

Abstract
We have generated mice transgenic for a human MDR3 mini-gene, under control of a hamster vimentin promoter. Expression of the MDR3 transgene was found in mesenchymal tissues, peripheral nerves, and the eye lens. These MDR3 transgenic mice have a slowed motor nerve conduction and dysmyelination of their peripheral nerves. An extensive dysmyelination in some transgenic strains results in a severe peripheral neuropathy with paresis of the hind legs. How expression of the MDR3 transgene causes these abnormalities is unknown. TheMDR3 gene encodes a large glycosylated plasma membrane protein with multiple transmembrane spanning domains, which are involved in the translocation of the phospholipid phosphatidylcholine through the hepatocyte canalicular membrane. The ability of the MDR3 P-glycoprotein to alter phospholipid distribution in the plasma membrane of Schwann cells may cause the damage. It is also possible, however, that the presence of a large glycoprotein in the cell membrane may be sufficient to severely disturb myelination of peripheral nerves.
Keywords: peripheral neuropathy, dysmyelination, vimentin promoter, transgenic mice, MDR3, P-glycoprotein
P-glycoproteins (P-gps) are large, glycosylated plasma membrane proteins that can function as ATP-dependent efflux pumps (for review, see Endicott and Ling, 1989; Schinkel and Borst, 1991; Gottesman and Pastan, 1993). P-gps are highly conserved and encoded by two genes in humans, MDR1 (Chen et al., 1986) andMDR3 (also called MDR2) (Chen et al., 1986; Van der Bliek et al., 1987, 1988), and three genes in mouse,mdr1 (or mdr1b), mdr3 (ormdr1a), and mdr2 (Gros et al., 1986a, 1988; Hsu et al., 1989; Devault and Gros, 1990).
The human MDR1 (and the related murine mdr1 and mdr3) P-gps can extrude a wide range of hydrophobic drugs from mammalian cells (Gros et al., 1986b; Ueda et al., 1987; Lincke et al., 1990). Increased levels of these proteins confer multidrug resistance (MDR) in cancer cells. Defense against naturally occurring xenobiotic (toxic) compounds may represent the main physiological function of these P-gps (Schinkel et al., 1994). In contrast, attempts to show that the humanMDR3 or the closely related (91% identity at the amino acid level) mouse mdr2 can confer MDR have been negative thus far (Gros et al., 1988; Van der Bliek et al., 1988; Buschman and Gros, 1991; Schinkel et al., 1991). To find a physiological function for this class of P-gps, we have generated mutant mice that are unable to make the mdr2 P-gp and transgenic mice that overproduce the MDR3 P-gp in many tissues. Mice homozygous for a disrupted mdr2 gene develop liver disease. A detailed analysis of these mice has shown that the mdr2 P-gp is essential for transport of the phospholipid phosphatidylcholine (PC) through the hepatocyte canalicular membrane into the bile (Smit et al., 1993). This indicated that this P-gp is a PC translocator, which was supported by the finding that this P-gp is able to transfer a PC analog through the membrane of yeast membrane vesicles in which it is incorporated (Ruetz and Gros, 1994). The MDR3 P-gp probably has the same function: it promotes the transfer of PC through the plasma membrane of fibroblasts (Smith et al., 1994) and can correct the liver defect in MDR3-transgenic mice lacking mdr2 P-gp (A. J. Smith and P. Borst, unpublished observations).
Here we describe the generation of mice transgenic for a humanMDR3 mini-gene driven by the vimentin promoter. These mice develop a peripheral neuropathy and a severe microphthalmia. The analysis of the abnormalities in the peripheral nervous system is presented in this paper.
MATERIALS AND METHODS
Vimentin expression construct. An expression plasmid was constructed containing the hamster vimentin promoter and polyadenylation sequences (described in Quax et al., 1983). Thevimentin promoter is subcloned as a 3.2 kb BamHI (blunted)–PstI fragment in theEcoRV–PstI sites of the Bluescript SK polylinker (Stratagene, LaJolla, CA). This 3.2 kb fragment directs tissue-specific (i.e., vimentin-like) expression in transgenic mice (Krimpenfort et al., 1988; Pieper et al., 1989). A 3′ vimentin HincII fragment, subcloned in pUC, was used for isolation of the polyadenylation sequences. From this plasmid, a ∼3 kbBclI–XbaI fragment (the restriction sites present in exon 9 and the pUC polylinker, respectively) was cloned in the BamHI–XbaI sites of the vector containing the vimentin promoter (described above). A unique cloning site was obtained by insertion of an HpaI linker (sequence: 5′-GTTAAC) into the SmaI site. This allows cloning of blunt-ended fragments on HpaI digestion. For excision of thevimentin fragment (plus insert) from the vector, the enzymesNotI and ClaI were used.
Modifications of the MDR3 cDNA. A full-lengthMDR3 cDNA [clone 3.27 (Van der Bliek et al., 1988)] was modified to increase the chance of high expression in transgenic mice. Putative mRNA destabilizing sequence elements, present in the 3′ untranslated region, were removed by introduction of an NotI site directly after the translation stop codon by PCR mutagenesis. Furthermore, intron sequences were introduced by replacing aBamHI–ApaI cDNA fragment with the corresponding genomic fragment [containing introns 9–13 (Lincke et al., 1991)]. This MDR3 mini-gene was excised by digestion withNotI, blunted with Klenow fragment of DNA polymerase I, and ligated into an HpaI-digested vimentin expression construct.
