

Abstract
The regulation of programmed cell death in the developing nervous system involves target-derived survival factors, afferent synaptic activity, and hormone- and cytokine-dependent signaling. Cultured immature cerebellar granule neurons die by apoptosis within several days in vitro unless maintained in depolarizing (high) concentrations of potassium (25 mmK+). Here we report that transforming growth factors (TGF)-β1, -β2, and -β3 accelerate apoptosis of these neurons when maintained in physiological (low) K+ medium (5 mm K+) as assessed by measures of viability, quantitative DNA fragmentation, and nuclear morphology. TGF-β-induced apoptosis of these neurons is not blocked by CNTF and LIF, cytokines that enhance neuronal survival when applied alone, or by IGF-I, which prevents apoptosis upon potassium withdrawal. In contrast, neurons that differentiate in high K+ medium for several days in vitroacquire resistance to TGF-β-mediated cell death. Granule neurons maintained in either low or high K+ medium produce latent, but not bioactive, TGF-β1 and -β2. Because neutralizing TGF-β antibodies fail to augment survival of low K+ neurons, the cerebellar neurons are apparently unable to activate latent TGF-β. Thus, apoptosis of low K+ neurons is not attributable to endogenous production of TGF-β. Taken together, our data suggest that TGF-β may limit the expansion of postmitotic neuronal precursor populations by promoting their apoptosis but may support survival of those neurons that have maturated, differentiated, and established supportive synaptic connectivity.
Keywords: apoptosis, cerebellum, depolarization, maturation-dependence, potassium, TGF-β
Cytokines produced in the CNS regulate immune effector mechanisms at a site that is shielded from the blood by the blood–brain barrier. However, local production of cytokines by brain parenchymal cells also may be essential for the development of both the nervous system and the maintenance of the state of differentiation and activation of neurons and glial cells. An example of such a dual system function is provided by transforming growth factor-β (TGF-β), a member of a multigene cytokine family (Massagué, 1990). TGF-β mediates immune regulatory functions in inflammatory processes (Fontana et al., 1984; Wrann et al., 1987) while functioning as a survival factor for embryonic motoneurons and dopaminergic and neonatal sensory neurons in vitro (Martinou et al., 1990; Chalazonitis et al., 1992; Poulsen et al., 1994; Krieglstein et al., 1995). TGF-β also promotes neurogenesis in cultures of hippocampal and olfactory neurons (Mahanthappa and Schwarting, 1993; Ishihara et al., 1994).In vivo, TGF-β exerts neurotrophic effects on lesioned neurons (Prehn et al., 1993a), rescues sympathetic neurons from death after destruction of the target cells (Blottner et al., 1996), and protects dopaminergic neurons fromN-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity and cortical neurons from sodium cyanide or glutamate (Prehn et al., 1993b;Krieglstein et al., 1995).
Three different isoforms of TGF-β with similar biological activities have been described in mammalian cells. As shown for TGF-β1, a biologically inactive form termed latent TGF-β complex is composed of three components, namely the mature TGF-β, the TGF-β latency-associated peptide, and the latent TGF-β binding protein (Kanzaki et al., 1990). In vitroactivation of TGF-β1 occurs by dissociation of the mature TGF-β1 from the complex. Thein vivo mechanisms of activation are not yet elucidated.
In light of their effects on cultured neurons, it is of relevance that TGF-β is produced in the CNS. Mouse embryos and adult rats express TGF-β1 mRNA in the meninges and the choroid plexus. Both postnatally and in the adults, TGF-β2 and -β3 mRNA are present in various regions, including choroid plexus, hippocampus, dentate gyrus, cerebellar granule neurons, and Purkinje cells (Heine et al., 1987; Unsicker et al., 1991; Constam et al., 1992). TGF-β receptor type I is expressed in the sensory retina and in the marginal zone of the brain (Iseki et al., 1995).
Cerebellar granule neurons are a well targeted model system for the study of neuronal apoptosis because these neurons survive for weeks when maintained in depolarizing concentrations of K+, but undergo apoptosis when cultured in physiological (low K+) conditions. Low K+ neurons express only low levels of glutaminase activity, few functional NMDA receptors, and few synapses, and thus are thought to reflect immature cells. In the present study, we report the following: (1) that cerebellar granule neurons maintained in low K+ medium undergo premature apoptosis when exposed to TGF-β; (2) that TGF-β-induced neuronal apoptosis is blocked by depolarization but not by cytokines such as ciliary neurotrophic factor (CNTF), human leukemia inhibitory factor (LIF), and recombinant human insulin-like growth factor I (IGF-I); and (3) that neurons maintained in low or high K+ medium produce TGF-β in latent bioinactive form, which apparently has no effect on neuronal survival.
MATERIALS AND METHODS
Materials. Recombinant rat CNTF and human LIF were purchased from Pepro Tech (Rocky Hill, NJ); DNase I, IGF-I, insulin, RNase A, terminal transferase, transferrin, biotinylated deoxyuridine triphosphate (dUTP), streptavidin-alkaline phosphatase conjugate, nitroblue tetrazolium chloride, 5-bromo-4-chloro-3-indolyl phosphate, and the cytotoxicity detection kit (for the measurement of lactate dehydrogenase) were from Boehringer Mannheim (Rotkreuz, Switzerland); recombinant human TGF-β1, human TGF-β2, human TGF-β3, polyclonal rabbit anti-TGF-β2, polyclonal rabbit pan-specific anti-TGF-β antibody, as well as both human TGF-β1 and TGF-β2immunoassay kits, were from R&D Systems (Abingdon, UK); the dishes, micro-well plates, and chamber slides were from Nunc (Roskilde, Denmark), and the medium and medium supplements from Gibco (Life Technologies, Basel, Switzerland); cytosine-1β-arabinofuranoside came from Fluka, bovine serum albumin from Serva (Buchs, Switzerland), and all other chemicals, including aprotinin, fluorescein diacetate, Hoechst 33258, rabbit IgG isotype control, poly-l-lysine sodium selenite,l-thyroxin, trypsin, and trypsin inhibitor were from Sigma (Buchs, Switzerland).
Cell culture. Cultures of rat cerebellar granule neurons were prepared from 7-d-old rat pups as described previously (Novelli et al., 1988; Marini and Paul, 1992; Weller et al., 1992). The cells were seeded at a density of 2.75 × 105cells/cm2 on poly-l-lysine (10 μg/ml)-coated plastic surfaces (either 35 mm dishes or 96-well plates) for survival, TGF-β assays, and fluorometric quantification of DNA fragmentation. Multichamber glass slides were used for staining of fixed-cell preparations. The serum-containing culture medium consisted of basal medium Eagle (BME) supplemented with 10% fetal calf serum, 2 mml-glutamine, and gentamycin (50 μg/ml). Cells maintained in depolarizing conditions were supplemented with 20 mmK+ at the time of seeding to achieve a final concentration of 25 mm. All serum-containing cultures were supplied with 10 μm cytosine arabinofuranoside (Ara-C) on day 1 in vitro (DIV1) to arrest the growth of non-neuronal cells. Ara-C was readded every tenth dayin vitro for long-term experiments. As shown by immunofluorescence analysis, >90% of the cells are neurons because they stain positive with the specific anti-neuronal filament antibody NF160, and ∼8% of the cells express glial fibrillary acidic protein, a marker for astrocytes (U. Malipiero and A. Fontana, personal communication). This is in accordance with previous reports (Kingsbury et al., 1985; Nicoletti et al., 1986). The serum-free culture medium (X1) consisted of BME supplemented with bovine serum albumin (1 mg/ml), aprotinin (1 μg/ml), glucose (2.5 mg/ml), 2 mml-glutamine, insulin (10 μg/ml), penicillin (50 IU/ml), streptomycin (50 μg/ml), 30 nm sodium selenite, transferrin (100 μg/ml), and 4 nml-thyroxin (Fischer, 1982; Piani et al., 1991). The cultures were maintained in the same medium throughout the experiment and were fed with 5 mm glucose on DIV7 and every third day thereafter.
