivo demonstrated that such BZR inverse agonists, administered systemically or into the basal forebrain, potentiate the increases in cortical ACh efflux that are induced by behavioral or cognitive activation (Moore et al., 1992, 1993, 1995a; Sarter and Bruno, 1994). The efficacy of this trans-synaptic modulation of cortical ACh efflux in subjects with cholinergic cell loss requires that the remaining cholinergic terminals in these subjects be capable of enhanced ACh release. Although some studies in animals and humans have provided indirect evidence for the assumption that the remaining cholinergic terminals in demented humans and in lesioned animals can increase their capacity for ACh synthesis and release (Kish et al., 1990; Pascual et al., 1991; Cossette et al., 1993; Holley et al., 1993), direct evidence for such plasticity has been lacking. The present experiment characterizes the in situ capacity of a diminished cholinergic system to respond to behavioral and pharmacological stimulation.
Loss of cholinergic inputs into the cortex was achieved after intracortical infusions of the immunotoxin 192 IgG-saporin (Wiley, 1992; Heckers et al., 1994; Torres et al., 1994; Wenk et al., 1994). Previous experiments supported the assumption that intracortical infusions of 192 IgG-saporin selectively lesion cholinergic inputs to the infusion area while sparing other fiber systems (Holley et al., 1994). Although intrabasalis infusions of the toxin may spare the cholinergic projections to the cortex (Heckers et al., 1994), the effects of intracortical infusions on cortical ACh efflux can be attributed to the cortical effects of this toxin. The interactions between the effects of the BZR-selective inverse agonist ZK 93,426 (Jensen et al., 1984; Sarter et al., 1990) and “activated” cortical ACh efflux in lesioned and sham-lesioned animals were tested. We attempted to “activate” ACh efflux using a pretrained stimulus consisting of the onset of darkness associated with a palatable food (Moore et al., 1992, 1993, 1995a; for other behavioral methods to activate ACh efflux, see Inglis et al., 1994; Inglis and Fibiger, 1995). Furthermore, the ability of the BZR partial inverse agonist FG 7142 to increase cortical ACh efflux in lesioned animals was tested. FG 7142 was found previously to stimulate basal ACh efflux in intact animals (Moore et al., 1995b). Thus, the experiments described below collectively assess the effects of partial loss of cortical cholinergic inputs on basal as well as on behaviorally and pharmacologically stimulated cortical ACh efflux.
EXPERIMENT 1: ZK 93,426 AND BEHAVIORALLY STIMULATED ACh EFFLUX
Materials and methods
Animals
Young adult (4–7 months of age) male F344/BNNia rats (National Institute of Aging Colony, Charles River, Wilmington, MA) were maintained in a temperature- and humidity-controlled environment on a 12 hr light/dark cycle (lights on at 6 A.M.) with food and water freely available. Animal care and experimentation were performed in accordance with protocols approved by the Ohio State University Institutional Laboratory Animal Care and Use Committee. All animals were handled extensively for several days before surgery.
Surgery
Surgery was performed under aseptic conditions on animals anesthetized with sodium pentobarbital (60 mg/kg, i.p.). A frozen stock solution of the cholinergic immunotoxin 192 IgG-saporin (0.75 μg/μl, in Dulbecco’s saline; a gift from Dr. R. G. Wiley, Veterans Administration Medical Center and Vanderbilt University, Nashville, TN) was prepared and purified as described previously (Wiley and Lappi, 1993). The stock solution was diluted with Dulbecco’s saline to a working concentration of 0.01 μg/μl. Animals received bilateral infusions of the toxin (n = 7) or its vehicle solution (n = 7) into frontoparietal cortex (three infusion sites/hemisphere; 0.5 μl/infusion) at the following coordinates, relative to Bregma: anterior (AP) −0.6 mm, lateral (L) ±3.0 mm, ventral (DV) −1.5 mm (from cortical surface); AP +2.4 mm, L ±3.0 mm, DV −1.5 mm; and AP +4.5 mm, L ±2.7 mm, DV −1.2 mm. This dose of 192 IgG-saporin, administered into cortex, has been shown to result in a specific, partial loss of acetylcholinesterase (AChE)-positive fibers in the infused areas (Holley et al., 1994). Animals were given injections of penicillin (30,000 U, i.m.) and 10% glucose in saline (2–3 ml, i.p.) at the conclusion of surgery.
Animal handling and habituation to the microdialysis testing chambers continued during the next 2 weeks, with training during the second week (see below). Two weeks after the initial surgery, each animal was implanted with a stainless steel microdialysis guide cannula (0.65 mm outer diameter; CMA Microdialysis, Acton, MA) at a 45° angle into the right frontoparietal cortex through the hole previously drilled for the middle toxin or vehicle infusion site. The cannula tip was lowered 1.5 mm below the dural surface and permanently fixed to the skull with stainless steel screws and dental cement. Anesthesia, surgical conditions, and care after surgery were identical to those described above for the initial surgery.
Behavioral training
Animals were handled and habituated to the microdialysis testing chambers (parabolic, clear plastic bowls; 35.0 cm height, 38.0 cm diameter; Carnegie Medicin, Stockholm, Sweden) and to injections (intraperitoneal) of normal saline during the 2 week period between intracortical infusions of 192 IgG-saporin or its vehicle solution and the implantation of microdialysis guide cannulae. During the second week, animals were trained to associate the onset of darkness in the testing room with the presentation of palatable food (a piece of fruit-flavored cereal), a manipulation that reliably increases cortical ACh efflux (Moore et al., 1992, 1993, 1995a). Between 9:00 and 10:00 A.M., the rats were placed in the testing bowls; 4–5 hr later, the lights in the testing room were extinguished, followed within 30 sec by the presentation of one piece of cereal. After 7 consecutive days of this training, the latency of the animals to consume the cereal was <30 sec. No difference in latency between lesioned and sham-lesioned control groups was observed during training or testing.
Microdialysis protocol
After guide cannula implantation, the animals were allowed a 3 d recovery period during which habituation and training continued. The first microdialysis session was performed on the fourth day after surgery. Each animal received three microdialysis sessions, with a nondialysis day between sessions. We have demonstrated previously the validity of this repeated dialysis design for studying the modulation of cortical (Moore et al., 1995b) and striatal (Johnson and Bruno, 1995) ACh efflux.
On each test day, the animals were habituated to the testing chamber for 30 min before insertion of the concentric microdialysis probe (0.5 mm outer diameter, 2.0 mm membrane tip, 6000–8000 MW cutoff; CMA Microdialysis). The probes were then perfused at a flow rate of 2.0 μl/min with an artificial cerebrospinal fluid (aCSF) containing (in mm): 126.5 NaCl, 27.5 NaHCO3, 2.4 KCl, 0.5 Na2SO4, 0.5 KH2PO4, 1.1 CaCl2, 0.8 MgCl2, and 4.9 d-glucose, and 0.5 μm neostigmine bromide (Sigma, St. Louis, MO). Collection of dialysates began 3 hr after probe insertion, a time point at which basal cortical ACh efflux is stable and dependent on axonal depolarization in the region surrounding the probe (Moore et al., 1992). Six consecutive 15 min baseline dialysate samples were then collected.
