

Abstract
γ-Aminobutyric acid (GABA)A receptor-mediated inhibitory synaptic transmission in visual cortex undergoes long-term potentiation (LTP), which is input-specific and associative. The present study, conducted under a blockade of ionotropic glutamate receptors, demonstrates an induction mechanism of LTP considerably different from those of associative LTP at excitatory synapses. Inhibitory responses of layer V cells evoked by layer IV stimulation were studied in developing rat visual cortex slices by using intracellular and whole-cell recording methods. LTP induction was prevented by the application of an antagonist for GABABreceptors but not for GABAA or metabotropic glutamate receptors. Inhibition of postsynaptic G-proteins, phospholipase C, inositol trisphosphate (IP3) receptors, or Ca2+increase prevented the generation of LTP, as did the blockade of GABAB receptors.
In rat cerebral cortex, GABAB receptor activation is not known to affect the IP3 level by itself. However, it facilitates IP3 formation induced by the activation of α1 adrenoceptors, which are believed to be located postsynaptically. Accordingly, I examined the involvement of these and other amine receptors, including histamine H1, muscarinic acetylcholine, and serotonin 5-HT2 receptors, all of which are coupled to IP3 formation. Only the blockade of α1 adrenoceptors or serotonin 5-HT2 receptors prevented LTP induction in most, but not all, of the cells. These results suggest that LTP induction requires the activation of postsynaptic GABAB receptors and that its effect is mediated at least partly by facilitation of the monoamine-induced IP3 formation, which then causes Ca2+ release from the internal stores in postsynaptic cells.
Keywords: long-term potentiation, inhibitory synaptic transmission, visual cortex, GABAA receptor, GABAB receptor, α1 adrenoceptor, serotonin 5-HT2 receptor, phospholipase C, inositol trisphosphate, G-protein, Ca2+
Long-term potentiation (LTP) of synaptic transmission is considered to be a cellular process underlying memory and learning. Although LTP has been found and analyzed mostly at glutamatergic excitatory synapses (Teyler and DiScenna, 1987), I have found recently that LTP also occurs in γ-aminobutyric acid (GABA)-mediated inhibitory synaptic transmission of visual cortical cells (Komatsu and Iwakiri, 1993). This synaptic modification could underlie the experience-dependent development of visual responsiveness in these cells, because it is induced more easily in developing than in mature animals (Komatsu, 1994). LTP at the inhibitory synapses has properties similar to those seen in most LTPs at excitatory synapses (Teyler and DiScenna, 1987). They occur specifically at synapses activated by conditioning stimulation. In addition, they require coactivation of more than a threshold number of presynaptic fibers for their induction and consequently are associative, which could be a basis of some form of learning.
Although LTPs at the excitatory and inhibitory synapses have similar basic properties, the induction mechanism seems different. At the excitatory synapses of hippocampal CA1 pyramidal cells, LTP is initiated by Ca2+ entry into postsynaptic cells through NMDA receptor channels (Collingridge et al., 1983; Lynch et al., 1983). The voltage dependence of these channels could explain the voltage dependence of LTP induction and, consequently, the associativity of LTP (Collingridge and Bliss, 1987; Gustafsson and Wigström, 1988;Madison et al., 1991). In contrast, our recent work has suggested that LTP induction at the inhibitory synapses is not dependent on postsynaptic membrane potential (Komatsu, 1994), indicating that it is based on other mechanisms. The present study was undertaken to determine which receptors are involved in the induction of LTP and to test whether postsynaptic cells participate in the induction.
MATERIALS AND METHODS
Slice preparation. As described previously (Komatsu, 1994), coronal slices (400 μm thick) of visual cortex were prepared from Sprague Dawley rats at postnatal days 15–25 and perfused with a medium containing (in mm): 124 NaCl, 5 KCl, 1.3 MgSO4, 4 CaCl2, 1.2 KH2PO4, 26 NaHCO3, and 10 glucose at 33°C. During recording experiments, the perfusate contained 100 μmdl-2-amino-5-phosphonovaleric acid (APV), an NMDA receptor antagonist, and 40 μm6,7-dinitroquinoxaline-2,3-dione (DNQX), a non-NMDA receptor antagonist. In experiments in which GABAA receptors were blocked by adding bicuculline methiodide during conditioning stimulation, 200 μmd-APV or 400 μmdl-APV was applied instead of 100 μmdl-APV to ensure that NMDA receptors were blocked even during conditioning stimulation.
Stimulation. Two pairs of bipolar stimulating electrodes (s1 and s2) were placed in layer IV (see Fig. 1A). Layers II–IV were surgically cut between the two stimulating electrodes to ensure that they activated separate groups of presynaptic fibers (Komatsu, 1994). Test stimulation was applied alternately to s1 and s2 at an interval of 5 sec. The intensity of test stimuli was adjusted to 1.5–2.0 times the threshold intensity (1.5–2.0 T) to evoke an inhibitory postsynaptic potential (IPSP) or current (IPSC). As a conditioning stimulation, 50 Hz, 1 sec stimulation was applied to one of the electrodes 10 times at an interval of 10 sec with an intensity of 5 T, unless otherwise mentioned.
Fig. 1.
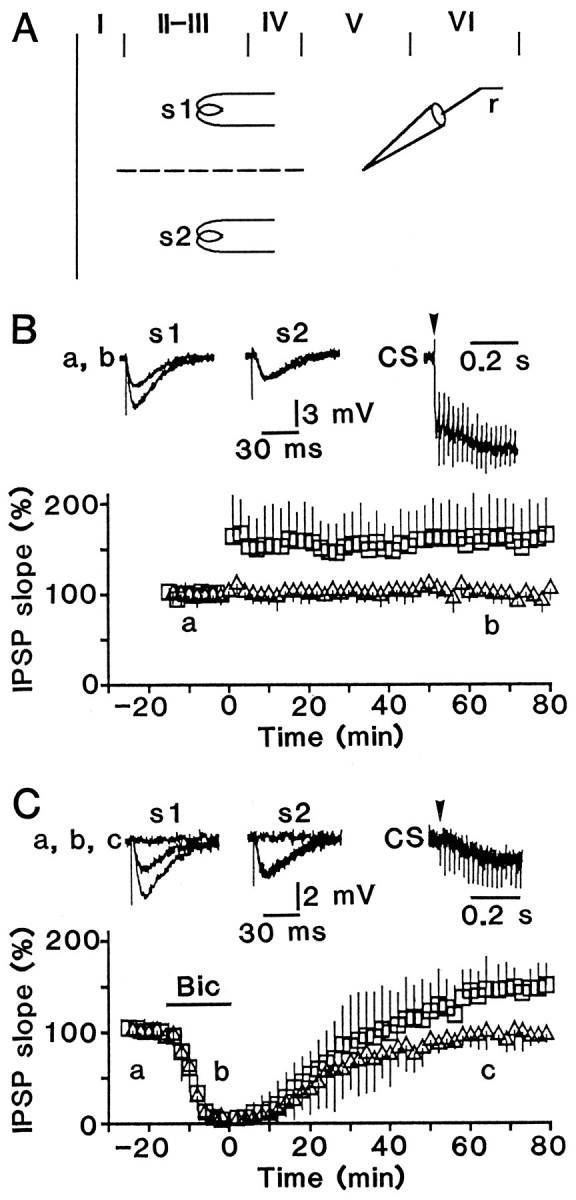
LTP is induced under blockade of GABAAreceptors. A, Experimental arrangement of stimulating (s1 and s2) and recording electrodes (r). The dashed line indicates a surgical cut between s1 and s2. B, LTP in control solution. Left traces (a,b) show superimposed average (n = 4) IPSPs intracellularly recorded from a cell before and after conditioning stimulation for conditioned (s1) and unconditioned (s2) pathways. Recorded time is indicated in the lower graph. Right trace(CS) shows a response evoked by conditioning stimulation, and the start is indicated by an arrowhead. Voltage calibration is the same for all traces. The lower graph plots the falling slope of IPSP (% of the mean baseline level) against the time after conditioning stimulation.Squares and triangles (mean ± SD for 13 tested cells) represent responses of conditioned and unconditioned pathways, respectively. There was no significant difference (p > 0.1) between the resting membrane potential before (57 ± 5 mV; n = 13) and 60 min after (57 ± 6 mV) conditioning stimulation. The effect of conditioning stimulation is illustrated similarly in the following figures. C, Similar to B, but conditioning stimulation was given during addition of 30 μm bicuculline methiodide to the perfusate, and the drug application period is indicated by a bar(Bic) in the lower graph (average for 6 cells). There was no significant difference (p > 0.6) between the resting membrane potential 20 min before (56 ± 4 mV; n = 6) and 60 min after (55 ± 4 mV) conditioning stimulation.