Assessment of viability and apoptosis. The cultures were monitored daily by phase-contrast microscopy. Neuronal viability was assessed by: (1) the ability of the cells to cleave and retain fluorescein diacetate (FDA) staining in their cytoplasm; and (2) the release of lactate dehydrogenase (LDH) into the culture medium, which indicates loss of membrane integrity and is an indicator of cell death. Briefly, the cells were labeled with FDA in Locke’s buffer [containing (in mm): 154 NaCl, 5.6 KCl, 2.3 CaCl2, 1 MgCl2, 3.6 NaHCO3, 5 HEPES, 20 glucose] for 10 min at 37°C and subsequently lysed in ice-cold 0.2% Triton X-100/10 mm EDTA/10 mm Tris-HCl. Fluorescence was measured with a Cytofluor 2350 spectrofluorometer (Millipore, Bedford, MA) at an excitation wavelength of 485 nm and an emission wavelength of 530 nm (Didier et al., 1990). LDH activity was measured as optical density at 492 nm and expressed according to the manufacturer’s directions: specific LDH release (%) = [experimental value − spontaneous release/maximum release − spontaneous release] × 100. Spontaneous release is defined as the amount of LDH released from untreated 25 mm K+cultures and the maximum release obtained after exposure of high K+-grown neurons to Triton X-100, 0.1% for 10 min at 37°C. Percentages of specific LDH release were converted into percentages of rescue [100 − specific release (%)], and the values obtained for the 5 mm K+controls were set to 0% rescue and the 25 mmK+ control values to 100%.
Apoptotic cell death was assessed by Hoechst 33258 nuclear staining to detect typical chromatin changes, quantitative determination of DNA fragmentation by fluorometry, and in situ DNA end labeling for the detection of DNA breaks in single cells (Gavrieli et al., 1992;Weller et al., 1994a,b; Yan et al., 1994; Weller et al., 1995a,b). Briefly, nuclear morphology and quantitative estimates of DNA fragmentation were investigated by Hoechst 33258 fluorescence at 460 nm in 5 mm HEPES/100 mm NaCl, pH 7.0, at a dye concentration of (5 μg/ml) for chromatin staining and (0.5 μg/ml) for quantification of DNA fragmentation. To ensure proper evaluation of the total amount of fragmented DNA, detached cells were harvested and centrifuged at 4000 rpm for 10 min. The recovered pellet was lysed, pooled with the lysate of adherents cells, and further processed. For in situ labeling of DNA breaks, the reaction mixture consisted of 0.25 units/μl terminal transferase and 20 μm biotin-16-dUTP in terminal deoxytransferase buffer (30 mm Tris-HCl, pH 7.2, 140 mm sodium cacodylate, 1 mm cobalt chloride). Streptavidin-alkaline phosphatase conjugate (1:500) in 100 mmTris-HCl/50 mm NaCl, pH 7.5, was chosen as a detection system, and as a substrate, 0.41 mmnitroblue tetrazolium chloride and 0.38 mm5-bromo-4-chloro-3-indolyl phosphate in 200 mmTris-HCl/10 mm MgCl2, pH 9.5, were chosen. As a negative control, cobalt chloride was omitted.
TGF-β assays. To assess the effects of TGF-β1, -β2, and -β3 on survival of granule neurons cultured in either high or low K+ medium, in serum-free as well as in serum-containing medium, the cytokines were added to the cultures on DIV1, readded on DIV4 at the beginning of the critical stimulus-dependent survival time for cerebellar granule neuronsin vitro (Gallo et al., 1987), and again on DIV7 at the time of feeding, and let throughout time in culture. When challenging the proapoptotic effect of TGF-β against either CNTF, LIF, or IGF-I, these cytokines were added simultaneously with TGF-β on DIV4 and a second time on DIV7. Commercial human TGF-β1and TGF-β2 immunoassays (ELISA) were used to test the production of TGF-β by cerebellar granule neurons. As described previously (Constam et al., 1992), latent TGF-β in the cell-free conditioned medium was activated by acid treatment in 0.12 m HCl for 1 hr at room temperature, followed by neutralization in 0.025 m Na-HEPES/0.12m NaOH, pH 7.0. The background level of TGF-β was determined in parallel in the culture medium and subtracted from samples of conditioned medium. In experiments investigating the effects of anti-TGF-β neutralizing antibodies on cell survival, the anti-TGF-β2 antibody, the pan-specific anti-TGF-β antibody, or the isotype control was administered at the time of seeding and readded every third day in culture, on DIV3, DIV6, and DIV9, until death of the untreated controls had occurred.
Statistics. EC50 values for TGF-β-mediated induction of apoptosis were determined by linear regression analysis. Individual data were compared by Scheffe’s F-test; multiple drug treatments were analyzed by two-way ANOVA and are presented as mean ± SEM. Unless indicated otherwise, data are representative of experiments run on triplicate culture dishes and repeated with at least two different batches of cells with similar results.
RESULTS
TGF-β1, -β2, and -β3impair the survival of immature cerebellar granule neurons grown in low K+ medium
Morphological differentiation began with neurite sprouting in either 5 or 25 mm K+ medium during the first day in culture. Neurons that had differentiated in high K+ survived for more than 4 weeks in culture, provided they were regularly supplied with 5 mm glucose. In contrast, cerebellar granule neurons grown for 5–6 d in low K+ medium displayed morphological and biochemical signs of apoptotic cell death, including reduction in FDA esterase activity, enhanced LDH release, and DNA fragmentation (Fig. 1A–C).
Fig. 1.

Depolarizing concentrations of K+ promote survival of cerebellar granule neurons by preventing apoptosis. Granule neurons were cultured as described in serum-containing medium either in 5 or 25 mmK+. A, Viability kinetics over time in culture, measured by FDA staining. B, Kinetics of LDH activity released in the culture medium, normalized to the value obtained on DIV3 for the 25 mmK+ control culture. C, Quantitative DNA fragmentation (% fragmented vs total DNA) measured by Hoechst 33258 (0.5 μg/ml) fluorometry is not normalized to control. Bars represent mean ± SEM of three independent determinations at *p < 0.05 versus respective high K+controls by ANOVA and Scheffe’s F-test.
