

Abstract
The ICE/CED-3 family of proteases has been implicated in playing a fundamental role in programmed cell death. Bcl-2 protein represses a number of apoptotic death programs, but the biochemical mechanism of its action is not known. We investigated the activation of ICE/CED-3 proteases induced by three apoptotic stimuli (staurosporine, ceramide, and serum withdrawal) in the neuronal cell line GT1-7 and in cells overexpressing Bcl-2. Rapid activation of a 17 kDa subunit of an activated member of the ICE/CED-3 family is demonstrated by affinity-labeling GT1-7 extracts from apoptotic controls cells with a biotinylated ICE/CED-3 inhibitor. This activation corresponds to an increased ICE/CED-3-like protease activity in extracts measured by a fluorogenic substrate assay. In a cell-free system, these extracts induce apoptotic morphological changes in intact nuclei. All three activities are readily inhibited by treatment of control extracts with ICE/CED-3-like protease inhibitors. Overexpressed Bcl-2 inhibits the activation of the 17 kDa protein, the ICE/CED-3-like protease activity in the fluorogenic assay, and the induction of apoptotic morphological changes in HeLa nuclei in the cell-free system, similar to results obtained with ICE/CED-3 protease inhibitors. At the mRNA level, overexpression of Bcl-2 did not alter expression of five members of the ICE/CED-3 family: CPP32, ICE, Mch 2, Nedd 2, and TX. Overexpression of Bcl-2 prevented the apoptosis-induced processing of pro-Nedd 2 to the cleaved form. These data suggest that Bcl-2 participates upstream from the function of ICE/CED-3 proteases and may inhibit apoptosis by preventing the post-translational activation of ICE/CED-3 proteases.
Keywords: apoptosis, programmed cell death, neural cells, Bcl-2, ICE/CED-3 proteases, ceramide, staurosporine, serum withdrawal
Apoptosis, a form of cell death morphologically distinct from necrosis, plays an essential role in the development and maintenance of homeostasis in multicellular organisms (Steller, 1995). Apoptosis may be the predominant form of cell death in various diseases (for reviews, see Thompson, 1995; Bredesen, 1995). In the last decade, significant progress has been made toward understanding the biochemical basis of cell death (Steller, 1995). One of the earliest apoptosis-related genes identified was bcl-2 (Tsujimoto et al., 1984). Bcl-2 functions in oncogenic cells by inhibiting apoptotic cell death rather than by causing an increase in cell proliferation (Hockenberry et al., 1990; Nuñez et al., 1990). The anti-apoptotic activity of Bcl-2 has since been demonstrated widely in various experimental paradigms (for review, see Reed, 1994).
Bcl-2 is a member of a large family of proteins sharing conserved sequence motifs (Boise et al., 1993; Reed, 1994). Protein–protein interactions between Bcl-2 family members in the form of homo- and heterodimerization seem to determine the susceptibility of a cell to a given apoptotic stimulus (Reed, 1994). It has been proposed that Bcl-2 functions via intracellularly generated reactive oxygen species (ROS) (Hockenberry et al., 1993; Kane et al., 1993; Wiedau-Pazos et al., 1996). Bcl-2, however, prevents apoptosis induced under very low oxygen conditions, suggesting that the mechanism of Bcl-2 action may not necessarily involve the regulation of ROS activity (Jacobson and Raff, 1995; Shimizu et al., 1995). Therefore, despite progress in defining some of the physiological roles of Bcl-2, the biochemical mechanism of its action remains unknown.
Demonstration of the close homology of the Caenorhabditis elegans protein CED-3 to the mammalian cysteine protease interleukin-1β-converting enzyme (ICE) (Yuan et al., 1993) led to the discovery of several proteases that are involved in apoptosis. A number of homologous cysteine proteases, collectively referred to as the ICE/CED-3 family, have since been identified, and their pro-apoptotic effects have been demonstrated (for review, see Martin and Green, 1995). It has been suggested that endogenous ICE/CED-3 family members are expressed constitutively in an inactive pro-form in normal cells and that on induction of apop-tosis, they are processed into an active form involving either self-proteolysis or cleavage by a related family member by a poorly understood mechanism (Martin and Green, 1995). Once activated, ICE/CED-3 proteases seem to cleave specific cellular proteins. A small number of cellular proteins have so far been identified as potential substrates for some of the ICE/CED-3 family proteases, but their role(s) in apoptosis remains unclear (Martin and Green, 1995). ICE and CPP32 show a nearly absolute requirement for an aspartyl residue at the P1 position of their respective substrates (Howard et al., 1991; Nicholson et al., 1995), which may be shared by the other family members.
In this communication, we demonstrate that the overexpression of Bcl-2 in the neural cell line GT1-7 inhibits apoptosis induced by various stimuli by preventing the generation of the activated forms of the ICE/CED-3 family proteases from inactive pro-forms that are constitutively expressed.
MATERIALS AND METHODS
Cell cultures. GT1-7 cells (Mellon et al., 1990) were retrovirally infected with the pBabe-puromycin vector and pBabe-puromycin-Bcl-2 as described (Mah et al., 1993). Cells were cultured on poly-l-lysine-coated tissue culture plates in DMEM containing 10% fetal bovine serum. Cell extracts were made from cells harvested in the linear growth phase, at ∼60-70% confluence. HeLa cells were grown in DMEM containing 10% fetal bovine serum.
Visualization of apoptotic nuclei. Control and treated cells (see figure legends) were incubated in DMEM containing 0.5 μg/ml Hoechst 33258 (Sigma, St. Louis, MO) for 20 min. Nuclei were visualized by ultraviolet epifluorescence and photographed at a 400× magnification on a Nikon Stereo inverted microscope. The reagents used in the treatments were obtained commercially: staurosporine from Sigma, C2-ceramide from Matreya (Pleasant Gap, PA), and VAD-FMK from Enzyme Systems Products (Dublin, CA).
