

Abstract
Overexpression of mutated human amyloid precursor protein (hAPP717V→F) under control of platelet-derived growth factor promoter (PDAPP minigene) in transgenic (tg) mice results in neurodegenerative changes similar to Alzheimer’s disease (AD). To clarify the pathology of these mice, we studied images derived from laser scanning confocal and electron microscopy and performed comparisons between PDAPP tg mice and AD. Similar to AD, neuritic plaques in PDAPP tg mouse contained a dense amyloid core surrounded by anti-hAPP- and anti-neurofilament-immunoreactive dystrophic neurites and astroglial cells. Neurons were found in close proximity to plaques in PDAPP tg mice and, to a lesser extent, in AD. In PDAPP tg mice, and occasionally in AD, neuronal processes contained fine intracellular amyloid fibrils in close proximity to the rough endoplasmic reticulum, coated vesicles, and electron-dense material. Extracellular amyloid fibrils (9–11 nm in diameter) were abundant in PDAPP tg and were strikingly similar to those observed in AD. Dystrophic neurites in plaques of PDAPP tg mouse and AD formed synapses and contained many dense multilaminar bodies and neurofilaments (10 nm). Apoptotic-like figures were present in the tg mice. No paired helical filaments have yet been observed in the heterozygote PDAPP tg mice. In summary, this study shows that PDAPP tg mice develop massive neuritic plaque formation and neuronal degeneration similar to AD. These findings show that overproduction of hAPP717V→F in tg mice is sufficient to cause not only amyloid deposition, but also many of the complex subcellular degenerative changes associated with AD.
Keywords: Alzheimer’s disease, amyloid precursor protein, transgenic, electron microscopy, confocal microscopy, neurodegeneration
The neurodegenerative process in Alzheimer’s disease (AD) is characterized by the progressive and irreversible deafferentation of the limbic system, association neocortex, and basal forebrain (Perry et al., 1977; Hyman et al., 1984; Wilcock et al., 1988; Hof et al., 1990; Palmer and Gershon, 1990; Masliah et al., 1993a), accompanied by neuritic plaque and tangle formation (for review, see Terry et al., 1994). The neuritic plaques in AD contain amyloid β protein (Aβ), which is derived from the amyloid β precursor protein (APP) (Selkoe et al., 1988; Selkoe, 1993). Embedded in the amyloid plaque core are dystrophic neurites, as well as reactive astrocytes and microglia (Terry et al., 1964; Terry and Wisniewski, 1970; Dickson et al., 1988; Wisniewski et al., 1989, 1991; Masliah et al., 1991c, 1993b; Frackowiak et al., 1992). The neuritic population in the plaque is mixed, being composed of spherical neurites that contain synaptic proteins and APP (type I), as well as fusiform neurites that contain cytoskeletal proteins and paired helical filaments (PHF; type II) (Dickson et al., 1988; Masliah et al., 1993b, 1994a; Wang and Munoz, 1995). The precise mechanisms by which neuritic plaques are formed and their relationship to the overall neurodegenerative process in AD are not yet clear. Increasing evidence indicates that errantly processed APP derivatives may be involved in the pathophysiological process that leads to neurodegeneration and plaque formation in AD (Sisodia et al., 1990; Golde et al., 1992; Seubert et al., 1992;Mattson et al., 1993; Selkoe, 1993).
To study the events involved in this process in vivo, several transgenic (tg) mice harboring the human APP (hAPP) gene have been developed (Higgins et al., 1994; Mucke et al., 1994; Games et al., 1995a,b). Some of these tg mouse models were designed based on the key observation that a number of APP mutations cosegregate with the familial form of AD and that these patients have, on autopsy, neuropathological alterations that are indistinguishable from sporadic AD (Chartier-Harlin et al., 1991; Goate et al., 1991; Murrell et al., 1991; Clark and Goate, 1993). Furthermore, it has been demonstrated that the APP717 mutations result in an overproduction of the highly amyloidogenic Aβ (1–42) relative to other Aβ peptides (Suzuki et al., 1994). Notably, one APP tg mouse model (PDAPP) was generated recently that exhibits AD-like neuropathology (Games et al., 1995a). This transgene used a platelet-derived growth factor-B chain (PDGF-B) promoter to drive a hAPP minigene (PDAPP) (Games et al., 1995a;Rockenstein et al., 1995) encoding alternatively spliced hAPP that contains the mutation V→F in position 717 that is associated with familial AD (Chartier-Harlin et al., 1991; Murrell et al., 1991; Clark and Goate, 1993). This resulted in an age- and brain region-dependent development of typical amyloid plaques, dystrophic neurites, loss of presynaptic terminals, astrocytosis, and microgliosis (Games et al., 1995a,b). More recent studies designed to characterize the transgene better have shown that the PDAPP minigene contains three modified hAPP introns that differ from the corresponding authentic hAPP gene introns by large deletions (introns 6 and 8) or insertion of four nucleotides (intron 7) (Rockenstein et al., 1995). Furthermore, PDAPP tg mice had four- to sixfold higher levels of total APP mRNA compared with non-tg mice or humans, whereas their endogenous mouse APP mRNA levels were reduced. This resulted in a high ratio of mRNA encoding mutated hAPP versus wild-type mouse APP (Rockenstein et al., 1995).
To clarify more fully the pathology of the heterozygous PDAPP tg mice at the ultrastructural level and to determine the fine ultrastructural differences and similarities between the tg mice and AD, we studied serially reconstructed images derived from the laser scanning confocal microscopy and electron microscopy.
MATERIALS AND METHODS
Mouse and human tissues. The heterozygous tg mice analyzed in this study were from the previously established line PDAPP-109 (n = 4, age 8 months; n = 3, age 12 months) (Games et al., 1995a). Age-matched non-tg littermates derived from the PDAPP (n = 4, age 8 months;n = 3, age 12 months) lines were used as controls. Mice were derived over several generations from hybrid backgrounds representing combinations of C57BL/6 + DBA + Swiss–Webster strains. The right hemibrain was processed for electron microscopy, and the left half was processed for double-immunolabeling/laser scanning confocal microscopy. For ultrastructural comparisons with the tg material, epoxy-embedded blocks from the frontal cortex biopsies of two clinically and histopathologically confirmed AD cases were examined for the present study. These were prepared in the 1960s at the Albert Einstein College of Medicine. The material was fixed in veronal- or chromate-buffered osmic acid; characteristics of these biopsies are described elsewhere (Terry et al., 1964). For double-immunolabeling/confocal microscopy comparisons with the tg material, autopsy tissue blocks from the frontal cortex of two AD cases from the Alzheimer Disease Research Center (ADRC) at the University of California-San Diego were placed in 2% paraformaldehyde–PBS, pH 7.4, at 4°C overnight and analyzed as described previously (Masliah et al., 1992, 1993b).
Tissue processing. Mice were anesthetized and perfused with cold saline followed by 2% glutaraldehyde + 2% paraformaldehyde in 0.15 m cacodylate buffer. Brains were removed and placed in the fixative for 24 hr at 4°C. After fixation, brains were divided sagittally and blocks (0.2 × 0.2 cm) were taken from the frontal cortex and hippocampal region of the right hemibrain for processing and embedding in epon for subsequent ultrastructural analysis as described previously (Masliah et al., 1991a, 1993b,c). The left hemibrain of tg and non-tg mice, as well as tissue blocks from the AD midfrontal cortex, was serially sectioned at 40 μm with the Vibratome 2000 (Technical Products International, St. Louis, MO), and sections were cryoprotected and stored at −20°C as described previously (Masliah et al., 1992, 1993b).
Ultrastructural analysis and immunogold labeling. The epoxy-embedded blocks from the murine and human cases were sectioned with the Reichert Ultracut-E ultramicrotome (Leica, Vienna, Austria), placed on 200-mesh copper grids and stained in saturated ethanol/uranyl acetate and bismuth subnitrate, as described previously (Masliah et al., 1993b,c). From each case, ∼10 sections (80 nm thick) were analyzed with a Zeiss EM10 Electron Microscope at 5000×, 10,000×, 20,000×, and 50,000× magnifications to compare the ultrastructural characteristics of neurons, synapses, and plaques between the tg mice and the AD material. From each case, a total of 60 micrographs (15 at each of the 4 magnifications indicated) were prepared. To determine the relationship among neuritic processes, cellular elements, and amyloid, additional series of 10 sections (each 80 nm thick) were divided and transferred onto formvar carbon-coated slot grids. Serial numbered electron micrographs of selected areas containing neuritic plaques were taken with a Zeiss EM10 electron microscope at a magnification of 3300× and 5000× for analysis as described previously (Masliah et al., 1993b). Further analysis of the characteristics of the amyloid fibrils in the plaque core was done by immunogold labeling. Briefly (Deftos et al., 1993), vibratome sections from control and PDAPP tg mice were fixed in 0.25% glutaraldehyde and 3% paraformaldehyde in 0.1 m cacodylate buffer, pH 7.4. Tissues were incubated in 100 mm glycine in sodium cacodylate buffer followed by brief fixation in 0.1% osmium tetraoxide. Sections were dehydrated in a graded series of ethanol solutions. From each section, the frontal cortex and hippocampus were dissected and placed in gelatin capsules containing a polymerized layer of LR white (medium grade, Ted Pella, Inc., Redding, CA). Fresh LR white was then added and polymerized for 24 hr in a 55°C oven. Blocks were sectioned on an Ultracut E, and thin sections (60–90 nm) were placed on nickel grids coated with a 0.5% paralodian film. Grids were incubated at room temperature (RT) for 30 min with blocking buffer (TBS + 0.8% BSA + 0.1% gelatin + 0.02% sodium azide), incubated for 12 hr at RT with the biotin-conjugated monoclonal antibody against amyloid β protein (1–5) (3D6, Athena Neurosciences, San Francisco, CA), followed by avidin–gold (5 nm, Zymed Laboratories, San Francisco, CA). Grids were then fixed in 2% glutaraldehyde and post-fixed with 2% uranyl acetate, followed by bismuth subnitrate. Immunogold-labeled grids were examined with a Zeiss EM10 electron microscope. Control experiments were done by incubating grids with avidin–gold in the absence of primary antibody.