Intracellular recording. IPSPs were recorded with microelectrode pipettes containing 2 m K-methylsulfate (90–150 mΩ). Cells with a stable resting membrane potential more hyperpolarized than −50 mV were selected for analysis. When the resting membrane potential was very hyperpolarized and consequently the amplitude of IPSP evoked by test stimulation was too small, the membrane potential was depolarized by current injection through the recording electrode to increase IPSP amplitude. In part of the experiments, intracellular recording was made with pipettes containing 3 m Cs-acetate (90–150 mΩ). In these cells, the resting membrane potential was depolarized gradually over a time period after penetration, and hyperpolarizing currents were injected when cells fired at the resting membrane potential. Therefore, the effect of antagonists on the synaptic responses was analyzed for cells that showed only a small change in the resting membrane potential during application of the antagonists. Input resistance was monitored throughout the experiments by injecting 0.1 nA hyperpolarizing current pulses of 300 msec duration, and it did not change significantly in association with LTP as described previously (Komatsu and Iwakiri, 1993). A conventional bridge circuit was used to record the membrane potential while current injection was made through the recording electrode.
Whole-cell recording. IPSCs were recorded with patch pipettes in the whole-cell mode (Axopatch 200A, Axon Instruments, Foster City, CA). Patch pipettes were pulled from thin-wall borosilicate glass and had a resistance of 3–6 mΩ. They were filled with a solution containing (in mm): 125 Cs-gluconate, 40 HEPES, 1 EGTA, 5 MgCl2, 2 Na-ATP, and 0.6 Na-GTP, pH 7.2 (with CsOH) for the control experiments to test the effect of guanosine 5′-O-(2-thiodiphosphate) (GDPβS) or guanosine 5′-O-(3-thiotriphosphate)(GTPγS) on LTP. When GTP was replaced with GDPβS or GTPγS, or when EGTA was replaced with 10 mmbis(2-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (BAPTA), the concentration of Cs-gluconate was adjusted so that the osmolarity of the solution was unchanged. Cs-gluconate was replaced with K-gluconate, pH 7.2 (with KOH) when the effect of baclofen was tested. Cells with a high seal resistance (>1 GΩ) and a series resistance < 30 mΩ (12–30 mΩ) were selected for analysis. The series resistance, monitored throughout the recording, was not compensated. Input resistance was 100–500mΩ at −50 mV. Unless otherwise mentioned, cells were voltage-clamped at +20 mV during the test stimulation to record a large amplitude of IPSCs by increasing the driving force on Cl−, which permeates GABAA receptor channels. They were also held at +20 mV during conditioning stimulation. LTP was induced in the same way at the depolarized (+20 mV) or hyperpolarized (−90 mV) membrane potential (Komatsu, 1994). Stable responses were recorded for a longer period at the depolarized membrane potential, as compared with the hyperpolarized membrane potential in our experimental conditions. In the experiments that examined the effects of GDPβS, GTPγS, BAPTA, heparin, or 1-[6-[[17β-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione (U73122) on LTP, tests were started 10 min after establishing whole-cell recording to allow diffusion of these compounds into the cell.
Data acquisition, statistical analysis, histology, and drugs.Data sampling was performed by a computer at an interval of 0.2 msec for most responses but at 10 sec for baclofen-induced outward currents. The statistical test used was Student’s t test. The laminar location of stimulation and recording electrodes was identified on histological sections stained with cresyl violet after the recording experiments (Komatsu, 1994). The compounds were obtained from the following sources: 2-hydroxysaclofen, (+)-α-methyl-4-carboxyphenylglycine [(+)-MCPG], and DNQX from Tocris Cookson (Bristol, UK); atropine, APV, baclofen, bicuculline methiodide, GDPβS, GTPγS, heparin, and pyrilamine from Sigma (St. Louis, MO); prazosin and ketanserin from Research Biochemicals International (Natick, MA); BAPTA from Dojindo Laboratories (Kumamoto, Japan); and U73122 from Wako Pure Chemical (Osaka, Japan).
RESULTS
LTP of inhibitory synaptic transmission was studied in visual cortical slices prepared from developing rats in which the LTP is easily induced (Komatsu, 1994). IPSPs or IPSCs evoked monosynaptically by layer IV stimulation were recorded from layer V cells while slices were perfused with a control solution containing non-NMDA and NMDA receptor antagonists to block excitatory synaptic transmission. One of the two pairs of stimulating electrodes placed in layer IV (s1 and s2 in Fig.1A) was used to test the effect of high-frequency conditioning stimulation; the other served as a control.
LTP occurs under blockade of GABAA receptors
To test whether activation of GABAA receptors, which mediate test IPSPs in these cells (Komatsu and Iwakiri, 1993), is necessary to induce LTP, a high dose of bicuculline methiodide (30 μm), a selective GABAA receptor antagonist, was added to the control solution. When test responses were abolished completely, conditioning stimulation was applied. After washout of the antagonist, LTP was manifested at a magnitude comparable to those in the control solution (Fig. 1B,C). This result suggests that substantial activation of GABAA receptors is unnecessary for LTP induction.
Although the application of bicuculline methiodide completely abolished test responses, the blockade of GABAA receptors might not be complete during conditioning stimulation, because a small hyperpolarizing response was still evoked by conditioning stimulation (CS, Fig. 1C). Therefore, the hyperpolarizing response was characterized. The addition of 30 μmbicuculline methiodide reduced responses evoked by high-frequency stimulation (Fig. 2A). The falling slope of the remaining response was ∼70 times smaller (0.038 ± 0.018 V/sec; n = 6) than that seen in the control solution (2.6 ± 0.88 V/sec; n = 6) and reached a peak at 243 ± 54 msec after starting the stimulation. The slow hyperpolarization was reduced to 33 ± 9% (n = 5) of control by the addition of 100 μm 2-hydroxysaclofen (Kerr et al., 1988), a GABAB receptor antagonist (Fig.2B), suggesting that it was mediated by GABAB receptors.
Fig. 2.

GABAA receptors are blocked completely by application of bicuculline methiodide. A, Responses evoked in a cell by high-frequency stimulation (50 Hz, 1 sec) before (left trace) and after (right trace) the addition of 30 μm bicuculline methiodide to the control solution. B, Responses evoked in another cell perfused with a solution containing 30 μm bicuculline methiodide by high-frequency stimulation before (left trace) and after (right trace) the addition of 100 μm2-hydroxysaclofen. The microelectrode contained K-methylsulfate inA and B. C, D, Similar toA and B, respectively. However, the microelectrodes contained Cs-acetate instead of K-methylsulfate, and 500 μm (+) MCPG was added instead of 2-hydroxysaclofen. These effects of antagonists accompanied no significant changes (p > 0.4) in input resistance or resting membrane potential. In these experiments, high-frequency stimulation (50 Hz, 1 sec) was applied with an intensity of 5 T at an interval of 1–3 min to avoid long-term effects on synaptic transmission. Time and voltage calibrations are common to A–D .
To ensure that the slow hyperpolarization is mediated by GABAB, but not by GABAA, receptors, cells were recorded with electrodes containing Cs-acetate instead of K-methylsulfate, because GABAB receptor-coupled K+ channels are known to be blocked by Cs+(Gähwiler and Brown, 1985). In the control solution without bicuculline methiodide, GABAA receptor-mediated IPSP was evoked in all cells (n = 6) in the same way as for the cells that were recorded with electrodes containing K-methylsulfate (Fig. 2A,C). However, no hyperpolarizing responses were recorded from any of the tested cells after bicuculline methiodide was added to the solution. Instead, high-frequency stimulation evoked small depolarizing responses (amplitude, 2.3 ± 0.37 mV; n = 6), which reached a peak after termination of the stimulation. This response seems to be mediated by metabotropic glutamate receptors (mGluRs), because it was reduced to one-fifth of control (20 ± 12%; n = 4) by the addition of high doses (500 μm) of (+)-MCPG (Eaton et al., 1993), an active isomer of an antagonist for mGluRs (Fig. 2D). Thus, it is concluded that 30 μm bicuculline methiodide is sufficient to block GABAA receptors during conditioning stimulation, and thereby the activation of GABAA receptors is not required to induce LTP.