DNA fragmentation. Cells resuspended in 100 μl of PBS were lysed by adding an equal volume of 2× lysis buffer (200 mmHEPES, pH 7.5, 2% Triton X-100, 400 mm NaCl, 20 mm EDTA). Samples were digested for 45 min at 37°C with 5 U of RNase A. Finally, samples were extracted with phenol-chloroform, and the DNA was precipitated in 70% ethanol. The pellets were resuspended in 10 μl of water and analyzed on a 2% agarose/Tris-acetate gel.
Preparation of GT1-7 cell extracts. After two washes in ice-cold PBS, cells were lysed in cell extraction buffer [100 mm HEPES, pH 7.5, 1% Triton X-100, 10 mm DTT, 1 mm phenylmethylsulfonyl fluoride (PMSF), 5 μg/ml leupeptin, 1 μg/ml pepstatin A, 1 mm EDTA]. Cell lysates were incubated on ice for 30 min with intermittent vortexing (four times, 2 sec each). The lysates were centrifuged at 20,000 × g for 15 min, and the supernatant was collected and used without further purification. The protein concentration of the supernatant was determined by Coomassie Plus Protein Assay (Pierce, Rockford, IL).
Labeling with the biotinylated inhibitor Tyr-Val-Lys-Asp-(acyloxy)methyl ketone, biotinylated at the lysine (BIN), of ICE/CED-3 proteases. In a 50 μl reaction, cell extracts (∼1 mg/ml protein) were incubated with 2 μm of a biotinylated ICE family protease inhibitor for 20 min at 25°C with constant shaking. Reactions were stopped by adding an equal volume of 2× sample buffer (20% glycerol, 3% SDS, 125 mm Tris, pH 6.8, 0.1% bromophenol blue, 2% β-mercaptoethanol). BIN was a generous gift from Dr. N. A. Thornberry (Merck Research Laboratories, Rahway, NJ).
Western blotting to visualize BIN-labeled proteins.BIN-labeled cell extracts were resolved on 15% SDS/polyacrylamide gels and electroblotted overnight to PVDF membranes (Tropix, Bedford, MA) in 20 mm Tris, pH 8, 150 mm glycine, and 20% methanol. After transfer of proteins, membranes were first incubated at room temperature in blocking buffer [PBS, 0.1% Tween-20, 0.2% I-Block (Tropix)] for 1 hr and then with streptavidin-alkaline phosphatase conjugate [1:20,000 dilution of Avidx-AP (Tropix) in blocking buffer] for 1 hr. The blots were washed in blocking buffer (four times, 5 min each) and then in assay buffer (0.1 mdiethanolamine, 1 mm MgCl2) (two times, 5 min each). The blots were finally incubated in chemiluminescent substrate solution [0.24 mm CSPD (Tropix) in assay buffer] for 5 min and exposed to x-ray film (Eastman Kodak, Rochester, NY).
Asp-Glu-Val-Asp-amino-4-trifluoromethyl coumarin (DEVD-AFC) hydrolysis assay. GT1-7 cellular extracts (1 mg/ml) were incubated at 37°C with the fluorogenic substrate z-DEVD-AFC (100 μm) (Enzyme Systems Products), and the increase in fluorescence versus time was monitored using a spectrofluorometer (λexcit = 400 nm, λemis = 505 nm). The rates of DEVD-AFC hydrolysis were calculated from the linear region of the graph.
HeLa nuclei assay in a cell-free system. HeLa cells were lysed with a Dounce homogenizer in nuclear buffer (NB) (10 mm PIPES, pH 7.4, 10 mm KCl, 2 mmMgCl2, 1 mm DTT, 10 μmcytochalasin B plus protease inhibitors) and pelleted through a 30% sucrose gradient at 800 × g for 10 min. The nuclear pellet was resuspended, washed once with NB, and stored at −20 at a concentration of 5 × 107 nuclei/ml.
GT1-7 cells were washed with PBS and lysed in buffer A (50 mm HEPES, pH 7.5, 50 mm KCl, 5 mmEGTA, 2 mm MgCl2, 1 mm DTT, 10 μm cytochalasin B, 1 mm PMSF, 5 μg/ml leupeptin, 1 μg/ml pepstatin A) by a combination of three freeze/thaw cycles and gentle homogenization in a Dounce homogenizer. Cell lysates were then centrifuged for 30 min at 20,000 × g. The supernatants were diluted to 8 mg/ml protein concentration in buffer B (10 mm HEPES, pH 7.5, 50 mm NaCl, 2 mm MgCl2, 5 mm EGTA, 1 mm DTT, 2 mm ATP, 10 mmphosphocreatine, 50 μg/ml creatine kinase). These extracts were used immediately or stored at −80°C for later use.
In a 30 μl reaction mixture, 1 × 106 HeLa nuclei were incubated at 37°C for 2 hr with 10 μl of diluted cytosolic extracts. Nuclei were stained with 5 μg/ml Hoechst 33342 and examined by fluorescence microscopy. Nuclei showing the characteristic margination and condensation of the chromatin were scored as apoptotic. Two hundred nuclei in four independent fields were scored for each data point.
RNA isolation and reverse transcription (RT). Total RNA was made from GT1-7 cells (1 × 106 to 1 × 107 cells) and mouse spleen (0.1 gm) using the acid guanidinium thiocyanate-phenol-chloroform extraction method (Chomczynski and Sacchi, 1987) (TriReagent, MRC, Cincinnati, OH). One hundred fifty picomoles of random deoxynucleotide hexamers (Pharmacia LKB Biotechnology, Piscataway, NJ) were annealed to 10–20 μg of RNA in RT buffer (50 mm Tris, pH 8.3, 3 mmMgCl2, 75 mm KCl) for 1 hr at 25°C in the presence of RNase inhibitor in a 20 μl reaction volume. The annealed samples were reverse-transcribed in a 40 μl volume containing RT buffer (10 mm DTT, 500 nm each dNTP, and 100 U of SuperScript II reverse transcriptase) (Life Technologies, Gaithersburg, MD) for 1 hr at 42°C. The sample was digested with 2 U of RNase A (Pharmacia) for 45 min at 37°C before use in the PCR. This cDNA mixture was used without further purification for PCR analysis.