Double-immunofluorescent labeling and confocal microscopy.To understand better the relationship between neuronal elements and amyloid in the plaques, vibratome sections from the murine and human cases were first washed in PBS, pH 7.4, blocked with normal serum, and incubated overnight at 4°C with a mixture of the rabbit polyclonal antibody against amyloid β protein (R1280, courtesy of Dr. Dennis Selkoe) (Joachim et al., 1989) and with the mouse monoclonal antibody against phosphorylated medium and high molecular weight neurofilaments (SMI312, Sternberger Monoclonals, Baltimore, MD) (Masliah et al., 1993a) as described previously (Masliah et al., 1993a,b). Sections were then incubated with a mixture of FITC-conjugated horse anti-mouse IgG (1:75) and Texas Red-conjugated goat anti-rabbit antibody (1:100, Vector Laboratories, Burlingame, CA). The double-immunolabeled sections were transferred to SuperFrost slides (Fisher Scientific, Tustin, CA) and mounted under glass coverslips with antifading media (VectaShield, Vector). All sections were processed simultaneously under the same conditions. The immunofluorescent labeling was done twice to assess the reproducibility of results. Additional comparisons of the spatial relationship among amyloid, dystrophic neurites, and astroglial elements between the PDAPP tg and AD plaques was done by double-immunolabeling with the following pairs of antibodies: (1) anti-glial fibrillary acidic protein (GFAP; Boehringer Mannheim, Indianapolis, IN)/anti-amyloid β protein (3D6), and (2) anti-hAPP (clone 8E5, Athena)/anti-amyloid β protein (3D6). After an overnight incubation at 4°C in the primary antibodies, the sections were incubated with a mixture of FITC-conjugated horse anti-mouse (1:75, Vector) and Texas Red avidin D (1:75, Vector). The double-immunolabeled sections were viewed with a Zeiss 63× (numerical aperture 1.4) objective on a Zeiss Axiovert 35 microscope with an attached laser confocal scanning system MRC 1024 (Bio-Rad, Wattford, UK) (Masliah et al., 1992, 1993a,b). From each case, five fields within the frontal cortex and five fields within the hippocampal CA1 region were imaged. Serial optical z-sections (0.2 μm in thickness) of the double-immunolabeled neuritic plaques were collected from each region using the dual-channel imaging capability of the laser confocal microscope (Masliah et al., 1992, 1993a,b). The Texas Red channel collected the amyloid deposits, and the FITC channel collected the corresponding images of the anti-neurofilament-, hAPP-, and GFAP-immunolabeled elements in the plaques. Each series of optical sections was scanned through a total of 20 μm, resulting in a complete series composed of ∼75-100 dual optical sections. The digitized video images were processed and stored on 650 MB rewritable optical disk cartridges. Serial optical sections of the neuritic plaques, obtained from the laser confocal microscope, were transferred to a Silicon Graphics Iris 4D/210VGX for visualization of the three-dimensional relationship among the dystrophic neurites and amyloid in the plaques (Masliah et al., 1993b).
RESULTS
General characteristics of the plaques
Analysis of a total of 250 neuritic plaques in the frontal cortex (layers 2–3 and 5) and hippocampus (pyramidal cell layer of CA1) of heterozygous PDAPP tg mice (n = 7) revealed that at least two general types of plaques could be identified: (1) those containing clusters of dystrophic neurites without an amyloid core (Fig. 1A), and (2) those containing extensive clusters of dystrophic neurites accompanied by one or more amyloid cores (Fig. 1B–D). Approximately 20% of the lesions were devoid of amyloid, whereas the other 80% were associated with amyloid deposits. At least three subtypes of amyloid-containing neuritic plaques were identified: (1) plaques with abundant dystrophic neurites and scant amyloid bundles (Figs.1B, 2B,E), (2) plaques with abundant dystrophic neurites, abundant amyloid deposition, and some glial cells (Fig. 1C), and (3) plaques with degenerated electrodense processes, dystrophic neurites, and very dense amyloid deposits (Fig. 1D) surrounded by glial cells. Another notable feature in the PDAPP tg mice was the presence of neurons in close association with the neuritic plaques (Figs.1A,C, 2B,3C,D, 6A–C). Similar to PDAPP tg, AD cases also displayed clusters of dystrophic neurites (Fig. 2C,F) with or without amyloid and occasional neuronal cells in close proximity to the neuritic plaques (Fig. 2C). However, AD neuritic plaques contained more cells with ultrastructural characteristics consistent with microglia (data not shown) than the neuritic plaques of heterozygous PDAPP tg mice. In contrast to the ultrastructural findings in the PDAPP tg mice, age- and line-matched non-tg littermates (n = 7) did not show neuritic alterations or amyloid formation. Only occasional hypertrophic astroglial cells were observed in the CA1 region of the hippocampus (Fig. 4D). Consistent with the ultrastructural findings, immunolabeling with the anti-neurofilament antibody (SMI312) showed a well preserved and organized neuritic structure (Fig. 4A) in the non-tg control mice.
Fig. 1.
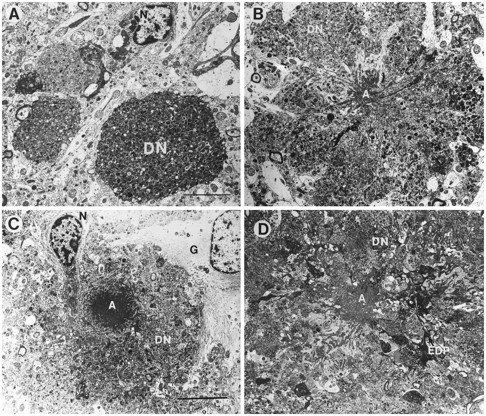
Types of neuritic plaques in the hippocampal CA1 pyramidal cell layer of PDAPP tg mice. A, Ultrastructural analysis revealed the presence of clusters of dystrophic neurites (DN) containing abundant electrodense multilaminar bodies. No amyloid was observed in this type of lesion. N indicates the nucleus of cells associated with the plaque. B, C, Classical neuritic plaques contained dystrophic neurites (DN), an amyloid core (A), astrocytic glial cells (G), and an associated neuronal cell (N). D, A third type of plaque presented degenerating and dystrophic neurites (DN) in the periphery, and abundant very dense amyloid cores (A) with prominent fusiform electrodense processes (EDP). Scale bar, 15 μm.
Fig. 2.
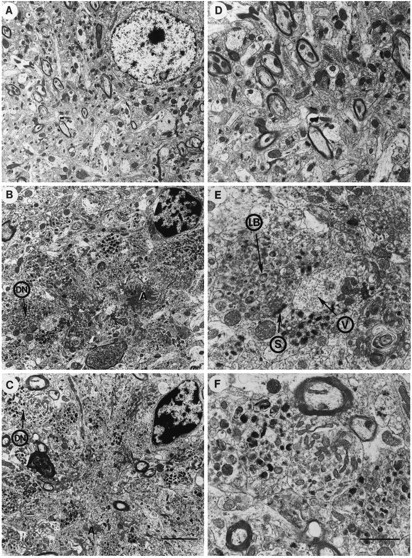
Comparison of neuritic plaques between PDAPP tg mice and AD. A, Low-power view of the frontal cortex in control non-tg mice shows preserved neuronal structure.B, Low-power view of a classical neuritic plaque in the frontal cortex of a PDAPP tg mouse shows abundant dystrophic neurites (DN), amyloid deposits (A), and an associated cell that displays chromatin clumps beneath the nuclear envelope. C, Low-power view of a classical neuritic plaque in the frontal cortex of an AD case that displayed abundant dystrophic neurites (DN), amyloid deposits (A), and an associated cell that also presents some nuclear chromatin aggregation. D, Higher-power view of the neuropil in the control non-tg mice shows preservation of the neuritic and synaptic structure. E, Higher-power view of the dystrophic neurites in the PDAPP tg mouse shows similar electrodense laminar bodies (LB) to the ones observed in AD. The dystrophic neurites made synaptic contacts (S) and contained abundant small vesicles (V).F, Higher-power view of a dystrophic neurite in AD that shows abundant laminar dense bodies and mitochondria. Scale bars:B, 10 μm; C, 1.5 μm.
Fig. 3.