Blockade of GABAB receptors prevents LTP induction
In contrast to the GABAA receptor antagonist, the addition of 100 μm 2-hydroxysaclofen to the perfusate during conditioning stimulation abolished the LTP (Fig.3A), but it had no direct effect on test responses. At this concentration, only short-term potentiation (STP) could be induced, which returned to the baseline level by 30 min after conditioning stimulation. When the antagonist was applied soon after conditioning stimulation, LTP occurred in all of the tested cells (Fig.3B), as it did in the control solution (Fig.1B), indicating that GABAB receptors are involved in the induction, but not in the maintenance, of LTP.
Fig. 3.

Application of GABAB receptor antagonists blocks LTP induction. A, Conditioning stimulation was given in the presence of 100 μm2-hydroxysaclofen. The drug application period is indicated by abar (Sac). Top traces(a–c) show superimposed test responses before and after conditioning stimulation. There was no significant difference (p > 0.3) between the resting membrane potential 20 min before (59 ± 6 mV; n = 9) and 40 min after (58 ± 7 mV) conditioning stimulation.B, 2-Hydroxysaclofen (100 μm) was applied soon after conditioning stimulation. There was no significant difference (p > 0.6) between the resting membrane potential before (60 ± 4 mV; n = 5) and 40 min after (61 ± 6 mV) conditioning stimulation. The time course was an average for nine (A) and five (B) tested cells. C, Displayed is the number of cells that showed LTP (filled bars), STP (shaded bars), or no change (open bars) for different doses of 2-hydroxysaclofen.D, Magnitude of LTP (mean ± SD) for cells that showed LTP (>15% increase from the baseline level at 30–40 min after conditioning stimulation). C, D, 2- Hydroxysaclofen was applied during conditioning stimulation, as shown in A.
The incidence of potentiation, either LTP or STP, decreased with increases in the dose of the antagonist (Fig. 3C). Among the potentiated cells, the ratio of LTP to STP decreased as well. This suggests that both STP and LTP require activation of GABABreceptors for their induction and that LTP requires stronger activation than STP. Although 2-hydroxysaclofen decreased the incidence of LTP, its magnitude was not different (p > 0.9) from that in the control solution when LTP occurred (Fig. 3D).
Application of an mGluR antagonist MCPG does not affect LTP
Because the control solution contained antagonists for ionotropic glutamate receptors, but not for mGluRs, I tested whether mGluRs are involved in LTP induction. The addition of high doses of (+)-MCPG (500 μm) had no effect on either the test responses or the induction of LTP (Fig. 4). LTP occurred in all of the tested cells (n = 6), and there was no significant difference (p > 0.8) in the magnitude of LTP between control and solution containing MCPG (compare Fig. 4 with 1B). The doses of MCPG used in this study seemed to be effective to block mGluRs, because MCPG greatly reduced slow depolarizing responses recorded under the blockade of responses mediated by GABAA, GABAB, non-NMDA, and NMDA receptors (Fig. 2D). Therefore, it is unlikely that substantial activation of MCPG-sensitive subtypes of mGluRs is necessary to induce LTP.
Fig. 4.

LTP is induced in the presence of an mGluR antagonist. The effect of conditioning stimulation was tested in six cells while 500 μm (+) MCPG was applied. There was no significant difference (p > 0.7) between the resting membrane potential 20 min before (59 ± 6 mV;n = 6) and 40 min after (59 ± 7 mV) conditioning stimulation.
LTP requires activation of G-proteins in postsynaptic cells
GABAB receptors are known to be present in both presynaptic terminals and postsynaptic cells (Newberry and Nicoll, 1984; Gähwiler and Brown, 1985; Howe et al., 1987; Connors et al., 1988; Harrison, 1990). Because GABAB receptors are coupled to G-proteins (Andrade et al., 1986; Holz et al., 1986;Thalmann, 1988), involvement of postsynaptic GABABreceptors was tested by loading GDPβS, an inhibitor of G-proteins, into postsynaptic cells with the blind-patch whole-cell recording method (Blanton et al., 1989).
Cells recorded with patch electrodes containing 0.6 mm GTP showed LTP of IPSCs similar to that of IPSPs (compare Fig.5A with 1B). When the electrode contained 1 mm GDPβS instead of GTP, conditioning stimulation elicited STP, but not LTP (Fig.5B), although there was no significant difference (p > 0.4) in the IPSC amplitude evoked by conditioning stimulation between control (1.2 ± 0.2 nA;n = 8) and GDPβS-loaded cells (1.1 ± 0.3 nA;n = 11). GDPβS decreased the incidence of the potentiation dose dependently (Fig. 5C), as did 2-hydroxysaclofen (Fig. 3C), and there was no significant difference (p > 0.7) in the magnitude of LTP between control and GDPβS-loaded cells when LTP occurred (Fig.5D). In addition, LTP induction was never found in cells loaded with 1 mm GTPγS, a nonhydrolyzable analog of GTP, which persistently activates G-proteins (Fig. 6). These results suggest that postsynaptic GABAB receptors are involved in the induction of LTP.
Fig. 5.

Postsynaptic loading of GDPβS blocks LTP.A, LTP of IPSCs recorded with patch electrodes containing 0.6 mm GTP. B, Effect of conditioning stimulation on GDPβS (1 mm)-loaded cells. The time course is an average for 8 (A) and 11 (B) tested cells. C, Dependence of LTP and STP incidence on GDPβS concentration. D, Magnitude of LTP for cells that showed LTP is displayed.
Fig. 6.

Postsynaptic loading of GTPγS blocks LTP. Effect of conditioning stimulation on GTPγS (1 mm)-loaded cells. The time course is an average for six tested cells. The IPSC amplitude evoked by conditioning stimulation for GTPγS-loaded cells (1.1 ± 0.5 nA) was not significantly different (p > 0.6) from that for the control cells.
To support this supposition, I tested whether postsynaptic GABAB receptors are commonly blocked by treatment of 2-hydroxysaclofen and GDPβS. GABAB receptors were activated by a bath application of baclofen, which is known to reduce transmitter release from presynaptic terminals and activate postsynaptic K+ channels via GABAB receptor activation (Newberry and Nicoll, 1984; Gähwiler and Brown, 1985;Howe et al., 1987; Harrison, 1990). The pre- and postsynaptic effects of the blockers were assessed, respectively, by their effects on IPSC reduction and outward K+ currents produced by baclofen during whole-cell voltage clamp with patch electrodes containing K+ instead of Cs+ (Fig.7A,B).
Fig. 7.
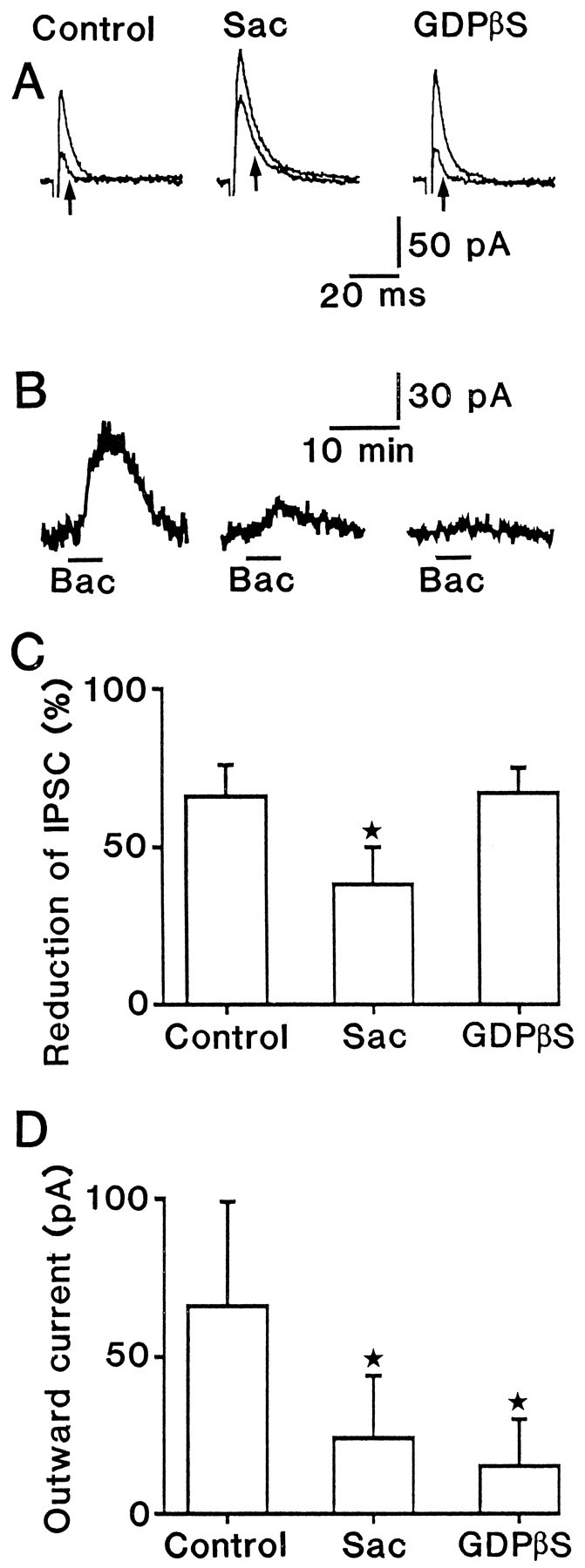
Effect of 2-hydroxysaclofen and GDPβS on presynaptic and postsynaptic baclofen-activated responses.A, Superimposed average (n = 4) IPSCs recorded from three cells before and during a bath application of 10 μm baclofen. The arrows indicate IPSCs evoked during a bath application of baclofen. Patch pipettes contained 0.6 mm GTP in the left and middle traces but 1 mm GDPβS in the right trace. Cells were perfused with control solution inleft and right traces but with a solution containing 100 μm 2-hydroxysaclofen in the middle trace. B, Outward currents produced by a bath application of 10 μm baclofen. The left,middle, and right traces were recorded from the same cells as shown in A, respectively. Thebar (Bac) indicates the application period of baclofen. C, D, Summary of the experiments illustrated in A and B. Number of cells is 9 for control, 10 for 2-hydroxysaclofen, and 10 for GDPβS, respectively. The asterisk indicates that the value is significantly different from the control value (p < 0.05). Cells were held at −40 mV.