Generation of transgenic mice. Fertilized mouse eggs were recovered from the oviducts of superovulated females (mated with males several hours earlier) of the mouse strain FVB. Approximately 2–8 ng of vimentin-MDR3 fragment, free of vector sequences, was microinjected into the pronucleus of fertilized eggs. Microinjected eggs were implanted into the oviducts of 1 d pseudopregnant (C57BL/6 × DBA) F1 foster mothers and carried to term. The presence of the transgene was determined by DNA (Southern) blot analysis of BamHI-digested genomic DNA isolated from mouse tail tips as described by Laird et al. (1991).
Cells and cell culture.MDR3-expressing fibroblasts were generated from mice transgenic for thevimentin-MDR3 mini-gene. V01 fibroblasts were obtained from a mouse heterozygous for thevimentin-MDR3 transgene, and V01V01 fibroblasts were obtained from a mouse that was homozygous for the transgene. Control fibroblast cell lines C and D were derived from nontransgenic FVB mice. Mouse ear fibroblasts were isolated using standard procedures and were immortalized by infection with SV40 virus (Bloemendal et al., 1980). Cells were grown in complete DMEM, i.e., supplemented with 2 mml-glutamine, penicillin (50 U/ml), streptomycin (50 μg/ml), and 10% (v/v) heat-inactivated fetal calf serum, in the presence of 5% CO2 at 37°C.
DNA and RNA analyses. Standard molecular–biological procedures were carried out as described (Sambrook et al., 1989). Total RNA from tissues was isolated by an acidic guanidinium isothiocyanate-phenol-chloroform extraction procedure (Chomczynski and Sacchi, 1987) or prepared by LiCl/urea precipitation (Auffray and Rougeon, 1980). RNA was analyzed by RNase protection as described byZinn et al. (1983) and modified by Baas et al. (1990). The plasmid construct for detection of MDR3 contains a 310 nucleotideHindII-TaqI fragment (Nooter et al., 1990). For detection of gapdh mRNA, a 146 bpBsteII-HindIII fragment from pmGAP was blunted and cloned in the SmaI site of pGEM-3Zf(−). To synthesize antisense RNA probes, we linearized the plasmid templates withBamHI (gapdh) and HindIII (MDR3) and transcribed them with T7 RNA polymerase.32P-labeled RNA transcripts were hybridized with 10 μg of total RNA from the tissue of interest. Protected probe was visualized by electrophoresis through a denaturing 6% acrylamide gel, followed by autoradiography.
Nerve conduction examination. Twelve immature (1–2 months old) and seven adult (>2 months old) animals were used for sciatic nerve conduction examinations. Of the immature mice, five were without paresis and transgenic (V01), two had paresis (V01V01), and five were normal control mice (strain used: FVB). Of the adult mice, two were without paresis (V01), two had paresis (V05), and three were normal. The animals were anesthetized with a mixture of midazolamhydrochloride (50 mg/kg), fluanizone (3.3 mg/kg), and fentanylcitrate (0.1 mg/kg) applied intraperitoneally. Compound muscle action potentials (CMAPs) were recorded from the small foot muscles after stimulation of the right sciatic nerve at the hip and at the knee using needle electrodes (Medelec/Teca “Sapphire” apparatus) (Low and McLeod, 1975). All measurements were performed by the same investigator without knowledge of the transgenic status of the animals.
Histological examination. Transgenic and control animals were anesthetized using pentobarbital and subsequently were transcardially perfused with PBS, followed by periodate-lysine-paraformaldehyde fixation (McLean and Nakane, 1974). Material was excised from the sciatic nerve that had not been electromyographed. The nerves were routinely embedded in epon. Transverse sections of 1 μm were prepared for light microscopy and were stained with p-phenylenediamine (1%). Sections for electron microscopy were contrasted using uranyl acetate and lead citrate.
RESULTS
Generation of MDR3 transgenic mice
To generate transgenic mice with MDR3 P-gp in many tissues, a human MDR3 mini-gene was constructed and subcloned in avimentin expression cassette (Fig.1A). Eight transgenic mice (V01–V08) were generated by introduction of vimentin–MDR3 DNA into mouse oocytes. DNA (Southern) analysis showed that all transgenic lines carried multiple copies of the injectedvimentin-MDR3 DNA fragment (not shown). Only one mouse, V07, did not transmit the transgene to its progeny. All other founders were capable of producing transgenic offspring; however, the offspring of founders V03, V05, and V08 could not be propagated, most likely because of progressive paresis of the hind legs (see below).
Fig. 1.