PCR analysis. Oligomers specific for murine ICE (Nett et al., 1992) and murine Nedd 2 (Kumar et al., 1994) were designed using the published sequences for the respective cDNAs. The cDNA sequence for murine CPP32 was kindly provided by Dr. E. S. Alnemri (Thomas Jefferson University). The oligomers specific for Mch 2 and TX were based on the reported human cDNA sequences (Faucheu et al., 1995; Fernandes-Alnemri et al., 1995a). The oligomers listed below were used in the PCR analysis.
CPP32:
Forward 5′-GAGCACTGGAATGTC-3′
Reverse 5′-ATGAAGAGTTTCGGC-3′
ICE:
Forward 5′-ACACGTCTTGCCCTCATTATCTGCA-3′
Reverse 5′-TGTCAGAAGTCTTGTGCTCTGGG-3′
Nedd 2:
Forward 5′-ATTCAGCACGTACTC-3′
Reverse 5′-TAGAGTAGTGTGGTC-3′
Mch 2:
Forward 5′-TTATCATCCAGGCATGT-3′
Reverse 5′-CATGAGCCGTTCACAGT-3′
TX:
Forward 5′-ATCATTGTCCAGGCCTGC-3′
Reverse 5′-CCAGGACACGTTGTGT-3′
The 50 μl PCR reaction mixture was composed of the following: 5 μl of the cDNA mixture, PCR buffer (20 mm Tris, pH 7.5, 2.5 mm MgCl2, 50 mm KCl), 200 μm each dNTP, 0.5 mm each of the forward and reverse primers, and 1.25 U of AmpliTaq DNA polymerase (Perkin-Elmer, Palo Alto, CA). The PCR program that was used consisted of 35 cycles of 1 min at 94°C, 1.5 min at 55°C, and 1.5 min at 72°C each, followed by 10 min at 72°C before refrigeration. An 8 μl aliquot of the PCR mixture was analyzed on an 8% nondenaturing polyacrylamide/Tris-borate gel.
The PCR fragments of interest were cloned into pCR II vector (Invitrogen, San Diego, CA); both strands of the insert were sequenced using the dideoxy chain termination technique (Sanger et al., 1977) (Sequenase, Version II, USB, Cleveland, OH). The identity of the PCR fragments generated for the different ICE/CED-3 family member messages was confirmed by comparing the sequences with reported cDNA or gene sequences.
Immunoblotting with the Nedd 2 antibody. BIN-labeled GT1-7 cell extracts were resolved by SDS-PAGE and immunoblotted using the same procedure used in the Western blotting. After protein transfer, immunoblots were first incubated at room temperature for 1 hr in blocking buffer. Blots were then incubated with the rabbit anti-Nedd 2 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) (1:350 dilution in blocking buffer) overnight at 4°C with gentle shaking. After washes in blocking buffer (three times, 5 min each), the blots were incubated for 1 hr at room temperature with anti-rabbit-AP (Tropix; 1:8000 dilution in blocking buffer). The membranes were then washed again in blocking buffer (four times, 5 min each), followed by washes in assay buffer (two times, 5 min each). Finally, the blots were incubated with the chemiluminescent substrate (see above) for 5 min and visualized by exposing the blots to x-ray film.
RESULTS
Bcl-2 blocks apoptosis induced by diverse stimuli, including staurosporine (Raff, 1992), ceramide (Obeid et al., 1993), and serum withdrawal (Raff, 1992) in various types of mammalian cells. Figure1 demonstrates that control GT1-7 cells (Fig.1A, panel 1) transfected with vector alone undergo apoptosis after treatment with staurosporine, as judged by two different criteria: nuclear fragmentation (Fig. 1A,panel 2) and formation of DNA ladders (Fig. 1B,lane 2). GT1-7 cells overexpressing Bcl-2, extensively characterized in this laboratory (Kane et al., 1995), showed complete protection from staurosporine-induced apoptosis, as evidenced by the lack of the fragmented nuclear morphology (Fig. 1A,panel 3) and DNA laddering (Fig. 1B, lane 3). Pretreatment of cells with the membrane-permeable, irreversible ICE/CED-3-like protease inhibitor Val-Ala-Asp-fluoromethyl ketone (VAD-FMK) (Pronk et al., 1996) was highly effective in preventing apoptosis in the control cells [Fig. 1, A(panel 4) and B (lane 4)]. Both Bcl-2 and VAD-FMK blocked apoptosis induced by either ceramide or serum withdrawal (data not shown). These results indicate that apoptosis in GT1-7 cells requires endogenous ICE/CED-3 protease activity and that Bcl-2 blocks apoptosis induced by diverse stimuli.
Fig. 1.

Induction of apoptosis and its inhibition by Bcl-2 and VAD-FMK in GT1-7 cells. A, Nuclear staining of cells with the fluorescent dye Hoechst 33258 after induction of apoptosis.Panel 1, Untreated, vector control cells; panel 2, vector control cells treated with staurosporine (0.5 μm, 18 hr); panel 3, cells overexpressing Bcl-2 treated with staurosporine (0.5 μm, 18 hr);panel 4, vector control cells preincubated with VAD-FMK (100 μm, 1 hr) before treatment with staurosporine (0.5 μm, 18 hr). Arrow in panel 2 indicates apoptotic nuclei. B, Agarose gel of low molecular weight DNA extracted from cells as indicated. Lane 1, Untreated, vector control cells; lane 2, vector control cells treated with staurosporine (0.5 μm, 18 hr); lane 3, cells overexpressing Bcl-2 treated with staurosporine (0.5 μm, 18 hr); lane 4, vector control cells preincubated with VAD-FMK (100 μm, 1 hr) before treatment with staurosporine (0.5 μm, 18 hr).