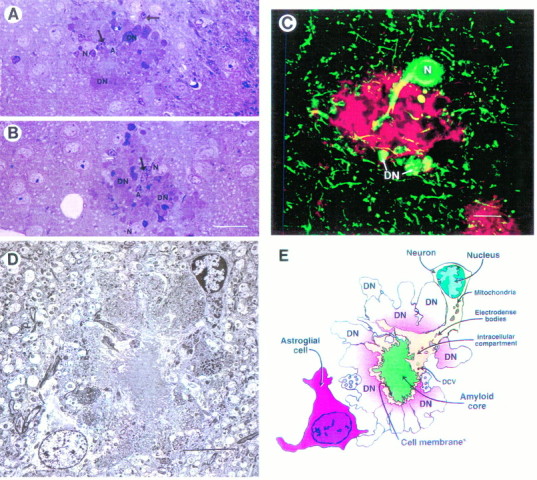
Cellular alterations associated with the neuritic plaque in the PDAPP tg mice. A, B, Semithin section (250 nm) of a neuritic plaque in the hippocampus (pyramidal cell layer) stained with toluidine blue demonstrates dystrophic neurites (DN), amyloid (A), neuronal cell involvement (N), and glial cells (arrows).C, The dystrophic neurites (DN) and a neuronal cell body (N) embedded in the plaque were strongly immunoreactive with SMI312, a monoclonal antibody that identifies phosphorylated neurofilaments (green). The amyloid component was immunolabeled with the polyclonal antibody against Aβ R1280 (red). This neuritic plaque was identified in the hippocampus and imaged with the LSCM as described previously (Masliah et al., 1993b). D, Electron micrograph of a classical neuritic plaque in the hippocampus shows that these lesions were composed of a prominent neuronal element accompanied by extensive neuritic dystrophy, amyloid deposits, and astroglial cell reaction. E, Diagrammatic representation of neuritic plaque illustrated in D. Scale bars: A, 30 μm; C, 10 μm; D, 25 μm.
Fig. 6.
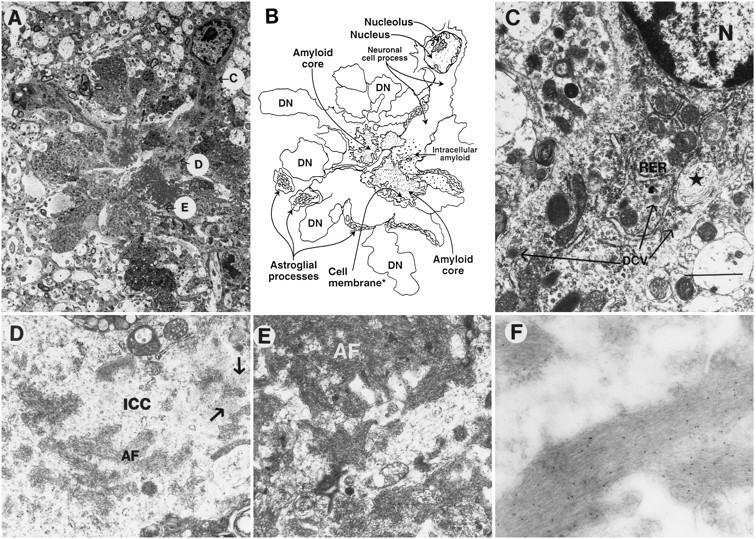
Subcellular neuronal alterations associated with the neuritic plaques in PDAPP tg mouse. Electron micrographs were taken from a lesion in the hippocampus. A, Low-power view of a neuritic plaque composed of a prominent neuronal element accompanied by extensive neuritic dystrophy, amyloid deposits, and astroglial cell reaction. The letters inside circles correspond to subsequent panels that identify various characteristics of the neuronal element in the plaque in greater detail. B, Diagrammatic representation of the neuritic plaque illustrated in A.C, Proximal to the nucleus (and distal to the amyloid core), the neuron displayed abundant RER, neurosecretory dense-core vesicles (long arrows), and peculiar elliptical bodies containing fibrilo-tubular material (*). Scale bar, 3 μm.D, Distal to the cell nucleus (and proximal to the amyloid core), the intracellular cytoplasmic compartments (ICC) of the neuronal process displayed amyloid fibrils (AF) and amorphous electrodense granular material (arrows) surrounded by a membrane-forming digitating processes (E). The neuronal cytoplasm surrounding the extracellular amyloid fibrils (AF) showed diffuse granular material and mitochondria. F, Immunogold labeling with a monoclonal antibody against β-amyloid (3D6) showed amyloid fibrils decorated with the 5 nm gold particles.
Fig. 4.

Comparison of the neuritic and glial components in the PDAPP tg model and AD. Sections were double-immunolabeled and imaged with the laser scanning confocal microscope. Images obtained in the Texas Red channel correspond to β-amyloid (3D6) and in the FITC channel (green) to phosphorylated neurofilament (SMI312) (A–C), GFAP (D–F), or hAPP (8E5) (G–I). A, Non-tg control mice displayed a well organized and preserved neuritic structure. The neuronal cell bodies (N) were not labeled with the anti-neurofilament antibody. PDAPP tg (B) and AD (C) cases displayed significant disruption of the neuritic structure and positive anti-SMI312 immunoreactivity in the dystrophic neurites (arrow) associated with the plaque.D, Non-tg control mice displayed occasional GFAP-immunoreactive astroglial cells. PDAPP tg (E) and AD (F) cases displayed hypertrophic GFAP-immunoreactive astroglial cells in the periphery of the plaque.G, Non-tg control mice were negative with the antibody specific for hAPP. PDAPP tg (H) and AD (I) cases displayed positive anti-hAPP immunoreactivity in the dystrophic neurites (arrow) associated with the plaque. Scale bar, 20 μm.
Amyloid deposits in plaques
Amyloid deposits in PDAPP tg mice were remarkably similar to those observed in AD (Fig. 5) and were usually associated with anti-neurofilament- and anti-hAPP-immunoreactive dystrophic neurites and neuronal cell bodies. In both PDAPP (Fig.5A–C) and AD (Fig.5D–F), the dense extracellular amyloid consisted of filaments ranging in diameter from 9 to 11 nm that were surrounded by cell membranes that were derived from the neuronal elements in the plaque (Figs. 3D,5A–C, 6A,B,D,E, 7B,E). Dense-core vesicles (neurosecretory type) ranging in diameter from 85 to 250 nm (Figs.5A,B, 7C–E) in the cytoplasmic compartment adjacent to the extracellular amyloid fibrils confirmed the neuronal origin of these cellular processes. In addition, the neuronal processes apposed to the extracellular amyloid fibrils contained fine granules (average size of grains was ∼5 nm in diameter) (Figs. 5A,C, 7E). Compared with biopsy material from the AD cases (Fig.5D,E), the plaques in the PDAPP mice (Fig.5A,C) contained more dense extracellular amyloid bundles and more abundant fine granular deposits in the cytoplasmic phase of the neuronal processes associated with the plaque (Figs.5A,C, 7D). Consistent with the double-immunolabeling results (Fig. 4B,E,H) and the ultrastructural findings, the amyloid fibrils in the PDAPP tg mice were recognized by the anti-β-amyloid antibody (3D6), as reflected by the labeling of the fibrils by colloidal gold (Fig.6F). Cellular processes near the amyloid core displayed fine intracellular amyloid fibrils adjacent to the rough endoplasmic reticulum (RER) (Fig. 7E) and coated vesicles (Fig. 7F,G), suggesting the possibility that neurons embedded in the plaque participate in amyloid synthesis. These subcellular organelles associated with intracellular amyloid formation in the PDAPP tg mice were similar to those described previously in amyloid-related cells (ARC) of the AD neuritic plaques (Roher et al., 1988; Frackowiak et al., 1992). However, whereas in AD the extracellular amyloid bundles contained several clear vesicles (Fig.5D,F) that have been associated with pinocytic functions of the ARCs (Roher et al., 1988), in PDAPP tg mice these were only occasionally observed (Fig. 5B) .
Fig. 5.
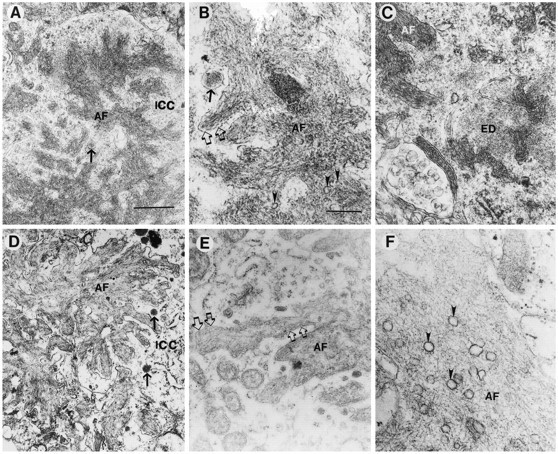
Comparison of the amyloid deposits in the PDAPP tg model and AD. Electron micrographs were obtained from the hippocampal region in the PDAPP tg mouse and from the frontal cortex in AD. Low-power (A) and high-power (B) view of amyloid fibrils (AF) in the PDAPP tg mouse showing dense deposits surrounded by a membrane (open arrows) and the neuronal intracytoplasmic compartment (ICC) containing electrodense granular material and dense-core neurosecretory vesicles (arrow). Occasional poorly defined clear vesicles (arrowheads) were associated with the amyloid fibrils in PDAPP mouse plaques.C, In the PDAPP tg mouse, the cytoplasmic component of neurons close to the extracellular amyloid fibrils (AF) showed the presence of amorphous electrodense material (ED). Low-power (D) and high-power (E) view of the amyloid deposits in AD showed abundant dense fibrils (AF) surrounded by a membrane (open arrows) and a cytoplasmic compartment (ICC) containing dense-core neurosecretory vesicles (arrows) and electrodense granular material.F, Clear vesicles were prominently associated with the amyloid fibrils (AF) in AD plaques (arrowheads). Scale bars: A, 1 μm;B, 100 nm.
