

Abstract
Herpes simplex viruses that lack ICP34.5 are neuroattenuated and are presently being considered for cancer and gene therapy in the nervous system. Previously, we documented the focal presence of the latency-associated transcripts (LATs) in the hippocampi of immunocompromised mice after intracranial (IC) inoculation of an ICP34.5-deficient virus called strain 1716. To characterize further the biological properties of strain 1716 in the CNS of immunocompetent mice, we determined the extent of viral gene expression in different cell types and regions of the CNS after stereotactic IC inoculation of this virus. At survival times of >30 d after inoculation, we found that (1) infectious virus was not detectable by titration and immunohistochemical studies; (2) neurons harbored virus as demonstrated by the detection of the LATs by in situ hybridization (ISH); (3) transcripts expressed during the lytic cycle of infection were not detected by ISH; and (4) subsets of neurons were selectively vulnerable to latent infection, depending on the site of inoculation. These results suggest that the absence of ICP34.5 does not abrogate latent infection of the CNS by strain 1716. Additional studies of strain 1716 in the model system described here will facilitate the elucidation of the mechanisms that regulate the selective vulnerability of CNS cells to latent viral infection and lead to the development of ICP34.5 mutant viruses as therapeutic vectors for CNS diseases.
Keywords: herpes simplex virus, ICP34.5, latency, neuron-specific gene expression, gene transfer, viral vectors
All herpes viruses are characterized by the ability to establish latent infections in their natural hosts. Herpes simplex virus type 1 (HSV-1) is a double-stranded DNA virus that has the capacity to form lytic and latent infections in neurons of the peripheral nervous system (PNS) and CNS of humans (their natural hosts) and numerous animals. Although lytic infection occurs in both neuronal and non-neuronal cells, latent infection occurs predominantly in neurons (Tenser et al., 1991). During latent infection of experimental animals, the genes of the viral lytic infection are transcriptionally silent, and only a small number of latency-associated transcripts (LATs) are expressed (for review, see Fraser et al., 1992). The contributions of viral and host factors, as well as the molecular mechanisms by which these factors influence the establishment, maintenance, and reactivation of latency, have been the subject of intense research during the past two decades. Many clinical and experimental reports suggest that there is differential replication and spread of HSV in the CNS, with a propensity for infection of the limbic system (Anderson and Field, 1983; Neeley et al., 1985; Stroop and Schaefer, 1986; McFarland and Hotchin, 1987; Roizman and Kaplan, 1992;Barnett et al., 1993). Although HSV DNA has been detected in the trigeminal ganglion and brainstem of humans (Fraser et al., 1981), only recently, with use of sensitive PCR techniques (Nicoll et al., 1993;Baringer and Pisani, 1994), has HSV DNA been detected in the human CNS rostral to the brainstem.
We have shown recently that strain 1716, a neuroattenuated form of HSV-1 strain 17+, can infect CNS neurons in immunocompromised nude mice after IC inoculation, as evidenced by the presence of LATs by in situ hybridization (ISH) studies (Kesari et al., 1995). Strain 1716 has a 759 bp deletion in both copies of the ICP34.5 gene (see Fig. 1) (MacLean et al., 1991). To gain further insights into the pathogenicity of strain 1716 and the vulnerability of cells to latent infection, the present study was undertaken to characterize the state of strain 1716 in immunocompetent mice (Balb/c) after stereotactic IC inoculation of this virus. At >30 days after inoculation, we found that (1) infectious virus was not detectable by titration and immunohistochemistry (IHC); (2) neurons harbored virus as demonstrated by the presence of the LATs by ISH; (3) transcripts indicative of a lytic infection (i.e., ICP4, TK, gC, VP5, ICP34.5) were not detected by ISH; and (4) neurons exhibited a selective vulnerability to latent infection depending on the site of inoculation. These studies suggest that although the replication of strain 1716 is restricted in the murine CNS, it can establish a latent infection in selectively vulnerable CNS neurons. This implies that the ICP34.5 gene product is not essential for the establishment or maintenance of latency in the CNS. Accordingly, further studies of the model system described here will facilitate the elucidation of mechanisms that regulate latent viral infection of selectively vulnerable CNS cells.
Fig. 1.

HSV-1 genome map. HSV-1 genome showing approximate location of the ICP34.5, 2 kb latency-associated transcripts (LATs), and neighboring genes. A, The 152 kb HSV-1 strain 17+ genome is shown that illustrates the unique long and short segments of the genome,UL and US(lines), bounded by internal (IR) and terminal repeat (TR) regions (open boxes). Hatch marks show location of the virion protein 5 (VP5), thymidine kinase (TK), and glycoprotein C (gC) genes. B, Expanded view of theUL/USregion of the genome showing the location of the ICP34.5, orfP, ICPO, and ICP4 genes. The transcripts in theBam SP region include the ICP34.5, ICP0, ICP4, mLAT, and L/STs. C, The location of the 2.0 kb LAT (2kb LATs) gene, which is expressed during acute and latent infection, and the primary 8.5 kb transcript (8.5kb LAT). D, The location of the proposed L/STs (Yeh and Schaffer, 1993).E, The location of the LAT-specificBstEII-BstEII probe used for in situ detection of HSV-specific latent gene expression and of one of the 759 bp deletions in strain 1716. Nucleotide positions are based on DNA sequence analysis of Perry and McGeoch (1988).
MATERIALS AND METHODS
Virus stocks. To produce virus stocks, subconfluent monolayers of baby hamster kidney 21 clone 13 (BHK) cells were infected with HSV strains 1716 or 1771 or with parental strain 17+. Strain 1716 has a mutation (deletion) in both copies of the ICP34.5 gene (MacLean et al., 1991) (Fig.1C). Strain 1771 has a single stop codon 9 bp downstream of the ATG in both copies of the ICP34.5 ORF (McKie et al., 1994). Virus was concentrated from the culture, titered on BHK cells by plaque assay, stored at −70°C in 0.5 ml aliquots of viral culture medium (MEM containing penicillin and streptomycin), and thawed rapidly just before use, as described previously (Spivack and Fraser, 1987;Valyi-Nagy et al., 1992). Virus FgDβ (Sodora et al., 1991) is deficient in glycoprotein D, and virus d27-1 (Rice and Knipe, 1990) is deficient in ICP27.