A, Schematic representation of the mini-gene construct used to generate MDR3 transgenic mice. White boxes represent parts of theMDR3 cDNA; intron sequences of the MDR3 gene are indicated by thin lines. TheMDR3 mini-gene is under the control of the hamstervimentin promoter and polyadenylation signal (thick lines). B, MDR3 mRNA levels in transgenic tissues. Total RNA was isolated from all major tissues and sciatic nerves of a V01 animal and analyzed by RNase protection. RNA isolated from transgenic (T) nerves and fibroblasts was compared with RNA isolated from wild-type (W) mice. The position of theMDR3-specific RNase protection probe is shown inA. The protected fragments representingMDR3, mdr2, and gapdh mRNA are indicated on the right. Because of the partial sequence homology of the MDR3-specific RNA probe with mouse mdr2 sequences, smaller fragments that representmdr2 mRNA were detected in RNA from liver, muscle, heart, and spleen. The expression pattern of this mdr2 mRNA is consistent with previous results (Croop et al., 1989; Teeter et al., 1990). An end-labeled DdeI digest of M13mp19 DNA was used as size marker (M); relevant sizes are indicated at the left.
MDR3 expression in transgenic tissues
To determine MDR3 mRNA levels, total RNA was isolated from all major tissues of the transgenic line V01 and analyzed by RNase protection (Fig. 1B). RNA from peripheral nerves was included in this analysis, because the transgenic mice had abnormal nerve Schwann cells (see below), a cell type in which thevimentin promoter is known to be active (Lazarides, 1982). An MDR3-specific RNA probe was used to discriminate between transgenic expression of MDR3 and the endogenous expression of the highly homologous mouse mdr2 mRNA. The highest level of MDR3 mRNA was found in RNA isolated from the sciatic nerves of V01 transgenic mice (Fig. 1B, lane T of nerve RNA). The MDR3 mRNA level in other tissues was lower and variable. Relatively high levels (comparable to a human liver RNA sample) were found in heart, muscle, brain, eye lens (not shown), and lung; lower levels were present in liver, stomach, intestine, spleen, salivary gland, and kidney.
Fibroblast cell lines were generated from the transgenic mice to determine whether the MDR3 mRNA in the transgenic mice could be translated into a full-size MDR3 P-gp. A high MDR3 mRNA level was found in the fibroblast cell line of founder V01 (Fig.1B, lane T of fibroblast RNA). Distinct plasma membrane staining was detected with the V01 and V01V01 cell lines using MDR3-specific polyclonal antibodies (Smit et al., 1994). In addition, on a Western blot of membranes isolated from these fibroblast cells, MDR3-specific polyclonal antibodies and the monoclonal antibody C219 (which recognizes all mammalian P-gps) reacted with a 140–170 kDa protein at a position similar to that of the endogenously synthesized mouse P-gps (Schinkel et al., 1993; Smit et al., 1994). Thus, transgenic ear fibroblast cells synthesize an MDR3 P-gp of the expected size that is routed to the plasma membrane. By inference we expect that in other tissues a full-length MDR3 P-gp is also synthesized.
Abnormalities in MDR3 transgenic mice
To screen for gross abnormalities, hematoxylin/eosin (H/E)-stained sections of all major tissues in offspring of founder lines V01, V03, V05, and V08 were analyzed by light microscopy. Visible aberrations were detected only in eyes, muscle, and peripheral nerves of the transgenic mice.
Eye abnormalities
The eyes were reduced in size in all transgenic lines (V01–V08). Further analysis of H/E-stained sections of formaldehyde-fixed eyes revealed that the eye lens was severely degenerated and in some cases almost absent. A detailed description of the eye abnormalities is presented elsewhere (Dunia et al., 1996).
Abnormalities in the peripheral nerve system
Some founders (V03, V05, and V08) generated transgenic offspring that developed a progressive flaccid paresis of the hind legs (see Table 1), eventually leading to a complete paresis of the hind legs. No transgenic lines could be established from this offspring because of the development of the paresis before adulthood. Viable and fertile offspring without paresis were generated from transgenic lines V01, V04, and V06. After crossing two transgenic V01 mice, however, ∼25% of the offspring showed paresis of the hindlegs. These mice were probably homozygous for the transgene (and are therefore called V01V01), which suggests that the severity of the paresis is related to the level of MDR3 expression.
Table 1.