Fig. 7.
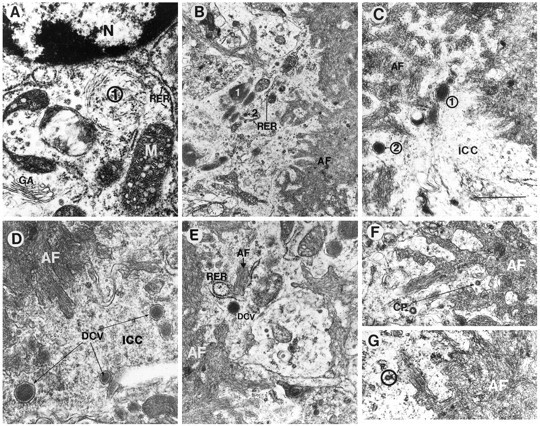
Additional neuronal subcellular characteristics associated with amyloid formation in the PDAPP tg mouse. Electron micrographs were obtained from the hippocampus and are derived from the neuritic plaque presented at the bottom of Figure 3D.A, Proximal to the nucleus (N) (and distal to the amyloid core), the neuron displayed, in addition to RER and elliptical inclusion bodies (1), a prominent Golgi apparatus (GA) and mitochondria (M). B, Distal to the cell nucleus (and proximal to the amyloid core), the cytoplasm of the neuronal process displayed the presence of compact electrodense organelles surrounded by a membrane (1) adjacent to RER, mitochondria, and neurosecretory vesicles (2).C, D, Dense-core neurosecretory vesicles (1, 2) were abundant in the intracytoplasmic compartment (ICC) of the neuron adjacent to the extracellular amyloid fibrils (AF). E, Intracellular amyloid fibrils (AF, black letterhead) adjacent to RER were present in the cytoplasmic compartment of the neurons near the extracellular amyloid fibrils (AF, white letterhead). Dense-core neurosecretory vesicles (DCV) were also present. F,G, Coated pits (CP) in the cytoplasmic compartment of the neurons were closely associated with the plasma membrane (circle) surrounding extracellular amyloid deposits (AF). Scale bar, 400 nm.
Plaque-associated neuronal alterations
In PDAPP tg mice, neuronal alterations were characterized by involvement of their processes in plaques (Figs. 3, 6) and by damage to their synaptic terminals (Fig. 8). The neuronal cell bodies in close proximity to the plaques formed neuritic processes that were associated with the amyloid core as well as with the dystrophic neurites (Fig.6). Serial section analysis of electron micrographs and optical sections revealed that neuronal cell bodies and their processes could be identified in >80% of the neuritic plaques (Figs. 1, 2, 3, 6). The neuronal origin of these cells was determined by the presence of the following: (1) immunoreactivity with antibodies against high and intermediate molecular weight phosphorylated neurofilaments (Fig.3B); (2) dense-core vesicles (85–250 nm in diameter) in the cell body and processes (Figs. 6C,7D,E); and (3) synapses between adjacent neuritic processes and the neuronal cell bodies (Fig.8A) or between neurites embedded in the plaque and dystrophic neurites (Figs. 2E,8D,E). Furthermore, the nucleus of these cells usually presented moderate chromatin aggregation within the nuclear envelope and a prominent nucleolus (Figs. 1C,6A).
Fig. 8.
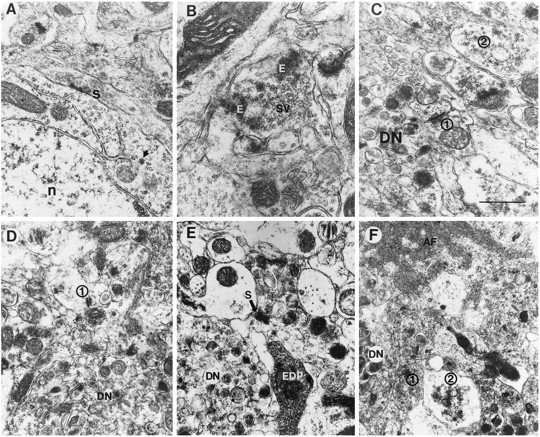
Synaptic alterations in PDAPP tg mice. Electron micrographs were obtained from the hippocampus. A, Neuronal cells associated with the plaques showed synaptic terminals (S) associated with the perikaryon (n, nucleus; arrow, neurosecretory vesicle).B, Presynaptic terminals in the periplaque region displayed amorphous electrodense material (E) in addition to synaptic vesicles (SV).C, D, Some synaptic terminals in the periplaque region showed decreased numbers of synaptic vesicles (1), whereas others were distended and contained abundant vesicles (2) (DN, dystrophic neurites). E, Axonal terminals in the midst of the plaque were distended and displayed accumulations of multilaminar, multivesicular, and electrodense bodies. Dystrophic neurites (DN) made occasional synaptic contacts (S) with neighboring neuritic processes. Other neuritic elements in the plaque were electrodense (EDP).F, Adjacent to the extracellular amyloid fibrils (AF), the axonal terminals were distended and contained abundant dense vesicles (1) or clear vesicles (2). A dystrophic neurite (DN) was also present. Scale bar, 1.5 μm.
Neurons embedded in the plaque appeared to be actively involved in biosynthesis of proteins as evidenced by the presence of a prominent Golgi apparatus (Fig. 7A) and RER (Figs.6C, 7A) in the perinuclear region. In addition, the neuritic processes embedded in the plaque contained abundant mitochondria, electrodense bodies surrounded by a membrane (suggesting a lysosomal origin), and dense-core vesicles (Figs. 6C,7B–E). The neurosecretory vesicles were occasionally distributed along the main neuritic process and were more abundant and enlarged in the distal end of the process adjacent to the amyloid deposits (Fig. 7B–E). In AD, similar neuronal cells were observed associated with neuritic plaques (Fig.2C); however, they were less prominent than in PDAPP tg mice.
Other neurodegenerative alterations observed in the PDAPP tg mice that were similar to AD (Masliah et al., 1991a, 1993b) included the damage to synaptic junctions. This was characterized by irregular enlargement of nerve terminals (Fig.8C,F), fewer synaptic vesicles (Fig. 8C,D), and abnormal accumulation of multilaminar bodies, dense-core vesicles (Fig.8E,F), and electrodense amorphous material (Fig. 8B). These synaptic alterations were more prominent inside the plaque than in the periplaque region.
Dystrophic neurites in the plaques
In addition to the neuritic alterations associated with amyloid formation described in the previous two sections, the neurites in the plaques of PDAPP tg mice also showed characteristic dystrophic changes. These dystrophic neurites contained abundant multivesicular, multilaminar dense bodies (Figs. 1A,2E, 9A–C) as well as dense-core vesicles and small clear synaptic vesicles (Figs.2E, 8C–E). Occasionally, synaptic contacts were observed between dystrophic neurites (Figs.2E, 8E). The ultrastructural characteristics of the dystrophic neurites in the plaques of the PDAPP tg mice were essentially identical to those observed in type I neurites (Masliah et al., 1993b) in the plaques of AD cases (Fig.2C,F), indicating that neurites in the plaques are probably undergoing reactive and neurodegenerative changes (Masliah et al., 1993b). Consistent with previous studies (Masliah et al., 1993b), the type I dystrophic neurites in the PDAPP and AD cases were immunoreactive with antibodies against hAPP (8E5) (Fig. 4, H and I, respectively).
Fig. 9.
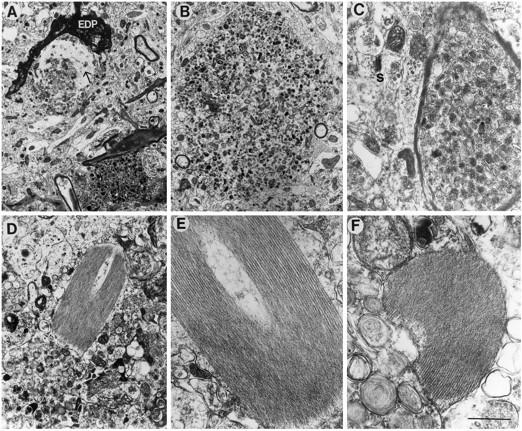
Neuritic alterations in PDAPP tg mice. Electron micrographs were obtained from the hippocampus. In PDAPP tg mice, the dystrophic unmyelinated (A, B) and myelinated neurites (C) contained abundant electrodense laminar and multivesicular bodies. Some neurites contained fine filaments (10 nm; arrow) and were surrounded by electrodense processes (EDP). Other neurites contained characteristic crystals displaying an array of symmetrically organized tubules (D–F). Scale bar, 2 μm.