To analyze the effect of TGF-β on survival of cerebellar granule cells, the cytokine (1 ng/ml) was added 24 hr after seeding to cultures maintained in low K+. As shown in Figure2, TGF-β2 interferes with neuronal survival. The proapoptotic effect became apparent on microscopic examination on DIV7, when isolated patches of dead cells emerged throughout the culture dish (Fig.3D), and was completed by DIV8 (Fig.3F) as attested by FDA staining and LDH release (Fig.2A,B). That is 24–48 hr before the time at which granule neurons undergo spontaneous apoptotic cell death when cultured at low potassium concentrations in the absence of TGF-β (Figs. 2 and3G). The TGF-β2-induced impairment of survival of cerebellar granule cells was concentration-dependent and was also seen when TGF-β1 and -β3 were added to the cultures. There was no significant difference between the EC50 values for the three TGF-β isoforms, which were 2.24 ± 0.17 ng/ml (n = 6), 2.14 ± 0.09 ng/ml (n = 6), and 1.87 ± 0.05 ng/ml (n = 6) for TGF-β1, -β2, and -β3, respectively (F-test; p = 0.03). The lower the concentrations of TGF-β added, the longer the time required for the induction of neuronal death. At concentrations of TGF-β exceeding 1 ng/ml, cell death occurred 48 hr before the death of untreated low K+ neurons. Even the lowest concentration tested (0.1 pg/ml) failed to improve survival. In addition, it was observed that a single application of TGF-β at the time of seeding or later, up to DIV4, accelerated apoptosis with the same kinetics. Furthermore, when treating the granule neurons with TGF-β2(10 ng/ml) at only one time point, on DIV7, a shift of cell death to a few hours (8 hr) before death of the untreated controls was noted.
Fig. 2.
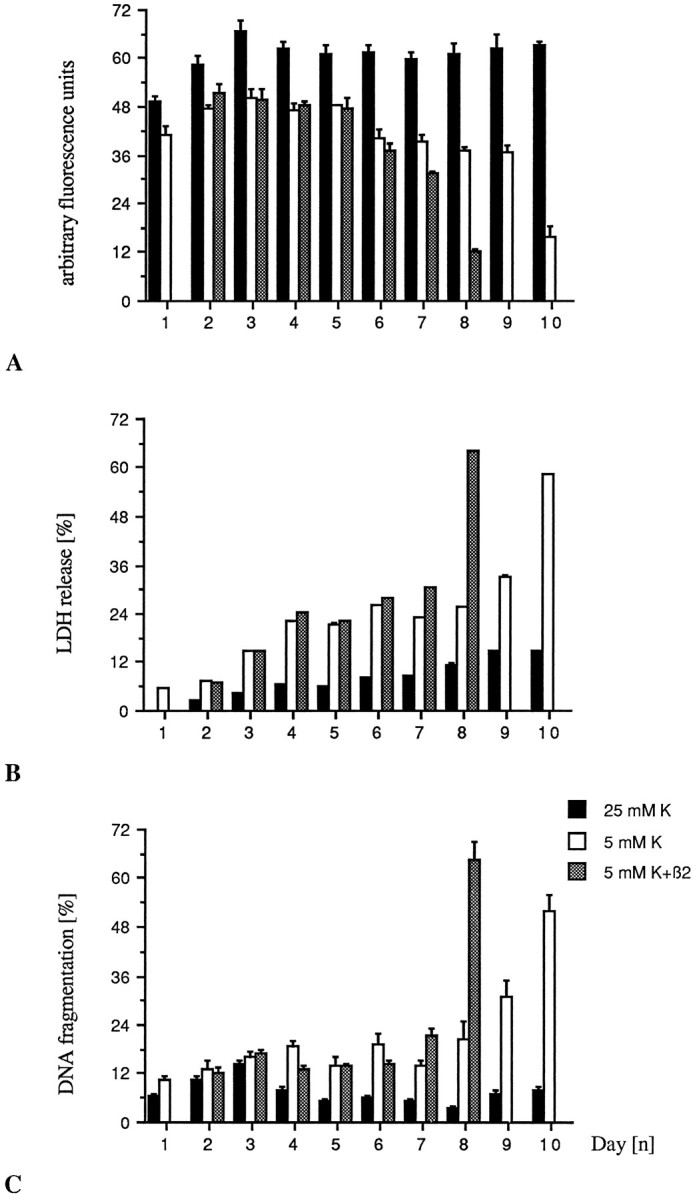
TGF-β induces premature cell death in granule neurons grown in 5 mm K+. Cells were cultured as described in Materials and Methods. TGF-β2 (1 ng/ml) additions were made on DIV1, DIV4, and again on DIV7. A, Viability as assessed by FDA staining. B, Specific LDH release normalized to the value obtained for the 25 mm K+control cultures at DIV1. C, DNA fragmentation as described. Bars represent mean ± SEM of three independent determinations.
Fig. 3.
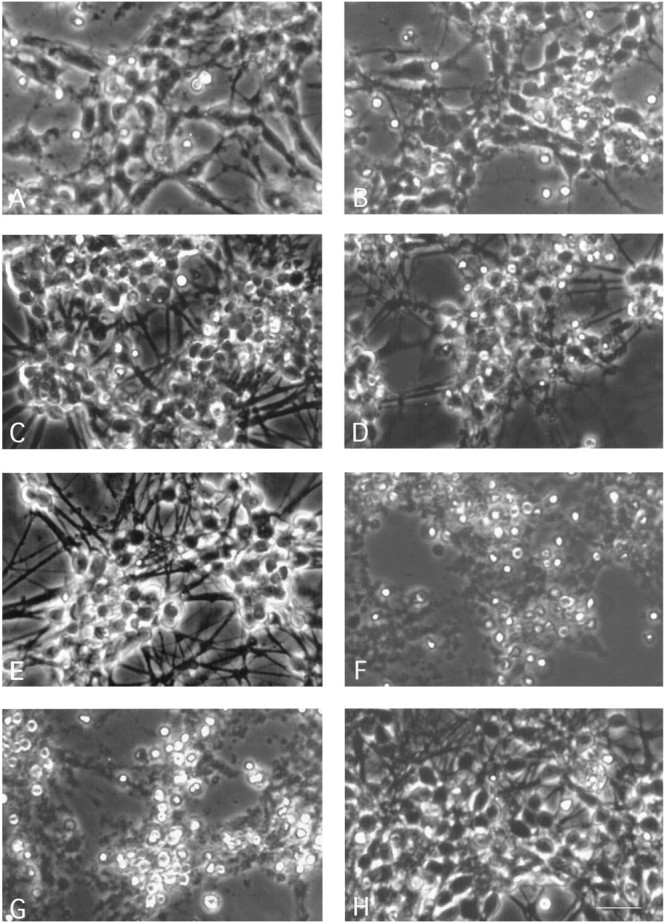
Acceleration of apoptosis by TGF-β2 becomes apparent 24 hr before death of the untreated control cultures maintained in low K+. Phase-contrast microscopy shows that untreated low K+ controls (A) and TGF-β2 (1 ng/ml)-treated cultures (B) are indistinguishable in the first 6 d in vitro. By DIV7, the TGF-β2-treated neurons (D) show patches of dead cells throughout the culture, whereas untreated controls (C) display the macroscopic features of homogeneous and progressive apoptotic degeneration: appearance of apoptotic cell bodies, reduced cell density, and a thinning of the neurite network. On DIV8, TGF-β2-treated neurons (F) are dead compared with untreated low K+ controls (E). On DIV10, apoptotic cell death is completed in low K+ controls (G) compared with neurons maintained in high K+ (H). Magnification, 580×; scale bar (shown in H), 20 μm.