Paresis in MDR3 transgenic mouse strains
| Founder | Apparent |
|---|---|
| V01 | − |
| V01V01 | ++ |
| V02 | − |
| V03 | + |
| V04 | − |
| V05 | + |
| V06 | − |
| V08 | ++ |
To determine the cause of the progressive paresis, the neuromuscular system was analyzed in detail. Light microscopical analysis of V05 muscle showed signs of reinnervation (e.g., type grouping) in affected animals, indicative of a neurogenic cause of the paresis. Electrophysiological analysis of the transgenic mice (Table2) showed that the motor nerve conduction velocities (MNCVs) were extremely low (range, 3.5–5.2 m/sec). The distal latencies were prolonged, and although the compound muscle action potential (CMAP) amplitudes were dispersed, it was reduced in three of the four groups of transgenic mice. In the V01 mice without visible signs of paresis, MNCVs were intermediate between the values of wild-type mice and mice with paresis (V01V01 and V05). These results are compatible with the muscular abnormalities that were detected. The MNCVs, distal latencies, and CMAP amplitudes in the normal adult controls were comparable to those obtained by other investigators in mice (Low and McLeod, 1975), with MNCVs of the 1- to 2-month-old normal mice somewhat lower than those of the adult animals. Light microscopical analysis of the spinal cord and the brain showed no apparent abnormalities (not shown), but examination of the sciatic nerves of paralyzed V01V01 and V05 transgenic mice showed nerve fibers with thin myelin sheaths and a diffuse loss of myelinated nerve fibers (Fig. 2C). Myelin loss was observed, but to a lesser extent in sciatic nerves of V01 mice without paresis (Fig.2B). The sciatic nerve of the V01 mice was analyzed at 11 d, 18 d, and 1 month. Alterations in the axon density and thickness of the myelin sheath were already detected at 11 d. These did not seem to progress during the first month of life, which suggests that the initial formation of myelin is defective, e.g., dysmyelination. These abnormalities were absent in control mice (Fig.2A). The nerves of the V01V01 transgenic mice contained macrophages with vacuolated cytoplasm and abundant phagosomes and lysosomes. Macrophages are often observed in biopsies showing nerve fiber and myelin degradation and regeneration (for review, see Perry and Brown, 1992). Macrophages were absent in controls. Electron microscopy revealed distended rough endoplasmic reticulum cisternae (RER) filled with electron-dense material in part of the Schwann cells (Fig. 3A–C). Apart from the RER abnormalities and the thin myelin sheaths, no abnormalities were observed in the Schwann cells. The periodicity of the myelin lamellae did not differ from that of controls.
Table 2.
Motor nerve conduction velocities (MNCVs) and compound muscle action potentials (CMAPs), measured in the hind legs of transgenic (V01 and V05) and wild-type mouse strains
| Mouse strain | Paresis | MNCV (m/sec) mean (range) | Distal latency (msec) mean (range) | CMAP amplitude (mV) mean (range) | |
|---|---|---|---|---|---|
| Wild type | Immature2_a n = 5 | No | 27.7 (15.2–36.6) | 1.2 (1.0–1.4) | 3.8 (2.0–6.1) |
| Wild type | Adult2_b n = 3 | No | 37.5 (31.9–40.6) | 1.2 (1.1–1.4) | 6.2 (1.6–10.3) |
| V01 | Immature n = 5 | No | 16.6 (11.5–22.0) | 1.6 (1.2 –1.9) | 3.3 (1.3–5.4) |
| V01 | Adult n = 2 | No | 12.6 (12.0–13.1) | 1.7 (1.1–2.2) | 1.6 (1.4–1.8) |
| V01V01 | Immature n = 2 | Yes | 3.9 (3.5–4.2) | 2.5 (2.5) | 0.3 (0.2–0.4) |
| V05 | Adult n = 2 | Yes | 4.4 (3.6–5.2) | 4.4 (3.8–4.9) | 0.9 (0.1–1.7) |
1–2 months of age.
Above 2 months of age.
Fig. 2.
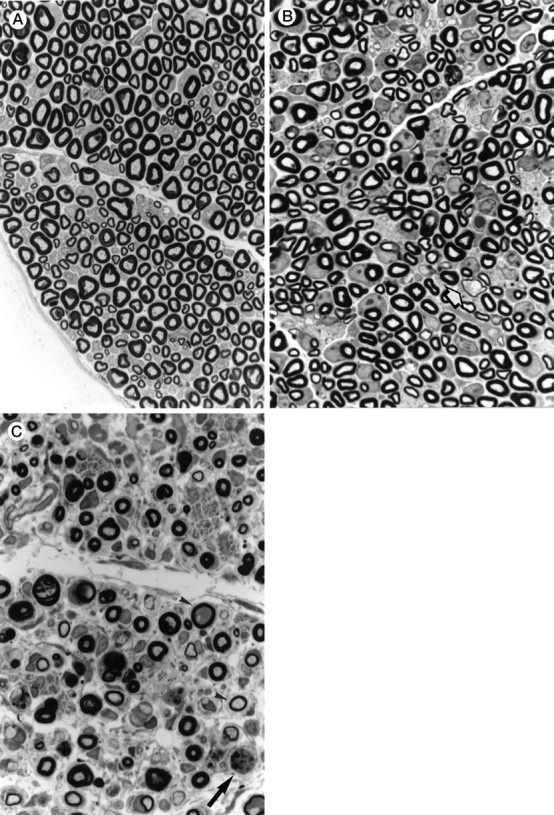
Light microscopy of an epon-embedded sciatic nerve stained with p-phenylenediamine (850×).A, Control. B, Section of a sciatic nerve from a V01 MDR3 transgenic mouse (11 d old) showing a decreased density of myelinated axons and several thinned myelin sheaths (arrow), compared with sections from control mice of the same age (not shown). C, Section of a sciatic nerve from a V01V01 MDR3 transgenic mouse, showing loss of myelinated nerve fibers, relatively thin myelin sheaths (arrowhead), and macrophage with myelin degradation products (arrow).
Fig. 3.