In the PDAPP tg mice, some dystrophic neurites in the plaque contained a dense cumulus of cytoskeletal filaments that was surrounded by multilaminated bodies (Fig. 10A,B). These filaments, ranging in diameter between 8 and 10 nm, were identical to neurofilaments. Consistent with these findings, double-immunolabeling studies confirmed that the dystrophic neurites in the plaques of the PDAPP and AD cases were immunoreactive with antibodies against phosphorylated middle and high molecular weight neurofilaments (Fig. 4, B and C, respectively). They formed a reticular matrix and were observed in both myelinated (Fig. 10A,B) and nonmyelinated (Fig. 9A) axons. Although clearly different from PHF of the type II dystrophic neurites in the plaques of AD cases (Masliah et al., 1993b), they were similar to neurofilament aggregates occasionally observed in the type I neurites of the AD plaques (Fig.10C,D). No classical AD-type PHF was observed in the dystrophic neurites of PDAPP tg mice (Fig. 10E,F). In addition to the neurofilamentous alterations observed in the neuritic plaques of PDAPP tg mice, detailed ultrastructural analysis revealed the presence of crystalline polygonal structures that were formed by symmetrical tubular elements (Fig. 9D–F).
Fig. 10.
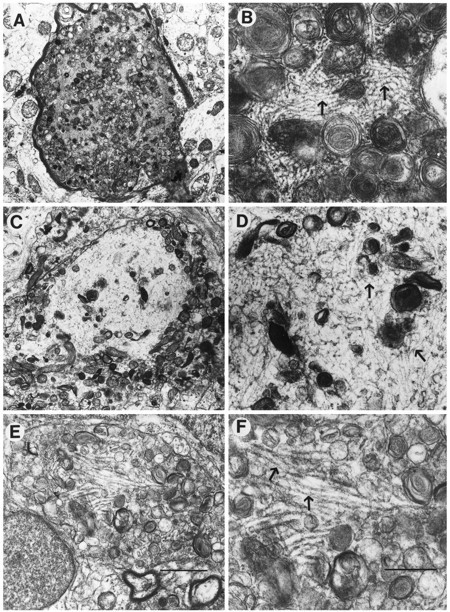
Comparison of cytoskeletal alterations between the PDAPP tg mouse and AD. Low-power (A) and high-power (B) views of a myelinated dystrophic neurite in the PDAPP tg containing abundant laminated bodies and filamentous material (10 nm in diameter; arrows). Low-power (C) and high-power (D) views of a dystrophic neurite in AD containing multilaminar bodies and neurofilamentous material similar to the one observed in the PDAPP mouse (arrows). Low-power (E) and high-power (F) views of a dystrophic neurite in AD containing classical paired helical filaments (arrows). Scale bars: E, 5 μm;F, 200 nm.
Other cellular alterations not associated with plaques
In addition to neuritic plaque formation, neurodegenerative changes in neocortical and hippocampal cells of the PDAPP tg mice included nuclear chromatin segmentation, formation of dense intranuclear and intracytoplasmic bodies, and intracellular vacuolization of the membranes (Fig. 11). Some cells contained RER and neurosecretory vesicles, suggesting a neuronal origin. However, in cells with extensive vacuolization it was not possible to identify ultrastructural markers indicative of cellular origin. These alterations were not observed in control non-tg littermates. Neurons of PDAPP tg mice that were not associated with plaques showed no neurofibrillary tangles (NFT) or evidence of intraneuronal PHF formation.
Fig. 11.
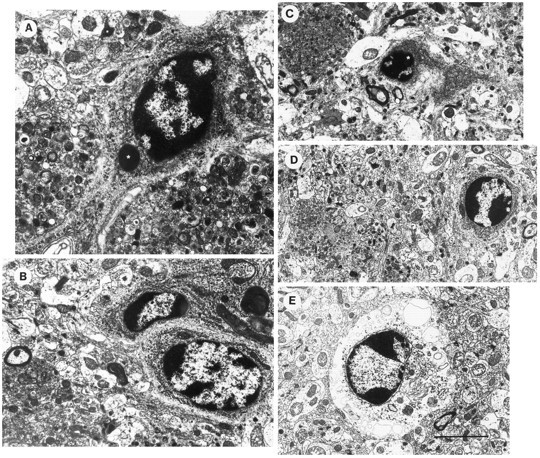
Apoptotic-like changes in neocortical cells of PDAPP tg mice. Electron micrographs were obtained from layer 5 of the frontal cortex. A–D, Neuronal nuclei contained chromatin segmentation and condensation. Some cells displayed dense intracytoplasmic or intranuclear inclusions surrounded by a membrane (*). These cells also contained RER and neurosecretory vesicles. E, Other cells displayed, in addition to the chromatin segmentation, cytoplasmic distention and vacuolization. Scale bar, 10 μm.
Glial cell alterations
Abundant glial cells were observed around the neuritic plaques (Figs. 1C, 3D,E, 12) as well as scattered in the neuropil of the neocortex and hippocampus of the PDAPP tg mice. The great majority of them contained intermediate filaments, consistent with an astroglial origin (Fig. 12). In both the PDAPP and the AD cases, these glial elements in the plaque strongly immunoreacted with antibodies against the astroglial marker (GFAP) (Fig. 4, E and F, respectively). Microglial cells were observed associated with neuritic plaques containing dense amyloid cores and in which the dystrophic neurites exhibited extensive degeneration characterized by the presence of abundant electrodense material. Compared with AD, neuritic plaques in PDAPP tg mice showed a more prominent astroglial reaction and a less abundant microglial component (not shown) (Terry et al., 1964; Terry and Wisniewski, 1970;Frackowiak et al., 1992).
Fig. 12.
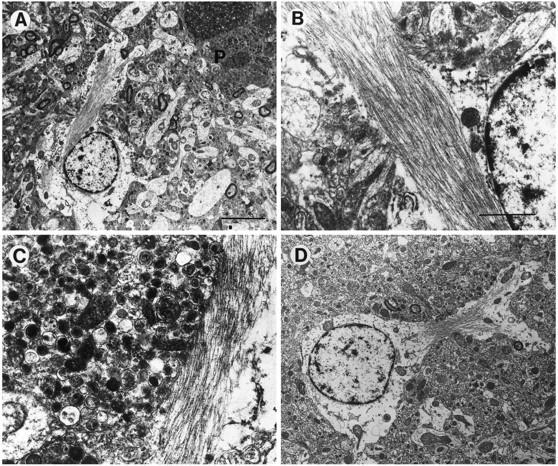
Glial cell alterations in PDAPP tg mice. Electron micrographs were obtained from the frontal cortex. A, Prominent astrocytic cells were observed associated with the neuritic plaques (P). These cells displayed some cytoplasmic swelling and contained abundant intermediate filaments.B, C, Higher-power view of the astrocytic processes in the plaque. D, View of an enlarged astrocyte distant from the plaque. Scale bars: A, 10 μm; B, 2 μm.
DISCUSSION
Neurodegeneration in PDAPP tg mice
The present study showed that PDAPP tg mice displayed subcellular neurodegenerative alterations strikingly similar to those observed in AD (Table 1). These include characteristic dystrophic neurites with electrodense multilaminar bodies, disruption of synaptic junctions, and intracellular amyloid and reactive gliosis. Furthermore, plaques in PDAPP tg mice displayed extracellular amyloid fibrils associated with cellular processes containing dense-core vesicles, coated vesicles, RER, and membranous recesses in the neuritic plaques (Table 1).
Table 1.
Ultrastructural similarities and differences between AD and PDAPP tg plaques
| Alzheimer’s disease | PDAPP tg | |
|---|---|---|
| Amyloid fibrils | ||
| Size | 9–11 nm | 9–11 nm |
| Electron density | Moderate | High |
| Pinocytic vesicles | Abundant | Occasional |
| Dystrophic neurites | ||
| Type I | ||
| Dense laminar bodies | Abundant | Abundant |
| Synaptic vesicles and contacts | Yes | Yes |
| Neurofilament accumulation | Yes | Yes |
| Type II | ||
| Paired helical filaments | Yes | None? |
| Cells associated with amyloid formation | ||
| Microglia | Abundant | Occasional |
| Neurons | Occasional | Abundant |
| Neurosecretory granules | Abundant | Abundant |
| Rough endoplasmic reticulum | Abundant | Abundant |
| Coated pits | Yes | Yes |
Whereas neuritic plaques in AD contained spherical (type I) and fusiform (type II) neurites (Masliah et al., 1993b), in PDAPP tg mice the neuritic component was mainly of the spherical type (type I). This is consistent with the finding that, to date, no PHF has been found in neuronal cell bodies or dystrophic neurites in the PDAPP tg mice. In fact, the plaques occasionally found in cognitively intact aged humans and macaques (Martin et al., 1994) display the presence of type I (but not type II) dystrophic neurites, amyloid fibrils, and astrocytic reaction, as is the case with the PDAPP tg mice. However, similar to AD, the plaques in the PDAPP tg mice appear to be more abundant and show more widespread neuritic dystrophy than the ones observed in aged humans and macaques. Furthermore, similar to ultrastructural findings in AD (Wisniewski and Terry, 1970, 1973; Wisniewski et al., 1970;Masliah et al., 1993b), the neuritic component of plaques in the PDAPP tg mice contained both dystrophic (i.e., mitochondria, lysosomes, and neurofilaments) and regenerative (i.e., clear and dense-core vesicles, mitochondria, ER, and lysosomes) alterations. Previous studies showed that similar ultrastructural alterations occurred in neurites of vitamin E-deficient rats (Lampert, 1967; Southam et al., 1991) and in grafts of peripheral nerves in the CNS (Campbell et al., 1992). Furthermore, consistent with previous studies in AD (Terry et al., 1964; Gonatas et al., 1967; Wisniewski et al., 1981; Masliah et al., 1991b, 1993b), several of the plaque neurites in PDAPP tg mice contained synaptic vesicles and formed synaptic contacts supporting an axonal role and origin of neuritic dystrophy. The neuritic plaque components associated with dense amyloid deposits were of the degenerative type, because they contained abundant electrodense deposits and multilaminar bodies consistent with alterations observed in other animal models of neurodegeneration (Lampert, 1967).