Survival impairment of immature cerebellar granule neurons exposed to TGF-β is attributable to accelerated apoptosis
To characterize the type of cell death induced by TGF-β, we daily monitored cellular morphology of cultures exposed to TGF-β2 under phase-contrast microscopy and quantified DNA fragmentation by Hoechst 33258 fluorometry. Nuclear morphology was investigated on DIV1, DIV3, DIV5, DIV7, and DIV9 by Hoechst staining. In parallel, DNA breaks were detected by in situ DNA end labeling. Increase of DNA fragmentation with time in culture is a characteristic feature of cerebellar granule neurons maintained in low K+ (Fig. 1C) compared with neurons grown in high K+. Phase-contrast optics revealed that the TGF-β-treated low K+ cerebellar neurons displayed morphological features of apoptotic cell death, namely, cell shrinkage and a concomitant appearance of highly refringent apoptotic bodies (Fig.3D). Progressive accumulation of DNA breaks, chromatin condensation, and nuclear fragmentation were detected in low K+ cultures (Fig.4A,C) compared with high K+-grown cultures (Fig. 4E,G). At the latest time point investigated, on DIV9, an increased number of single neurons staining for DNA breaks could be observed in TGF-β-treated cultures compared with control cultures (Fig. 4A,B). Similarly, an increase of condensed nuclei in the TGF-β-treated neurons was clearly visualized after staining with Hoechst 33258 on DIV9, compared with untreated low K+ controls (Fig. 4C,D). In contrast to low K+conditions, in situ end DNA labeling of neurons maintained in high K+ showed no significant increase, and chromatin condensation as revealed by Hoechst staining was scarcely detected (Fig. 4E,G). No quantitative difference could be seen between TGF-β-treated high K+ neurons and untreated controls (Fig. 4F,H). Thus, based on cellular morphology, chromatin condensation, and detection of DNA breaks in single cells, the survival impairment observed after exposure of the granule neurons to TGF-β can be attributed to an acceleration of the apoptotic process.
Fig. 4.
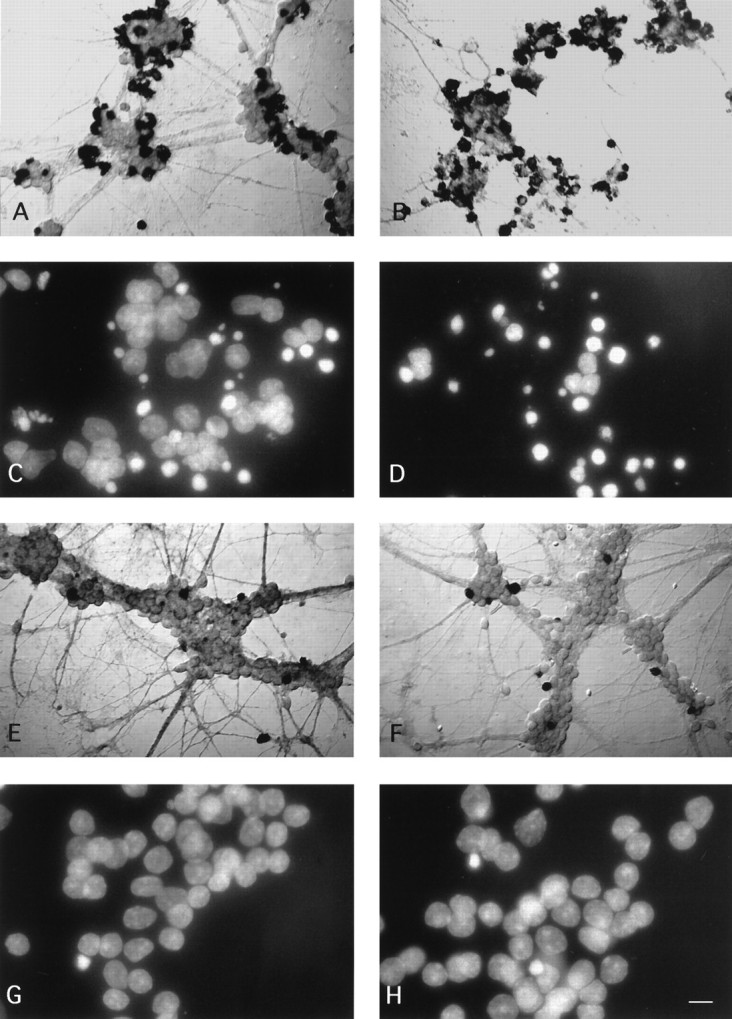
TGF-β accelerates 5 mm K+-mediated apoptotic cell death of cerebellar granule neurons in vitro. Granule neurons were cultured and treated as described in Materials and Methods. All neuron preparations presented here were fixed and processed on DIV9. Nomarsky optics of cultures stained for in situ DNA end breaks shows that there is substantially more DNA fragmentation in low K+ than in high K+-grown neurons. The percentages of stained cells increase in low K+-grown neurons exposed to TGF-β2, whereas there is no difference between treated and untreated high K+ cultures.A, Low K+ control; B, low K+ exposed to TGF-β2 (1 ng/ml); E, high K+ control;F, high K+ exposed to TGF-β2 (1 ng/ml). For A andB, magnification is 480×; scale bar (shown inH), 13 μm. For E and F, magnification is 320×; scale bar (shown in H), 20 μm. Visualization under UV illumination of nuclei stained with the fluorescent dye Hoechst 33258 (5 μg/ml) reveals more condensed chromatin in low K+ than in high K+ culture conditions. TGF-β2 accelerates the rate of chromatin condensation in low K+-grown neurons, but it does not affect neurons maintained in high K+ culture conditions. C, Low K+ control;D, low K+ exposed to TGF-β2 (1 ng/ml); G, high K+ control; H, high K+ exposed to TGF-β2 (1 ng/ml). Magnification, 1100×; scale bar (shown in H), 6 μm.
Depolarized cerebellar granule neurons are resistant to the proapoptotic effects of TGF-β
As outlined above, the survival of cerebellar granule cells is greatly prolonged in the presence of depolarizing concentrations of potassium (25 mm). In these conditions, none of the three isoforms of TGF-β negatively modulated neuronal survival (Fig. 5A,B) at any concentration tested and all time points assessed over 4 weeks in culture. The protection afforded by high K+-induced depolarization was not dependent on the presence of serum. Twenty-five millimolar K+serum-free (X1) and serum-containing cultures were set in parallel and exposed to TGF-β2 (10 ng/ml) on DIV1, DIV4, and again on DIV7. At early time points such as DIV7 and DIV10, TGF-β2-treated and untreated cultures in both culture conditions did not display any difference in regard to survival (data not shown). When maintaining high K+-grown neurons in a serum-containing medium over 4 weeks in vitro, no significant difference could be observed either between TGF-β2-treated and untreated cultures (data not shown).
Fig. 5.