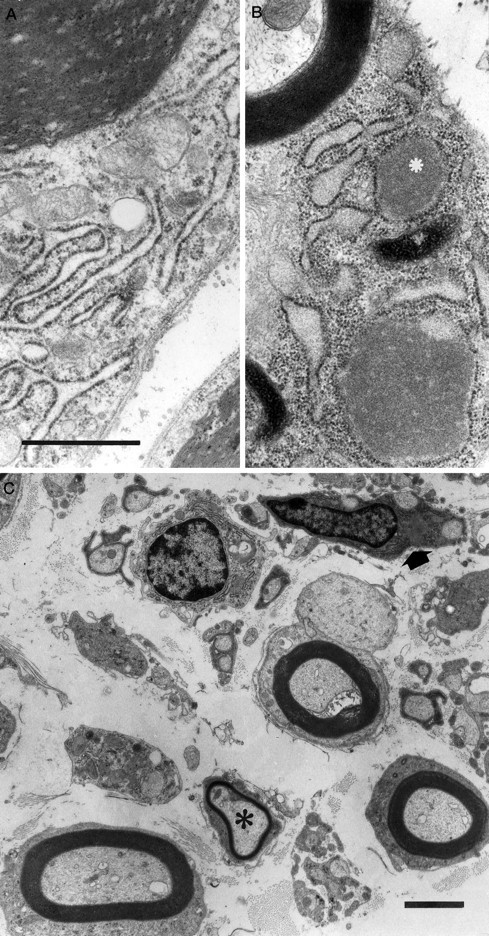
Electron microscopy of Schwann cells of myelinated axons. A, Control. Observe the slender RER cisterns. B, Section of a sciatic nerve from a V01V01MDR3 transgenic mouse showing widened RER cisterns filled with homogeneous electron dense material (asterisk). Note the presence of normal myelin lamellae. Scale bar, 1 μm. C, Electron micrograph of a sciatic nerve of a V01V01 MDR3 transgenic mouse showing an axon with a thinned myelin sheath (asterisk) and a Schwann-cell (arrow) with two axons, with distended RER cisterns filled with electron-dense material within the Schwann-cell cytoplasm. Scale bar, 2 μm.
DISCUSSION
Our results show that mice transgenic for an MDR3 P-gp mini-gene develop a paresis. The absence of primary abnormalities in muscle, skeleton, and joints, the reduced nerve conductance velocities, and the severe pathological abnormalities in the sciatic nerves indicate that the primary cause of the paresis is the dysmyelination of the peripheral nerves. The vimentin promoter directs gene expression in Schwann cells (Lazarides, 1982), the major cellular component of peripheral nerves. This explains the high level ofMDR3 mRNA found in sciatic nerves. It is also the Schwann cell that is abnormal, which suggests that the overproduction of MDR3 P-gp in Schwann cells is the cause of dysmyelination and therefore the paresis.
In humans, alterations in the genes encoding the peripheral myelin protein 22 (PMP22), protein zero (P0), and connexin 32 (Cx32) (for review, see Patel and Lupski, 1994) are associated with hereditary motor and sensory neuropathy type 1 (HMSN), a dominant progressive neuropathy with signs of demyelination and remyelination. In all three cases, the Schwann cells are abnormal, resulting in dysmyelination and strongly reduced MNCVs. Two types of mutations affecting those membrane-associated proteins are found in HMSN. First, in humanPMP22, P0, and Cx32, mutations were found in the coding region, potentially altering the structure of the encoded protein. Mutations in mouse pmp22 were also found in the neurological mouse mutant Trembler(Low and McLeod, 1975; Suter et al., 1992). Alternatively, duplication or deletion of large chromosomal regions was found, suggesting that gene dosage effects play a role. For example, a majority of HMSN patients have a duplication of a large DNA segment encompassing thePMP22 gene. The structure of the PMP22 gene is not altered in this case. In addition, deletion of one copy ofPMP22 is also associated with a peripheral neuropathy [hereditary neuropathy with liability to pressure palsies (Chance et al., 1993)]. A gene dosage effect is also found for another protein, the proteolipid protein (PLP) in the central nervous system. Duplication of the PLP gene is associated with Pelizaeus-Merzbacher disease, also characterized by dysmyelination (Malcolm, 1994; Readhead et al., 1994). These results suggest that the amounts of some myelin proteins have to remain between narrow limits: a twofold reduction or an increase of these proteins can result in a progressive dysmyelination and neuropathy. Recently, this has been elegantly demonstrated by the generation of mice with inactivated alleles of integral myelin proteins PMP22 and P0 (Adlkofer et al., 1995; Martini et al., 1995). These findings illustrate that alterations in the primary sequence or the amount of proteins localized in the membrane of Schwann cells can result in a peripheral neuropathy.