In addition to neuritic plaque formation, neurodegenerative changes in neocortical and hippocampal cells of the PDAPP tg mice included nuclear chromatin segmentation, formation of dense intranuclear and intracytoplasmic bodies, and vacuolization. These alterations are similar to apoptotic changes observed in experimental models after treatment with excitotoxins (Pollard et al., 1994; Portera-Cailliau et al., 1995) or adrenalectomy (Sloviter et al., 1993). In contrast to these experimental animal models, previous ultrastructural studies have not described extensive apoptotic changes in cells in AD (Terry et al., 1964; Terry and Wisniewski, 1970). However, recent studies utilizing the in situ labeling technique have suggested that both neurons and glial cells undergo apoptosis in AD (Forloni et al., 1993;Su et al., 1994; Dragunow et al., 1995; Lassman et al., 1995; Smale et al., 1995), as well as in another Aβ tg animal model (LaFerla et al., 1995).
Cytoskeletal alterations in PDAPP tg mice
Consistent with findings in AD, PDAPP tg mice also showed cytoskeletal alterations characterized by abnormal deposits of neurofilamentous material in neuronal cell bodies, as well as in neuritic plaques. Similar alterations have been described in dystrophic neurites in AD (Wisniewski and Terry, 1973; Gheuens et al., 1991), as well as in rodents and humans treated with antimitotic agents (Wisniewski and Terry, 1967; Schochet et al., 1968; Shelansky, 1969;Nakagawa et al., 1987) and in rabbits treated with aluminum (Terry and Pena, 1965). In addition to the neurofilamentous deposits, PDAPP tg mice displayed the formation of crystals formed by symmetrical tubular elements arranged in an angular manner. These crystals had some ultrastructural similarities to Hirano bodies; however, additional electron microscopic and immunocytochemical studies will be necessary to confirm the origin of these structures. Hirano bodies are crystalloid structures that preferentially occur in the CA1 area of the hippocampus in AD, amyotrophic lateral sclerosis, Parkinsonism/dementia complex, and other disorders (Hirano, 1994). Because Hirano bodies contain actin, neurofilaments, and tau immunoreactivity, they are considered part of the spectrum of cytoskeletal alterations that occur in AD (Hirano, 1994). These crystals have also been described in rodents treated with colchicine and in aged primates (Hirano, 1994). Taken together, these findings suggest that overexpression of mutated hAPP or Aβ in tg mice is associated with neuritic cytoskeletal alterations. These changes could be secondary to the neurodegenerative process associated with a limited sprouting capacity in partially denervated neurons, consistent with the observation that sprouting neurites expressing GAP-43 and APP also display neurofilament immunoreactivity (Cras et al., 1991; Gheuens et al., 1991; Masliah et al., 1991b, 1993a).
Amyloid fibril similarities between PDAPP tg mice and AD
Another prominent feature in the PDAPP tg mice, occasionally observed in AD, was the presence of neuronal elements in close association with the plaques, suggesting the possibility that extracellular amyloid was derived from these structures. Neuronal processes embedded in plaques contained discrete amyloid bundles associated with their RER, as well as large membranous electrodense bodies, coated vesicles, and dense-core neurosecretory vesicles that were also closely associated with amyloid deposits. Moreover, these subcellular neuronal alterations were more prominent and abundant around the amyloid core. Similar subcellular findings have been reported in amyloid-producing cells in AD (Roher et al., 1988;Wisniewski et al., 1991). The role of the dense-core vesicles in this process is uncertain, but previous studies have shown that these vesicles are abundant in dystrophic neurites of AD (Weiler et al., 1990; Munoz, 1991) and are rich in chromogranin, indicating that these cellular processes are of axonal origin and that neurosecretory granules could be involved in the process of amyloid formation.
The precise sequence of events that lead to plaque formation remains to be resolved. Nevertheless, the findings reported here, together with observations from other animal models (Cork et al., 1990) as well as AD (Masliah et al., 1993b; Heinonen et al., 1995; Masliah, 1995), suggest that plaque formation initiates with local synaptic alterations induced by possible abnormalities in APP processing and Aβ formation. The overproduction of the amyloidogenic Aβ (1–42) peptide that occurs as a result of the APP717 mutation in the PDAPP tg mouse is likely also to be critical to the rapid development of plaques seen in this animal model of AD (Suzuki et al., 1994). These events might then be followed by dystrophic neurite formation and extracellular amyloid deposition. These dystrophic neurites might represent regenerative and sprouting terminals and may eventually degenerate to become electrodense processes associated with plaques containing abundant dense amyloid. Supporting this contention, recent studies in PDAPP tg mice of various ages have shown that synaptic and neuritic alterations in the limbic system occur early and are closely followed by microdeposits of β-amyloid (Games et al., 1995a,b). Neuronal alterations and amyloid deposition in the neocortex appear at a later time point. Further studies in PDAPP tg mice of various ages are underway to clarify the progression of the neurodegenerative changes and plaque formation. An alternative explanation could be that neuronal uptake of extracellular amyloid fibrils, followed by disruption of the subcellular neuronal organelles triggers the cascade of events. However, this seems to be a less likely possibility because the ultrastructural characteristics of the cellular processes embedded in the plaque were different from the subcellular alterations previously identified in cells phagocytizing amyloid (Wisniewski et al., 1991; Frackowiak et al., 1992). It has been shown both in vitro and in vivo that cells which phagocytize amyloid contain nondegraded fibrils within phagosomes, whereas in cells which produce amyloid, fibrils appear first in altered RER deep infoldings of cell membranes (Wisniewski et al., 1991;Frackowiak et al., 1992). The neuronal processes embedded in the amyloid core of PDAPP tg mice contained abundant RER, neurosecretory vesicles, electrodense granular material, and coated vesicles, suggesting active synthesis and secretion of amyloidogenic elements rather than phagocytosis. These subcellular alterations in PDAPP tg mice imply that neurons play an important role in plaque formation and are not simply trapped in the midst of the lesions.
It has been postulated that microglial cells play an important role in amyloid formation in the AD plaque (Terry et al., 1964; Wisniewski and Terry, 1970; Roher et al., 1988; Wisniewski et al., 1989, 1991; Masliah et al., 1991c; Frackowiak et al., 1992). Although microglial cells were observed in the plaques of PDAPP tg mice, they were not as prominent as those seen in AD. These differences might be related to differences in the stage and duration of the lesions in humans versus tg mice. Furthermore, it is possible that the currently ongoing analysis of aged PDAPP tg mice will reveal more prominent astroglial alterations. Interestingly, astroglial reaction around plaques was of similar prominence in PDAPP tg mice and AD cases.
In conclusion, this study shows that PDAPP tg mice share several critical subcellular alterations with AD that make them a valuable model in which to study mechanisms of neurodegeneration and plaque formation. Furthermore, marked neuronal overexpression of a mutant hAPP (hAPP717V→F) can result in the accumulation of excess Aβ protein, and this accumulation may contribute to neurodegeneration and plaque formation seen in the PDAPP tg mice.
Footnotes
This work was supported by Athena Neurosciences, Inc. Additional support was provided by National Institutes of Health Grants AG10689 and AG5131 (E.M.) and AG11385 (L.M.). This work was also partially supported by Grant RR04050. We thank Dr. Robert Terry for his valuable comments, and Karen Khan, Ferdie Soriano, Tony Carr, Terry Guido, and Albert Maya for their technical assistance.
Correspondence should be addressed to Dr. E. Masliah, Department of Neurosciences, University of California-San Diego, La Jolla, CA 92093-0624.