2-Hydroxysaclofen (100 μm) decreased IPSC reduction to approximately one-half of control and outward currents to one-third of control (Fig. 7C,D). GDPβS (1 mm) decreased the outward currents to one-fourth of control, but it did not affect IPSC reduction. The observation that postsynaptic, but not presynaptic, GABAB receptors were substantially blocked by both drugs is consistent with the above supposition.
LTP requires IP3-induced Ca2+ release in postsynaptic cells
To elucidate further the postsynaptic mechanism, I tested the effect of postsynaptic loading of a Ca2+ chelator BAPTA on LTP, because the induction of LTP at glycinergic inhibitory synapses, as well as most excitatory synapses, requires a Ca2+increase in postsynaptic cells (Lynch et al., 1983; Malenka et al., 1988; Korn et al., 1992). No indication of LTP was demonstrated when cells were recorded with a patch electrode containing 10 mmBAPTA instead of 1 mm EGTA (Fig.8A), suggesting that the LTP at these inhibitory synapses also requires a postsynaptic Ca2+increase for the induction.
Fig. 8.
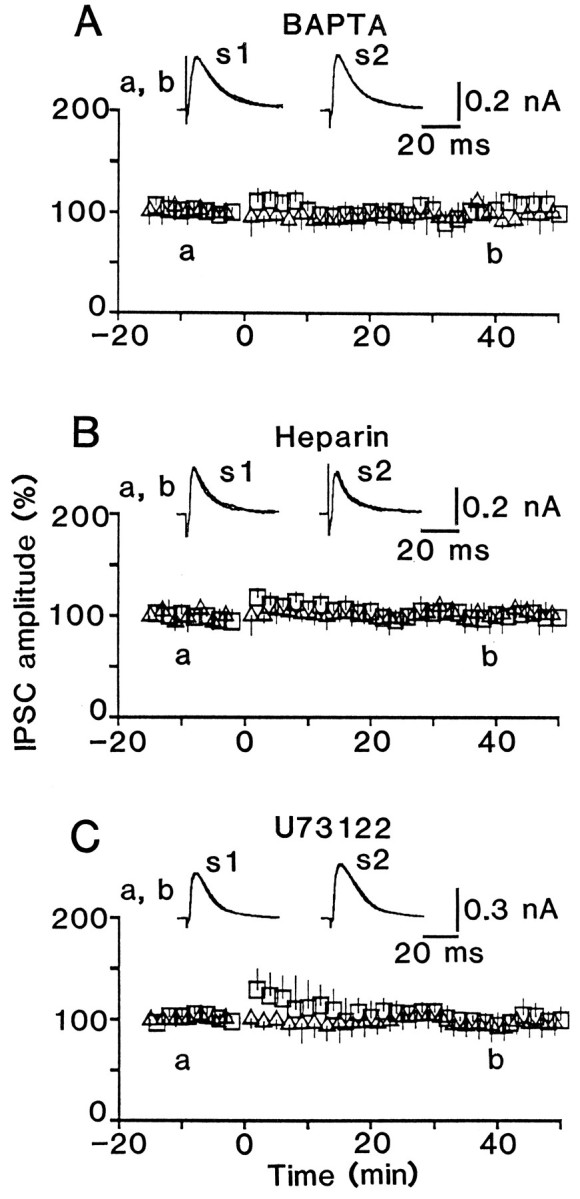
LTP is blocked by postsynaptic loading of BAPTA, heparin, or U73122. A, Effect of conditioning stimulation on BAPTA (10 mm)-loaded cells.B, C, Similar to A, but for heparin (2 mg/ml)- and U73122 (20 μm)-loaded cells, respectively. The time course is an average for six tested cells inA–C. The IPSC amplitude evoked by conditioning stimulation for BAPTA (1.0 ± 0.4 nA), heparin (1.1 ± 0.2 nA), or U73122-loaded (0.9 ± 0.4 nA) cells was not significantly different (p > 0.1) from that for the control cells.
It is unlikely that Ca2+ influx through voltage-dependent channels is involved in the induction of LTP, because LTP was generated consistently by conditioning stimulation applied during voltage clamp at either −90 or +20 mV (Komatsu, 1994). Thus, it would be expected that the LTP is abolished by the blockade of Ca2+ release from internal stores. This possibility was supported by experiments in which cells were loaded with heparin, an antagonist for inositol trisphosphate (IP3) receptors (Hill et al., 1987; Worley et al., 1987), activation of which releases Ca2+ from internal stores (Berridge, 1993). As shown in Figure 8B, no indication of LTP was demonstrated when heparin (2 mg/ml) was added to the patch pipette solution. Furthermore, LTP induction was prevented by postsynaptic loading of 20 μm U73122 (Smith et al., 1990), an inhibitor of phospholipase C, activation of which produces IP3 (Fig. 8C). Therefore, it is likely that LTP induction requires IP3-induced Ca2+ release from internal stores in postsynaptic cells.
α1 Adrenoceptors and serotonin 5-HT2receptors participate in LTP induction
The observation that a blockade of GABAB receptors and postsynaptic G-proteins similarly prevented the generation of LTP suggests that the GABAB receptors convey signals for initiation of the LTP to postsynaptic cells. If so, postsynaptic GABAB receptor activation might form IP3 via the activation of phospholipase C. On the contrary, biochemical studies have demonstrated that application of GABAB receptor agonists alone does not affect the IP3 level in rat cerebral cortical slices (Crawford and Young, 1988). However, the activation of GABAB receptors facilitates α1adrenoceptor-mediated IP3 formation, whereas it depresses histamine H1 receptor mediated-IP3 formation or does not affect muscarinic receptor-mediated IP3 formation (Crawford and Young, 1988, 1990). Thus, I tested the effects of antagonists for these amine receptors on the LTP by intracellular recording.
The incidence of LTP was greatly reduced by bath application of an α1 adrenoceptor antagonist, prazosin, whereas LTP occurred even in the presence of high doses of an H1receptor antagonist, pyrilamine, or a muscarinic receptor antagonist, atropine, in almost all of the tested cells, as in the control solution (Fig. 9). When LTP occurred in the presence of these antagonists, their magnitude and time course were both similar to those in the control solution (compare Fig.10A–C with 1B). These results are consistent with the hypothesis that GABABreceptor activation participates in the LTP induction by facilitating α1 adrenoceptor-mediated IP3 formation in postsynaptic cells.
Fig. 9.

Incidence of LTP is reduced by the application of prazosin or ketanserin, but not of pyrilamine or atropine. Displayed is the number of cells that showed LTP (filled bars), STP (shaded bars), or no change (open bars) for control and antagonists of different amine receptors. The antagonists were present in the perfusate throughout the recording period. Separate control experiments demonstrated that the application of these antagonists at the concentration used here had no significant effects (p > 0.2) on test IPSPs.
Fig. 10.