Viral inoculation. Four- to six-week-old Balb/c mice obtained from HSD (Indianapolis, IN) were anesthetized with ketamine/xylazine (87 mg/kg ketamine, 13 mg/kg xylazine). After the heads of the mice were cleansed with 70% ethanol, a Hamilton syringe with a 30 gauge removable needle was used to inject the appropriate amount of virus (105-106 PFU in 5 μl) into the brain at coordinates that corresponded to different anatomical locations (i.e., cortex, hippocampus, caudato-putamen, posterior fossa, olfactory bulb), using a small animal stereotactic apparatus (Kopf Instruments, Tujunga, CA) as described previously (Kesari et al., 1995). The injection was performed for 3 min. The needle was left in place for 3 min after the injection, after which time it was withdrawn slowly over 1 min, as described previously (Kesari et al., 1995). In pilot studies, injection of trypan blue dye was used to confirm the location of the injection sites and to assess leakage. For corneal infections, mice corneas were scarified and inoculated with 1 × 106 PFU of virus, as described previously (Spivack et al., 1995) .
Titration of virus from brain. Mice that were intracranially inoculated into the caudato-putamen with 1 × 106 PFU (in 5 μl) of strain 17+ or 5 × 105 PFU (in 5 μl) of strain 1716 were killed by lethal injection of anesthesia. The brains were removed from mice that were killed on different days after viral inoculation (days 0, 1, 3, 5, 30), quick-frozen in liquid nitrogen, and stored at −70°C. The samples from the different time points were thawed rapidly in a 37°C water bath, and the tissue was homogenized in viral culture medium at a 10% w/v ratio using a Pyrex Ten Broeck tissue grinder (Bellco Glass, Vineland, NJ). The homogenates were centrifuged at 3,000 × g for 10 min at 4°C, the supernatant was diluted logarithmically in media, and the viral titer of each sample was determined by plaque assay on BHK cells (Spivack and Fraser, 1987). When stock virus is used for this procedure, there is always a consistent drop in the viral titer of approximately two log-fold compared with the expected titer (approximately one log is lost in the freeze–thaw step, and one log is lost in the homogenization steps).
IHC procedures. The whole bodies of the mice were transcardially perfused with PBS and fixed with 4% paraformaldehyde (0.1 m PBS, pH 7.4). The brains and trigeminal ganglia were then dissected for histological and IHC analysis. The methods for tissue processing and light microscopic IHC analysis were similar to those described elsewhere (Trojanowski et al., 1993). Rabbit polyclonal antisera to HSV-1, which detects the major glycoproteins present in the viral envelope and at least one core protein (Dako, Carpinteria, CA), was used at a dilution of 1:2000 to detect replicating virus (Adams et al., 1984). Viral antigen-expressing cells were detected by the indirect avidin-biotin immunoperoxidase (Vectastain ABC Kit, Vector Labs, Burlingame, CA) method with 3, 3′-diaminobenzadine as the chromagen. The spread of virus in all animals was monitored by screening every tenth coronal section through the entire brain with the HSV antibody.
ISH for HSV-1-specific gene expression. Sections of perfused and fixed tissue were mounted on slides, and ISH was performed to detect viral gene expression, as described previously (Deatly et al., 1988; Valyi-Nagy et al., 1994). The LAT probeBstEII-BstEII subfragment (0.9kb) ofBamHI B (Fig. 1E), the 3.4 kb BamHI fragment from plasmid LE578 containing the TK probe, theBamHI- Y fragment from plasmid pRB113 containing the ICP4 probe, the EcoRI-BamHI fragment I/I (KOS) from plasmid pBR322 containing the gC probe, the BamHI fragment a′ (KOS) from plasmid pBR322 containing the VP5 probe, and theBamHI SP fragment from plasmid pRB115 containing the ICP0, ICP4, and ICP34.5 probes were isolated from restriction digests by gel electrophoresis and purified by GeneClean (Bio 101, La Jolla, CA (see Fig. 1) (Valyi-Nagy et al., 1991, 1992). DNA probes were nick-translated and separated from unincorporated nucleotides by passage through Sephadex G-50 spin columns (Pharmacia, Piscataway, NJ) (Deatly et al., 1988). The specific activities of the probes were ∼1 × 108-5 × 108 cpm/μg DNA. Serial tissue sections were hybridized with one of the following35S-labeled HSV probes: LAT, ICP4, TK, gC, or VP5. In all ISH experiments, sets of slides from each animal were exposed for 3, 5, 7, and 10 d.
RESULTS
Recovery of infectious virus after IC inoculation of strain 1716 in immunocompetent Balb/c mice
To determine whether infectious virus was present after IC inoculation of strain 1716, we performed experiments in immunocompetent Balb/c mice. Accordingly, 15 mature (6 week old) Balb/c mice were stereotactically inoculated in the brain with 5 × 105PFU of strain 1716, and three of these mice were killed at each of five different times (i.e., at 0, 1, 3, 5, and 30 d after inoculation). As in nude mice, titerable strain 1716 virus could not be recovered 3 d after inoculation, and there were no signs of any obvious disease during the subsequent 30 d period (Kesari et al., 1995) (Fig. 2). Although this does not formally exclude the possibility of a low-level persistent viral infection, viral antigens could not be detected on any sections of brains from latently infected mice that were used for IHC (see below). In contrast, the recovery of infectious strain 17+ virus after IC inoculation increased with time and exceeded the input inoculum (Fig. 2). Strain 17+-infected mice showed the characteristic symptoms of encephalitis: i.e., ruffled fur, hunching, limb paralysis, lethargy, and death.
Fig. 2.

Quantification of infectious virus in Balb/c mouse brain after IC inoculation. To establish the growth characteristic of strain 1716 and parental strain 17+ in the CNS, Balb/c mice were injected intracranially with either 5 × 105 PFU of 1716 (squares) or 1 × 106 PFU of 17+ (circles). At the times indicated, mice were killed, and the brains were frozen in liquid nitrogen and stored at −70°C. Specimens were thawed rapidly and homogenized, and viral titration was performed in triplicate on BHK cells. Mice were killed at the times shown and processed as described in Materials and Methods. Each point is the mean of either three mice (for strain 1716) or two mice (for strain 17+); error bars represent SEM.