Thornberry and colleagues (1994) synthesized and used the tetrapeptide BIN as a potent and irreversible inhibitor of ICE in human monocytes. BIN reacts covalently with the catalytic site cysteine and affinity-labels the larger of the two subunits of activated ICE/CED-3 proteases. The advantages of using BIN are that (1) it is an irreversible inhibitor and affinity-labels all ICE/CED-3 family proteases, and (2) on labeling activated ICE/CED-3 members, the biotinylated subunit containing the catalytic site cysteine can be visualized in a Western blot with a streptavidin-based probe.
Figure 2 demonstrates that in extracts made from GT1-7 cells treated with staurosporine or ceramide, or after serum withdrawal, BIN affinity-labels a 17 kDa protein (p17). p17 was not detected in samples pretreated with VAD-FMK (Fig. 2) or in samples not labeled with BIN (data not shown). In cells overexpressing Bcl-2, p17 labeling was not detected, and the cells were resistant to apoptosis induced by these treatments. Incubation of the apoptotic, control GT1-7 cell extracts with inhibitors of ICE/CED-3-like proteases, e.g., iodoacetamide and the cowpox viral protein Crm-A (Ray et al., 1992;Gagliardini et al., 1994), before labeling with BIN inhibited the labeling of p17. Inhibitors that have no significant effects on ICE/CED-3-like proteases, including E-64,l-1-chloro-3[4-tosylamido]−4-phenyl-2-butanone (TPCK),l-1-chloro-3-[4-tosylamido]−7-amino-2-heptanone (TLCK), PMSF, leupeptin, and pepstatin A, did not affect the labeling of p17 (data not shown).
Fig. 2.

Induction of p17 by different apoptotic stimuli and its inhibition by Bcl-2. Western blots of BIN-labeled cell extracts made from vector control and Bcl-2-overexpressing cells treated as indicated. CNTL, Untreated cells; STS, staurosporine (0.5 μm, 18 hr); CER, C2-ceramide (50 μm, 18 hr);SFM, serum-free medium (48 hr);VAD-FMK/STS, preincubated with VAD-FMK (100 μm, 1 hr) before staurosporine treatment.
Figure 3A shows the results of a time-course experiment in which extracts prepared from GT1-7 cells treated with staurosporine for various periods of time were BIN-labeled and analyzed by Western blotting procedures. Extracts from control cells showed rapid induction of p17, as early as 1 hr after staurosporine treatment, whereas this protein was not detected in extracts made from staurosporine-treated, Bcl-2-expressing cells after BIN-labeling.
Fig. 3.
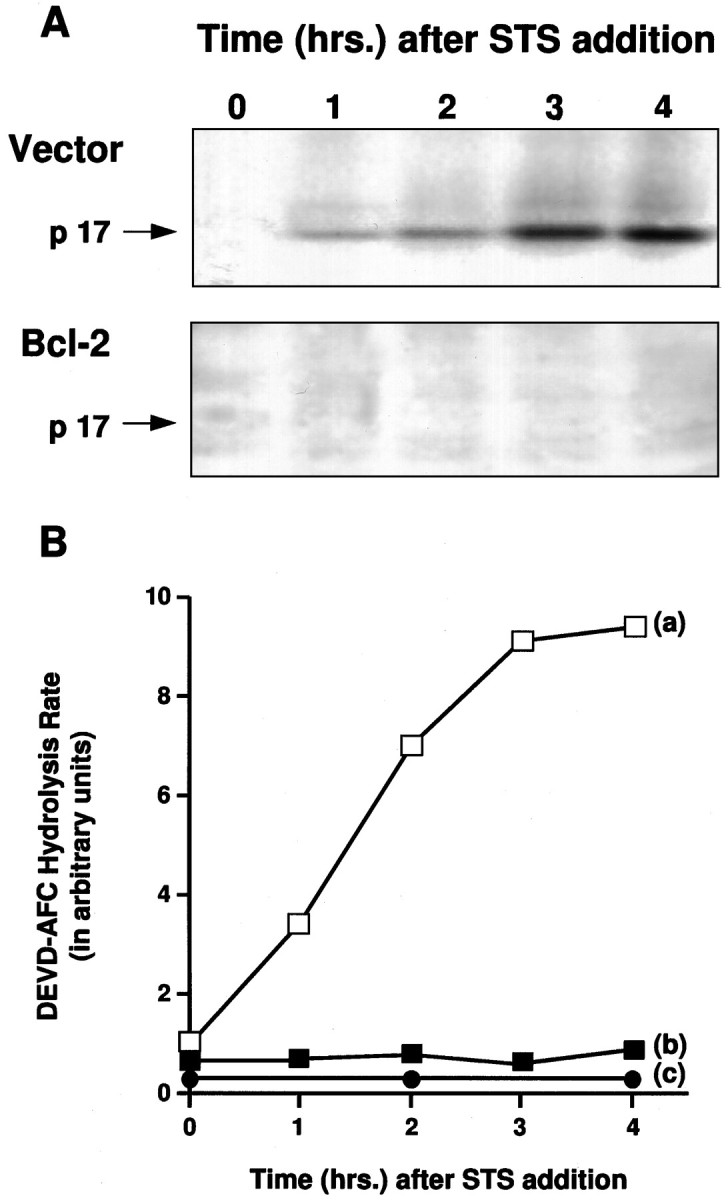
Time course of p17 induction and DEVD-AFC hydrolysis during apoptosis. A, Western blot of BIN-labeled cell extracts from vector control and Bcl-2-overexpressing cells at the indicated times after staurosporine (0.5 μm) treatment. B, ICE/CED-3-like proteolytic activity measured by DEVD-AFC hydrolysis at the indicated times after staurosporine (0.5 μm) treatment. a, Vector control cells; b, Bcl-2-overexpressing cells;c, extracts from staurosporine-treated vector control cells preincubated with VAD-FMK (100 μm, 15 min) before measurement of DEVD-AFC hydrolysis rates.