Time course of LTP in the presence of amine receptor antagonists. Average time course of LTP for cells that showed LTP in the presence of prazosin (A), pyrilamine (B), atropine (C), and ketanserin (D). Number of cells: A, 7;B, 7; C, 9; D, 8. The magnitude of LTP in the presence of ketanserin, but not of other antagonists, was significantly (p < 0.05) different from that in the control solution.
Although prazosin reduced LTP incidence, the blockade seemed incomplete, because a 10-fold higher concentration of the antagonist failed to reduce further the incidence (36% at 1 μm; 38% at 10 μm). This observation prompted us to test the involvement of serotonin 5-HT2 (5-HT2A and 5-HT2C) receptors, which also are known to contribute to IP3 formation in developing and mature rat cerebral cortex (Kendall and Nahorski, 1985; Balduini et al., 1991). A 5-HT2 receptor antagonist ketanserin (Leysen et al., 1982) reduced the LTP incidence to 50% at 1 μm and to 40% at 5 μm (Fig. 9). When LTP occurred, in contrast, the magnitude was slightly larger in ketanserin than in the control solution, but their time courses were similar (compare Fig.10D with 1B). Because further elevation of the concentration decreased the IPSP itself, I did not examine whether the LTP incidence was reduced even more at higher concentrations.
I further studied whether α1 and 5-HT2receptors are involved in the induction or the maintenance of LTP. LTP was generated only in a few cells when prazosin (10 μm) and ketanserin (5 μm) were applied simultaneously during conditioning stimulation, but it occurred in all of seven tested cells when the same drugs were applied soon after conditioning stimulation (Fig. 11). This indicates that α1 and 5-HT2 receptors participate in the induction, but not the maintenance, of the LTP.
Fig. 11.
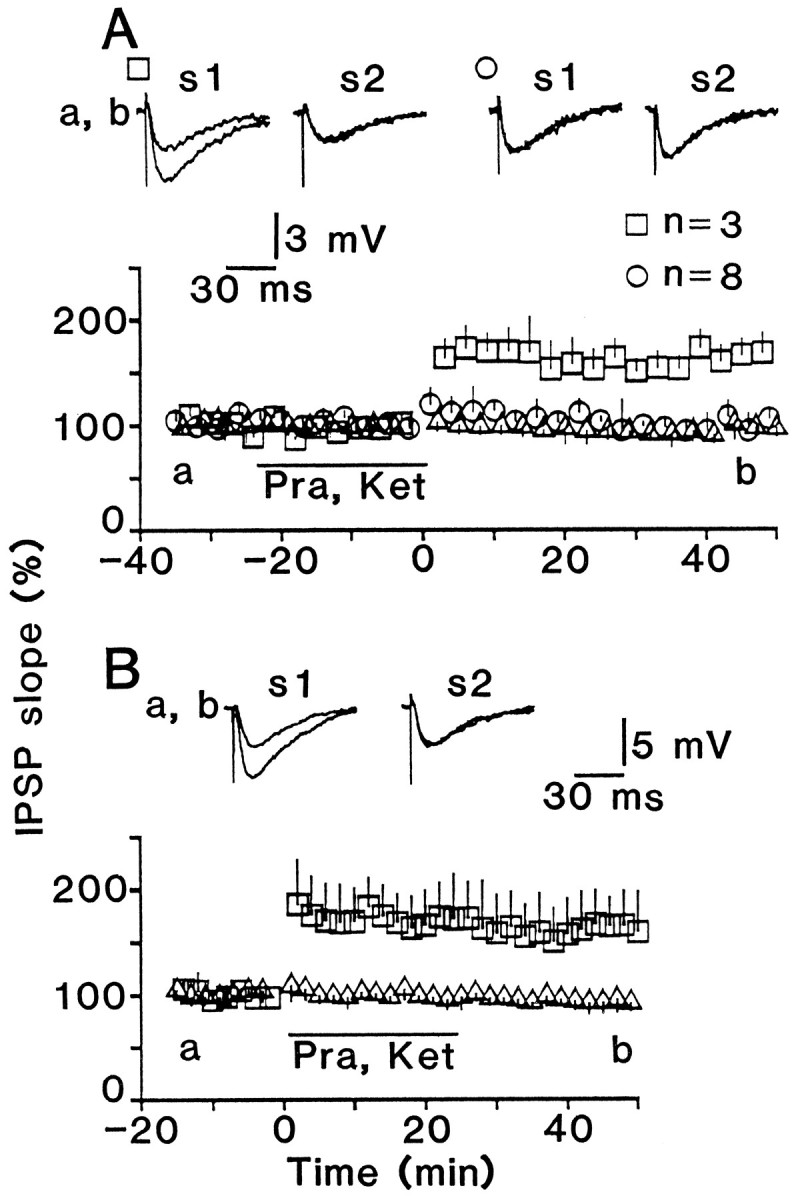
α1 Adrenoceptors and 5-HT2 receptors are involved in the induction, but not the maintenance, of LTP. A, Conditioning stimulation was applied in the presence of 10 μm prazosin and 5 μm ketanserin. Squares andcircles represent responses of conditioned pathway for cells that showed LTP (n = 3) and no LTP (n = 8), respectively. B, The same antagonists were applied soon after conditioning stimulation. The application period is indicated by a bar(Pra, Ket).
In the experiments above, in which conditioning stimulation was applied with an intensity (5 T) strong enough to consistently produce LTP in the control solution, LTP still occurred in some of the cells in the presence of high doses of prazosin and ketanserin. In contrast, when a weak conditioning stimulation was applied, with an intensity of 1.6–1.8 T, LTP was still evoked in approximately one-half of cells in the control solution, but no LTP was demonstrated in the presence of either prazosin (1 μm) or ketanserin (1 μm; Fig. 12). This suggests that the activation of both α1 and 5-HT2 receptors is required to initiate LTP when a relatively small number of inhibitory presynaptic fibers are activated.
Fig. 12.

Blockade of either α1 adrenoceptors or 5-HT2 receptors abolishes LTP evoked by weak conditioning stimulation. Displayed is the number of cells that showed LTP (filled bar), STP (shaded bars), or no change (open bars) in response to weak conditioning stimulation in control, prazosin (1 μm), or ketanserin solution (1 μm).
DISCUSSION
The main findings of the present study are (1) LTP of GABAA receptor-mediated inhibitory synaptic transmission requires the activation of GABAB receptors for the induction, but not of GABAA receptors themselves; (2) a similar blockade of LTP is produced by the inhibition of G-proteins, phospholipase C, IP3 receptors, or Ca2+increase in postsynaptic cells; and (3) α1 adrenoceptors and 5-HT2 receptors, known to be coupled to IP3formation, are implicated in the LTP induction. These results suggest that LTP induction requires postsynaptic activities mediated by G-protein-coupled receptors, probably including GABAB and monoamine receptors.
Functional roles of GABAB receptors
The present study demonstrated a new functional role of GABAB receptors. Their well known roles are the reduction of transmitter release caused by the activation of presynaptic receptors and hyperpolarization caused by the activation of postsynaptic receptors (Connors et al., 1988; Dutar and Nicoll, 1988;Deisz and Prince, 1989; Thompson and Gähwiler, 1989; Davies et al., 1990). In addition, these transient effects can either facilitate or depress induction of LTP and long-term depression (LTD) at excitatory synapses by modulating NMDA receptor-mediated responses (Olpe and Karlsson, 1990; Davies et al., 1991; Mott and Lewis, 1991;Wagner and Alger, 1995). In contrast to the indirect involvement of GABAB receptors in plasticity of the excitatory synapses, this study revealed that GABAB receptors are indispensable for the homosynaptic LTP at GABAergic synapses.
Postsynaptic mechanisms for the induction of LTP
LTP was prevented by the inhibition of G-proteins, phospholipase C, IP3 receptors, or Ca2+ increase in postsynaptic cells, indicating that postsynaptic mechanisms participate in the generation of LTP. Because GDPβS prevented the induction of LTP in the same way as the GABAB receptor antagonists, it is likely that the activation of GABAB receptors initiates the process in the postsynaptic cells. In addition, some mGluRs could be involved, because they are known to be linked to IP3formation (Nakanishi, 1992). However, MCPG-sensitive subtypes of mGluRs seem unnecessary for the LTP induction, because the induction was not prevented by high doses of MCPG, which effectively blocks mGluR-mediated Ca2+ increase in rat visual cortex (Haruta et al., 1994). If the mGluRs are involved in the plasticity at the inhibitory synapses, they can contribute to LTD, because their activation is known to produce LTD at hippocampal inhibitory synapses (Liu et al., 1993).