Presence of LAT-expressing, antigen-negative cells in the CNS of mice infected with strain 1716
To determine whether strain 1716 could be detected at times when latent infection occurs (>30 d after inoculation), five mature (6-week-old) Balb/c mice were stereotactically inoculated with 5 × 105 PFU of strain 1716 and were killed at day 45 after inoculation. Brains were harvested and processed as described in Materials and Methods. IHC, using a polyclonal antibody specific to HSV, revealed that HSV-antigen staining was absent throughout the brains of these mice, indicating a lack of a persistent, low-level viral infection (Fig. 3A), but ISH on serial sections for the LATs, using a 35S-labeled LAT-specificBstEII-BstEII probe (Fig. 1E), revealed the presence of a large number of cells with a strong, nuclear-localized ISH signal (LAT+ cells; Fig.3B). This signal was predominantly in neurons (as determined by morphology and location of these cells in neuron-rich gray matter areas). The expression levels of LATs varied among cells in a given area (Fig. 3C). As a positive control, inoculation of 1 × 106 PFU of strain 17+ virus revealed the presence of antigen-positive and LAT+ cells at 3 d after inoculation (Fig. 3D–F). The number of LAT+ cells is higher in the CNS after IC inoculation with strain 1716 (Fig. 3C) compared with the number of LAT+ cells that can be found in the CNS by peripheral (via eye) inoculation of wild-type strain 17+ virus (quantitation not shown) (Fig. 3G) (Deatly et al., 1988). Because the deletion in strain 1716 encompasses four genes, we used another engineered virus strain, whose only deficiency is in ICP34.5 (strain 1771), to determine whether the absence of ICP34.5 alone would allow for LAT expression during latency (McKie et al., 1994). Strain 1771 does not synthesize ICP34.5 protein and should have a minimal effect on the LATs and other neighboring transcripts. Also, the LD50 of strain 1771, like strain 1716, is >106PFU (McKie et al., 1994). The same profile of gene expression for strain 1716 is observed for strain 1771 at day 45 after inoculation, implying that this phenomena is attributable to a specific deficiency of ICP34.5 and not to an effect of the deletions in orfP, LAT, or L/STs.
Fig. 3.

Detection of replicating and latent virus by IHC and ISH. Balb/c mice were stereotactically inoculated into the caudato-putamen with either 5 × 105 PFU of strain 1716 (5 mice, day 45, postinoculation) or 1 × 106 PFU of strain 17+ (3 mice, day 3, postinoculation). They were killed, and the brains were removed and processed as described in Materials and Methods. A, Absence of HSV antigen immunostaining in a representative region of the brain of a strain 1716-infected mouse using a polyclonal anti-HSV-1 antibody.B, LAT+ cells were present by ISH in serial sections of all strain 1716-infected mice. C, In strain 1716-infected brains, LATs were localized to the nucleus of neurons as during PNS latency. D, E, HSV-1 antigen-positive neurons and to a lesser extent glial cells were found throughout the brains of 17+-infected mice at day 3 after IC inoculation. F, LAT+ cells were found in sections of 17+-infected mice probed by ISH.G, A small number of LAT+ cells also were found in the brainstems of 17+-infected mice after corneal inoculation. Scale bar: 800 μm in A, B, F; 80 μm inC; 1.6 mm in D; 160 μm inE; 60 μm in G.
To determine whether the large number of LAT+ cells observed is specific for ICP34.5-deficient viruses or for the inoculation route, we performed similar inoculations with two other attenuated viruses that are deficient in other viral genes: FgDβ (five mice) and d27-1 (three mice). With both viruses, mice showed symptoms of disease for the first week, and none of five FgDβ- and one of three d27-1-infected mice died during this time. The survivors were killed at day 30, and ISH for LAT was performed on sections of brains. Only a few LAT+ cells (<5 cells/section) were found in the FgDβ-infected mice, and none were detected in d27-1-infected mice. This suggests that the latency phenomena described above is specific for ICP34.5-deficient viruses and not for the inoculation route.
Lytic cycle viral transcripts are not detectable by ISH in LAT+ cells
Because others have speculated about and reported on the presence of a low level of α gene expression in the mouse PNS during latency (Kosz-Vnenchak et al., 1993; Kramer and Coen, 1995), and to exclude the possibility of a persistent infection, we determined the extent of viral gene expression in the CNS (Fig. 4). We used ISH to document that the three classes of viral transcripts were not expressed in strain 1716-infected mice at latent times. Tissue sections from strain 17+-infected mice were used as positive controls (Fig. 4F–O). In all ISH experiments, serial tissue sections were exposed for 3–10 d. Using nick-translated35S-labeled DNA probes for the LAT, ICP4, TK, VP5, gC, and ICP34.5 genes to examine serial sections of brains from strain 1716-infected mice, we showed that these brains were positive for the LATs but negative for the lytic transcripts [ICP4, TK (not shown), VP5, gC, ICP34.5, and the L/STs] (Fig. 4A–E); however, this does not exclude the possibility of a low level of IE or E gene expression that is beyond the detection level of our ISH technique.
Fig. 4.
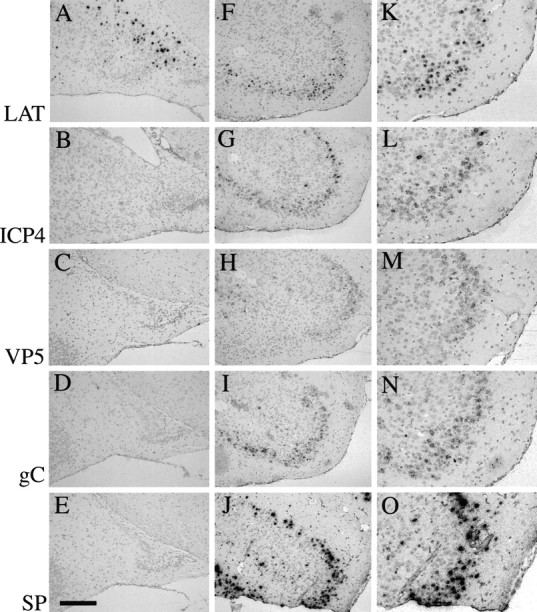
Absence of productive cycle HSV-1 gene expression in latently infected mice by in situ hybridization. Balb/c mice were stereotactically inoculated in the caudato-putamen with either 5 × 105 PFU of strain 1716 (3 mice, day 45, postinoculation) or 1 × 106 PFU of strain 17+ (3 mice, day 3, postinoculation). They were killed, and the brains were removed and processed as described in Materials and Methods. A–E, Representative coronal sections showing that strain 1716-infected mouse brains were positive for LAT transcripts (A) but negative for ICP4 (B), VP5 (C), gC (D), and SP transcripts (E). F–O, Strain 17+-infected mice were positive for LAT (F, K ), ICP4 (G, L), VP5 (H, M ), gC (I, N ), and SP transcripts (J, O). Scale bar: 320 μm in A–J; 160 μm inK–O).