After the last baseline collection, each animal received an injection of the BZR-selective inverse agonist ZK 93,426 (1.0 or 5.0 mg/kg, i.p.; Lot Pe6642K1, Schering AG, Berlin, Germany) or its vehicle solution, 10% Cremephor EL (CEL) (BASF, Ludwigshafen, Germany) in saline. Only one dose was administered per microdialysis session, and each animal received all three doses, counterbalanced across sessions. Fifteen minutes after injection of drug or vehicle solution, the lights in the testing room were extinguished, and the animal was presented with a piece of fruit-flavored cereal. Six additional 15 min dialysate samples were then collected while the lights remained off.
ACh analysis
ACh in each dialysate was quantitated by HPLC with electrochemical detection (Potter et al., 1983; Tyrefors and Carlsson, 1990). ACh and choline were separated by a C18 carbon polymer column (530 × 1 mm; Bioanalytical Systems, West Lafayette, IN) using a mobile phase, pH 8.5, containing 30 mmNaH2PO4 and 20 ppm of the microbicide ProClin (Rohm and Haas, Philadelphia, PA). Postcolumn derivatization of ACh and choline was achieved by an immobilized enzyme reactor column (Bioanalytical Systems) containing covalently bound AChE and choline oxidase. The hydrogen peroxide generated by the enzymatic degradation of ACh and choline was detected by a platinum working electrode (+500 mV) coupled to an LC–4C electrochemical detector (Bioanalytical Systems). Detector output was recorded and analyzed using INJECT software (Interactive Microware, State College, PA). The peak corresponding to ACh was quantitated by integration of the peak area and comparison with a four-point external standard curve bounding the expected range of ACh values. The detection limit for ACh by this method was 20 fmol/20 μl dialysate injection.
Histology
Three days after the last microdialysis session, animals were given a sublethal dose of sodium pentobarbital and transcardially perfused with 0.2% heparin in saline followed by 10% formalin. The brains were removed, blocked rostral to the cerebellum, and stored in 10% formalin at 4°C with transfer to 30% sucrose phosphate buffer at least 3 d before sectioning. Sections from each brain were processed for AChE staining according to modifications of the method ofTago et al. (1986).
Statistical analysis
Individual baseline ACh efflux (picomoles/minute) for each subject in each dialysis session was defined as the median value of the six baseline collections for that session. Comparisons of basal efflux between the two conditions (sham- or 192 IgG-saporin-lesioned), as a function of microdialysis session, were conducted using ANOVA.
Consistent with previous findings (Moore et al., 1992, 1993, 1995a), ACh efflux values returned to basal levels by the second collection period after darkness/cereal. Hence, an overall three-way ANOVA, with TIME (two levels) and DOSE (three levels) as within-subjects factors and LESION CONDITION (two levels) as a between-subjects factor, was performed. Time analysis was restricted to the final baseline collection and the first collection after darkness/cereal to assess the interaction of drug dose with the darkness/cereal manipulation. All efflux values for these two time points were expressed as percentage changes from the median baseline for each animal in each dialysis session to control for interanimal and intersession variability in basal ACh efflux. Post hoc analyses consisted of pairedt tests involving dose and time, with statistical significance defined as p < 0.05.
Results
Basal ACh efflux
Figure 1 illustrates that compared with sham-lesioned control animals, rats that received intracortical infusions of 192 IgG-saporin exhibited reduced basal cortical ACh efflux. This reduction was demonstrated statistically by a main effect of LESION CONDITION on basal efflux (F(1,12) = 8.07; p < 0.02). There was, however, no significant effect of DIALYSIS SESSION on basal ACh efflux (F(2,24) = 2.71; p > 0.08), nor was there a CONDITION × SESSION interaction (F(2,24) = 1.04; p > 0.3), indicating that basal efflux for both groups of animals was relatively stable across the three dialysis sessions. Given this lack of a session effect, overall basal efflux across the three sessions is summarized in the right-hand column in Figure 1, which shows that basal ACh efflux in 192 IgG-saporin-lesioned animals was decreased by an average of 47% relative to sham-lesioned controls.
Fig. 1.

Mean ± SEM basal ACh efflux in frontoparietal cortex for each dialysis session in 192 IgG-saporin-lesioned (n = 7) and sham-lesioned rats (n = 7). When collapsed across sessions (ALL), median baseline efflux averaged 0.17 ± 0.03 pmol/min for control animals and 0.09 ± 0.01 pmol/min for lesioned animals, a decrease of 47%.
Effects of ZK 93,426 and darkness/cereal on ACh efflux
As shown in Figures 2 and 3, theoverall darkness/cereal manipulation increased cortical ACh efflux levels in both lesioned and control animals, as evidenced by a highly significant effect of TIME (F(1,12) = 21.44; p = 0.001) in a LESION CONDITION × DOSE × TIME ANOVA. This effect was short-lived, because significant elevations in cortical ACh efflux did not extend beyond the first collection interval after darkness/cereal. The lack of a main LESION effect (F(1,12) = 0.73; p > 0.4) or a LESION × TIME interaction (F(1,12) = 0.05; p > 0.8) indicated that both the relative magnitude and the temporal dynamics of the increase in cortical ACh efflux in the lesioned animals did not differ from sham-lesioned controls.
Fig. 2.

Effect of systemic preadministration of ZK 93,426 or its vehicle on the darkness/cereal-induced increase in cortical ACh efflux (mean ± SEM) in sham-lesioned control animals (n = 7). The darkness/cereal stimulus, presented 15 min after systemic injection of vehicle solution, increased efflux by 98 ± 33% over baseline efflux during the first collection interval. This effect was potentiated by systemic administration of ZK 93,426 15 min before the darkness/cereal stimulus. Peak increases during the first collection period after darkness/cereal averaged 122 ± 33% at the 1.0 mg/kg dose and 200 + 66% at the 5.0 mg/kg dose. last bsln, Last baseline.
Fig. 3.

Effect of systemic preadministration of ZK 93,426 or its vehicle on the darkness/cereal-induced increase in cortical ACh efflux (mean ± SEM) in 192 IgG-saporin-lesioned animals (n = 7). The darkness/cereal stimulus, presented 15 min after systemic injection of vehicle solution, increased efflux by 91 ± 32% over baseline efflux during the first collection interval. This effect was potentiated by systemic administration of ZK 93,426 at the 1.0 mg/kg dose (peak = 156 ± 47%) but not the 5.0 mg/kg dose (peak = 94 ± 31%). last bsln, Last baseline.