Other receptors possibly involved in the LTP induction are amine receptors, linked to IP3 formation (Nahorski, 1988). The present study demonstrated involvement of α1adrenoceptors in the LTP induction. These receptors are known to be present in visual cortical layer V (Sargent Jones et al., 1985;Parkinson et al., 1988) and believed to be located entirely postsynaptically (Nicoll et al., 1990). In addition, it is known that GABAB receptor activation alone does not affect the IP3 level, but it facilitates α1adrenoceptor-mediated IP3 formation in rat cerebral cortex (Crawford and Young, 1990). These results strongly suggest that activation of postsynaptic GABAB receptors initiates the LTP by potentiating α1 receptor-mediated IP3formation.
In addition, 5-HT2 (5-HT2A/5-HT2C) receptors, located in all visual cortical layers (Pazos and Palacios, 1985; Pazos et al., 1985; Dyck and Cynader, 1993), also participated in the LTP induction, suggesting that GABAB receptors can also facilitate 5-HT2 receptor-mediated IP3formation. On the contrary, it is reported that GABABreceptor agonists depress the 5-HT2 receptor-mediated inositol phosphate formation in mouse cerebral cortex (Godfrey et al., 1988), although no studies on this have been performed on rats. Thus, 5-HT2 receptors could contribute to LTP induction either presynaptically or postsynaptically independent of the action of GABAB receptors. However, the effect of GABA on inositol phosphate formation seems to vary considerably in different species (Godfrey et al., 1988; Crawford and Young, 1990). Furthermore, the inositol phosphate formation was measured by using adult cerebral cortex without area or layer specifications. Therefore, the relationship between the activation of GABAB and 5-HT2 receptors is uncertain at present.
Even simultaneous application of high doses of prazosin and ketanserin failed to block LTP in some of the cells, whereas the inhibition of IP3-induced Ca2+ release in postsynaptic cells completely abolished the generation of LTP. This might be attributable to an insufficient dose of ketanserin, because the dose was limited to a range producing no depressive effect on IPSP itself. Alternatively, some other receptors for neuropeptides, coreleased with GABA (Jones and Hendry, 1986) and linked to IP3 formation (Nahorski, 1988), could be involved in the LTP induction. Furthermore, the possibility remains that GABAB receptor activation alone could form IP3 in developing visual cortex. To fully understand the mechanisms of the LTP, more knowledge is required on the relationship between GABAB receptors and IP3 formation and the elucidation of processes activated by IP3-induced Ca2+ release is also important.
Comparison with LTP at hippocampal CA1 excitatory synapses
LTP at visual cortical inhibitory synapses has similar properties to those at CA1 pyramidal cell excitatory synapses (Komatsu, 1994). Both are input-specific and associative. Despite this similarity, their induction mechanism is considerably different, as demonstrated in this study. LTP induction at excitatory synapses of hippocampal CA1 requires the activation of voltage-dependent and Ca2+-permeable NMDA receptor channels or voltage-gated Ca2+ channels in postsynaptic cells (Collingridge et al., 1983; Lynch et al., 1983;Mayer et al., 1984; Nowak et al., 1984; MacDermott et al., 1986; Grover and Teyler, 1990; Aniksztejn and Ben-Ari, 1991). In addition, it has been reported that mGluRs also participate in LTP (Bashir et al., 1993;Aiba et al., 1994), although the issue is controversial (Chinestra et al., 1993; Conquet et al., 1994; Manzoni et al., 1994; Selig et al., 1995). Because the occlusion of postsynaptic G-proteins by GTPγS does not prevent the induction of LTP (Goh and Pennefather, 1989), postsynaptic mGluRs may not be involved in LTP induction, although the receptor contributes to the LTP. In contrast, the LTP shown in this study required the activation of G-protein-coupled receptors, but not voltage-gated channels, in postsynaptic cells.
Both the application of the GABAB receptor antagonist and postsynaptic loading of GDPβS reduced the incidence, but not the magnitude, of LTP. In addition, our previous study demonstrated that intense conditioning stimulation elicited LTP more frequently, although the magnitude of LTP did not depend on the intensity (Komatsu, 1994), suggesting that the associativity of LTP is attributable to the presence of a threshold level at some of the steps after the activation of G-protein-coupled receptors in postsynaptic cells. Once this threshold is exceeded, LTP seems to be generated with a similar magnitude. Therefore, in associative LTPs, coactivation of presynaptic fibers could be assessed by postsynaptic responses mediated by either voltage-dependent channels or G-protein-coupled receptors.
In hippocampal CA1, both LTP and LTD of excitatory synaptic transmission require a postsynaptic Ca2+ increase because of NMDA receptor activation. It is proposed that the level of Ca2+ transients determines the direction of the modification (Lisman, 1985; Malenka and Nicoll, 1993). At inhibitory synapses, in contrast, LTP requires Ca2+ release from internal stores, whereas LTD requires Ca2+ influx through NMDA receptor channels (Komatsu and Iwakiri, 1993), suggesting that the source of Ca2+ is critical for determining the direction.
Relevance of LTP at inhibitory synapses to plasticity of visual responses
Selective responsiveness of visual cortical cells develops under the influence of visual experience in rats, as it does in cats (Benevento et al., 1992; Maffei et al., 1992). LTP of inhibitory synaptic transmission in visual cortex is induced most easily in developing rats (Komatsu, 1994), and GABAB binding levels peak during postnatal development and decline to the adult level in rat neocortex (Turgeon and Albin, 1994). Therefore, it is tempting to study the consequence of a blockade of GABAB receptors on the development of response selectivity in visual cortical cells.
Noradrenaline contributes to the ocular dominance plasticity of cortical cells through β receptors and serotonin through 5-HT1 and 5-HT2 receptors (Shirokawa and Kasamatsu, 1986; Gu and Singer, 1995). This plasticity may be ascribed primarily to changes at excitatory synapses (Hubel et al., 1977; Shatz and Stryker, 1978). Because LTP at inhibitory synapses may contribute to plasticity of orientation and direction selectivity rather than the ocular dominance preference, it is likely that the monoamines participate in either of these kinds of plasticity via different subsets of receptors.
When weak conditioning stimulation was applied, a blockade of either α1 adrenoceptor or 5-HT2 receptor completely prevented the induction of LTP. Visual inputs far more frequently may produce inhibitory synaptic activities similar to those evoked by weak rather than by strong conditioning stimulation, suggesting that the LTP usually can be induced when the inhibitory synapses are activated in conjunction with simultaneous activities of adrenergic and serotonergic cells. Because locus ceruleus and raphe cells maintain high-frequency spike activities during awake, but not sleep, states (McGinty and Harper, 1976; Sakai and Jouvet, 1980; Cespuglio et al., 1981), it is likely that potentiation at inhibitory synapses effectively occurs when animals are looking attentively at their visual environment.
Footnotes
This work was supported by Grants-in-Aid for Scientific Research 06260235, 07680898, and 07279104 from the Japanese Ministry of Education, Science, Sports, and Culture. I thank Dr. T. Kurotani for his comments on this manuscript.
Correspondence should be addressed to Dr. Y. Komatsu, Division of Higher Nervous Control, Research Institute of Environmental Medicine, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-01, Japan.