To measure the enzymatic activity of ICE/CED-3 proteases activated on apoptosis in GT1-7 cells, we used the fluorogenic substrate DEVD-AFC (Thornberry et al., 1992; Nicholson et al., 1995). The rate of hydrolysis of DEVD-AFC increased rapidly in extracts made from staurosporine-treated control GT1-7 cells (Fig. 3B,a), whereas DEVD-AFC hydrolysis stayed at the same low level in extracts from staurosporine treated Bcl-2-expressing GT1-7 cells (Fig.3B,b). No significant level of DEVD-AFC hydrolysis was detected in extracts made from control GT1-7 cells that had been preincubated with ICE/CED-3 protease inhibitor VAD-FMK (Fig.3B,c).
Cell-free systems have been used to reconstitute apoptotic eventsin vitro (Earnshaw, 1995). Two hallmarks of apoptosis, nuclear fragmentation and DNA laddering, as shown in Figure 1, occurredin vitro when cytosolic extracts prepared from cells undergoing apoptosis were added to intact nuclei isolated from healthy cells (Lazebnik et al., 1993). Figure 4Ademonstrates that although cytosols from untreated GT1-7 cells had very little effect on HeLa nuclei, cytosols prepared from control GT1-7 cells treated with staurosporine induced a fragmented morphology, characteristic of apoptosis, in ∼38% of the HeLa nuclei. Conversely, cytosols prepared from staurosporine-treated GT1-7 cells overexpressing Bcl-2 had no significant morphological effect on HeLa nuclei (Fig.4B). Preincubation of the apoptotic GT1-7 cytosols prepared from the vector controls with either VAD-FMK or BIN dramatically reduced their ability to induce the morphological changes in isolated HeLa nuclei (Fig. 4A).
Fig. 4.

Cell-free apoptosis and its inhibition by BIN, VAD-FMK, and Bcl-2. A, Generation of apoptotic HeLa nuclei by incubation with the following: 1, buffer;2, extract from untreated vector control cells;3–7, extracts from vector control cells treated with staurosporine (0.5 μm, 2 hr); 4–7, staurosporine-treated vector control cell extracts incubated with BIN or VAD-FMK at the indicated concentrations. B, Generation of apoptotic HeLa nuclei by incubation with the following:1, buffer; 2, extract from untreated Bcl-2-overexpressing GT1-7 cells; 3, extract from Bcl-2-overexpressing cells treated with staurosporine (0.5 μm, 2 hr).
Our results show that ICE/CED-3-like proteases are activated rapidly in control GT1-7 cells on the onset of apoptosis. In cells overexpressing Bcl-2, this activation is inhibited. The data, however, do not distinguish between the regulation of ICE/CED-3 protease activity by Bcl-2 at the level of transcription, translation, or post-translational processing. To explore the possibility that overexpression of Bcl-2 inhibited or significantly altered the expression of ICE/CED-3 proteases at the mRNA level, total RNA was prepared from control and Bcl-2-expressing GT1-7 cells and analyzed by qualitative RT-PCR (Fig.5). No significant difference was observed between control and Bcl-2-expressing cells in the expression of five ICE/CED-3 family members. CPP32, Mch 2, and Nedd 2 are expressed in both control and Bcl-2-expressing cells. Only a splice variant form of ICE was found to be expressed in both the control and Bcl-2-expressing cells. The expected fragment for TX was not amplified in either control or Bcl-2-expressing cells, but was present in the control spleen RNA, suggesting possible absence of TX in neural cells.
Fig. 5.

Qualitative RT-PCR analysis of five ICE/CED-3 family members. Total RNA was isolated from untreated (−) and staurosporine-treated (+) vector control and Bcl-2-overexpressing cells. Spleen RNA and cDNA were used as controls. Arrowsand numbers on the right indicate the size and position of the amplification products. The 570 bp band amplified by the ICE primers from GT1-7 RNA is an alternately spliced variant of ICE, containing intron 4, found in GT1-7 cells.
To study the regulation of ICE/CED-3 proteases by Bcl-2 at the translational level, antibodies specific for each ICE/CED-3 family member can be used in immunoprecipitation or immunoblotting procedures. We tested commercially available antibodies and found that the only one that gave clean signals on immunoblots was an anti-Nedd 2, C-terminal-specific polyclonal antibody. This antibody recognized pro-Nedd 2 (MW ∼50 kDa) in extracts made from control and Bcl-2-expressing GT1-7 cells (Fig. 6). In extracts made from apoptotic control cells after staurosporine treatment, the antibody recognized a protein band of ∼10-12 kDa (open arrow), in addition to the 50 kDa pro-form. The appearance of the 10–12 kDa band was accompanied by a concomitant decrease in the level of pro-Nedd 2, indicating that the 10–12 kDa band was the smaller subunit of processed Nedd 2. Although the 50 kDa pro-Nedd 2 band was detected in extracts made from Bcl-2-expressing cells treated with staurosporine, the smaller band associated with the processing of pro-Nedd 2 in apoptosing cells was not detected, indicating that expression of Bcl-2 inhibited the apoptosis-induced processing of pro-Nedd 2.
Fig. 6.

Inhibition of the processing of pro-Nedd 2 by Bcl-2. Immunoblots of staurosporine (0.5 μm)-treated GT1-7 cell extracts made at the indicated times using an anti-Nedd 2 antibody. Vector, Extracts from vector control GT1-7 cells; Bcl-2, extracts from Bcl-2-overexpressing GT1-7 cells. Pro-Nedd 2 (∼50 kDa) is indicated by a filled arrow; open arrow indicates a protein band (∼10–12 kDa) that appears to correspond to the C-terminal, smaller subunit of processed Nedd 2.