REFERENCES
- 1.Bonhomme V, Hans P, Collette J, Moonen G. Neuron-specific enolase as a marker of in vitro neuronal damage. II. Investigation of the astrocyte protective effect against kainate-induced neurotoxicity. J Neurosurg Anesthesiol. 1993;5:117–120. doi: 10.1097/00008506-199304000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Campbell G, Lieberman AR, Anderson PN, Turmaine M. Regeneration of adult rat CNS axons into peripheral nerve autografts: ultrastructural studies of the early stages of axonal sprouting and regenerative axonal growth. J Neurocytol. 1992;21:755–787. doi: 10.1007/BF01237903. [DOI] [PubMed] [Google Scholar]
- 3.Chartier-Harlin M-C, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J, Mullan M. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the β-amyloid precursor protein gene. Nature. 1991;353:844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- 4.Clark RF, Goate AM. Molecular genetics of Alzheimer’s disease. Arch Neurol. 1993;50:1164–1172. doi: 10.1001/archneur.1993.00540110044004. [DOI] [PubMed] [Google Scholar]
- 5.Cork LC, Masters C, Beyreuther K, Price DL. Development of senile plaques: relationships of neuronal abnormalities and amyloid deposits. Am J Pathol. 1990;137:1383–1392. [PMC free article] [PubMed] [Google Scholar]
- 6.Cras P, Kawai M, Lowery D, Gonzalez-DeWhitt P, Greenberg B, Perry G. Senile plaque neurites in Alzheimer’s disease accumulate amyloid precursor protein. Proc Natl Acad Sci USA. 1991;88:7552–7556. doi: 10.1073/pnas.88.17.7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deftos LJ, Burton DW, Brandt DW. Parathyroid hormone-like protein is a secretory product of atrial myocytes. J Clin Invest. 1993;92:727–735. doi: 10.1172/JCI116643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen SC. Alzheimer’s disease: a double immunohistochemical study of senile plaques. Am J Pathol. 1988;132:86–101. [PMC free article] [PubMed] [Google Scholar]
- 9.Dragunow M, Faull RL, Lawlor P, Beilharz EJ, Singleton K, Walker EB, Mee E. In situ evidence for DNA fragmentation in Huntington’s disease striatum and Alzheimer’s disease temporal lobes. NeuroReport. 1995;6:1053–1057. doi: 10.1097/00001756-199505090-00026. [DOI] [PubMed] [Google Scholar]
- 10.Forloni G, Chiesa R, Smiroldo S, Verga L, Salmona M, Tagliavini F, Angeretti N. Apoptosis mediated neurotoxicity induced by chronic application of β amyloid fragment 25–35. NeuroReport. 1993;4:523–526. doi: 10.1097/00001756-199305000-00015. [DOI] [PubMed] [Google Scholar]
- 11.Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce β-amyloid fibrils. Acta Neuropathol. 1992;84:225–233. doi: 10.1007/BF00227813. [DOI] [PubMed] [Google Scholar]
- 12.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemes J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995a;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 13.Games D, Carr T, Guido T, Khan K, Soriano F, Tan H, McConlogue L, Lieberburg I, Schenk D, Masliah E. Progression of Alzheimer-type neuropathology in PDAPP 717 V→F transgenic mice. Soc Neurosci Abstr. 1995b;21:258. [Google Scholar]
- 14.Gheuens J, Cras P, Perry G, Boons J, Ceuterick-de Groote C, Lubke U, Mercken M, Tabaton M, Ganbetti PL, Vandermeeren M. Demonstration of a novel neurofilament associated antigen with the neurofibrillary pathology of Alzheimer and related diseases. Brain Res. 1991;558:43–52. doi: 10.1016/0006-8993(91)90712-5. [DOI] [PubMed] [Google Scholar]
- 15.Goate A, Chartier-Harlin M-C, Mullan M, Brown J, Crawford F, Fidani L, Guiffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 16.Golde TE, Estus S, Younkin LH, Selkoe DJ, Younkin SG. Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science. 1992;255:728–730. doi: 10.1126/science.1738847. [DOI] [PubMed] [Google Scholar]
- 17.Gonatas NK, Anderson WW, Evangelista I. The contribution of altered synapses in the senile plaque: an electron microscopic study in Alzheimer’s disease. J Neuropathol Exp Neurol. 1967;26:25–39. doi: 10.1097/00005072-196701000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Heinonen O, Soininen H, Sorvari H, Kosunene O, Paljarvi L, Koivisto E, Riekkinen PJ. Loss of synaptophysin-like immunoreactivity in the hippocampal formation is an early phenomenon in Alzheimer’s disease. Neuroscience. 1995;64:375–384. doi: 10.1016/0306-4522(94)00422-2. [DOI] [PubMed] [Google Scholar]
- 19.Higgins LS, Holtzman DM, Rabin J, Mobley WC, Cordell B. Transgenic mouse brain histopathology resembles early Alzheimer’s disease. Ann Neurol. 1994;35:598–607. doi: 10.1002/ana.410350514. [DOI] [PubMed] [Google Scholar]
- 20.Hirano A. Hirano bodies and related neuronal inclusions. Neuropathol Appl Neurobiol. 1994;20:3–11. doi: 10.1111/j.1365-2990.1994.tb00951.x. [DOI] [PubMed] [Google Scholar]
- 21.Hof PR, Cox K, Morrison JH. Quantitative analysis of a vulnerable subset of pyramidal neurons in Alzheimer’s disease. I. Superior frontal and inferior temporal cortex. J Comp Neurol. 1990;301:44–54. doi: 10.1002/cne.903010105. [DOI] [PubMed] [Google Scholar]
- 22.Hyman BT, VanHoesen GW, Damasio AR, Barnes CL. Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science. 1984;225:1168–1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- 23.Joachim CL, Mori H, Selkoe DJ. Amyloid beta-protein deposition in tissues other than brain in Alzheimer’s disease. Nature. 1989;341:226–230. doi: 10.1038/341226a0. [DOI] [PubMed] [Google Scholar]
- 24.LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G. The Alzheimer’s A beta peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nature Genet. 1995;9:21–30. doi: 10.1038/ng0195-21. [DOI] [PubMed] [Google Scholar]
- 25.Lampert P. A comparative electron microscopic study of reactive, degenerating, regenerating, and dystrophic axons. J Neuropathol Exp Neurol. 1967;26:345–368. doi: 10.1097/00005072-196707000-00001. [DOI] [PubMed] [Google Scholar]
- 26.Lassman H, Bancher C, Breitschopf H, Wegiel J, Bobinski M, Jellinger K, Wisniewski HM. Cell death in Alzheimer’s disease evaluated by DNA fragmentation in situ. Acta Neuropathol. 1995;89:35–41. doi: 10.1007/BF00294257. [DOI] [PubMed] [Google Scholar]
- 27.Martin LJ, Pardo CA, Cork LC, Price DL. Synaptic pathology and glial responses to neuronal injury precede the formation of senile plaques and amyloid deposits in the aging cerebral cortex. Am J Pathol. 1994;145:1358–1381. [PMC free article] [PubMed] [Google Scholar]
- 28.Masliah E, Hansen L, Albright T, Mallory M, Terry RD. Immunoelectron microscopic study of synaptic pathology in Alzheimer’s disease. Acta Neuropathol. 1991a;81:428–433. doi: 10.1007/BF00293464. [DOI] [PubMed] [Google Scholar]
- 29.Masliah E, Mallory M, Hansen L, Alford M, Albright T, DeTeresa R, Terry RD, Baudier J, Saitoh T. Patterns of aberrant sprouting in Alzheimer’s disease. Neuron. 1991b;6:729–739. doi: 10.1016/0896-6273(91)90170-5. [DOI] [PubMed] [Google Scholar]
- 30.Masliah E, Mallory M, Hansen L, Alford M, Albright T, Terry R, Shapiro P, Sundsmo M, Saitoh T. Immunoreactivity of CD45, a protein phosphotyrosine phosphatase, in Alzheimer’s disease. Acta Neuropathol. 1991c;83:12–20. doi: 10.1007/BF00294425. [DOI] [PubMed] [Google Scholar]
- 31.Masliah E, Ellisman M, Carragher B, Mallory M, Young S, Hansen L, DeTeresa R, Terry RD. Three-dimensional analysis of the relationship between synaptic pathology and neuropil threads in Alzheimer’s disease. J Neuropathol Exp Neurol. 1992;51:404–414. doi: 10.1097/00005072-199207000-00003. [DOI] [PubMed] [Google Scholar]
- 32.Masliah E, Mallory M, Hansen L, Alford M, DeTeresa R, Terry R. An antibody against phosphorylated neurofilaments identifies a subset of damaged association axons in Alzheimer’s disease. Am J Pathol. 1993a;142:871–882. [PMC free article] [PubMed] [Google Scholar]
- 33.Masliah E, Mallory M, Deerinck T, DeTeresa R, Lamont S, Miller A, Terry RD, Carragher B, Ellisman M. Re-evaluation of the structural organization of neuritic plaques in Alzheimer’s disease. J Neuropathol Exp Neurol. 1993b;52:619–632. doi: 10.1097/00005072-199311000-00009. [DOI] [PubMed] [Google Scholar]
- 34.Masliah E, Mallory M, Ge N, Godson C, Saitoh T. Phorbol ester-induced neuritic alterations in the rat neocortex: structural and immunocytochemical studies. Mol Chem Neuropathol. 1993c;20:125–145. doi: 10.1007/BF02815367. [DOI] [PubMed] [Google Scholar]
- 35.Masliah E, Honer WG, Mallory M, Voigt M, Kushner P, Terry RD. Topographical distribution of synaptic-associated proteins in the neuritic plaques of Alzheimer’s disease hippocampus. Acta Neuropathol. 1994a;87:135–142. doi: 10.1007/BF00296182. [DOI] [PubMed] [Google Scholar]
- 36.Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R. Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci Lett. 1994b;174:67–72. doi: 10.1016/0304-3940(94)90121-x. [DOI] [PubMed] [Google Scholar]