Granule neurons grown in 25 mm K+ are resistant to the proapoptotic effects of all three TGF-β isoforms. The neurons grown in either 5 or 25 mm K+conditions were exposed on DIV1, DIV4, and again on DIV7 to 10 ng/ml of either TGF-β1, TGF-β2, or TGF-β3. Neuronal survival as assessed by FDA staining (A) and LDH release (B) was measured upon death of the TGF-β3-treated low K+ cultures 36 hr before death of the untreated low K+ controls, whereas degeneration in either the TGF-β1- or TGF-β2-treated neurons was not yet completed. As determined by linear regression from the concentration–dependence curves, TGF-β3 scores the highest biological activity of the three isoforms with an EC50 = 1.87 ng/ml. Bars represent mean ± SEM of quadruplicate at *p < 0.05 versus respective 25 mmK+ controls by ANOVA and Scheffe’s F-test.
CNTF, LIF, and IGF-I do not antagonize the proapoptotic effect of TGF-β on the survival of immature low K+ cerebellar granule neurons
IGF-I has recently been claimed to block apoptosis after potassium withdrawal of cerebellar granule neurons that have differentiated in high K+ (D’Mello et al., 1993). We found that the neuropoietic cytokines CNTF and LIF delay apoptotic cell death of the granule neurons when maintained in culture in low K+ concentrations (Fig. 6). However, when CNTF (10 ng/ml), LIF (10 ng/ml), and IGF-I (25 ng/ml) were administered to low K+-grown neurons together with TGF-β2 (1 ng/ml) on DIV4 and DIV7, the cultures exposed to TGF-β2 underwent premature cell death regardless of cotreatment with CNTF, LIF, or IGF-I (Fig. 7A,B). Thus, TGF-β-induced apoptosis is dominant to the survival-promoting properties of CNTF and LIF.
Fig. 6.

CNTF and LIF, but not IGF-I, delay cell death in low K+ medium. Cultures were treated with the factors on DIV1, DIV4, and DIV7. Neuronal viability was assessed by LDH release on DIV3, DIV6, DIV8, and every day thereafter until death of the CNTF- and LIF-treated cultures occurred. IGF-I-treated cultures died at the same time as the untreated low K+controls on DIV10. CNTF and LIF were found to prolong survival over 2 din vitro. Neuronal survival is expressed as 100 − LDH release [%]. LDH values were calculated as described in Materials and Methods and normalized to the value obtained for the 25 mm K+ control on DIV3, set as 0% release. Bars represent mean ± SEM of two independent determinations.
Fig. 7.

CNTF, LIF, and IGF-I do not reverse the proapoptotic effect of TGF-β. Low K+-grown neurons were treated on DIV4 and DIV7 simultaneously with TGF-β2 (1 ng/ml) and one of the following cytokines: CNTF (10 ng/ml), LIF (10 ng/ml), or IGF-I (25 ng/ml). Assessment of neuronal survival by FDA staining (A) and measurement of LDH release (B) were performed upon death of the cultures exposed to TGF-β2, which occurred, regardless of the factor added, on DIV9, i.e., 24 hr before death of the untreated low K+ controls. At that time point (DIV9), the survival-promoting effect exerted by CNTF and LIF in the absence of TGF-β2 and measured in terms of LDH release represents an increase of viability of 13.7 and 7.9% for CNTF and LIF, respectively, over untreated control. LDH values were normalized to the value obtained for the untreated 25 mm K+ control, set as 0% release. Bars represent mean of triplicate ± SEM of a representative experiment.
Latent TGF-β produced by cerebellar granule neurons lacks autocrine effects
Previous work from our laboratory has shown that latent TGF-β2 is produced by cerebellar granule neurons maintained in low K+ serum-free culture conditions (Constam et al., 1994). In the present serum-containing experimental setting, we found production of latent TGF-β1 and TGF-β2 by either low or high K+-grown neurons (Fig.8). There was significant production of TGF-β1 regardless of the potassium concentration, and the maximum of secretion occurred during the first day in vitro. In contrast, secretion of TGF-β2 maintains over time in vitro, and high K+ neurons produce slightly more TGF-β2 than low K+neurons. Neither TGF-β1 nor TGF-β2 could be detected before transient acidification of the cell supernatants, which indicates presence only of the latent, but not of the mature, biologically active TGF-β. Because a biological effect of the TGF-β precursor has been reported (Oberhammer et al., 1991), it has been assumed that the physiological mechanisms that activate TGF-β might function both in vivoand in vitro (Lucas et al., 1990). We examined then whether the induction of apoptosis in low K+ could relate to neuron-derived TGF-β acting in an autocrine or paracrine way. Neutralizing antibodies to TGF-β2 (10 μg/ml) and to TGF-β1, -β2, and -β3 (pan-specific) (10 μg/ml) were administered to low K+-grown neurons. Survey of neuronal viability, using FDA staining and measurement of LDH release, failed to reveal any specific promotion of survival of either antibody (Fig. 9A,B).
Fig. 8.

Cerebellar granule neurons maintained in either high or low K+ medium secrete latent TGF-β1 and TGF-β2. Cell-free conditioned media were transiently acidified as described in Materials and Methods, and both activated and nonactivated media were tested in triplicate for TGF-β1 and TGF-β2 in a quantitative sandwich enzyme immunoassay at the respective dilutions of 1:7 and 1:4. The detection limit of both immunoassays does not allow quantification of concentrations <60 pg/ml. A, Neurons grown in either low or high K+ concentrations produce equal amounts of latent TGF-β1, mostly in the first 24 hr of culture, with a slight increase over time in vitro. Naturally active TGF-β1 was not detected by the present technique. B, Neurons grown in either low or high K+ culture conditions release latent TGF-β2 with a continuous production over time in culture and a major contribution by high K+neurons. Naturally active TGF-β2 also was not detected.
Fig. 9.

Antibodies to TGF-β do not prevent apoptotic cell death of cerebellar granule neurons. The neurons were set in culture in either 5 or 25 mmK+, as described. Upon seeding on DIV0 and every third day thereafter, on DIV3, DIV6, and DIV9, the 5 mm K+-grown neurons were exposed to either an anti-TGF-β2 (10 μg/ml) antibody (a-β2), a pan-specific anti-TGF-β (10 μg/ml) antibody (a-πβ), or an isotype IgG control (10 μg/ml). Viability checkings by FDA staining (A) and measurement of LDH release (B) were performed every third day. Bars represent mean of six different wells ± SEM of a representative experiment.
DISCUSSION
Approximately half of all neuronal cells are eliminated during the normal development and maturation of the mammalian CNS (Johnson and Deckwerth, 1993; Raff et al., 1993). The key regulatory mechanisms that control programmed neuronal death appear to involve target-derived growth factors. Deprivation of those factors leads to apoptosis. Another pathway of induction of apoptosis may involve direct actions of cytokines on neurons. Both CNTF and LIF promote apoptosis of sympathetic neurons in vitro (Kessler et al., 1993).