Systemic administration of ZK 93,426 before the onset of darkness potentiated the trend toward increased cortical ACh efflux produced by the darkness/cereal manipulation, as shown by a significant DOSE × TIME interaction (F(2,24) = 3.69;p < 0.01). Post hoc analyses revealed that this interaction was generated by the effects of the BZR inverse agonist and was not produced by the vehicle injections. Vehicle-induced ACh efflux was not significantly different from the last baseline values for either sham or lesioned rats (both p values > 0.05). Peak increases in ACh efflux, after ZK 93,426, reached 200 ± 66% (mean ± SEM) above median baseline at the 5.0 mg/kg dose in the control group and 156 ± 47% at the 1.0 mg/kg dose in the lesioned group. Post hoc analyses revealed that these were significant increases relative to the last baseline period (t6 = 2.75, p < 0.05 for controls; t6 = 3.62, p < 0.02 for lesioned animals) and relative to the increases seen during darkness/cereal presentation after vehicle solution administration (t6 = 2.63, p < 0.05 for controls; t6 = 3.31, p < 0.02 for lesioned animals). The potency of ZK 93,426 was not significantly different between the two groups, as shown by the lack of a LESION × DOSE (F(2,24) = 0.68; p > 0.5) or LESION × DOSE × TIME (F(2,24) = 2.56; p > 0.09) interaction.
Histology
Cortical infusions of 192 IgG-saporin were associated with marked reductions in AChE-positive fiber staining in the cortex (Fig.4) and basal forebrain (Fig. 5C,D) relative to sham-lesioned controls. Quantitative estimates of the degree of this loss of AChE-positive fibers were on the order of 40–60%, a range similar to that observed previously using the same dose of cortically administered 192 IgG-saporin (Holley et al., 1994). All animals included in the study were confirmed to have microdialysis probe placement in the frontoparietal cortex, with no systematic differences in placement between the two groups (Fig. 5A,B).
Fig. 4.

Representative coronal brain sections (stained for AChE-positive fibers) from a sham-lesioned animal (A) and a 192 IgG-saporin-lesioned animal (B). Cortical infusions of 192 IgG-saporin resulted in a 40–60% reduction of AChE-positive fiber density in the medial and dorsal parietal areas. The arrow in B marks the transition from an extensively depleted dorsal cortical area (near the toxin infusion site) to a less depleted more ventral zone. Magnification, 10×.
Fig. 5.
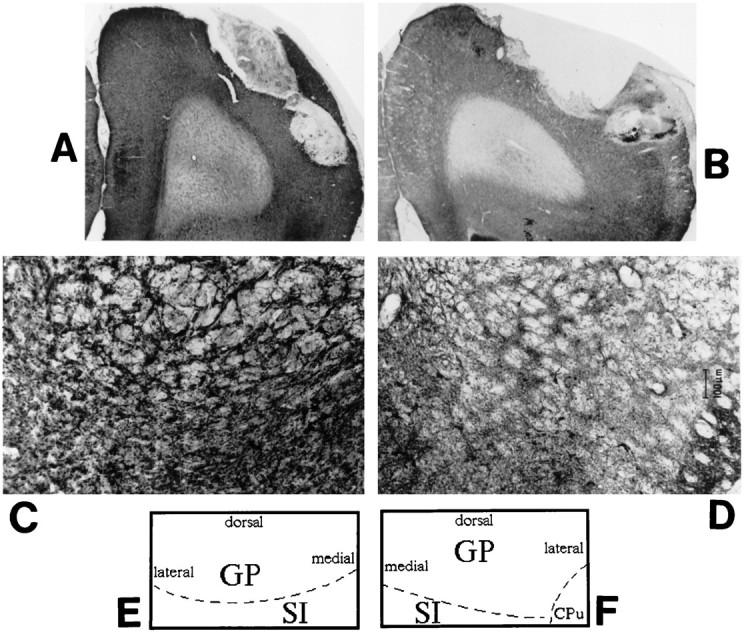
Representative coronal brain sections (stained for AChE) from a sham-lesioned animal (A) and a 192 IgG-saporin-lesioned animal (B) demonstrating representative placement of the microdialysis probe in the frontoparietal cortex. Again, the lesioned cortex shows a marked reduction in AChE-positive fiber staining. C andD show coronal sections through the basal forebrain area of representative control and lesioned animals, respectively. AChE-positive fiber staining is markedly reduced in the lesioned basal forebrain, implying a loss of cholinergic neurons in the basal forebrain after cortical infusion of 192 IgG-saporin. Eand F schematically illustrate the anatomical zones present in C and D. GP, Globus pallidus; SI, substantia innominata;CPu, caudate putamen. Magnification: A,B, 5×; C, D, 25×.
Discussion
The above results demonstrate that cortical infusions of the immunotoxin 192 IgG-saporin produce marked reductions inbasal cortical ACh efflux as measured by in vivomicrodialysis. Thus, ACh efflux correlates well with previous static histochemical markers after cortical infusion of similar doses of this toxin, supporting a relationship between changes in cholinergic fiber density and cortical ACh efflux. This reduction in basal ACh efflux in saporin-treated rats was seen on each of the three microdialysis sessions. These results are consistent with our previous studies using the repeated perfusion/dialysis design, demonstrating that basal or dynamic ACh efflux does not interact with the microdialysis session (Moore et al., 1992, 1993, 1995a,b,c; Johnson and Bruno, 1995).
Despite the significant reduction in basal levels of cortical ACh efflux, however, the ability of the BZR-selective inverse agonist ZK 93,426 to potentiate basal efflux or that after the vehicle injection plus darkness/cereal presentation was of similar relative magnitude in lesioned animals and sham-lesioned controls. Thus, BZR-selective inverse agonists are capable of potentiating the effects of a behavioral stimulus on cortical ACh efflux, even in a compromised system.
EXPERIMENT 2: BASAL ACh EFFLUX AND FG 7142
Materials and methods
In the second experiment, the effects of an additional BZR ligand, the partial inverse agonist FG 7142, on cortical ACh efflux were determined in 192 IgG-saporin- and sham-lesioned rats. Unlike ZK 93,426, FG 7142 stimulates cortical ACh efflux under basalconditions (i.e., in the absence of a manipulation such as the darkness/cereal association) (Moore et al., 1995b). Furthermore, FG 7142 increases dopaminergic transmission in the nucleus accumbens (McCullough and Salamone, 1992), an effect that interacts with the ability of this drug to enhance cortical ACh efflux (Moore et al., 1995c). Therefore, FG 7142 was used to assess the ability of cortical ACh efflux in 192 IgG-saporin-lesioned rats to respond to pharmacological stimulation in the absence of an entrained stimulus. In separate dialysis sessions, the neuronal origin of ACh efflux in lesioned and control animals was investigated by perfusing the voltage-gated Na+-channel blocker tetrodotoxin (TTX) through the dialysis probe before FG 7142 administration.
Animals and surgery
Young adult male F344/BNNia rats were housed and handled as described above in Experiment 1. Animals were infused with 192 IgG-saporin (n = 4) or its vehicle solution (n = 4) using the surgical conditions, toxin dose, and stereotaxic coordinates described above. During the next 2 weeks, these animals were handled further and habituated to the microdialysis testing bowls and saline injections (intraperitoneal). These animals were not trained in the darkness/fruit cereal association, however, and thus were not subjected to this manipulation during microdialysis testing sessions. Two weeks after the cortical infusion of toxin or vehicle solution, these animals were implanted with microdialysis guide cannulae into the right frontoparietal cortex, again as described above.