Distribution of LAT+ cells is dependent on the site of inoculation
Four- to six-week-old old Balb/c mice were stereotactically inoculated with 5 × 105 PFU of strain 1716 and separated into groups on the basis of the site of inoculation (olfactory bulb, cortex, caudato-putamen, hippocampus, and posterior fossa). Mice were killed at day 45 after inoculation, and the brains were harvested and processed for IHC and ISH. Screening of sections using the HSV polyclonal antibody revealed that there were no antigen-positive cells in any of these mice; however, ISH using the LAT probe revealed the presence of LAT+ cells, and the distribution of these cells was dependent on the site of inoculation (Fig. 5).
Fig. 5.

Diagrams of the distribution of LAT+cells in the CNS after IC inoculation in different locations. Schematic of the injection sites (B–F in A) and diagrams (B–F) of representative coronal sections of mouse brain showing the distribution of LAT+ cells at day 45 postinoculation of strain 1716 into olfactory bulb (B), cortex (C), caudato-putamen (D), hippocampus (E), and posterior fossa (F).
In mice inoculated in the olfactory bulb, LAT+ cells could be detected in the hippocampus, caudato-putamen, thalamus, and brainstem (Figs. 5B, 6). In mice inoculated in the cortex, LAT+ cells could be detected in the olfactory bulb, cortex, caudato-putamen, cerebellum, and brainstem (Fig. 5C). In mice inoculated in the caudato-putamen, abundant LAT+ cells were found in the olfactory bulb, hippocampus, cortex, caudato-putamen, and brainstem (Figs.5D, 7). In mice inoculated in the hippocampus, LAT+ cells could be detected in the cortex, caudato-putamen, hippocampus, cerebellum, and brain stem (Fig.5E). In mice inoculated in the posterior fossa, LAT+ cells could be detected in the caudato-putamen, brainstem (including the trigeminal nuclei), and trigeminal ganglion of the PNS (Figs. 5F, 8). Table 1is a semi-quantitative summary of the distribution of LAT+cells after inoculation at the different sites. In all cases, LAT+ cells were present in the brainstem, and very few cells were detected in the cerebellum after posterior fossa inoculation (none were detected in the Purkinje cell layer). Interestingly, in the olfactory bulb injections, no LAT+ cells were found in the bulb itself. This suggests that there is a selectivity to latent infection beyond the simple issue of access of virus to cells. Furthermore, even in LAT+ areas, not all cells were positive, although they had access to the inoculum.
Fig. 6.

Distribution of LAT+ cells in the CNS after IC inoculation of the olfactory bulb. Three Balb/c mice that were stereotactically inoculated into the olfactory bulb with 5 × 105 PFU of strain 1716 were killed at day 45 postinoculation. ISH for LATs revealed the presence of LATs in the subependymal region (A), thalamus (B), and brainstem (C). Scale bar, 320 μm inA–C.
Fig. 7.

Distribution of LAT+ cells in the CNS after IC inoculation of the caudato-putamen. Three Balb/c mice that were stereotactically inoculated into the caudato-putamen with 5 × 105 PFU of strain 1716 were killed at day 45 postinoculation. ISH for LATs revealed the presence of LATs in the olfactory system (A), cortex (B, E), caudato-putamen (C), hippocampus (D, E), and brainstem (F). Scale bar: 320 μm in A–C, F; 800 μm in D, E.
Fig. 8.

Distribution of LAT+ cells in the CNS after IC inoculation of the posterior fossa. Two Balb/c mice that were stereotactically inoculated into the posterior fossa with 5 × 105 PFU of strain 1716 were killed at day 45 postinoculation. ISH for LATs revealed the presence of LATs in the caudato-putamen (A), brainstem (B), and trigeminal ganglion (C). Scale bar, 320 μm inA–C.
Table 1.
Summary of the distribution of LAT+ cells in the CNS after IC inoculation of Balb/c mice at different anatomical locations
| Injection site | Distribution of LAT+cells | |||||||
|---|---|---|---|---|---|---|---|---|
| Olf | Crtx | Stm | Dcp | Hipp | Bstm | Cere | TG | |
| Olfactory bulb | − | + | ++ | + | ++ | ++ | − | − |
| Cortex | + | + | ++ | ++ | − | +++ | + | − |
| Caudato-putamen | + | ++++ | +++ | +++ | ++++ | + | − | − |
| Hippocampus | − | ++++ | ++ | + | +++ | +++ | + | − |
| Posterior fossa | − | − | + | + | − | ++ | + | + |
Two sections from each area were used to count and average the number of LAT+ cells. Olf, Olfactory bulb and tract; Ctx, cortex; Stm, striatum; Dcp, diencephalon; Hipp, hippocampus; Bstm, brainstem; Cere, cerebellum; TG, trigeminal ganglion. +, <10 cells/section; ++, 10–25 cells/section; +++, 25–50 cells/section, ++++, >50 cells/section.
DISCUSSION
The studies presented in this report document the detection of latent virus (as defined by LAT expression in the absence of lytic viral gene expression) in the CNS of immunocompetent mice after IC inoculation of an ICP34.5-deficient HSV-1. This is a particularly important finding, because viruses of this type are being considered for use in gene and cancer therapy in the CNS of humans. The CNS latency model presented here will be useful for studying many aspects of HSV biology in the CNS in natural and therapeutic settings.
Previous reports have shown that strain 1716 is neuroattenuated (MacLean et al., 1991; McKie et al., 1994; Kesari et al., 1995).Spivack et al. (1995) studied the latency and replication characteristics of strain 1716 in the PNS using a mouse eye model. They report that strain 1716 does form a latent infection, as shown by the expression of LATs (detected by ISH), but cannot be reactivated by the explant co-cultivation method (Spivack et al., 1995). Although the deletion in strain 1716 encompasses at least four potential genes (ICP34.5, LATs, L/STs, and orfP), the neurovirulence phenotype seems to be the result of the ICP34.5 gene (McKie et al., 1994). Likewise, the latency phenotype described here is also attributable to the specific absence of ICP34.5, because a stop codon insertion into the ICP34.5 ORF (strain 1771) had the same phenotype as strain 1716.