REFERENCES
- 1.Aiba A, Chen C, Herrup K, Rosenmund C, Stevens CF, Tonegawa S. Reduced hippocampal long-term potentiation and context-specific deficit in associative learning in mGluR1 mutant mice. Cell. 1994;79:365–375. doi: 10.1016/0092-8674(94)90204-6. [DOI] [PubMed] [Google Scholar]
- 2.Andrade R, Malenka RC, Nicoll RA. A G-protein couples serotonin and GABAB receptors to the same channels in hippocampus. Science. 1986;234:1261–1265. doi: 10.1126/science.2430334. [DOI] [PubMed] [Google Scholar]
- 3.Aniksztejn L, Ben-Ari Y. Novel form of long-term potentiation produced by a K+ channel blocker in the hippocampus. Nature. 1991;349:67–69. doi: 10.1038/349067a0. [DOI] [PubMed] [Google Scholar]
- 4.Balduini W, Candura SM, Costa LG. Regional development of carbachol-, glutamate-, norepinephrine-, and serotonin-stimulated phosphoinositide metabolism in rat brain. Dev Brain Res. 1991;62:115–120. doi: 10.1016/0165-3806(91)90196-p. [DOI] [PubMed] [Google Scholar]
- 5.Bashir ZI, Bortolotto ZA, Davies CH, Berretta N, Irving AJ, Seal AJ, Henley JM, Jane DE, Watkins JC, Collingridge GL. Induction of LTP in the hippocampus needs synaptic activation of glutamate metabotropic receptors. Nature. 1993;363:347–350. doi: 10.1038/363347a0. [DOI] [PubMed] [Google Scholar]
- 6.Benevento LA, Bakkum BW, Port JD, Cohen RS. The effect of dark-rearing on the electrophysiology of the rat visual cortex. Brain Res. 1992;572:198–207. doi: 10.1016/0006-8993(92)90470-t. [DOI] [PubMed] [Google Scholar]
- 7.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 8.Blanton MG, Lo Turco JJ, Kriegstein AR. Whole-cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- 9.Cespuglio R, Faradji H, Gomez ME, Jouvet M. Single unit recordings in the nuclei raphe dorsalis and magnus during the sleep–waking cycle of semi-chronic prepared cats. Neurosci Lett. 1981;24:133–138. doi: 10.1016/0304-3940(81)90236-6. [DOI] [PubMed] [Google Scholar]
- 10.Chinestra P, Aniksztejn L, Diabira D, Ben-Ari Y. (RS)-α-methyl-4-carboxyphenylglycine neither prevents induction of LTP nor antagonizes metabotropic glutamate receptors in CA1 hippocampal neurons. J Neurophysiol. 1993;70:2684–2689. doi: 10.1152/jn.1993.70.6.2684. [DOI] [PubMed] [Google Scholar]
- 11.Collingridge GL, Bliss TVB. NMDA receptors: their role in long-term potentiation. Trends Neurosci. 1987;10:288–293. [Google Scholar]
- 12.Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral–commissural pathway of the rat hippocampus. J Physiol (Lond) 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Connors BW, Malenka RC, Silva LR. Two inhibitory postsynaptic potentials and GABAA and GABAB receptor-mediated responses in neocortex of rat and cat. J Physiol (Lond) 1988;406:443–468. doi: 10.1113/jphysiol.1988.sp017390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franz-Bacon K, Reggiani A, Matarese V, Conde F, Collingridge GL, Crepel F. Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature. 1994;372:237–244. doi: 10.1038/372237a0. [DOI] [PubMed] [Google Scholar]
- 15.Crawford MLA, Young JM. GABAB receptor-mediated inhibition of histamine H1-receptor-induced inositol phosphate formation in slices of rat cerebral cortex. J Neurochem. 1988;51:1441–1447. doi: 10.1111/j.1471-4159.1988.tb01109.x. [DOI] [PubMed] [Google Scholar]
- 16.Crawford MLA, Young JM. Potentiation by γ-aminobutyric acid of α1-agonist-induced accumulation of inositol phosphates in slices of rat cerebral cortex. J Neurochem. 1990;54:2100–2109. doi: 10.1111/j.1471-4159.1990.tb04916.x. [DOI] [PubMed] [Google Scholar]
- 17.Davies CH, Davies SN, Collingridge GL. Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. J Physiol (Lond) 1990;424:513–531. doi: 10.1113/jphysiol.1990.sp018080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davies CH, Starkey SJ, Pozza MF, Collingridge GL. GABAB autoreceptors regulate the induction of LTP. Nature. 1991;349:609–611. doi: 10.1038/349609a0. [DOI] [PubMed] [Google Scholar]
- 19.Deisz RA, Prince DA. Frequency-dependent depression of inhibition in guinea-pig neocortex in vitro by GABAB receptor feedback on GABA release. J Physiol (Lond) 1989;412:513–541. doi: 10.1113/jphysiol.1989.sp017629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dutar P, Nicoll RA. A physiological role for GABAB receptors in the central nervous system. Nature. 1988;332:156–158. doi: 10.1038/332156a0. [DOI] [PubMed] [Google Scholar]
- 21.Dyck RH, Cynader MS. Autoradiographic localization of serotonin receptor subtypes in cat visual cortex: transient regional, laminar, and columnar distributions during postnatal development. J Neurosci. 1993;13:4316–4338. doi: 10.1523/JNEUROSCI.13-10-04316.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eaton SA, Jane DE, Jones PLStJ, Porter RHP, Pook PCK, Sunter DC, Udvarhelyi PM, Roberts PJ, Salt TE, Watkins JC. Competitive antagonism at metabotropic glutamate receptors by (S)-4-carboxyphenylglycine and (RS)-α-methyl-4-carboxyphenylglycine. Eur J Pharmacol. 1993;244:195–197. doi: 10.1016/0922-4106(93)90028-8. [DOI] [PubMed] [Google Scholar]
- 23.Gähwiler BH, Brown DA. GABAB-receptor-activated K+ current in voltage-clamped CA3 pyramidal cells in hippocampal cultures. Proc Natl Acad Sci USA. 1985;82:1558–1562. doi: 10.1073/pnas.82.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Godfrey PP, Grahame-Smith DG, Gray JA. GABAB receptor activation inhibits 5-hydroxytryptamine-stimulated inositol phospholipid turnover in mouse cerebral cortex. Eur J Pharmacol. 1988;152:185–188. doi: 10.1016/0014-2999(88)90854-0. [DOI] [PubMed] [Google Scholar]
- 25.Goh JW, Pennefather PS. A pertussis toxin-sensitive G-protein in hippocampal long-term potentiation. Science. 1989;244:980–983. doi: 10.1126/science.2543072. [DOI] [PubMed] [Google Scholar]
- 26.Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- 27.Gu Q, Singer W. Involvement of serotonin in developmental plasticity of kitten visual cortex. Eur J Neurosci. 1995;7:1146–1153. doi: 10.1111/j.1460-9568.1995.tb01104.x. [DOI] [PubMed] [Google Scholar]
- 28.Gustafsson B, Wigström H. Physiological mechanisms underlying long-term potentiation. Trends Neurosci. 1988;11:156–162. doi: 10.1016/0166-2236(88)90142-7. [DOI] [PubMed] [Google Scholar]
- 29.Harrison NL. On the presynaptic action of baclofen at inhibitory synapses between cultured rat hippocampal neurones. J Physiol (Lond) 1990;422:433–446. doi: 10.1113/jphysiol.1990.sp017993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haruta H, Kamishita T, Hicks TP, Takahashi MP, Tsumoto T. Induction of LTD, but not LTP, through metabotropic glutamate receptors in visual cortex. NeuroReport. 1994;5:1829–1832. doi: 10.1097/00001756-199409080-00036. [DOI] [PubMed] [Google Scholar]
- 31.Hill TD, Berggren PO, Boynton AL. Heparin inhibits inositol trisphosphate-induced calcium release from permeabilized rat liver cells. Biochem Biophys Res Commun. 1987;149:897–901. doi: 10.1016/0006-291x(87)90492-x. [DOI] [PubMed] [Google Scholar]
- 32.Holz GG, IV, Rane SG, Dunlap K. GTP-binding proteins mediate transmitter inhibition of voltage-dependent calcium channels. Nature. 1986;319:670–672. doi: 10.1038/319670a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Howe JR, Sutor B, Zieglgansberger W. Baclofen reduces postsynaptic potentials of rat cortical neurones by an action other than its hyperpolarizing action. J Physiol (Lond) 1987;384:539–569. doi: 10.1113/jphysiol.1987.sp016469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hubel DH, Wiesel TN, LeVay S. Plasticity of ocular dominance columns in monkey striate cortex. Philos Trans R Soc Lond [Biol] 1977;278:131–163. doi: 10.1098/rstb.1977.0050. [DOI] [PubMed] [Google Scholar]
- 35.Jones EG, Hendry SH. Co-localization of GABA and neuropeptides in neocortical neurons. Trends Neurosci. 1986;9:71–76. [Google Scholar]
- 36.Kendall DA, Nahorski SR. 5-Hydroxytryptamine-stimulated inositol phospholipid hydrolysis in rat cerebral cortex slice: pharmacological characterization and effects of antidepressants. J Pharmacol Exp Ther. 1985;233:473–479. [PubMed] [Google Scholar]