Microdialysis protocol
Each animal received three microdialysis sessions, beginning on the fourth day after cannula implantation, with a nondialysis day between sessions. The order of dose administration for half of the animals (two lesion plus two control) was vehicle, FG 7142, and TTX/FG 7142 in the first through third sessions, respectively. The remaining half (two lesion plus two control) received TTX/vehicle, vehicle, and FG 7142 in the three respective sessions. This design allowed us to determine the effect of TTX on both basal andstimulated efflux as well as to investigate the possibility of session-dependent differences in TTX sensitivity of cortical ACh efflux.
On the testing days, animals were placed in the dialysis testing bowls 30 min before probe insertion. Three hours after probe insertion and perfusion with aCSF (2.0 μl/min; see above for composition), six consecutive 15 min baseline dialysates were collected. During non-TTX sessions, FG 7142 (8.0 mg/kg, i.p.) (Research Biochemicals International, Natick, MA) or its vehicle solution (10% CEL in saline) was administered immediately after the last baseline period, and four additional 15 min dialysates were then collected. During TTX sessions, 1.0 μm TTX (Sigma) was infused through the microdialysis probe beginning after the last baseline period, followed 45 min later by injection of FG 7142 or its vehicle solution. Again, four additional postinjection 15 min dialysates were collected. HPLC analysis and histological procedures were carried out as described in Experiment 1.
Statistical analysis
Individual and group baseline cortical ACh efflux values were defined and calculated as described in Experiment 1. Again, individual median baselines were used in an ANOVA comparing basal efflux between the two lesion conditions as a function of dialysis session.
To assess the effects of systemic administration of FG 7142, three-way ANOVAs with TIME and DOSE as within-subjects factors and LESION CONDITION as a between-subjects factor were performed. Because of the “inverted U-shaped” nature of the response function over time, two separate ANOVAs were conducted. The first ANOVA compared the first two collection intervals after FG 7142 with the last baseline. The second ANOVA compared the final two collection intervals after FG 7142. As in Experiment 1, efflux values for this ANOVA were expressed as percentage changes from median baseline for each animal in each dialysis session. Administration of TTX through the dialysis probe consistently reduced ACh efflux to below detectable limits (i.e., <20 fmol/20 μl dialysate) in all cases; therefore, these data were treated descriptively.
Results
Basal efflux
As in Experiment 1, animals infused with 192 IgG-saporin exhibited marked reductions in cortical ACh efflux (data not shown graphically). Basal efflux across sessions for animals in this experiment averaged 0.15 ± 0.02 pmol/min for sham-lesioned animals and 0.07 ± 0.01 pmol/min for 192 IgG-saporin-lesioned animals. This difference was significant (LESION CONDITION, F(11,6) = 6.13;p < 0.05).
Effects of FG 7142 and TTX on ACh efflux
Figure 6 summarizes the effects of vehicle and FG 7142 on cortical ACh efflux in sham- and saporin-treated rats. Even in the absence of the darkness/cereal manipulation, the BZR partial inverse agonist FG 7142 stimulated cortical ACh efflux in both groups of animals. An overall ANOVA, comparing the last baseline and the first two collection intervals after drug treatment, revealed significant main effects of TIME (F2,12 = 9.13;p < 0.05) and DOSE (F(1,6) = 9.14; p < 0.05), indicating that the increases in cortical ACh efflux after FG 7142 was greater than that seen after administration of its vehicle solution alone. A significant TIME × DOSE interaction (F2,12 = 11.89;p < 0.01) indicated that the duration of the increase in cortical ACh efflux after FG 7142 was greater than that after injection of its vehicle solution. Post hoc comparisons between vehicle- and FG-treated rats revealed differences at 15–30 min (p < 0.005) but not at 0–15 min intervals. The lack of a significant LESION CONDITION × TIME (F2,12 = 0.82; p > 0.4) or LESION CONDITION × DOSE (F(1,6) = 1.38;p > 0.2) interaction supported the similarity in the responsiveness of lesioned and control animals over the time points and doses analyzed.
Fig. 6.

Effect of the BZR partial inverse agonist FG 7142 on basal (i.e., no darkness/cereal) cortical ACh efflux in 192 IgG-saporin-lesioned (n = 4) and sham-lesioned animals (n = 4). ACh efflux (mean ± SEM) was enhanced significantly by systemic administration of FG 7142, relative to vehicle injections, with peak increases occurring in the second 15 min collection period in both groups of animals.
An overall ANOVA, comparing the last baseline and the final two collection intervals after drug treatment revealed no significant main effects (all p values > 0.05). The significant TIME × DOSE interaction (F2,12 = 3.90;p < 0.05) suggested that the duration of the increase in cortical ACh efflux after FG 7142 was greater than that after injection of its vehicle solution. Post hoc comparisons between vehicle- and FG-treated rats revealed a difference at the 45–60 min interval (p < 0.05). Nevertheless, the nonsignificant interaction between LESION CONDITION × DOSE × TIME confirmed that there were no obvious differences in the responses of sham- and saporin-treated rats.
Administration of TTX through the dialysis probe, before injection of FG 7142, decreased cortical ACh efflux to below detectable amounts (i.e., 20 fmol/injection) by the second period after TTX in all cases, indicating that basal and stimulated efflux measured in the two groups of animals were dependent on axonal depolarization in the region surrounding the dialysis probe. Because the percentage decreases after TTX plus FG 7142 (−70% and −64% from baseline in sham- and saporin-treated rats, respectively) are artificially constrained by the detection limits for ACh, these data generally underestimate the TTX dependency. Thus, the analysis of these data was limited to a descriptive treatment.
Histology
Again, all animals included in this experiment were confirmed to have microdialysis probe placement in the frontoparietal cortex (similar to those seen in Fig. 5A,B). The decrease in AChE-positive fiber staining in lesioned animals relative to controls was similar to that shown in the representative sections in Figures 4and 5C,D.
Discussion
These data demonstrate that rats depleted of cortical ACh with intracortical infusions of 192 IgG-saporin still exhibit increases in ACh efflux, similar in magnitude to those seen in intact animals, after systemic administration of the BZR partial inverse agonist FG 7142. Because FG 7142 produced these increases in the absence of any behavioral stimulus (e.g., darkness/palatable food), this provides additional evidence for the ability of pharmacological manipulations targeting GABAergic transmission in the basal forebrain to stimulate cortical ACh efflux in animals that have undergone significant but subtotal deafferentation of cortical cholinergic inputs.
Basal and FG 7142-stimulated cortical ACh efflux in both groups of animals were reduced to nondetectable levels by the presence of TTX in the dialysis perfusion medium, indicating that ACh efflux after cortical infusions of 192 IgG-saporin is dependent on axonal depolarization in the region surrounding the dialysis probe. This TTX effect was the same whether it was administered in the first or third microdialysis session, demonstrating that repeated insertion and perfusion of the probe does not seem to alter the depolarization dependency of cortical ACh efflux.