Functions of ICP34.5
The ICP34.5 gene encodes a 263 amino acid acidic protein that has homology to the cellular genes MyD116 and GADD34 (Chou et al., 1990;McGeoch and Barnett, 1991; Chou and Roizman, 1994). It is thought to act by inhibiting the cessation of protein synthesis associated with programmed cell death in some nonpermissive, dividing cells (i.e., in SK-N-SH human neuroblastoma cells) by regulating the interferon-inducible PKR pathway (Chou and Roizman, 1992, 1994; Chou et al., 1995); however, the specific mechanism by which the absence of ICP34.5 confers the neuroattenuation property is not clear. We show here that although the absence of ICP34.5 precludes high titer viral replication in the CNS, it does not preclude latent infection. This is in agreement with the model of the establishment of latency in which latency is established early or before the establishment of the lytic replication cycle (Steiner et al., 1990; Speck and Simmons, 1991,1992). Furthermore, we show that ICP34.5, orfP, and L/STs are not required for the establishment or maintenance of latency in CNS after IC inoculation. This is in agreement with parallel studies of ICP34.5-deficient viruses in the PNS (Whitley et al., 1993; Spivack et al., 1995). We also speculate, on the basis of the fact that the phenomena described here is specific for ICP34.5-deficient viruses, that ICP34.5 may play a role in the establishment of latency in the IC inoculation model.
Distribution of LAT+ cells
The mechanisms for the selective vulnerability of neurons to various pathogens or disease processes is not well understood (for review, see Johnson, 1980; Schreiber and Baudry, 1995), and the selective distribution of LAT+ cells in the brain as detected in these studies may be attributable to several factors. These include the connectivity of the inoculated areas, diffusion or leakage of the initial inoculum, possible low-level replication that is beyond our ISH or IHC detection level, differences in the level of LAT gene expression in different neurons, migration and turnover of cells, and other unknown viral and host factors. Intra-axonal transport of virus is the major mechanism of virus spread in the PNS and CNS (Cook and Stevens, 1973; Ugolini et al., 1987). After primary infection in humans, the virus travels from peripheral epithelial cells of the skin to the trigeminal ganglion (PNS) and then into the brainstem and rostral CNS. The possibility that the virus could travel back from the CNS to the PNS has been a subject of speculation, but no experimental evidence has ever demonstrated this conclusively (Sekizawa and Openshaw, 1984; Roizman and Kaplan, 1992; Whitley and Lakeman, 1995). The finding of LAT+ cells in the trigeminal ganglion after the posterior fossa injections, to our knowledge, is the first experimental report of HSV-1 traveling from CNS to PNS to establish latency. This is presumed to be attributable to intra-axonal transport after infection of the brainstem trigeminal nuclei, but we cannot formally exclude the unlikely possibility of spread via the cerebrospinal fluid (Boerman et al., 1992). In pilot experiments to confirm anatomical location, leakage of the inoculum was minimal, as assessed by injection of trypan blue dye. Certainly, CSF spread or diffusion alone would not account for the differences in the distribution of LAT+ cells in the different inoculation sites.
We used immunocompetent mice because of evidence that a low level of persistent infection may occur with some neuroattenuated strains in immunocompromised mice (Valyi-Nagy et al., 1992, 1994). The titration data (Fig. 2), in addition to the IHC and ISH (Figs. 3, 4) data for viral antigens and acute transcripts, respectively, suggest that it is unlikely that strain 1716 replicates at a low level in immunocompetent mice during latent times. Infectious strain 1716 was recovered during the first 3 d after inoculation, however, and this suggests that there may be limited replication during this early time (Fig. 2), which may contribute to the distribution pattern of the LAT+cells seen at latent times.
In contrast to these experimental results showing latent virus in many different areas of the CNS (Figs. 5, 6, 7, 8), it is interesting to note that there seems to be a preference for HSV to infect the limbic system in humans (Roizman and Kaplan, 1992). Also, human CNS lytic infection involves neurons and glial cells (Schmidbauer et al., 1989; Roizman and Kaplan, 1992). Although LAT+ cells were not present throughout the whole brain in neuronal and non-neuronal cells, we cannot exclude the possibility that viral DNA is present in these cells in the absence of any detectable gene expression (at least as detectable by ISH), as suggested by studies using in situPCR techniques (Ramakrishnan et al., 1994a,b; Mehta et al., 1995). Many host cell factors, such as the ability to activate viral genes (Mitchell, 1995), may be crucial to the selective vulnerability of cells to latent infection. Thus our ISH data may under-represent the actual distribution of HSV in these studies.
Another speculative factor accounting for the distribution pattern is based on the fact that there are populations of cells in the adult CNS that divide, migrate, and differentiate (e.g., subependymal cells) (Reynolds and Weiss, 1992; Gage et al., 1995). Infection of these cells will give a pattern unexpected by the simple issue of connectivity. This hypothesis will have to be addressed in the future, using marker studies.
The simplest interpretation of the LAT+ cell distribution data is that the different distribution patterns of LAT+cells reflect differences in the neuronal connectivity of these regions or access of virus to cells. In areas with LAT+ neurons, however, not all neurons were LAT+, and at the inoculation site (e.g., olfactory bulb and cerebellum), neurons often were not LAT+ (Table 1). Also, the level of LAT expression is variable among individual neurons in a given region (Figs. 3, 6, 7, 8). Thus, viral and neuronal factors may act synergistically or independently to determine the expression of the LATs, which may in turn indicate the selective vulnerability of subsets of CNS neurons to latent infection.