DISCUSSION
Our data show that overexpression of Bcl-2 in the hypothalamic neural cell line GT1-7 protects cells from apoptosis in response to various death-inducing stimuli. Overexpression of Bcl-2 has been examined in trigeminal neurons (Garcia et al., 1992; Greenlund et al., 1995), and it delays but does not block apoptosis induced by trophic factor deprivation. Survival of ciliary ganglion neurons after withdrawal of ciliary neurotrophic factor is not altered by expression of Bcl-2 (Allsopp et al., 1993). PC12 cells transfected with Bcl-2 are resistant to apoptosis induced by a myriad of stimuli (Mah et al., 1993). Moreover, transgenic mice overexpressing Bcl-2 in the nervous system demonstrate decreased neuronal cell deaths during periods of naturally occurring programmed neuronal death (Martinou et al., 1994). Thus, it seems that Bcl-2 does not inhibit neuronal apoptosis uniformly, suggesting more than one cell-death pathway.
We examined whether the ability of Bcl-2 to block apoptosis in GT1-7 cells is directly or indirectly related to interaction of Bcl-2 with the ICE/CED-3-like family of proteases. Our data show that ICE/CD-3 proteases are activated rapidly on induction of apoptosis and that Bcl-2 blocks this activation. Using an affinity-labeling technique that labels the catalytic site cysteine, we have identified a 17 kDa protein that is rapidly induced in extracts of GT1-7 cells undergoing apoptosis and appears to be larger subunit of the enzymatically active form of an ICE/CED-3 family member. On the basis of the molecular weight of p17 (Nicholson et al., 1995; Tewari et al., 1995; Duan et al., 1996), BIN labeling, processing pattern, and reaction with ICE/CED-3 inhibitors, the most probable precursor for the generation of p17 seems to be CPP32. At the present time, however, we cannot rule out the possibility that p17 may be formed partially from some other member(s) of the ICE/CED-3 family of proteases.
What is clear from our studies is that the overexpression of Bcl-2 in GT1-7 cells blocks the formation of p17, suggesting a direct or indirect interaction with the ICE/CED-3-like molecule. Using a cell-free system, we have reconstituted apoptotic events and found that the ICE/CED-3-like enzymatic activity level in apoptotic cytosols was 10-fold higher than in control extracts and was drastically reduced by pretreatment with either BIN or VAD-FMK. Overexpression of Bcl-2 in GT1-7 cells displayed a similar inhibition of enzyme activity, further suggesting an interaction of Bcl-2 with the ICE/CED-3 family of proteases.
Overexpression of Bcl-2 did not alter the mRNA levels for five members of the ICE/CED-3 family. RT-PCR analysis revealed the presence of an alternately spliced variant of ICE mRNA. To date, the structure of the full-length mRNA from which the 570 bp ICE fragment was amplified has not been established. Alternate splicing for ICE mRNA in GT1-7 cells could represent an important mechanism that regulates the biological activity of proteins in a tissue-specific manner. Differential splicing of mRNAs for members of the ICE/CED-3 family, e.g., Ich 1 (Wang et al., 1994), ICE (Alnemri et al., 1995), Mch 2 (Fernandes-Alnemri et al., 1995a), and Mch 3 (Fernandes-Alnemri et al., 1995b), have been reported. The significance of the differential splicing of ICE mRNA and the possible cell specificity in neurons is under further investigation.
The biochemical mechanism(s) by which Bcl-2 inhibits these proteases is not known. Our data suggest that Bcl-2 may block apoptosis by preventing processing of the pro-forms of these proteases into the active forms. This idea is supported by our results using the Nedd 2 antibody. We demonstrate for the first time the rapid conversion of pro-Nedd 2 into a shorter, cleaved form on induction of apoptosis in control cells, whereas this conversion is blocked in Bcl-2-overexpressing cells. This interpretation is also favored by the fact that in extracts from Bcl-2-overexpressing cells, p17 is not labeled. In addition, ceramide-induced apoptosis in T cell lines also demonstrates that Bcl-2 inhibits activation of ICE/CED-3 family proteases (Martin et al., 1995). Consistent with our observation in neural cells, Chinnaiyan et al. (1996) recently reported that in Jurkat cells, Bcl-2 and Bcl-x function upstream of YAMA/CPP32 and ICE-LAP3/Mch3 in inhibiting apoptosis. Although the activation of ICE/CED-3 family proteases has been shown to be a prerequisite for Fas-induced cell death (Los et al., 1995), conflicting data regarding the ability of Bcl-2 to block this death pathway have been reported (Martin et al., 1995; Armstrong et al., 1996; Chinnaiyan et al., 1996). Granzyme-B has been shown to activate ICE/CED-3 family proteases and induce cell death that is not inhibited by Bcl-2 (Darmon et al., 1995; Quan et al., in press). Our findings are important in that they suggest that the ICE/CED-3 family of proteases may be activated in all apoptotic paradigms and that Bcl-2 may participate upstream from the action of ICE/CED-3 proteases to inhibit apoptosis by blocking the post-translational activation of ICE/CED-3 proteases. Moreover, our results are consistent with the earlier conclusions based on genetic studies in C. elegans regarding the ordering of the action of ced-9 and ced-3, the nematode homologs of Bcl-2 and ICE family proteins (Ellis and Horvitz, 1986; Yuan et al., 1993; Hengartner and Horvitz, 1994). Understanding the mechanism of resistance to apoptosis may prove to be important in understanding neurodegenerative disease states, in which neurons are susceptible to death, and in the development of therapeutics that modulate the apoptotic pathways.
Both authors contributed equally.
This work was supported by Grants AG 12282 and NS 25554 from National Institutes of Health and by a grant from the American Health Assistance Foundation to D.E.B. We are grateful for fellowship support from the Mathers Charitable Fund, the Joseph Drown Foundation, and the Vivorx Corporation. We are grateful to the following people for their generous gifts: Dr. P. Mellon for GT1-7 cells, Dr. H. Land for pBabe-puromycin vector DNA, Dr. M. Cleary for the cDNA for human Bcl-2 cDNA, Dr. N. A. Thornberry for the biotinylated ICE inhibitor (BIN), and Dr. E. S. Alnemri for making the sequence of murine CPP32 available before its publication.
Correspondence should be addressed to Celik Kayalar, 311 4th Avenue, #607, San Diego, CA 92101, or Anu Srinivasan, Idun Pharmaceuticals, 11085 North Torrey Pines Road, La Jolla, CA 92037.