In the present study, we found that TGF-β1, TGF-β2, and TGF-β3 precipitate apoptosis of cerebellar granule neurons maintained low K+ culture conditions. The first detectable signs of apoptosis were observed with a significant delay after TGF-β exposure, that is, 24 hr before death. The observation that TGF-β applied, on the latest, on DIV4 accelerates apoptosis with the same kinetics as when the exposure starts at the time of seeding, indicates that TGF-β exerts its effect from DIV5 onward, at a time when the apoptotic process is already at work. The mechanisms underlying the proapoptotic effects of TGF-β on immature low K+ granule neurons are not yet understood. It is of note that the proapoptotic effects of TGF-β on immature low K+ granule neurons strikingly contrast with its effects on mature high K+neurons. TGF-β fails to induce apoptosis of high K+ cultured neurons.
Depolarizing concentrations of potassium are thought to mimic in vitro the first synaptic afferences received in vivo by the cerebellar granule neurons from the mossy fibers. In vitro high K+ is known to promote differentiation in cerebellar granule neurons, measured in terms of K+-evoked glutamate release (Gallo et al., 1982;Didier et al., 1989), elevated glutaminase activity (Moran and Patel, 1989), establishment of mature synapses (Didier et al., 1992), and regulation of the functional expression of glutamate receptors (Balàsz et al., 1992; Resink et al., 1994) and of glucose transporters (Maher and Simpson, 1994). In cortical neurons, the survival-promoting effect induced by K+depolarization is attributable to the enhanced expression of brain-derived neurotrophic factor mRNA (Ghosh et al., 1994). The K+ effects are mediated through influx of Ca2+ into the cytosol, and depolarization results in a fundamental alteration in Ca2+ homeostatic mechanisms (Tymianski et al., 1994). But bioelectric activity per se is not the critical factor; the effect is not even specific for K+ (Gallo et al., 1987). It appears that a rise in [Ca2+]i up to a critical level is necessary and sufficient to maintain neuronal survival (Pearson et al., 1992). Moreover, high K+ specifically stimulates the oxidative metabolism (Erecinska et al., 1991; Peng et al., 1994).
TGF-β2 and TGF-β3suppress the ability of an embryonic chick eye tissue extract to promote survival but do not interfere with the neurotrophic effect of several purified survival factors such as CNTF, basic fibroblast growth factor, and nerve growth factor (Flanders et al., 1991). In our paradigm, however, it is unlikely that TGF-β interferes with production or effect of neurotrophic factors, because TGF-β does not impair viability of the low K+ granule neurons during the first 6 d in culture, and the proapoptotic effect in low K+ conditions cannot be overcome by the neurotrophins CNTF and LIF, which enhance neuronal survival when applied alone (Fig. 6).
Induction of an apoptotic type of cell death by TGF-β has been documented for other cell types (Weller and Fontana, 1996), including different types of transformed cell lines (Lin and Chou, 1992; Lotem and Sachs, 1992; Yanagihara and Tsumuraya, 1992), T and B lymphocytes (Weller et al., 1994a; Lømo et al., 1995), and hepatocytes in vitro and in vivo (Oberhammer et al., 1992). This appears to be the first instance of TGF-β-mediated apoptosis in postmitotic cells.
Both TGF-β1 and -β2 are produced by cerebellar granule neurons in vitro in their latent form. However, no correlation can be drawn between the secretion kinetics of TGF-β and the viability of the neurons. The amounts of endogenous latent TGF-β accumulating over time in culture yields on DIV9 concentrations of ≤500 pg/ml that match those of exogenous active TGF-β required for the induction of apoptosis. Although some target cells of TGF-β appear to be able to activate latent TGF-β in vitro (Lucas et al., 1990), this is not the case in cerebellar granule cell cultures because neutralizing antibodies to mature bioactive TGF-β fail to delay apoptosis of neurons maintained in low K+. In vivo, however, other cerebellar cell types may provide the proteases required for the activation of TGF-β.
Franklin and Johnson (1992) postulated in their “Ca2+ set-point” hypothesis the in vivo existence of four steady-state levels of [Ca2+]i that affect neuronal survival and neurotrophic factor dependence. In the first set-point, levels of [Ca2+]i are too low to support essential Ca2+-dependent pathways and cause neuronal death. Such a mechanism may lie behind the proapoptotic effect of TGF-β observed in low K+ neurons. The binding protein that is associated with latent TGF-β displays several binding sites for Ca2+ (Kanzaki et al., 1990). TGF-β is able to regulate the functional expression of intracellular ryanodine-sensitive Ca2+ channels (Giannini et al., 1992) and thereby to control Ca2+ release mechanisms (Neylon et al., 1994). TGF-β prevents Ca2+ overloading in rat hippocampal neurons in response to NMDA and Ca2+ ionophores (Prehn et al., 1994). Recent work by Baffy et al. (1995) established that modulation of the mitogenic effect of PDGF by TGF-β involves the inhibition of the intracellular Ca2+ mobilization induced by PDGF. Therefore, it is tempting to speculate that TGF-β may act by sequestering Ca2+ into a subset of internal Ca2+ stores and accelerate apoptotic death of granule neurons maintained in low K+ by lowering the [Ca2+]i.
The development of the cerebellum has been characterized extensivelyin vivo. There is a well documented cell loss within the maturing granule cell layer during the first weeks of postnatal life. Large numbers of granule neurons in both the mitotic and postmitotic regions of the external granule layer (EGL) undergo DNA fragmentation, with a maximum at postnatal day 7 (Wood et al., 1993). Apoptosis is hypothesized to match the population of granule neurons to the number of Purkinje cells, their target-cells. To comply with the hypothesis that TGF-β may play a role in the regulation of apoptosis of cerebellar neurons during development, these cytokines have to be produced in a time- and location-dependent way. Immature postnatal cerebellar granule neurons express TGF-β2 mRNA between postnatal days P3 and P13, with a peak of expression around P10, mostly in the EGL, where both proliferating and postmitotic granule neurons stain positive for TGF-β2 mRNA (Constam et al., 1994). The TGF-β2 and TGF-β3 protein expression pattern localizes mostly in zones composed of differentiating neurons, radial glial cells, and their processes, whereas no staining is found in areas where cell proliferation occurs (Flanders et al., 1991). Temporal and spatial regulation of TGF-β receptors type I expression coincides also with tissue differentiation (Iseki et al., 1995).
Transgenic mice overexpressing TGF-β1 in astrocytes have provided clues about the in vivo effects of overexpression of TGF-β1 on the function and structure of the CNS. These mice develop hydrocephalus and die before 3 weeks of age (Galbreath et al., 1995). Their cerebellum is considerably smaller than that of control mice. These transgenic mice express TGF-β1 at an embryonic time point (E12.5) that coincides with the early stages of neural stem cell proliferation and differentiation in the CNS. It has therefore been postulated that elevated TGF-β1 at this stage will result in a generalized arrest of cell proliferation.
Aberrant expression of TGF-β has been associated with neuropathological conditions of the adult CNS as well. TGF-β is detected in the supernatant of glioblastoma cells (Wrann et al., 1987) and observed in neuritic plaques in Alzheimer‘s disease (van der Wal et al., 1993). Nevertheless, TGF-β is thought to play a vital role in the response of the CNS to trauma. The upregulation of TGF-β1 mRNA observed in activated astrocytes at a site of injury and in reactive microglia cells in the zones of neural degeneration is suggestive of a role of TGF-β in tissue repair processes (Logan et al., 1992; Morgan et al., 1993; Pasinetti et al., 1993). Neurons may benefit from the ability of TGF-β to stimulate growth factor production, e.g., NGF in astrocytes, in vitroand in vivo (Lindholm et al., 1990; Saad et al., 1991), to upregulate growth factor receptors (Lefebvre et al., 1992), and to interfere with pathological changes in cerebral bood flow (Pfister et al., 1992).