Latency in the CNS
There are several operational definitions of HSV latency, including (1) the ability to persist in the host in the absence of clinical manifestation; (2) the ability to reactivate and recover infectious virus from a latent state; (3) the presence of viral genome in the absence of viral antigens or RNA; and (4) the expression of LATs in the absence of lytic viral gene expression and proteins (Roizman and Sears, 1993). In these studies, we used the fourth definition as a criterion for latent HSV-1 infection. Although there are several good animal models for studying the mechanism of viral latency in the PNS (for review, see Fraser et al., 1991), the current models for studying viral latency in the CNS are of limited use because of the low number of CNS cells that are latently infected after peripheral or intracranial inoculation of wild-type viruses (Rock and Fraser, 1983;Deatly et al., 1988; Martin and Suzuki, 1989; Lynas et al., 1993). Latent viral DNA has been detected in the rostral CNS, but high levels of LAT expression were not detectable in other experimental models of latency (Rock and Fraser, 1983; Ramakrishnan et al., 1994a,b). In the model described here, we detected a large number of cells expressing the LATs by ISH in mice infected with strain 1716 but not with two other attenuated viruses. The ability of strain 1716 to form a latent infection in many types of neurons in the CNS after direct IC inoculation suggests that sensory neurons of the PNS are not unique with respect to the ability of HSV to form a latent infection and to produce the LATs. Because sensory neurons are more easily accessible than CNS neurons during a natural infection in man and experimental animals, additional studies of the model system described here will facilitate the elucidation of the mechanisms of latent CNS infection. In addition, this model is amenable to many neurobiological techniques for studying the physiology of acutely and latently infected neuronsin vivo.
Uses of neuroattenuated HSV
The use of neuroattenuated HSV-1 strains has been proposed for gene therapy and cancer therapy in the CNS (Kesari et al., 1995;Lawrence et al., 1995). This study suggests two important uses of strain 1716 as a vector for gene transfer and gene therapy studies. The ability of strain 1716 to form a latent infection in a large number of CNS cells after IC inoculation suggests its potential to transfer genes into postmitotic neurons of the adult mouse. This would be an efficient technique to transfer and express proteins in neurons in vivo to study function. Thus, it is necessary to determine whether different neuroattenuated strains replicate to high titers in the CNS, cause morbidity or death, or produce latent infection (Kesari et al., 1995). The long-term sequelae of latency in the CNS is an important facet of the HSV life cycle to study, especially in reference to the use of HSV as a vector in the CNS. In our studies of nude mice and immunocompetent mice of several different backgrounds, the animals survived for >6 months after IC inoculation without any signs of morbidity (Kesari et al., 1995) (our unpublished observations). Thus, these studies suggest that strain 1716 (and possibly other neuroattenuated viruses) has potential as a highly efficient gene transfer vector for CNS neurons for the treatment of diffuse CNS diseases.
Footnotes
This work was supported in part by grants from the Albert R. Taxin Brain Tumor Center, the National Institute of Neurological Disorders and Stroke (NS29390), the National Institute of Mental Health (MH10915), and the National Cancer Institute (CA-36245). We thank G. Cohen (Philadelphia) for FgDβ and D. Knipe (Boston) ford27-1.
Correspondence should be addressed to Nigel W. Fraser, Wistar Institute, Room 317, 3601 Spruce Street, Philadelphia, PA 19104.
REFERENCES
- 1.Adams RL, Springall DR, Levene MM, Bushell TE. The immunocytochemical detection of herpes simplex virus in cervical smears: a valuable technique for routine use. J Pathol. 1984;143:241–247. doi: 10.1002/path.1711430403. [DOI] [PubMed] [Google Scholar]
- 2.Anderson JR, Field HJ. The distribution of herpes simplex type 1 antigen in mouse central nervous system after different routes of inoculation. J Neurol Sci. 1983;60:181–195. doi: 10.1016/0022-510x(83)90061-8. [DOI] [PubMed] [Google Scholar]
- 3.Baringer JR, Pisani P. Herpes simplex virus genomes in human nervous system tissue analyzed by polymerase chain reaction. Ann Neurol. 1994;36:823–829. doi: 10.1002/ana.410360605. [DOI] [PubMed] [Google Scholar]
- 4.Barnett EM, Cassel MD, Perlman S. Two neurotropic viruses, herpes simplex virus type 1 and mouse hepatitis virus, spread along different neural pathways from the main olfactory bulb. Neuroscience. 1993;57:1007–1025. doi: 10.1016/0306-4522(93)90045-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boerman RH, Peters ACB, Bloem BR, Raap AK, Ploeg MVD. Spread of herpes simplex virus to the cerebrospinal fluid and the meninges in experimental mouse encephalitis. Acta Neuropathol. 1992;83:300–307. doi: 10.1007/BF00296793. [DOI] [PubMed] [Google Scholar]
- 6.Chou J, Roizman B. The γ134.5 gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programmed cell death in neuronal cells. Proc Natl Acad Sci USA. 1992;89:3266–3270. doi: 10.1073/pnas.89.8.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chou J, Roizman B. Herpes simplex virus 1 γ134.5 gene function, which blocks the host response to infection, maps in the homologous domain of the genes expressed during growth arrest and DNA damage. Proc Natl Acad Sci USA. 1994;91:5247–5251. doi: 10.1073/pnas.91.12.5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chou J, Kern ER, Whitley RJ, Roizman B. Mapping of herpes simplex virus neurovirulence to gamma 1 34.5, a gene nonessential for growth in culture. Science. 1990;250:1262–1266. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 9.Chou J, Chen J-J, Gross M, Roizman B. Association of an Mr 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF2α and premature shutoff of protein synthesis after infection with γ34.5-mutants of herpes simplex virus 1. Proc Natl Acad Sci USA. 1995;92:10516–10520. doi: 10.1073/pnas.92.23.10516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook ML, Stevens JG. Pathogenesis of herpetic neuritis and ganglionitis in mice: evidence for intra-axonal transport of infection. Infect Immun. 1973;7:272–288. doi: 10.1128/iai.7.2.272-288.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deatly AM, Spivack JG, Lavi E, O’Boyle DR, II, Fraser NW. Latent herpes simplex virus type 1 transcripts in peripheral and central nervous system tissues of mice map to similar regions of the viral genome. J Virol. 1988;62:749–756. doi: 10.1128/jvi.62.3.749-756.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraser NW, Lawrence NC, Wroblewska Z, Gilden DH, Koprowski H. Herpes simplex type I DNA in human brain tissue. Proc Natl Acad Sci USA. 1981;78:6461–6465. doi: 10.1073/pnas.78.10.6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fraser NW, Spivack JG, Wroblewska Z, Block T, Deshmane SL, Valyi-Nagy T, Natarajan R, Gesser R (1991) A review of the molecular mechanism of HSV-1 latency. Curr Eye Res 10[Suppl]:1–14. [DOI] [PubMed]
- 14.Fraser NW, Block TM, Spivack JG. The latency-associated transcripts of herpes simplex virus: RNA in search of function. Virology. 1992;191:1–8. doi: 10.1016/0042-6822(92)90160-q. [DOI] [PubMed] [Google Scholar]
- 15.Gage FH, Ray J, Fisher LJ. Isolation, characterization, and use of stem cells from the CNS. Annu Rev Neurosci. 1995;18:159–192. doi: 10.1146/annurev.ne.18.030195.001111. [DOI] [PubMed] [Google Scholar]
- 16.Johnson RT. Selective vulnerability of neural cells to viral infections. Brain. 1980;103:447–472. doi: 10.1093/brain/103.3.447. [DOI] [PubMed] [Google Scholar]
- 17.Kesari S, Randazzo BP, Valyi-Nagy T, Huang QS, Brown SM, MacLean AR, Lee VM-Y, Trojanowski JQ, Fraser NW. Therapy of experimental human brain tumors using a neuroattenuated herpes simplex virus mutant. Lab Invest. 1995;73:636–648. [PubMed] [Google Scholar]
- 18.Kosz-Vnenchak M, Jacobson J, Coen DM, Knipe DM. Evidence for a novel regulatory pathway for herpes simplex virus gene expression in trigeminal ganglion neurons. J Virol. 1993;67:5383–5393. doi: 10.1128/jvi.67.9.5383-5393.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kramer MF, Coen DM. Quantification of transcripts from the ICP4 and thymidine kinase genes in mouse ganglia latently infected with herpes simplex virus. J Virol. 1995;69:1389–1399. doi: 10.1128/jvi.69.3.1389-1399.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lawrence MS, Ho DY, Dash R, Sapolsky RM. Herpes simplex virus vectors overexpressing the glucose transporter gene protect against seizure-induced neuron loss. Proc Natl Acad Sci USA. 1995;92:7247–7251. doi: 10.1073/pnas.92.16.7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lynas C, Hill TJ, Maitland N, Love S. Latent infection with the MS strain of herpes simplex virus type 2 in the mouse following intracerebral inoculation. J Neurol Sci. 1993;120:107–114. doi: 10.1016/0022-510x(93)90033-u. [DOI] [PubMed] [Google Scholar]
- 22.MacLean AR, Ul-Fareed M, Robertson L, Harland J, Brown SM. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the “a” sequence. J Gen Virol. 1991;72:631–639. doi: 10.1099/0022-1317-72-3-631. [DOI] [PubMed] [Google Scholar]
- 23.Martin JR, Suzuki S. Targets of infection in a herpes simplex-reactivation model. Acta Neuropathologica. 1989;77:402–411. doi: 10.1007/BF00687375. [DOI] [PubMed] [Google Scholar]
- 24.McFarland DJ, Hotchin J. Contrasting patterns of virus spread and neuropathology following microinjection of herpes simplex virus into the hippocampus or cerebellum of mice. J Neurol Sci. 1987;79:255–265. doi: 10.1016/0022-510x(87)90233-4. [DOI] [PubMed] [Google Scholar]
- 25.McGeoch DJ, Barnett BC. Neurovirulence factor. Nature. 1991;353:609. doi: 10.1038/353609b0. [DOI] [PubMed] [Google Scholar]
- 26.McKie EA, Hope RG, Brown SM, MacLean AR. Characterization of the herpes simplex virus type 1 strain 17+ neurovirulence gene RL1 and its expression in a bacterial system. J Gen Virol. 1994;75:733–741. doi: 10.1099/0022-1317-75-4-733. [DOI] [PubMed] [Google Scholar]
- 27.Mehta A, Maggioncalda J, Bagasra O, Thikkavarapu S, Saikumari P, Valyi-Nagy T, Fraser N, Block T. In situ DNA PCR and RNA hybridization detection of herpes simplex virus sequences in trigeminal ganglia of latently infected mice. Virology. 1995;206:633–640. doi: 10.1016/s0042-6822(95)80080-8. [DOI] [PubMed] [Google Scholar]
- 28.Michell WJ. Neurons differentially control expression of a herpes simplex virus type 1 immediate-early promoter in transgenic mice. J Virol. 1995;69:7942–7950. doi: 10.1128/jvi.69.12.7942-7950.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neeley SP, Cross AJ, Crow TJ, Johnson JA, Taylor GR. Herpes simplex virus encephalitis. J Neurol Sci. 1985;71:325–337. doi: 10.1016/0022-510x(85)90071-1. [DOI] [PubMed] [Google Scholar]
- 30.Nicoll JAR, Love S, Kinrade E. Distribution of herpes simplex virus DNA in the brains of human long-term survivors of encephalitis. Neurosci Lett. 1993;157:215–218. doi: 10.1016/0304-3940(93)90740-c. [DOI] [PubMed] [Google Scholar]
- 31.Perry LJ, McGeoch DJ. The DNA sequences of the long repeat region and adjoining parts of the long unique region in the genome of herpes simplex virus type 1. J Gen Virol. 1988;69:2831–2846. doi: 10.1099/0022-1317-69-11-2831. [DOI] [PubMed] [Google Scholar]
- 32.Ramakrishnan R, Fink D, Jiang G, Desai P, Glorioso J, Levine M. Competitive quantitative PCR analysis of herpes simplex virus type 1 DNA and latency-associated transcript RNA in latently infected cells of the rat brain. J Virol. 1994a;68:1864–1873. doi: 10.1128/jvi.68.3.1864-1873.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramakrishnan R, Levine M, Fink D. PCR-based analysis of herpes simplex virus type 1 latency in the rat trigeminal ganglion established with a ribonucleotide reductase-deficient mutant. J Virol. 1994b;68:7083–7091. doi: 10.1128/jvi.68.11.7083-7091.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 35.Rice SA, Knipe DM. Genetic evidence for two distinct transactivation functions of the herpes simplex virus a protein ICP27. J Virol. 1990;64:1704–1715. doi: 10.1128/jvi.64.4.1704-1715.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rock DL, Fraser NW. Detection of HSV-1 genome in the central nervous system of latently infected mice. Nature. 1983;302:523–525. doi: 10.1038/302523a0. [DOI] [PubMed] [Google Scholar]