How does expression of the MDR3 transgene result in a peripheral neuropathy? In view of the mutations identified in HMSN, it is possible that overproduction of a large membrane protein, the MDR3 P-gp, in the Schwann cell could directly interfere with the function(s) or amount of proteins normally present in the plasma membrane, e.g., P0, PMP22, and/or Cx32. This could be accomplished by steric hindrance or by (non)specific binding to the membrane proteins needed for normal myelination. A second possibility is that a high production of MDR3 P-gp in the RER of Schwann cell interferes with the normal processing and sorting mechanisms and thereby reduces the amount of myelin proteins that is in the membrane. The presence of electron-dense deposits in the RER of the Schwann cells could indicate that the processing and sorting mechanism is overloaded. A third explanation is that the abnormalities are caused by the PC translocator activity of the MDR3 P-gp. In the absence of bile salts, the activity of the mdr2 P-gp does not lead to net transport of PC from the cell (Oude Elferink et al., 1995), but it can still be expected to cause an abnormal distribution of PC between the outer and inner leaflet of the cell membrane and to an increased movement of PC between the leaflets (Smith et al., 1994). This could interfere with the formation of a normal myelin sheath by Schwann cells.
In summary, high expression of MDR3 in Schwann cells results in a progressive paresis attributable to dysmyelination of the peripheral nerve system. There are some similarities with HMSN found in humans, but the presence of electron-dense material in the RER makes this a mouse model with a distinct pathology. The transgenic MDR3 mice may be useful for analyzing the processes that interfere with myelination. A more detailed developmental analysis will be required to show whether MDR3 overexpression affects the initial steps in myelin formation, e.g., compaction, or whether it results in progressive deterioration of normal myelinated neurons. In the latter case, these mice can be a useful tool for analyzing the effects of neurotrophic factors on nerve regeneration.
Footnotes
Received August 11, 1995; revised June 11, 1996; accepted July 30, 1996.
This work was supported in part by Grants NKI 88-6 and NKI 92-41 of the Dutch Cancer Society to P.B. We thank G. J. van Bruggen for assisting with the nerve conduction measurements and Professor Dr. J. M. B. V. de Jong and Professor Dr. F. G. I. Jennekens for help in the initial analysis of the transgenic mice. We acknowledge H. Eelderink and H. Veltman for histological preparations of the peripheral nerves.
Correspondence should be addressed to Dr. Piet Borst, The Netherlands Cancer Institute, Division of Molecular Biology, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands.
REFERENCES
- 1.Adlkofer K, Martini R, Aguzzi A, Zielasek J, Toyka KV, Suter U. Hypermyelination and demyelination peripheral neuropathy in Pmp22-deficient mice. Nature Genet. 1995;11:275–280. doi: 10.1038/ng1195-274. [DOI] [PubMed] [Google Scholar]
- 2.Auffray C, Rougeon F. Purification of mouse immunoglobulin heavy-chain messenger RNAs from total myeloma tumor RNA. Eur J Biochem. 1980;107:303–314. doi: 10.1111/j.1432-1033.1980.tb06030.x. [DOI] [PubMed] [Google Scholar]
- 3.Baas F, Jongsma APM, Broxterman HJ, Arceci RJ, Housman D, Scheffer GL, Riethorst A, van Groenigen M, Nieuwint AW, Joenje H. Non-P-glycoprotein mediated mechanism for multidrug resistance precedes P-glycoprotein expression during in vitro selection for doxorubicin resistance in a human lung cancer cell line. Cancer Res. 1990;50:5392–5398. [PubMed] [Google Scholar]
- 4.Bloemendal H, Lenstra JH, Dodemont H, Ramaekers FCS, Groeneveld AA, Dunia I, Benedetti EL. SV40-transformed hamster lens epithelial cells: a novel system for the isolation of cytoskeletal messenger RNAs and their translation products. Exp Eye Res. 1980;31:513–525. doi: 10.1016/s0014-4835(80)80010-8. [DOI] [PubMed] [Google Scholar]
- 5.Buschman E, Gros P. Functional analysis of chimeric genes obtained by exchanging homologous domains of the mouse mdr1 and mdr2 genes. Mol Cell Biol. 1991;11:595–603. doi: 10.1128/mcb.11.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chance PF, Alderson MK, Leppig KA, Lensch MW, Matsunami N, Smith B, Swanson PD, Odelberg SJ, Disteche CM, Bird TD. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143–151. doi: 10.1016/0092-8674(93)90058-x. [DOI] [PubMed] [Google Scholar]
- 7.Chen CJ, Chin JE, Ueda K, Clark DP, Pastan I, Gottesman MM, Roninson IB. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell. 1986;47:381–389. doi: 10.1016/0092-8674(86)90595-7. [DOI] [PubMed] [Google Scholar]