Taken together, these data suggest that TGF-β may limit the expansion of neuronal precursor populations by promoting their apoptosis, but may support survival of those neurons that have maturated, differentiated, and established supportive synaptic connectivity.
Footnotes
This work was supported by the Swiss National Science Foundation (Project No. 3100-042900.95/1). We thank Dr. M. Hoechli at the Laboratory for Electron Microscopy, University of Zürich, for excellent assistance in microscopy techniques and digital image processing.
Correspondence should be addressed to Dr. Adriano Fontana, Section of Clinical Immunology, Department of Internal Medicine, University Hospital, Häldeliweg 4, CH-8044 Zürich, Switzerland.
REFERENCES
- 1.Baffy G, Sharma K, Shi W, Ziyadeh FN, Williamson JR. Growth arrest of a murine mesanglial cell line by transforming growth factor β1 is associated with inhibition of mitogen-induced Ca2+mobilization. Biochem Biophys Res Commun. 1995;210:378–383. doi: 10.1006/bbrc.1995.1672. [DOI] [PubMed] [Google Scholar]
- 2.Balàsz R, Resink A, Hack N, Van der Walk JBF, Kumar KN, Michaelis E. NMDA treatment and K+-induced depolarization selectively promote the expression of a NMDA-preferring class of the ionotropic glutamate receptor in cerebellar granule cells. Neurosci Lett. 1992;137:109–113. doi: 10.1016/0304-3940(92)90310-4. [DOI] [PubMed] [Google Scholar]
- 3.Blottner D, Wolf N, Lachmund A, Flanders KC, Unsicker K. TGF-β rescues target-deprived preganglionic sympathetic neurons in the spinal cord. Eur J Neurosci. 1996;8:202–210. doi: 10.1111/j.1460-9568.1996.tb01181.x. [DOI] [PubMed] [Google Scholar]
- 4.Chalazonitis A, Kalberg J, Twardzik DR, Morrison RS, Kessler JA. Transforming growth factor-β has neurotrophic actions on sensory neurons in vitro and is synergistic with nerve growth factor. Dev Biol. 1992;152:121–132. doi: 10.1016/0012-1606(92)90162-a. [DOI] [PubMed] [Google Scholar]
- 5.Constam DB, Philipp J, Malipiero UV, ten Dijke P, Schachner M, Fontana A. Differential expression of transforming growth factor-β1, -β2 and -β3 by glioblastoma cell lines, astrocytes and microglia. J Immunol. 1992;148:1404–1410. [PubMed] [Google Scholar]
- 6.Constam DB, Schmid P, Aguzzi A, Schachner M, Fontana A. Transient production of TGF-β2 by postnatal cerebellar neurons and its effect on neuroblast proliferation. Eur J Neurosci. 1994;6:766–778. doi: 10.1111/j.1460-9568.1994.tb00988.x. [DOI] [PubMed] [Google Scholar]
- 7.Didier M, Roux P, Piechaczyk M, Verrier B, Bockaert J, Pin JP. Cerebellar granule cell survival and maturation induced by K+ and NMDA correlate with c-fos protooncogene expression. Neurosci Lett. 1989;107:55–62. doi: 10.1016/0304-3940(89)90790-8. [DOI] [PubMed] [Google Scholar]
- 8.Didier M, Heaulme M, Soubrie P, Pin J-P. Rapid, sensitive, and simple method for quantification of both neurotoxic and neurotrophic effects of NMDA on cultured cerebellar granule cells. J Neurosci Res. 1990;27:25–35. doi: 10.1002/jnr.490270105. [DOI] [PubMed] [Google Scholar]
- 9.Didier M, Roux P, Piechaczyk M, Mangeat P, Devilliers G, Bockaert J, Pin JP. Long-term expression of the c-fos protein during the in vitro differentiation of cerebellar granule cells induced by potassium and NMDA. Mol Brain Res. 1992;12:249–258. doi: 10.1016/0169-328x(92)90091-o. [DOI] [PubMed] [Google Scholar]
- 10.D’Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erecinska M, Nelson D, Chance B. Depolarization-induced changes in cellular energy production. Proc Natl Acad Sci USA. 1991;88:7600–7604. doi: 10.1073/pnas.88.17.7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer G. Cultivation of mouse cerebellar cells in serum free, hormonally defined media: survival of neurons. Neurosci Lett. 1982;28:325–329. doi: 10.1016/0304-3940(82)90079-9. [DOI] [PubMed] [Google Scholar]
- 13.Flanders KC, Lüdecke G, Engels S, Cissel DS, Roberts AB, Kondaiah P, Lafyatis R, Sporn M, Unsicker K. Localization and actions of transforming growth factor-βs in the embryonic nervous system. Development. 1991;113:183–191. doi: 10.1242/dev.113.1.183. [DOI] [PubMed] [Google Scholar]
- 14.Fontana A, Hengartner H, de Tribolet N, Weber E. Glioblastoma cells release interleukin-1 and factors inhibiting interleukin-2 mediated effects. J Immunol. 1984;132:1837–1844. [PubMed] [Google Scholar]
- 15.Franklin JL, Johnson EM. Suppression of programmed neuronal death by sustained elevation of cytoplasmic calcium. Trends Neurosci. 1992;15:501–508. doi: 10.1016/0166-2236(92)90103-f. [DOI] [PubMed] [Google Scholar]
- 16.Galbreath E, Kim S-J, Park K, Brenner M, Messing A. Overexpression of TGF-β1 in the central nervous system of transgenic mice resulting in hydrocephalus. J Neuropathol Exp Neurol. 1995;54:334–349. doi: 10.1097/00005072-199505000-00007. [DOI] [PubMed] [Google Scholar]
- 17.Gallo V, Ciotti MT, Coletti A, Aloisi F, Levi G. Selective release of glutamate from cerebellar granule cells differentiating in culture. Proc Natl Acad Sci USA. 1982;79:7919–7923. doi: 10.1073/pnas.79.24.7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallo V, Kingsbury A, Balasz R, Jørgensen OS. The role of depolarization in the survival and differentiation of cerebellar granule cells in culture. J Neurosci. 1987;7:2203–2213. doi: 10.1523/JNEUROSCI.07-07-02203.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1622. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- 21.Giannini G, Clementi E, Ceci R, Marziali G, Sorrentino V. Expression of a ryanodine receptor-Ca2+ channel that is regulated by TGF-β. Science. 1992;257:91–94. doi: 10.1126/science.1320290. [DOI] [PubMed] [Google Scholar]
- 22.Heine UI, Munoz EF, Flanders KC, Ellingsworth LR, Lam HYP, Thompson NL, Roberts AB, Sporn MB. Role of transforming growth factor-β in the development of the mouse embryo. J Cell Biol. 1987;105:2861–2876. doi: 10.1083/jcb.105.6.2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iseki S, Osumi-Yamashita N, Miyazono K, Franzén P, Ichijo H, Ohtani H, Hayashi Y, Eto K. Localization of transforming growth factor-β type I and type II receptors in mouse development. Exp Cell Res. 1995;219:339–347. doi: 10.1006/excr.1995.1237. [DOI] [PubMed] [Google Scholar]