- 37.Roizman B, Kaplan LJ. Herpes simplex viruses, central nervous system, and encephalitis. In: Roos RP, editor. Molecular neurovirology. Humana; Totowa, NJ: 1992. pp. 3–23. [Google Scholar]
- 38.Roizman B, Sears AE. Herpes simplex viruses and their replication. In: Roizman B, Whitley RJ, Lopez C, editors. The human herpes viruses. Raven; New York: 1993. pp. 11–68. [Google Scholar]
- 39.Schmidbauer M, Budka H, Ambros P. Herpes simplex virus (HSV) DNA in microglial nodular brainstem encephalitis. J Neuropathol Exp Neurol. 1989;48:645–652. doi: 10.1097/00005072-198911000-00006. [DOI] [PubMed] [Google Scholar]
- 40.Schreiber SS, Baudry M. Selective neuronal vulnerability in the hippocampus: a role for gene expression. Trends Neurosci. 1995;18:446–451. doi: 10.1016/0166-2236(95)94495-q. [DOI] [PubMed] [Google Scholar]
- 41.Sekizawa T, Openshaw H. Encephalitis resulting from reactivation of latent herpes simplex virus in mice. J Virol. 1984;50:263–266. doi: 10.1128/jvi.50.1.263-266.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sodora DL, Eisenberg RJ, Cohen GH. Characterization of a recombinant herpes simplex virus which expresses a glycoprotein D lacking asparagine-linked oligosaccharides. J Virol. 1991;65:4432–4441. doi: 10.1128/jvi.65.8.4432-4441.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Speck PG, Simmons A. Divergent molecular pathway of productive and latent infection with a virulent strain of herpes simplex virus type 1. J Virol. 1991;65:4001–4005. doi: 10.1128/jvi.65.8.4001-4005.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Speck PG, Simmons A. Synchronous appearance of antigen-positive and latently infected neurons in spinal ganglia of mice infected with a virulent strain of herpes simplex virus. J Gen Virol. 1992;73:1281–1285. doi: 10.1099/0022-1317-73-5-1281. [DOI] [PubMed] [Google Scholar]
- 45.Spivack JG, Fraser NW. Detection of herpes simplex type 1 transcripts during latent infection in mice. J Virol. 1987;61:3841–3847. doi: 10.1128/jvi.61.12.3841-3847.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spivack JG, Fareed MU, Valyi-Nagy T, Nash TC, O’Keefe JS, Gesser RM, McKie EA, MacLean AR, Fraser NW, Brown SM. Replication, establishment of latent infection, expression of the latency-associated transcripts and explant reactivation of herpes simplex virus type 1 gamma 34.5 mutants in a mouse eye model. J Gen Virol. 1995;76:321–332. doi: 10.1099/0022-1317-76-2-321. [DOI] [PubMed] [Google Scholar]
- 47.Steiner I, Spivack JG, Deshmane SL, Ace CI, Preston CM, Fraser NW. A herpes simplex virus type 1 mutant containing a non-transinducing Vmw65 protein establishes latent infection in vivo in the absence of viral replication and reactivates efficiently from explanted trigeminal ganglia. J Virol. 1990;64:1630–1638. doi: 10.1128/jvi.64.4.1630-1638.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stroop WG, Schaefer DC. Production of encephalitis restricted to the temporal lobes by experimental reactivation of herpes simplex virus. J Infect Dis. 1986;153:721–731. doi: 10.1093/infdis/153.4.721. [DOI] [PubMed] [Google Scholar]
- 49.Tenser RB, Edris W, Hay K, De Galan B. Expression of herpes simplex virus type 2 latency associated transcript in neurons and nonneurons. J Virol. 1991;65:2745–2750. doi: 10.1128/jvi.65.5.2745-2750.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trojanowski J, Mantione R, Lee J, Seid D, You T, Inge L, Lee V. Neurons derived from a human teratocarcinoma cell line establishes molecular and structural polarity following transplantation into the rodent brain. Exp Neurol. 1993;122:283–294. doi: 10.1006/exnr.1993.1128. [DOI] [PubMed] [Google Scholar]
- 51.Ugolini G, Kuypers HGJM, Simmons A. Retrograde transneuronal transfer of herpes simplex virus type 1 (HSV1) from motor neurones. Brain Res. 1987;422:242–256. doi: 10.1016/0006-8993(87)90931-0. [DOI] [PubMed] [Google Scholar]
- 52.Valyi-Nagy T, Deshmane SL, Spivack JG, Steiner I, Ace CI, Preston CM, Fraser NW. Investigation of herpes simplex virus type 1 (HSV-1) gene expression and DNA synthesis during the establishment of latent infection by an HSV-1 variant, in 1814, that does not replicate in mouse trigeminal ganglia. J Gen Virol. 1991;72:641–649. doi: 10.1099/0022-1317-72-3-641. [DOI] [PubMed] [Google Scholar]
- 53.Valyi-Nagy T, Deshmane SL, Raengsakulrach B, Nicosia M, Gesser RM, Wysocka M, Dillner A, Fraser NW. Herpes simplex virus type 1 mutant strain in 1814 establishes a unique, slowly progressing infection in SCID mice. J Virol. 1992;66:7336–7345. doi: 10.1128/jvi.66.12.7336-7345.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valyi-Nagy T, Gesser RM, Raengsakulrach B, Deshmane SL, Randazzo BP, Dillner AJ, Fraser NW. A thymidine kinase-negative HSV-1 strain establishes a persistent infection in SCID mice that features uncontrolled peripheral replication but only marginal nervous system involvement. Virology. 1994;199:484–490. doi: 10.1006/viro.1994.1150. [DOI] [PubMed] [Google Scholar]
- 55.Whitley RJ, Lakeman F. Herpes simplex virus infections of the central nervous system: therapeutic and diagnostic considerations. Clin Infect Dis. 1995;20:414–420. doi: 10.1093/clinids/20.2.414. [DOI] [PubMed] [Google Scholar]
- 56.Whitley RJ, Kern ER, Chatterjee S, Choi J, Roizman B. Replication, establishment of latency, and induced reactivation of herpes simplex virus gamma 1 34.5 deletion mutants in rodent models. J Clin Invest. 1993;91:2837–2843. doi: 10.1172/JCI116527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yeh L, Schaffer P. A novel class of transcripts expressed with late kinetics in the absence of ICP4 spans the junction between the long and short segments of the herpes simplex virus type 1 genome. J Virol. 1993;67:7373–7382. doi: 10.1128/jvi.67.12.7373-7382.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]