GENERAL DISCUSSION
These experiments revealed two important findings regarding the modulation of cortical ACh efflux in rats after intracortical infusions of the selective cholinotoxin 192 IgG-saporin. First, infusions of 192 IgG-saporin produced a partial deafferentation of cortical cholinergic innervation as evidenced by decreases in the density of AChE-positive fibers in the cortex and basal levels of extracellular ACh. Second, despite this deafferentation, cortical ACh efflux in lesioned rats was modulated by environmental and pharmacological stimuli in a manner similar to that demonstrated in intact rats. The discussion below focuses on the use of ACh efflux as a measure of cortical cholinergic transmission, the nature of the 192 IgG-saporin-induced deafferentation, and the development of cholinomimetic treatments for age- and dementia-associated cognitive impairments.
Validity of ACh efflux
There is significant evidence suggesting that ACh efflux, measured by microdialysis techniques, is a valid measure of cortical cholinergic transmission. Electrical stimulation of the basal forebrain, the site of origin of cortical cholinergic afferents (Butcher and Woolf, 1986), increases cortical ACh efflux (Kurosawa et al., 1989; Rasmusson et al., 1992) in anesthetized animals. Local injections of scopolamine and oxotremorine into the nucleus basalis increase and decrease, respectively, cortical ACh efflux in anesthetized rats (Bertorelli et al., 1991). Trans-synaptic activation of cortical cholinergic neurons by electrical stimulation of the pedunculopontine tegmentum increases cortical ACh efflux (Szerb et al., 1994). Finally, intrabasalis infusions of BZR ligands that either positively or negatively modulate GABA-mediated Cl− flux (for review, see Bruno and Miller, 1995) predictably decrease or increase, respectively, cortical ACh efflux in awake rats (Moore et al., 1993, 1995a). These changes in ACh efflux are almost completely blocked by co-perfusion with TTX, suggesting a dependence of Na+-gated depolarization around the area of the dialysis probe.
The present results extend these observations by demonstrating that ACh efflux remains a valid measure of cholinergic transmission after partial deafferentation of these cortical afferents. Basal cortical ACh efflux was reduced commensurate with decreases in AChE histochemistry. Although we did not quantify these histochemical results here, we have reported previously that intracortical infusions of this same dose of 192 IgG-saporin reduce the number of cortical AChE-positive fibers by ∼40% (Holley et al., 1994). This reduction in fiber density is similar to the 47% decrease in basal cortical ACh efflux reported in Experiment 1 of the present study. A previous report in anesthetized rats demonstrated that ibotenic acid-induced lesions of the nucleus basalis reduced basal cortical ACh efflux, yet administration of 100 mm K+ still enhanced efflux (Herrera-Marschitz et al., 1990).
Cortical cholinergic transmission after partial deafferentation
Several recent studies have characterized the neurochemical effects of 192 IgG-saporin-induced deafferentation of cortical cholinergic inputs. Reductions in staining after intracortical 192 IgG-saporin seem to be selective for AChE-positive fibers (Holley et al., 1994). Intraventricular injections of the immunotoxin decrease choline acetyltransferase activity and high-affinity choline uptake (HACU) and increase the density of M1 binding sites (that were not related to an increase in sensitivity to M1-related transduction mechanisms) and c-junmRNA in the parietal and occipital cortices in layer IV (Robner et al., 1994, 1995). Similar to the present study, Robner et al. (1994, 1995)reported a K+-stimulated release of [3H]-ACh from cortical slices of lesioned rats that was, however, less than that seen in slices from intact animals.
Despite the obvious loss of cortical cholinergic afferents in the present study, the residual ACh-containing neurons exhibited a normalpattern of stimulated ACh efflux (both in duration and percentage change from baseline) to the environmental and behavioral stimuli. Exposure to the previously trained darkness/cereal stimulus tended to transiently increase cortical ACh efflux in lesioned as well as in intact rats. Although this increase was not significant in the present study, we previously reported a significant activation of cortical cholinergic transmission in intact rats after this same darkness/cereal stimulus (Moore et al., 1993, 1995a). Close inspection of the data presented in Figures 2 and 3 reveals that the lack of a significant effect of vehicle plus darkness/cereal reflected unanticipated increases in ACh efflux during the last baseline periods in these groups. The basis for these increases in efflux during the last baseline period is not apparent, because drug condition was a within-subject factor and drug order was counterbalanced. Importantly, increases in ACh efflux after the vehicle injections in Experiment 1 (85–90%) were quite similar to those reported in our previous study (95–100%; Moore et al., 1993). Clearly, the lesion also did not affect the ability of BZR inverse agonists to trans-synapticallypotentiate the effects of the darkness/cereal stimulus on ACh release (as in Experiment 1 with ZK 93,426) or to increase basal release (as in Experiment 2 with the more efficacious FG 7142).
Although the reactivity of residual neurons remainedqualitatively similar to cortical cholinergic neurons in intact rats, the magnitude of these effects wasquantitatively reduced. Basal cortical ACh efflux was depressed by 47% in the lesioned group, and although the various manipulations consistently stimulated efflux above baseline values, theabsolute amounts of ACh efflux were always less than those obtained from sham-lesioned controls. The relationship between the magnitude of the deafferentation and the ability of BZR inverse agonists to enhance cortical ACh efflux merits further study. For instance, will these BZR ligands potentiate stimulated ACh efflux after larger deafferentations than those studied here? Defining the limits of this trans-synaptic modulation will have important implications for any potential clinical applications after more severe cholinergic denervations (see below).
The fact that basal ACh efflux was reduced to approximately the same extent as was the density of AChE-positive fiber density (Holley et al., 1994) suggests that presynaptic compensations either did not occur in the 3 week interval between lesion and testing or were insufficient to maintain basal ACh efflux at control levels. The literature, however, suggests a potential for plasticity within the damaged cortical cholinergic system. Although intracortical administration of 192 IgG-saporin produces a long-lasting decrease in HACU in rats, vesamicol binding (a measure of newly synthesized ACh) seems comparable in lesioned and control animals (Holley et al., 1993). Similar discrepancies between decreases in HACU and vesamicol binding have been reported in brain tissue from Alzheimer’s disease patients (Kish et al., 1990). Finally, we do not know whether postsynaptic compensations develop sufficiently to offset the effects of this partial deafferentation on cortical information processing. Although there are reports of increased density of M1 binding sites and expression of c-jun mRNA in cortex afterintraventricular administration of 192 IgG-saporin, the functional consequences of these changes are unknown. In this regard, we have reported enduring performance deficits in these 192 IgG-saporin-treated animals in operant tasks of sustained (McGaughy et al., 1996) and divided (Turchi et al., 1995) attention.