REFERENCES
- 1.Allsop TE, Tschopp J, Jongeneel CV. The proto-oncogene bcl-2 can selectively rescue neurotrophic-factor dependent neurons from apoptosis. Cell. 1993;73:295–307. doi: 10.1016/0092-8674(93)90230-n. [DOI] [PubMed] [Google Scholar]
- 2.Alnemri ES, Fernandes-Alnemri T, Litwack G. Cloning and expression of four novel isoforms of human interleukin-1β-converting enzyme with different apoptotic activities. J Biol Chem. 1995;270:4312–4317. doi: 10.1074/jbc.270.9.4312. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong RC, Aja T, Xiang J, Gaur S, Krebs JF, Hoang K, Bai X, Korsmeyer SJ, Karanewsky DS, Fritz LC, Tomaselli KJ. Fas-induced activation of the cell death-related protease CPP32 is inhibited by Bcl-2 and by ICE family protease inhibitors. J Biol Chem. 1996;271:16850–16855. doi: 10.1074/jbc.271.28.16850. [DOI] [PubMed] [Google Scholar]
- 4.Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB. bcl-x, a bcl2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 5.Bredesen DE. Neural apoptosis. Ann Neurol. 1995;38:839–851. doi: 10.1002/ana.410380604. [DOI] [PubMed] [Google Scholar]
- 6.Chinnaiyan AM, Orth K, O’Rourke K, Duan H, Poirier GG, Dixit VM. Molecular ordering of the cell death pathway. J Biol Chem. 1996;271:4573–4576. doi: 10.1074/jbc.271.9.4573. [DOI] [PubMed] [Google Scholar]
- 7.Chomczynski P, Sacchi N. Single step method of RNA isolation by acid guanidinium isothiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 8.Darmon AJ, Nicholson DW, Bleackley RC. Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature. 1995;377:446–448. doi: 10.1038/377446a0. [DOI] [PubMed] [Google Scholar]
- 9.Duan H, Chinnaiyan AM, Hudson PL, Wing JP, He W, Dixit VM. ICE-LAP3, a novel mammalian homologue of the Caenorhabditis elegans cell death protein Ced-3 is activated during Fas- and tumor necrosis factor-induced apoptosis. J Biol Chem. 1996;271:1621–1625. doi: 10.1074/jbc.271.3.1621. [DOI] [PubMed] [Google Scholar]
- 10.Earnshaw WC. Apoptosis: lessons from in vitro systems. Trends Cell Biol. 1995;5:217–220. doi: 10.1016/s0962-8924(00)89006-6. [DOI] [PubMed] [Google Scholar]
- 11.Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans . Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- 12.Faucheu C, Diu A, Chan AWE, Blanchet A, Miossec C, Herve F, Collard-Dutilleul V, Gu Y, Aldape RA, Lippke JA, Rocher C, Su MS, Livingston DJ, Hercend T, Lalanne J. A novel human protease similar to the interleukin-1β-converting enzyme induces apoptosis in transfected cells. EMBO J. 1995;14:1914–1922. doi: 10.1002/j.1460-2075.1995.tb07183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandes-Alnemri T, Litwack G, Alnemri ES. Mch2, a new member of the apoptotic Ced-3/ICE cysteine protease gene family. Cancer Res. 1995a;55:2737–2742. [PubMed] [Google Scholar]
- 14.Fernandes-Alnemri T, Takahashi A, Armstrong R, Krebs J, Fritz L, Tomaselli KJ, Wang L, Yu Z, Croce CM, Salvesen G, Earnshaw WC, Litwack G, Alnemri ES. Mch3, a novel human apoptotic cysteine protease highly related to CPP32. Cancer Res. 1995b;55:6045–6052. [PubMed] [Google Scholar]
- 15.Gagliardini V, Fernandez P, Lee RKK, Drexler HCA, Rotello RJ, Fishman MC, Yuan J. Prevention of vertebrate neuronal death by the crmA gene. Science. 1994;263:826–828. doi: 10.1126/science.8303301. [DOI] [PubMed] [Google Scholar]
- 16.Garcia I, Martinou I, Tsujimoto Y, Martinou JC. Prevention of programmed cell death of sympathetic neurons by bcl-2 proto-oncogene. Science. 1992;258:302–304. doi: 10.1126/science.1411528. [DOI] [PubMed] [Google Scholar]
- 17.Greenlund LJS, Krosmeyer SJ, Johnson EM., Jr Role of Bcl-2 in the survival and function of developing and mature sympathetic neurons. Neuron. 1995;15:649–661. doi: 10.1016/0896-6273(95)90153-1. [DOI] [PubMed] [Google Scholar]
- 18.Hengartner MO, Horvitz RH. C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell. 1994;75:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- 19.Hockenberry D, Nuñez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 20.Hockenberry D, Oltvai ZN, Xin X, Milliman C, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 21.Howard AD, Kostura MJ, Thornberry N, Ding GJF, Limjuco G, Weidner J, Salley JP, Hogquist KA, Chaplin DP, Mumford RA, Schmidt JA, Tocci MJ. IL-1 converting enzyme requires aspartic acid residues for processing the IL-1β precursor at two distinct sites and does not cleave 31 kDa IL-1α. J Immunol. 1991;147:2964–2969. [PubMed] [Google Scholar]
- 22.Jacobson MD, Raff MC. Programmed cell death and Bcl-2 protection in very low oxygen. Nature. 1995;374:814–816. doi: 10.1038/374814a0. [DOI] [PubMed] [Google Scholar]
- 23.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Örd T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 24.Kane DJ, Örd T, Anton R, Bredesen DE. Expression of Bcl-2 inhibits necrotic neural cell death. J Neurosci Res. 1995;40:269–275. doi: 10.1002/jnr.490400216. [DOI] [PubMed] [Google Scholar]
- 25.Kumar S, Kinoshita M, Noda M, Copeland NG, Jenkins NA. Induction of apoptosis by the mouse Nedd2 gene which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene Ced-3 and the mammalian IL-1β-converting enzyme. Genes Dev. 1994;8:1613–1626. doi: 10.1101/gad.8.14.1613. [DOI] [PubMed] [Google Scholar]