- 37.Kerr DIB, Ong J, Johnston GAR, Abbenante J, Prager RH. 2-Hydroxysaclofen: an improved antagonist at central and peripheral GABAB receptors. Neurosci Lett. 1988;92:92–96. doi: 10.1016/0304-3940(88)90748-3. [DOI] [PubMed] [Google Scholar]
- 38.Komatsu Y. Age-dependent long-term potentiation of inhibitory synaptic transmission in rat visual cortex. J Neurosci. 1994;14:6488–6499. doi: 10.1523/JNEUROSCI.14-11-06488.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Komatsu Y, Iwakiri M. Long-term modification of inhibitory synaptic transmission in developing visual cortex. NeuroReport. 1993;4:907–910. doi: 10.1097/00001756-199307000-00017. [DOI] [PubMed] [Google Scholar]
- 40.Korn H, Oda Y, Faber DS. Long-term potentiation of inhibitory circuits and synapses in the CNS. Proc Natl Acad Sci USA. 1992;89:440–443. doi: 10.1073/pnas.89.1.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leysen JE, Niemegeers CJE, Van Nueten JM, Laduron PM. [3H]ketanserin (R 41,468), a selective 3H-ligand for serotonin2 receptor binding sites. Mol Pharmacol. 1982;21:301–314. [PubMed] [Google Scholar]
- 42.Lisman JE. A mechanism for memory storage insensitive to molecular turnover: a bistable autophosphorylating kinase. Proc Natl Acad Sci USA. 1985;82:3055–3057. doi: 10.1073/pnas.82.9.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y-B, Disterhoft JF, Slater NT. Activation of metabotropic glutamate receptors induces long-term depression of GABAergic inhibition in hippocampus. J Neurophysiol. 1993;69:1000–1004. doi: 10.1152/jn.1993.69.3.1000. [DOI] [PubMed] [Google Scholar]
- 44.Lynch G, Larson J, Kelso S, Barrionuevo G, Schotler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305:719–720. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- 45.MacDermott AB, Mayer ML, Westbrook GL. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- 46.Madison DV, Malenka RC, Nicoll RA. Mechanisms underlying long-term potentiation of synaptic transmission. Annu Rev Neurosci. 1991;14:379–397. doi: 10.1146/annurev.ne.14.030191.002115. [DOI] [PubMed] [Google Scholar]
- 47.Maffei L, Berardi N, Domenici L, Parisi V, Pizzorusso T. Nerve growth factor (NGF) prevents the shift in ocular dominance distribution of visual cortical neurons in monocularly deprived rats. J Neurosci. 1992;12:4651–4662. doi: 10.1523/JNEUROSCI.12-12-04651.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- 49.Malenka RC, Kauer JA, Zucker RS, Nicoll RA. Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science. 1988;242:81–84. doi: 10.1126/science.2845577. [DOI] [PubMed] [Google Scholar]
- 50.Manzoni OJ, Weisskopf MG, Nicoll RA. MCPG antagonizes metabotropic glutamate receptors but not long-term potentiation in the hippocampus. Eur J Neurosci. 1994;6:1050–1054. doi: 10.1111/j.1460-9568.1994.tb00599.x. [DOI] [PubMed] [Google Scholar]
- 51.Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 52.McGinty DJ, Harper RM. Dorsal raphe neurons: depression of firing during sleep in cats. Brain Res. 1976;101:569–575. doi: 10.1016/0006-8993(76)90480-7. [DOI] [PubMed] [Google Scholar]
- 53.Mott DD, Lewis DV. Facilitation of the induction of long-term potentiation by GABAB receptors. Science. 1991;252:1718–1720. doi: 10.1126/science.1675489. [DOI] [PubMed] [Google Scholar]
- 54.Nahorski SR. Inositol polyphosphates and neuronal calcium homeostasis. Trends Neurosci. 1988;11:444–448. doi: 10.1016/0166-2236(88)90196-8. [DOI] [PubMed] [Google Scholar]
- 55.Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- 56.Newberry NR, Nicoll RA. Direct hyperpolarizing action of baclofen on hippocampal pyramidal cells. Nature. 1984;308:450–452. doi: 10.1038/308450a0. [DOI] [PubMed] [Google Scholar]
- 57.Nicoll RA, Malenka RC, Kauer JA. Functional comparison of neurotransmitter receptor subtypes in mammalian central nervous system. Physiol Rev. 1990;70:513–565. doi: 10.1152/physrev.1990.70.2.513. [DOI] [PubMed] [Google Scholar]
- 58.Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 59.Olpe H-R, Karlsson G. The effects of baclofen and two GABAB-receptor antagonists on long-term potentiation. Naunyn Schmiedebergs Arch Pharmacol. 1990;342:194–197. doi: 10.1007/BF00166964. [DOI] [PubMed] [Google Scholar]
- 60.Parkinson D, Coscia E, Daw NW. Identification and localization of adrenergic receptors in cat visual cortex. Brain Res. 1988;457:70–78. doi: 10.1016/0006-8993(88)90058-3. [DOI] [PubMed] [Google Scholar]
- 61.Pazos A, Palacios JM. Quantitative autoradiographic mapping of serotonin receptors in the rat brain. I. Serotonin-1 receptors. Brain Res. 1985;346:205–230. doi: 10.1016/0006-8993(85)90856-x. [DOI] [PubMed] [Google Scholar]
- 62.Pazos A, Cortes R, Palacios JM. Quantitative autoradiographic mapping of serotonin receptors in the rat brain. II. Serotonin-2 receptors. Brain Res. 1985;346:231–249. doi: 10.1016/0006-8993(85)90857-1. [DOI] [PubMed] [Google Scholar]
- 63.Sakai K, Jouvet M. Brain stem PGO–on cells projecting directly to the cat dorsal lateral geniculate nucleus. Brain Res. 1980;194:500–505. doi: 10.1016/0006-8993(80)91231-7. [DOI] [PubMed] [Google Scholar]
- 64.Sargent Jones L, Gauger LL, Davis JN, Slotkin TA, Bartolome JV. Postnatal development of brain alpha1-adrenergic receptors: in vitro autoradiography with [125I]HEAT in normal rats and rats treated with alpha-difluoromethylornithine, a specific irreversible inhibitor of ornithine decarboxylase. Neuroscience. 1985;15:1195–1202. doi: 10.1016/0306-4522(85)90262-3. [DOI] [PubMed] [Google Scholar]
- 65.Selig DK, Lee HK, Bear MF, Malenka RC. Reexamination of the effects of MCPG on hippocampal LTP, LTD, and depotentiation. J Neurophysiol. 1995;74:1075–1082. doi: 10.1152/jn.1995.74.3.1075. [DOI] [PubMed] [Google Scholar]
- 66.Shatz CJ, Stryker MP. Ocular dominance in layer IV of the cat’s visual cortex and the effects of monocular deprivation. J Physiol (Lond) 1978;281:267–283. doi: 10.1113/jphysiol.1978.sp012421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shirokawa T, Kasamatsu T. Concentration-dependent suppression by β-adrenergic antagonists of the shift in ocular dominance following monocular deprivation in kitten visual cortex. Neuroscience. 1986;18:1035–1046. doi: 10.1016/0306-4522(86)90115-6. [DOI] [PubMed] [Google Scholar]
- 68.Smith RJ, Sam KM, Justen JM, Bundy GL, Bala GA, Bleasdale JE. Receptor-coupled signal transduction in human polymorphonuclear neutrophils: effects of a novel inhibitor of phospholipase C-dependent processes on cell responsiveness. J Pharmacol Exp Ther. 1990;253:688–697. [PubMed] [Google Scholar]
- 69.Teyler TJ, DiScenna P. Long-term potentiation. Annu Rev Neurosci. 1987;10:131–161. doi: 10.1146/annurev.ne.10.030187.001023. [DOI] [PubMed] [Google Scholar]
- 70.Thalmann RH. Evidence that guanosine triphosphate (GTP)-binding proteins control a synaptic response in brain: effect of pertussis toxin and GTPγS on the late inhibitory postsynaptic potential of hippocampal CA3 neurons. J Neurosci. 1988;8:4589–4602. doi: 10.1523/JNEUROSCI.08-12-04589.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thompson SM, Gähwiler BH. Activity-dependent disinhibition. III. Desensitization and GABAB receptor-mediated presynaptic inhibition in the hippocampus in vitro. J Neurophysiol. 1989;61:524–533. doi: 10.1152/jn.1989.61.3.524. [DOI] [PubMed] [Google Scholar]
- 72.Turgeon SM, Albin RL. Postnatal ontogeny of GABAB binding in rat brain. Neuroscience. 1994;62:601–613. doi: 10.1016/0306-4522(94)90392-1. [DOI] [PubMed] [Google Scholar]
- 73.Wagner JJ, Alger BE. GABAergic and developmental influence on homosynaptic LTD and depotentiation in rat hippocampus. J Neurosci. 1995;15:1577–1586. doi: 10.1523/JNEUROSCI.15-02-01577.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Worley PF, Baraban JM, Supattapone S, Wilson VS, Snyder SH. Characterization of inositol trisphosphate receptor binding in brain. J Biol Chem. 1987;262:12132–12136. [PubMed] [Google Scholar]