Therapeutic implications for cognition enhancement
The potential therapeutic implications of selective BZR inverse agonists as trans-synaptic enhancers of cortical cholinergic transmission have been reviewed elsewhere (Sarter and Bruno, 1994,1996a,b; Sarter et al., 1996). Our previous work indicates that selective BZR inverse agonists such as ZK 93,426 satisfy a necessary requirement of any efficacious cognition enhancer. Namely, it enhances cortical ACh efflux only when the basal forebrain cholinergic system has already been activated (Sarter and Bruno, 1994). The present results on the ability of ZK 93,426 to potentiate the effects of darkness/cereal on ACh efflux add an additional necessary requirement of an effective pharmacotherapy: namely, the ability to enhance release in a partially deafferented system. Although this is encouraging, several issues surrounding the therapeutic efficacy of this strategy remain unresolved. First, will these selective BZR inverse agonists enhance cortical ACh efflux that is being activated by more “cognitively charged” tasks than the darkness/cereal stimulus used in this experiment? Our preliminary observations suggest that cortical ACh efflux in intact rats is activated during performance in an operant task of sustained attention (Sarter et al., 1996). Given that partially deafferented animals exhibit performance deficits in this task (McGaughy et al., 1996), we are currently determining whether administration of selective BZR inverse agonists will both enhance performance and increase ACh efflux in these lesioned animals. Second, we must determine how the capacity of these ligands to potentiate ACh efflux interacts with the extent of the ACh lesion. Constraints on the ability of these drugs to enhance cortical ACh efflux after larger deafferentations may limit their therapeutic efficacy to earlier stages of the disease process. Finally, all of our microdialysis studies have used acute administration of these BZR-selective inverse agonists. It will be important to determine the ability of these ligands to modulate cortical ACh efflux after chronic administration.
In summary, these experiments demonstrate that BZR inverse agonists stimulate basal ACh efflux (in the case of FG 7142) and potentiate the effects of darkness/cereal on ACh efflux (in the case of ZK 93,426) after partial deafferentation of cortical cholinergic inputs. The time course and magnitude (as a percentage increase from baseline) of these effects were similar in lesioned and sham-lesioned rats. Studies of the effects of these ligands on cortical ACh efflux and cognitive performance in animals selectively deafferented with 192 IgG-saporin may provide important insights into the pharmacotherapy of clinical populations impaired by reductions in cortical cholinergic transmission.
Footnotes
This research was supported in part by Public Health Service Grants AG10173 (M.S., J.P.B.) and NS32938 (M.S.). M.S. was supported by a Research Scientist Award (MH01072).
Correspondence should be addressed to Dr. John P. Bruno, Department of Psychology, The Ohio State University, Columbus, OH 43210.
REFERENCES
- 1.Bertorelli R, Forloni GL, Consolo S. Modulation of cortical in vivo acetylcholine release by the basal nuclear complex: role of the pontomesencephalic tegmental area. Brain Res. 1991;563:353–356. doi: 10.1016/0006-8993(91)91562-f. [DOI] [PubMed] [Google Scholar]
- 2.Bowen DM, Smith CB, White P, Davidson AN. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain. 1976;99:457–496. doi: 10.1093/brain/99.3.459. [DOI] [PubMed] [Google Scholar]
- 3.Bruno JP, Miller JA. Inhibition of GABAergic transmission: interactions with other transmitter systems. In: Sarter M, Nutt DJ, Lister RG, editors. Benzodiazepine receptor inverse agonists. Wiley; New York: 1994. pp. 41–82. [Google Scholar]
- 4.Butcher LL, Woolf NJ. Central cholinergic systems: synopsis of anatomy and overview of physiology and pathology. In: Scheibel AB, Wechsler AF, editors. The biological substrates of Alzheimer’s disease. Academic; New York: 1986. pp. 73–86. [Google Scholar]
- 5.Cossette P, Umbriaco D, Zamar N, Hamel E, Descarries L. Recovery of choline acetyltransferase activity without sprouting of the residual acetylcholine innervation in adult rat cerebral cortex after lesion of the nucleus basalis. Brain Res. 1993;630:195–206. doi: 10.1016/0006-8993(93)90657-9. [DOI] [PubMed] [Google Scholar]
- 6.Decker MW, McGaugh JL. The role of interactions between the cholinergic system and other neuromodulatory systems in learning and memory. Synapse. 1991;7:151–168. doi: 10.1002/syn.890070209. [DOI] [PubMed] [Google Scholar]
- 7.DeKosky ST, Harbaugh RE, Schmitt FA, Bakay RA, Chui HC, Knopman DS, Reeder TM, Shetter AG, Senter HJ, Markesberry WR. Cortical biopsy in Alzheimer’s disease: diagnostic accuracy and neurochemical, neuropathological, and cognitive correlations. Ann Neurol. 1992;32:625–632. doi: 10.1002/ana.410320505. [DOI] [PubMed] [Google Scholar]
- 8.Drachman DA. Central cholinergic system and memory. In: Lipton MA, DiMascio A, Killam KF, editors. Psychopharmacology: a generation of progress. Raven; New York: 1978. pp. 651–661. [Google Scholar]
- 9.Heckers S, Ohtake T, Wiley RG, Lappi DA, Geula C, Mesulam MM. Complete and selective cholinergic denervation of rat neocortex and hippocampus but not amygdala by an immunotoxin against the p75 NGF receptor. J Neurosci. 1994;14:1271–1289. doi: 10.1523/JNEUROSCI.14-03-01271.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herrera-Marschitz H, Goiny M, Utsumi H, Ferre S, Hakansson L, Nordberg A, Ungerstedt U. Effect of unilateral nucleus basalis lesion on cortical and striatal acetylcholine and dopamine release monitored in vivo with microdialysis. Neurosci Lett. 1990;110:172–179. doi: 10.1016/0304-3940(90)90807-l. [DOI] [PubMed] [Google Scholar]
- 11.Holley LA, Miller JA, Chmielewski PA, Dudchenko P, Sarter M. Interactions between the effects of basal forebrain lesions and chronic treatment with MDL 26,479 on learning and markers of cholinergic transmission. Brain Res. 1993;610:181–193. doi: 10.1016/0006-8993(93)91399-d. [DOI] [PubMed] [Google Scholar]
- 12.Holley LA, Wiley RG, Lappi DA, Sarter M. Cortical cholinergic deafferentation following the intracortical infusion of 192 IgG-saporin: a quantitative histochemical study. Brain Res. 1994;663:277–286. doi: 10.1016/0006-8993(94)91274-2. [DOI] [PubMed] [Google Scholar]