- 26.Lazebnik YA, Cole S, Cooke CA, Nelson WG, Earnshaw WC. Nuclear events of apoptosis in vitro in cell-free mitotic extracts: a model system for the analysis of the active phase of apoptosis. J Cell Biol. 1993;123:7–22. doi: 10.1083/jcb.123.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Los M, Van de Craen M, Penning LC, Schenk H, Westendorp M, Baeuerle PA, Droge W, Krammer PH, Flers W, Schulze-Osthoff K. Requirement of an ICE/CED-3 protease for FAS/APO-1 mediated apoptosis. Nature. 1995;375:81–83. doi: 10.1038/375081a0. [DOI] [PubMed] [Google Scholar]
- 28.Mah SP, Zhong LT, Liu Y, Roghani A, Edwards RH, Bredesen DE. The proto-oncogene bcl -2 inhibits apoptosis in PC12 cells. J Neurochem. 1993;60:1183–1186. doi: 10.1111/j.1471-4159.1993.tb03275.x. [DOI] [PubMed] [Google Scholar]
- 29.Martin SJ, Green DR. Protease activation during apoptosis: death by a thousand cuts. Cell. 1995;82:1–4. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 30.Martin SJ, Takayama S, McGahon AJ, Miyashita T, Corbeil J, Kolesnick RN, Reed JC, Green DR. Inhibition of ceramide-induced apoptosis by Bcl-2. Cell Death Differ. 1995;2:253–257. [PubMed] [Google Scholar]
- 31.Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertoni P, Talbot D, Catsicas S, Pietra C, Huarte J. Over-expression of Bcl-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 32.Mellon PL, Windle JJ, Goldsmith PC, Padula CA, Roberts JL, Weiner RI. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron. 1990;5:1–10. doi: 10.1016/0896-6273(90)90028-e. [DOI] [PubMed] [Google Scholar]
- 33.Nett MA, Cerretti DP, Berson DR, Seavitt J, Gilbert DJ, Jenkins NA, Copeland NG, Black RA, Chaplin DD. Molecular cloning of the murine IL-1β converting enzyme cDNA. J Immunol. 1992;149:3254–3259. [PubMed] [Google Scholar]
- 34.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnick YA, Munday NA, Raju SM, Smulson ME, Yamin T, Yu VL, Miller DK. Identification and inhibition of the ICE/Ced-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 35.Nuñez G, London L, Hockenberry D, Alexander M, McKearn JP. Deregulated Bcl-2 gene expression selectively prolongs survival of growth factor-deprived hemopoetic cell lines. J Immunol. 1990;144:3602–3610. [PubMed] [Google Scholar]
- 36.Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed cell death induced by ceramide. Science. 1993;259:1769–1771. doi: 10.1126/science.8456305. [DOI] [PubMed] [Google Scholar]
- 37.Pronk GJ, Ramer K, Amiri P, Williams LT. Requirement of ICE-like protease for induction of apoptosis and ceramide generation of REAPER. Science. 1996;271:810–812. doi: 10.1126/science.271.5250.808. [DOI] [PubMed] [Google Scholar]
- 38.Quan LT, Tewari M, O’Rourke K, Dixit VM, Snipas SJ, Poirier GG, Ray C, Pickup DJ, Salvesen GS (1996) Proteolytic activation of the cell death protease Yama/CPP32 by granzyme B. Proc Natl Acad Sci USA, in press. [DOI] [PMC free article] [PubMed]
- 39.Raff MC. Social controls on cell survival and cell death. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- 40.Ray CA, Black RA, Kronheim SR, Greenstreet TA, Sleath PR, Salvesen GS, Pickup DJ. Viral inhibition of inflammation: cowpox virus encodes an inhibitor of the interleukin-1β-converting enzyme. Cell. 1992;69:597–604. doi: 10.1016/0092-8674(92)90223-y. [DOI] [PubMed] [Google Scholar]
- 41.Reed JC. Bcl-2 and the regulation of programmed cell death. J Cell Biol. 1994;124:1–6. doi: 10.1083/jcb.124.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimizu S, Eguchi Y, Kosaka H, Kamiike W, Matsuda H, Tsujimoto Y. Prevention of hypoxia-induced cell death by Bcl-2 and Bcl-xL. Nature. 1995;374:811–813. doi: 10.1038/374811a0. [DOI] [PubMed] [Google Scholar]
- 44.Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 45.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32β, a mammalian homolog of CED-3, is a Crm A-inhibitable protease that cleaves the death substrate poly(ADP)ribose polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 46.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 47.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, Elliston KO, Ayala JM, Casano FJ, Chin J, Ding GJF, Egger LA, Gaffney EP, Limjuco G, Palyha OC, Raju SM, Rolando AM, Salley JP, Yamin T, Lee TD, Shively JE, MacCross M, Mumford RA, Schmidt JA, Tocci MJ. A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 48.Thornberry NA, Peterson EP, Zhao JJ, Howard AD, Griffin PR, Chapman KT. Inactivation of interleukin-1β-converting enzyme by peptide (acyloxy)methyl ketones. Biochemistry. 1994;33:3934–3940. doi: 10.1021/bi00179a020. [DOI] [PubMed] [Google Scholar]
- 49.Tsujimoto Y, Finger L, Yunis J. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226:1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- 50.Wang L, Miura M, Bergeron L, Zhu H, Yuan J. Ich-1, an ICE/Ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell. 1994;78:739–750. doi: 10.1016/s0092-8674(94)90422-7. [DOI] [PubMed] [Google Scholar]
- 51.Wiedau-Pazos M, Trudell JR, Altenbach C, Kane DJ, Hubbell WL and Bredesen DE (1996) Free Radic Res Commun, in press. [DOI] [PubMed]
- 52.Yuan J, Shaman S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1β-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]