- 8.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 9.Croop JM, Raymond M, Haber D, Devault A, Arceci RJ, Gros P, Housman DE. The three mouse multidrug resistance (mdr) genes are expressed in a tissue-specific manner in normal mouse tissues. Mol Cell Biol. 1989;9:1346–1350. doi: 10.1128/mcb.9.3.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Devault A, Gros P. Two members of the mouse mdr gene family confer multidrug resistance with overlapping but distinct drug specificities. Mol Cell Biol. 1990;10:1652–1663. doi: 10.1128/mcb.10.4.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunia I, Smit JJM, van der Valk MA, Bloemendal H, Borst P, Benedetti EL. Human multidrug resistance 3-P-glycoprotein expression in transgenic mice induces lens membrane alterations leading to cataract. J Cell Biol. 1996;132:701–716. doi: 10.1083/jcb.132.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Endicott JA, Ling V. The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu Rev Biochem. 1989;58:137–171. doi: 10.1146/annurev.bi.58.070189.001033. [DOI] [PubMed] [Google Scholar]
- 13.Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- 14.Gros P, Croop J, Housman D. Mammalian multidrug resistance gene: complete cDNA sequence indicates strong homology to bacterial transport proteins. Cell. 1986a;47:371–380. doi: 10.1016/0092-8674(86)90594-5. [DOI] [PubMed] [Google Scholar]
- 15.Gros P, Neriah YB, Croop JM, Housman DE. Isolation and expression of a cDNA (mdr) that confers multidrug resistance. Nature. 1986b;323:728–731. doi: 10.1038/323728a0. [DOI] [PubMed] [Google Scholar]
- 16.Gros P, Raymond M, Bell J, Housman D. Cloning and characterization of a second member of the mouse mdr gene family. Mol Cell Biol. 1988;8:2770–2778. doi: 10.1128/mcb.8.7.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu SI, Lothstein L, Horwitz SB. Differential overexpression of three mdr gene family members in multidrug-resistant J774.2 mouse cells: evidence that distinct P-glycoprotein precursors are encoded by unique mdr genes. J Biol Chem. 1989;264:12053–12062. [PubMed] [Google Scholar]
- 18.Krimpenfort PJ, Schaart G, Pieper FR, Ramaekers FC, Cuypers HT, van den Heuvel RM, Vree Egberts WT, van Eys GJ, Berns A, Bloemendal H. Tissue-specific expression of a vimentin-desmin hybrid gene in transgenic mice. EMBO J. 1988;7:941–947. doi: 10.1002/j.1460-2075.1988.tb02899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laird PW, Zijderveld A, Linders K, Rudnicki MA, Jaenish R, Berns A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazarides E. Intermediate filaments: a chemically heterogeneous, developmentally regulated class of proteins. Annu Rev Biochem. 1982;51:219–250. doi: 10.1146/annurev.bi.51.070182.001251. [DOI] [PubMed] [Google Scholar]
- 21.Lincke CR, Van der Bliek AM, Schuurhuis GJ, Van der Velde-Koerts T, Smit JJM, Borst P. Multidrug resistance phenotype of human BRO melanoma cells transfected with a wild-type human mdr1 complementary DNA. Cancer Res. 1990;50:1779–1785. [PubMed] [Google Scholar]
- 22.Lincke CR, Smit JJM, Van der Velde Koerts T, Borst P. Structure of the human MDR3 gene and physical mapping of the human MDR locus. J Biol Chem. 1991;266:5303–5310. [PubMed] [Google Scholar]
- 23.Low PA, McLeod JG. Hereditary demyelinating neuropathy in the Trembler mouse. J Neurol Sci. 1975;26:565–574. doi: 10.1016/0022-510x(75)90057-x. [DOI] [PubMed] [Google Scholar]
- 24.Malcolm DE. Proteolipid protein gene dosage effect in Pelizaeus-Merzbacher disease. Nature Genet. 1994;6:333–334. doi: 10.1038/ng0494-333. [DOI] [PubMed] [Google Scholar]
- 25.Martini R, Zielasek J, Toyka KV, Giese P, Schachner M. Protein zero (P0)-deficient mice show myelin degeneration in peripheral nerves characteristic of inherited human neuropathies. Nature Genet. 1995;11:281–286. doi: 10.1038/ng1195-281. [DOI] [PubMed] [Google Scholar]
- 26.McLean IW, Nakane PK. Periodate-lysine-paraformaldehyde fixative: a new fixative for immunoelectron microscopy. J Histochem Cytochem. 1974;22:1077–1083. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- 27.Nooter K, Sonneveld P, Janssen A, Oostrum R, Boersma T, Herweijer H, Valerio D, Hagemeijer A, Baas F. Expression of the mdr3 gene in prolymphocytic leukemia: association with cyclosporin-A-induced increase in drug accumulation. Int J Cancer. 1990;45:626–631. doi: 10.1002/ijc.2910450409. [DOI] [PubMed] [Google Scholar]
- 28.Oude Elferink RPJ, Ottenhoff R, van Wijland M, Smit JJM, Schinkel AH, Groen AK. Regulation of biliary lipid secretion by mdr2 P-glycoprotein in the mouse. J Clin Invest. 1995;95:31–38. doi: 10.1172/JCI117658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel PI, Lupski JR. Charcot-Marie-Tooth disease: a new paradigm for the mechanism of inherited disease. Trends Genet. 1994;10:128–133. doi: 10.1016/0168-9525(94)90214-3. [DOI] [PubMed] [Google Scholar]