- 13.Inglis FM, Day JC, Fibiger HC. Enhanced acetylcholine release in hippocampus and cortex during the anticipation and consumption of a palatable meal. Neuroscience. 1994;62:1049–1056. doi: 10.1016/0306-4522(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 14.Inglis FM, Fibiger HC. Increases in hippocampal and frontal cortical acetylcholine release associated with presentation of sensory stimuli. Neuroscience. 1995;66:81–86. doi: 10.1016/0306-4522(94)00578-s. [DOI] [PubMed] [Google Scholar]
- 15.Jensen LH, Petersen EN, Breastrup C, Honore T, Kehr W, Seidelmann D, Schmiechen R. Evaluation of the β-carboline ZK 93,426 as a benzodiazepine receptor antagonist. Psychopharmacology (Berl) 1984;83:249–256. doi: 10.1007/BF00464789. [DOI] [PubMed] [Google Scholar]
- 16.Johnson BJ, Bruno JP. Dopaminergic modulation of striatal acetylcholine release in rats depleted of dopamine as neonates. Neuropharmacology. 1995;34:191–203. doi: 10.1016/0028-3908(94)00144-h. [DOI] [PubMed] [Google Scholar]
- 17.Kish SJ, Distefano LM, Dozic S, Robitaille Y, Rajput A, Deck JHN, Hornykiewicz O. [3H]Vesamicol binding in human brain cholinergic deficiency disorders. Neurosci Lett. 1990;117:347–352. doi: 10.1016/0304-3940(90)90689-7. [DOI] [PubMed] [Google Scholar]
- 18.Kurosawa M, Sato A, Sato Y. Stimulation of the nucleus basalis of Meynert increases acetylcholine release in the cerebral cortex in rats. Neurosci Lett. 1989;98:45–50. doi: 10.1016/0304-3940(89)90371-6. [DOI] [PubMed] [Google Scholar]
- 19.McCullough LD, Salamone JD. Anxiogenic drugs beta-CCE and FG 7142 increase extracellular dopamine levels in nucleus accumbens. Psychopharmacology (Berl) 1992;109:382–397. doi: 10.1007/BF02245888. [DOI] [PubMed] [Google Scholar]
- 20.McGaughy J, Kaiser T, Sarter M. Behavioral vigilance following the infusions of 192 IgG-saporin into the basal forebrain: selectivity of the behavioral impairment and relation to cortical AChE-positive fiber density. Behav Neurosci. 1996;110:247–265. doi: 10.1037//0735-7044.110.2.247. [DOI] [PubMed] [Google Scholar]
- 21.Miller JA. Evidence for a modified model of agonist and inverse agonist actions at the GABAA receptor. In: Sarter M, Nutt DJ, Lister RG, editors. Benzodiazepine receptor inverse agonists. Wiley; New York: 1994. pp. 25–41. [Google Scholar]
- 22.Moore H, Sarter M, Bruno JP. Age-dependent modulation of in vivo cortical acetylcholine release by benzodiazepine receptor ligands. Brain Res. 1992;596:17–29. doi: 10.1016/0006-8993(92)91527-l. [DOI] [PubMed] [Google Scholar]
- 23.Moore H, Sarter M, Bruno JP. Bidirectional modulation of stimulated cortical acetylcholine release by benzodiazepine receptor ligands. Brain Res. 1993;627:267–274. doi: 10.1016/0006-8993(93)90330-p. [DOI] [PubMed] [Google Scholar]
- 24.Moore H, Sarter M, Bruno JP. Bidirectional modulation of cortical acetylcholine efflux by infusion of benzodiazepine receptor ligands into the basal forebrain. Neurosci Lett. 1995a;189:31–34. doi: 10.1016/0304-3940(95)11444-2. [DOI] [PubMed] [Google Scholar]
- 25.Moore H, Stuckman S, Sarter M, Bruno JP. Stimulation of cortical acetylcholine efflux by FG 7142 measured with repeated microdialysis sampling. Synapse. 1995b;21:324–331. doi: 10.1002/syn.890210407. [DOI] [PubMed] [Google Scholar]
- 26.Moore H, Fadel J, Sarter M, Bruno JP. Interactions between benzodiazepine and dopamine receptors in the modulation of cortical acetylcholine release. Soc Neurosci Abstr. 1995c;21:763.2. [Google Scholar]
- 27.Muir JL, Dunnett SB, Robbins TW, Everitt BJ. Attentional functions of the forebrain cholinergic systems: effects of intraventricular hemicholinium, physostigmine, basal forebrain lesions and intracortical grafts on a multiple-choice serial reaction time task. Exp Brain Res. 1992;89:611–622. doi: 10.1007/BF00229886. [DOI] [PubMed] [Google Scholar]
- 28.Muir JL, Everitt BJ, Robbins TW. AMPA-induced excitotoxic lesions of the basal forebrain: a significant role for the cortical cholinergic system in attentional function. J Neurosci. 1994;14:2313–2326. doi: 10.1523/JNEUROSCI.14-04-02313.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmer AM, Francis PT, Benton SJ, Sims NR, Mann DMA, Neary D, Snowdon JS, Bowen DM. Presynaptic serotonergic dysfunction in patients with Alzheimer’s disease. J Neurochem. 1987a;48:8–15. doi: 10.1111/j.1471-4159.1987.tb13120.x. [DOI] [PubMed] [Google Scholar]
- 30.Palmer AM, Francis PT, Bowen DM, Benton JS, Neary D, Mann DMA, Snowdon JS. Catecholaminergic neurons assessed ante-mortem in Alzheimer’s disease. Brain Res. 1987b;414:365–375. doi: 10.1016/0006-8993(87)90018-7. [DOI] [PubMed] [Google Scholar]
- 31.Pascual J, Fontan A, Zarranz JJ, Berciano J, Florez J, Pazos A. High-affinity choline uptake carrier in Alzheimer’s disease: implications for the cholinergic hypothesis of dementia. Brain Res. 1991;552:170–174. doi: 10.1016/0006-8993(91)90676-m. [DOI] [PubMed] [Google Scholar]
- 32.Perry EK, Perry RH, Blessed G, Tomlinson BE. Necropsy evidence of central cholinergic deficits in senile dementia. Lancet. 1977;1:189. doi: 10.1016/s0140-6736(77)91780-9. [DOI] [PubMed] [Google Scholar]
- 33.Perry EK, Johnson M, Kerwin JM, Piggott MA, Court JA, Shaw PJ, Ince PG, Brown A, Perry RH. Convergent cholinergic activities in aging and Alzheimer’s disease. Neurobiol Aging. 1992;13:393–400. doi: 10.1016/0197-4580(92)90113-c. [DOI] [PubMed] [Google Scholar]
- 34.Potter PE, Meek JL, Neff NH. Acetylcholine and choline in neuronal tissue measured by HPLC with electrochemical detection. J Neurochem. 1983;41:133–194. doi: 10.1111/j.1471-4159.1983.tb13668.x. [DOI] [PubMed] [Google Scholar]
- 35.Rasmusson D, Clow K, Szerb JC. Frequency-dependent increase in cortical acetylcholine release evoked by stimulation of the nucleus basalis magnocellularis in the rat. Brain Res. 1992;594:1501–1554. doi: 10.1016/0006-8993(92)91041-c. [DOI] [PubMed] [Google Scholar]
- 36.Robbins TW, Everitt BJ, Marston HM, Wilkinson J, Jones GH, Page KJ. Comparative effects of ibotenic acid- and quisqualic acid-induced lesions of the substantia innominata on attentional function in the rat: further implications for the role of the cholinergic neurons of the nucleus basalis in cognitive processes. Behav Brain Res. 1989;35:221–240. doi: 10.1016/s0166-4328(89)80143-3. [DOI] [PubMed] [Google Scholar]