- 37.Masliah E. Mechanisms of synaptic dysfunction in Alzheimer’s disease. Histol Histopathol. 1995;10:509–519. [PubMed] [Google Scholar]
- 38.Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the β-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- 39.Mucke L, Masliah E, Johnson WB, Ruppe MD, Rockenstein EM, Forss-Petter S, Pietropaolo M, Mallory M, Abraham CR. Synaptotrophic effects of human amyloid β protein precursors in the cortex of transgenic mice. Brain Res. 1994;666:151–167. doi: 10.1016/0006-8993(94)90767-6. [DOI] [PubMed] [Google Scholar]
- 40.Munoz DG. Chromogranin A-like immunoreactive neurites are major constituents of senile plaques. Lab Invest. 1991;64:826–832. [PubMed] [Google Scholar]
- 41.Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science. 1991;254:97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- 42.Nakagawa Y, Nakamura S, Kase Y, Noguchi T, Ishihara T. Colchicine lesions in the rat hippocampus mimic the laterations of several markers in Alzheimer’s disease. Brain Res. 1987;408:57–64. doi: 10.1016/0006-8993(87)90358-1. [DOI] [PubMed] [Google Scholar]
- 43.Palmer AM, Gershon S. Is the neuronal basis of Alzheimer’s disease cholinergic or glutamatergic? FASEB J. 1990;2745:2752. doi: 10.1096/fasebj.4.10.2165009. [DOI] [PubMed] [Google Scholar]
- 44.Perry EK, Perry RH, Blessed G, Tomlinson BE. Neurotransmitter enzyme abnormalities in senile dementia: CAT and GAD activities in necropsy tissue. J Neurol Sci. 1977;34:247–265. doi: 10.1016/0022-510x(77)90073-9. [DOI] [PubMed] [Google Scholar]
- 45.Pollard H, Charriaut-Marlangue C, Cantagrel S, Represa A, Robain O, Moreau J, Ben-Ari Y. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience. 1994;63:7–18. doi: 10.1016/0306-4522(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 46.Portera-Cailliau C, Hedreen JC, Price DL, Koliatsos VE. Evidence for apoptotic cell death in Huntington’s disease and excitotoxic animal models. J Neurosci. 1995;15:3775–3787. doi: 10.1523/JNEUROSCI.15-05-03775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rockenstein EM, McConlogue L, Tan H, Power M, Masliah E, Mucke L. Levels and alternative splicing of amyloid β protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer’s disease. J Biol Chem. 1995;270:28257–28267. doi: 10.1074/jbc.270.47.28257. [DOI] [PubMed] [Google Scholar]
- 48.Roher A, Gray EG, Paula-Barbosa M (1988) Alzheimer’s disease: coated vesicles, coated pits and the amyloid-related cells. Proc R Soc Lond [Biol]232:367–373. [DOI] [PubMed]
- 49.Schochet SS, Lampert PW, Earle KM. Neuronal changes induced by intrathecal vincristine sulfate. J Neuropathol Exp Neurol. 1968;27:645–658. [PubMed] [Google Scholar]
- 50.Selkoe DJ. Physiological production of the β-amyloid protein and the mechanisms of Alzheimer’s disease. Trends Neurosci. 1993;16:403–409. doi: 10.1016/0166-2236(93)90008-a. [DOI] [PubMed] [Google Scholar]
- 51.Selkoe DJ, Podlisny MB, Joachim CL, Vickers EA, Lee G, Fritz LC, Oltersdorf T. Beta-amyloid precursor protein of Alzheimer’s disease occurs as 110- to 135-kilodalton membrane-associated proteins in neural and nonneural tissue. Proc Natl Acad Sci USA. 1988;85:7341–7345. doi: 10.1073/pnas.85.19.7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, McCormack R, Wolfert R, Selkoe D, Lieberburg I, Schenk D. Isolation and quantification of soluble Alzheimer’s β-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 53.Shelansky ML. Neurofibrillary degeneration. Arch Neurol. 1969;20:199–206. doi: 10.1001/archneur.1969.00480080099012. [DOI] [PubMed] [Google Scholar]
- 54.Sisodia SS, Koo EH, Beyreuther K, Unterbeck A, Price DL. Evidence that β-amyloid protein in Alzheimer’s disease is not derived by normal processing. Science. 1990;248:492–494. doi: 10.1126/science.1691865. [DOI] [PubMed] [Google Scholar]
- 55.Sloviter RS, Dean E, Neubort S. Electron microscopic analysis of adrenalectomy-induced hippocampal granule cell degeneration in the rat: apoptosis in the adult central nervous system. J Comp Neurol. 1993;330:337–351. doi: 10.1002/cne.903300305. [DOI] [PubMed] [Google Scholar]
- 56.Smale G, Nichols NR, Brady DR, Finch CE, Horton WE., Jr Evidence for apoptotic cell death in Alzheimer’s disease. Exp Neurol. 1995;133:225–230. doi: 10.1006/exnr.1995.1025. [DOI] [PubMed] [Google Scholar]
- 57.Southam E, Thomas PK, King RHM, Goss-Sampson MA, Muller DPR. Experimental vitamin E deficiency in rats. Morphological and functional evidence of abnormal axonal transport secondary to free radical damage. Brain. 1991;114:915–936. doi: 10.1093/brain/114.2.915. [DOI] [PubMed] [Google Scholar]
- 58.Su JH, Anderson AJ, Cummings BJ, Cotman CW. Immunohistochemical evidence for apoptosis in Alzheimer’s disease. NeuroReport. 1994;5:2529–2533. doi: 10.1097/00001756-199412000-00031. [DOI] [PubMed] [Google Scholar]
- 59.Suzuki N, Cheung TT, Cai X-D, Odaka A, Otvos L, Jr, Eckman C, Golde TE, Younkin SG. An increased percentage of long amyloid β protein secreted by familial amyloid β protein precursor (beta APP717) mutants. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 60.Terry RD, Pena C. Experimental production of neurofibrillary degeneration. II. Electron microscopy: phosphatase histochemistry and electron probe analysis. J Neuropathol Exp Neurol. 1965;24:200–210. doi: 10.1097/00005072-196504000-00003. [DOI] [PubMed] [Google Scholar]
- 61.Terry RD, Wisniewski HM. The ultrastructure of the neurofibrillary tangle and the senile plaque. In: Wolstenholme GEW, O’Connor M, editors. Ciba Foundation symposium on Alzheimer’s disease and related conditions. J. & A. Churchill; London: 1970. pp. 145–168. [Google Scholar]
- 62.Terry RD, Gonatas NK, Weiss M. Ultrastructural studies in Alzheimer’s presenile dementia. Am J Pathol. 1964;44:269–297. [PMC free article] [PubMed] [Google Scholar]
- 63.Terry RD, Peck A, DeTeresa R, Schechter R, Horoupian DS. Some morphometric aspects of the brain in senile dementia of the Alzheimer type. Ann Neurol. 1981;10:184–192. doi: 10.1002/ana.410100209. [DOI] [PubMed] [Google Scholar]
- 64.Terry RD, Hansen L, Masliah E. Structural alterations in Alzheimer’s disease. In: Terry RD, Katzman R, editors. Alzheimer’s disease. Raven; New York: 1994. pp. 179–196. [Google Scholar]
- 65.Toggas SM, Masliah E, Rockenstein EM, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- 66.Wang D, Munoz DG. Qualitative and quantitative differences in senile plaque dystrophic neurites of Alzheimer’s disease and normal aged brain. J Neuropathol Exp Neurol. 1995;54:548–556. doi: 10.1097/00005072-199507000-00009. [DOI] [PubMed] [Google Scholar]
- 67.Weiler R, Lassmann H, Fischer P, Jellinger K, Winkler H. A high ratio of chromogranin A to synaptin/synaptophysin is a common feature of brains in Alzheimer’s and Pick’s disease. FEBS Lett. 1990;263:337–339. doi: 10.1016/0014-5793(90)81408-g. [DOI] [PubMed] [Google Scholar]
- 68.Wilcock GK, Esiri MM, Bowen DM, Hughes AO. The differential involvement of subcortical nuclei in senile dementia of Alzheimer’s type. J Neurol Neurosurg Psychiatry. 1988;51:842–849. doi: 10.1136/jnnp.51.6.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wisniewski H, Terry RD. Experimental colchicine encephalopathy. I. Introduction of neurofibrillary degeneration. Lab Invest. 1967;17:577–587. [PubMed] [Google Scholar]
- 70.Wisniewski H, Terry RD. An experimental approach to the morphogenesis of neurofibrillary degeneration and the argyrophilic plaque. In: Wolstenholme GEW, O’Connor M, editors. Ciba Foundation symposium on Alzheimer’s disease and related conditions. J. & A. Churchill; London: 1970. pp. 223–248. [Google Scholar]
- 71.Wisniewski H, Terry RD. Re-examination of the pathogenesis of the senile plaque. In: Zimmerman HM, editor. Progress in neuropathology, Vol. 2. Grune & Stratton; New York: 1973. pp. 1–26. [Google Scholar]
- 72.Wisniewski H, Johnson AB, Paine CS, Kay WJ, Terry RD. Senile plaques and cerebral amyloidosis in aged dogs. Lab Invest. 1970;23:287–296. [PubMed] [Google Scholar]
- 73.Wisniewski HM, Sinatra RS, Iqbal IY. Neurofibrillary and synaptic pathology in aged brain. In: Johnson J, editor. Aging and cell structure, Vol. 1. Plenum; New York: 1981. pp. 105–142. [Google Scholar]
- 74.Wisniewski HM, Wegiel J, Wang KC, Kujawa M, Lach B. Ultrastructural studies of the cells forming amyloid fibers in classical plaques. Can J Neurol Sci. 1989;16:535–542. doi: 10.1017/s0317167100029887. [DOI] [PubMed] [Google Scholar]
- 75.Wisniewski HM, Barcikowska M, Kida E. Phagocytosis of β/A4 amyloid fibrils of the neuritic neocortical plaques. Acta Neuropathol. 1991;81:588–590. doi: 10.1007/BF00310142. [DOI] [PubMed] [Google Scholar]