- 30.Perry VH, Brown MC. Role of macrophages in peripheral nerve degeneration and repair. Bioessays. 1992;14:401–406. doi: 10.1002/bies.950140610. [DOI] [PubMed] [Google Scholar]
- 31.Pieper FR, Schaart G, Krimpenfort PJ, Henderik JB, Moshage HJ, van de Kemp A, Ramaekers FC, Berns A, Bloemendal H. Transgenic expression of the muscle-specific intermediate filament protein desmin in nonmuscle cells. J Cell Biol. 1989;108:1009–1024. doi: 10.1083/jcb.108.3.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quax W, Vree Egberts W, Hendriks W, Quax-Jeuken Y, Bloemendal H. The structure of the vimentin gene. Cell. 1983;35:215–223. doi: 10.1016/0092-8674(83)90224-6. [DOI] [PubMed] [Google Scholar]
- 33.Readhead C, Schneider A, Griffiths I, Nave K-A. Premature arrest of myelin formation in transgenic mice with increased proteolipid protein gene dosage. Neuron. 1994;12:583–595. doi: 10.1016/0896-6273(94)90214-3. [DOI] [PubMed] [Google Scholar]
- 34.Ruetz S, Gros P. Phosphatidylcholine translocase: a physiological role for the mdr2 gene. Cell. 1994;77:1071–1081. doi: 10.1016/0092-8674(94)90446-4. [DOI] [PubMed] [Google Scholar]
- 35.Sambrook J, Fritsch EF, Maniatis T. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. Molecular cloning: a laboratory manual, 2nd Ed. . [Google Scholar]
- 36.Schinkel AH, Borst P. Multidrug resistance mediated by P-glycoproteins. Semin Cancer Biol. 1991;2:213–226. [PubMed] [Google Scholar]
- 37.Schinkel AH, Roelofs MEM, Borst P. Characterization of the human MDR3 P-glycoprotein and its recognition by P-glycoprotein-specific monoclonal antibodies. Cancer Res. 1991;51:2628–2635. [PubMed] [Google Scholar]
- 38.Schinkel AH, Arceci RJ, Smit JJM, Wagenaar E, Baas F, Dollé M, Tsuruo T, Mechetner EB, Roninson IB, Borst P. Binding properties of monoclonal antibodies recognizing external epitopes of the human MDR1 P-glycoprotein. Int J Cancer. 1993;55:478–484. doi: 10.1002/ijc.2910550326. [DOI] [PubMed] [Google Scholar]
- 39.Schinkel AH, Smit JJM, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, Mol CAAM, van der Valk MA, Robanus-Maandag EC, te Riele HPJ, Berns AJM, Borst P. Disruption of the mouse mdr1a P-glycoprotein gene leads to deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 40.Smit JJM, Schinkel AH, Oude Elferink RPJ, Groen AK, Wagenaar E, van Deemter L, Mol CAAM, Ottenhoff R, van der Lugt NMT, van Roon MA, van der Valk MA, Offerhaus GJA, Berns AJM, Borst P. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451–462. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 41.Smit JJM, Schinkel AH, Mol CAAM, Majoor D, Mooi WJ, Jongsma APM, Lincke CR, Borst P. The tissue distribution of the human MDR3 P-glycoprotein. Lab Invest. 1994;71:638–649. [PubMed] [Google Scholar]
- 42.Smith AJ, Timmermans-Hereijgers JLPM, Roelofsen B, Wirtz KWA, van Blitterswijk WJ, Smit JJM, Schinkel AH, Borst P. The human MDR3 P-glycoprotein promotes translocation of phosphatidylcholine through the plasma membrane of fibroblasts from transgenic mice. FEBS Lett. 1994;354:263–266. doi: 10.1016/0014-5793(94)01135-4. [DOI] [PubMed] [Google Scholar]
- 43.Suter U, Welcher AA, Özcelik T, Snipes GJ, Kosaras B, Francke U, Billings-Gagliardi S, Sidman RL, Shooter EM. Trembler mouse carries a point mutation in a myelin gene. Nature. 1992;356:241–244. doi: 10.1038/356241a0. [DOI] [PubMed] [Google Scholar]
- 44.Teeter LD, Becker FF, Chisari FV, Li D, Kuo MT. Overexpression of the multidrug resistance gene mdr3 in spontaneous and chemically induced mouse hepatocellular carcinomas. Mol Cell Biol. 1990;10:5728–5735. doi: 10.1128/mcb.10.11.5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ueda K, Cardarelli C, Gottesman MM, Pastan I. Expression of a full-length cDNA for the human “MDR1” gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc Natl Acad Sci USA. 1987;84:3004–3008. doi: 10.1073/pnas.84.9.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van der Bliek AM, Baas F, Ten Houte de Lange T, Kooiman PM, Van der Velde-Koerts T, Borst P. The human mdr3 gene encodes a novel P-glycoprotein homologue and gives rise to alternatively spliced mRNAs in liver. EMBO J. 1987;6:3325–3331. doi: 10.1002/j.1460-2075.1987.tb02653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van der Bliek AM, Kooiman PM, Schneider C, Borst P. Sequence of mdr3 cDNA encoding a human P-glycoprotein. Gene. 1988;71:401–411. doi: 10.1016/0378-1119(88)90057-1. [DOI] [PubMed] [Google Scholar]
- 48.Zinn K, Dimaio D, Maniatis T. Identification of two distinct regulatory regions adjacent to the human β-interferon gene. Cell. 1983;34:865–879. doi: 10.1016/0092-8674(83)90544-5. [DOI] [PubMed] [Google Scholar]