- 37.Robner S, Perez-Polo JR, Wiley RG, Schliebs R, Bigl V. Differential expression of immediate early genes in distinct layers of rat cerebral cortex after selective immunolesion of the forebrain cholinergic system. J Neurosci Res. 1994;38:282–293. doi: 10.1002/jnr.490380306. [DOI] [PubMed] [Google Scholar]
- 38.Robner S, Schliebs R, Hartig W, Bigl V. 192 IgG-saporin-induced selective lesion of cholinergic basal forebrain system: neurochemical effects on cholinergic neurotransmission in rat cerebral cortex and hippocampus. Brain Res Bull. 1995;38:371–381. doi: 10.1016/0361-9230(95)02002-9. [DOI] [PubMed] [Google Scholar]
- 39.Sahakian BJ, Owen AM, Morant NJ, Eagger SA, Boddington S, Crayton L, Crockford HA, Crooks M, Hill K, Levy R. Further analysis of the cognitive effects of tetrahydroaminoacridine (THA) in Alzheimer’s disease: assessment of attentional and mnemonic function using CANTAB. Psychopharmacology (Berl) 1993;110:395–401. doi: 10.1007/BF02244644. [DOI] [PubMed] [Google Scholar]
- 40.Sarter M, Bruno JP. Cognitive functions of cortical acetylcholine: lessons from studies on the transsynaptic modulation of activated efflux. Trends Neurosci. 1994;17:217–221. doi: 10.1016/0166-2236(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 41.Sarter M, Bruno JP (1996a) Transsynaptic stimulation of cortical acetylcholine and attention: a rational approach for the development of cognition enhancers. Behav Brain Res, in press. [DOI] [PubMed]
- 42. Sarter M, Bruno JP. Cortical acetylcholine, attention, and neuropharmacological and cognitive principles of a rational development of treatment strategies for cognitive disorders. Brioni JE, Decker MW. Alzheimer’s disease: molecular aspects and pharmacological treatments 1996b. Wiley; New York: in press. [Google Scholar]
- 43.Sarter M, Steckler T. Spontaneous exploration of a 6-arm radial tunnel maze by basal forebrain lesioned rats: effects of the benzodiazepine receptor antagonist β-carboline ZK 93426. Psychopharmacology (Berl) 1989;98:193–202. doi: 10.1007/BF00444691. [DOI] [PubMed] [Google Scholar]
- 44.Sarter M, Schneider HH, Stephens DN. Treatment strategies in senile dementia: antagonist β-carbolines. Trends Neurosci. 1988;11:13–17. doi: 10.1016/0166-2236(88)90042-2. [DOI] [PubMed] [Google Scholar]
- 45.Sarter M, Bruno JP, Dudchenko P. Activating the damaged basal forebrain cholinergic system: tonic stimulation versus signal amplification. Psychopharmacology (Berl) 1990;101:1–17. doi: 10.1007/BF02253710. [DOI] [PubMed] [Google Scholar]
- 46.Sarter M, McGaughy J, Holley LA, Dudchenko PA. Behavioral facilitation and cognition enhancement. In: Sarter M, Nutt DJ, Lister RG, editors. Benzodiazepine receptor inverse agonists. Wiley; New York: 1994. pp. 213–242. [Google Scholar]
- 47.Sarter M, Bruno JP, Givens BS, Moore H, McGaughy J, McMahon K (1996) Neuronal mechanisms of drug effects on cognition: cognitive activity as a necessary intervening variable. Cog Brain Res, in press. [DOI] [PubMed]
- 48.Sillito AM, Murphy PC. The cholinergic modulation of cortical function. In: Jones EG, Peters A, editors. Cerebral cortex, Vol 6. Plenum; New York: 1987. pp. 161–185. [Google Scholar]
- 49.Sims NR, Bowen DM, Allen SJ, Smith CCT, Neary D, Thomas DJ, Davison AN . Presynaptic cholinergic dysfunction in patients with dementia. J Neurochem. 1983;40:503–509. doi: 10.1111/j.1471-4159.1983.tb11311.x. [DOI] [PubMed] [Google Scholar]
- 50.Soares JC, Gershon S. THA-historical aspects, review of pharmacological properties and therapeutic effects. Dementia. 1995;6:255–234. doi: 10.1159/000106951. [DOI] [PubMed] [Google Scholar]
- 51.Szerb JC, Clow K, Rasmusson DD. Pharmacological but not physiological modulation of cortical acetylcholine release by cholinergic mechanism in the nucleus basalis magnocellularis. Can J Physiol Pharmacol. 1994;72:893–898. doi: 10.1139/y94-126. [DOI] [PubMed] [Google Scholar]
- 52.Tago H, Kimura H, Maeda T. Visualization of detailed acetylcholinesterase fiber and neuron staining in rat brain by a sensitive histochemical procedure. J Histochem Cytochem. 1986;34:1431–1438. doi: 10.1177/34.11.2430009. [DOI] [PubMed] [Google Scholar]
- 53.Torres EM, Perry TA, Blokland A, Wilkinson LS, Wiley RG, Lappi DA, Dunnett SB. Behavioural histochemical and biochemical consequences of selective immunolesions in discrete regions of the basal forebrain cholinergic system. Neuroscience. 1994;63:95–122. doi: 10.1016/0306-4522(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 54.Traub M, Friedman SB. The implication of current therapeutic approaches for cholinergic hypothesis of dementia. Dementia. 1992;3:189–192. [Google Scholar]
- 55.Turchi J, Bruno JP, Sarter M. Cortical acetylcholine and processing capacity: effects of cortical cholinergic deafferentation on crossmodal divided attention in rats. Soc Neurosci Abstr. 1995;21:763.9. doi: 10.1016/s0926-6410(97)00027-x. [DOI] [PubMed] [Google Scholar]
- 56.Tyrefors N, Carlsson A. Improvements in the separation and detection of acetylcholine and choline using liquid chromatography and electrochemical detection. J Chromatogr. 1990;502:337–339. [Google Scholar]
- 57.Voytko ML, Olton DS, Richardson RT, Gorman LK, Tobin JR, Price DL. Basal forebrain lesions in monkeys disrupt attention but not learning and memory. J Neurosci. 1994;14:167–186. doi: 10.1523/JNEUROSCI.14-01-00167.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wenk GK, Stoehr JD, Quintana G, Mobley S, Wiley RG. Behavioral, biochemical, histological, and electrophysiological effects of 192 IgG-saporin injections into the basal forebrain of rats. J Neurosci. 1994;14:5986–5995. doi: 10.1523/JNEUROSCI.14-10-05986.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Whitehouse PJ, Price DL, Struble RG, Coyle JT, DeLong MR. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- 60.Wiley RG. Neural lesioning with ribosome-inactivating proteins: suicide transport and immunolesioning. Trends Neurosci. 1992;15:285–290. doi: 10.1016/0166-2236(92)90078-m. [DOI] [PubMed] [Google Scholar]
- 61.Wiley RG, Lappi DA. Preparation of anti-neuronal immunotoxins for selective neural immunolesioning. In: Floris G, editor. Neuroscience protocols. Elsevier; Amsterdam: 1993. pp. 275–324. [Google Scholar]