- 24.Ishihara A, Saito H, Abe K. Transforming growth factor-β1 and -β2promote neurite sprouting and elongation of cultured rat hippocampal neurons. Brain Res. 1994;639:21–25. doi: 10.1016/0006-8993(94)91759-0. [DOI] [PubMed] [Google Scholar]
- 25.Johnson EM, Deckwerth TL. Molecular mechanisms of developmental neuronal death. Annu Rev Neurosci. 1993;16:31–46. doi: 10.1146/annurev.ne.16.030193.000335. [DOI] [PubMed] [Google Scholar]
- 26.Kanzaki T, Olofsson A, Morén A, Wernstedt C, Hellman U, Miyazono K, Claesson-Welsh L, Heldin C-H. TGF-β1 binding protein: a component of the large latent complex of TGF-β1with multiple repeat sequences. Cell. 1990;61:1051–1061. doi: 10.1016/0092-8674(90)90069-q. [DOI] [PubMed] [Google Scholar]
- 27.Kessler JA, Ludlam WH, Freidin MM, Hall DH, Michaelson MD, Spray DC, Dougherty M, Batter DK. Cytokine-induced programmed death of cultured sympathetic neurons. Neuron. 1993;11:1123–1132. doi: 10.1016/0896-6273(93)90225-g. [DOI] [PubMed] [Google Scholar]
- 28.Kingsbury AE, Gallo V, Woodhams PL, Balàsz R. Survival, morphology and adhesion properties of cerebellar interneurones cultured in chemically defined and serum-supplemented medium. Dev Brain Res. 1985;17:17–25. doi: 10.1016/0165-3806(85)90128-2. [DOI] [PubMed] [Google Scholar]
- 29.Krieglstein K, Suter-Crazzolara C, Fischer WH, Unsicker K. TGF-β superfamily members promote survival of midbrain dopaminergic neurons and protect them against MPP+ toxicity. EMBO J. 1995;14:736–742. doi: 10.1002/j.1460-2075.1995.tb07052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lefebvre PP, Martin D, Staecker H, Weber T, Moonen G, Van De Water TR. TGFβ1 expression is initiated in adult auditory neurons by sectioning of the auditory nerve. NeuroReport. 1992;3:295–298. doi: 10.1097/00001756-199204000-00001. [DOI] [PubMed] [Google Scholar]
- 31.Lin J-K, Chou C-K. In vitro apoptosis in the human hepatoma cell line induced by transforming growth factor β1. Cancer Res. 1992;52:385–388. [PubMed] [Google Scholar]
- 32.Lindholm D, Hengerer B, Zafra F, Thoenen H. Transforming growth factor-β1 stimulates expression of nerve growth factor in the rat CNS. NeuroReport. 1990;1:9–12. doi: 10.1097/00001756-199009000-00003. [DOI] [PubMed] [Google Scholar]
- 33.Logan A, Frautschy SA, Gonzalez A-M, Sporn MB, Baird A. Enhanced expression of transforming growth factor β1 in the rat brain after a localized cerebral injury. Brain Res. 1992;587:216–225. doi: 10.1016/0006-8993(92)91000-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lømo J, Blomhoff HK, Beiske K, Stokke T, Smeland EB (1995) TGF-β1 and cyclic AMP promote apoptosis in resting human B lymphocytes. J Immunol 1634–1643. [PubMed]
- 35.Lotem J, Sachs L. Hematopoietic cytokines inhibit apoptosis induced by transforming growth factor β1 and cancer chemotherapy compounds in myeloid leukemic cells. Blood. 1992;80:1750–1757. [PubMed] [Google Scholar]
- 36.Lucas C, Bald LN, Fendly BM, Mora-Worms M, Figari IS, Patzer EJ, Palladino MA. The autocrine production of transforming growth factor-β1 during lymphocyte activation. J Immunol. 1990;145:1415–1422. [PubMed] [Google Scholar]
- 37.Mahanthappa NK, Schwarting GA. Peptide growth factor control of olfactory neurogenesis and neuron survival in vitro: roles of EGF and TGF-βs. Neuron. 1993;10:293–305. doi: 10.1016/0896-6273(93)90319-m. [DOI] [PubMed] [Google Scholar]
- 38.Maher F, Simpson IA. Modulation of expression of glucose transporters GLUT3 and GLUT1 by potassium and N -methyl-d-aspartate in cultured cerebellar granule neurons. Mol Cell Neurosci. 1994;5:369–375. doi: 10.1006/mcne.1994.1044. [DOI] [PubMed] [Google Scholar]
- 39.Marini AS, Paul SM. N -methyl-d-aspartate receptor mediated neuroprotection in cerebellar granule cells requires new RNA and protein synthesis. Proc Natl Acad Sci USA. 1992;89:6555–6559. doi: 10.1073/pnas.89.14.6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinou J-C, Le Van Thai A, Valette A, Weber MJ. Transforming growth factor β1 is a potent survival factor for rat embryo motorneurons in culture. Dev Brain Res. 1990;52:175–181. doi: 10.1016/0165-3806(90)90233-o. [DOI] [PubMed] [Google Scholar]
- 41.Massagué J. The transforming growth factor-β family. Annu Rev Cell Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- 42.Moran J, Patel AJ. Effect of potassium depolarization on phosphate-activated glutaminase activity in primary cultures of cerebellar granule neurons and astroglial cells during development. Dev Brain Res. 1989;46:97–105. doi: 10.1016/0165-3806(89)90146-6. [DOI] [PubMed] [Google Scholar]
- 43.Morgan TE, Nichols NR, Pasinetti GM, Finch CE. TGF-β1 mRNA increases in macrophage/microglial cells of the hippocampus in response to deafferentation and kainic acid-induced neurodegeneration. Exp Neurol. 1993;120:291–301. doi: 10.1006/exnr.1993.1063. [DOI] [PubMed] [Google Scholar]
- 44.Neylon CB, Bryant SM, Little PJ, Bobik A. Transforming growth factor β1 regulates the expression of ryanodine-sensitive Ca2+ oscillations in cardiac myocytes. Biochem Biophys Res Commun. 1994;204:678–684. doi: 10.1006/bbrc.1994.2513. [DOI] [PubMed] [Google Scholar]
- 45.Nicoletti F, Wroblewski JT, Novelli A, Alho H, Guidotti A, Costa E. The activation of inositol phospholipid metabolism as a signal-transducing system for excitatory amino acids in primary cultures of cerebellar granule cells. J Neurosci. 1986;6:1905–1911. doi: 10.1523/JNEUROSCI.06-07-01905.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Novelli A, Reilly JA, Lysko PG, Henneberry RC. Glutamate becomes neurotoxic via the N -methyl-d-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- 47.Oberhammer F, Sedivy R, Jirtle R, Schulte-Hermann R. Role of pro and mature TGF-β1 in cell death (apoptosis) of hepatocytes. Naunyn Schmiedebergs Arch Pharmacol Suppl. 1991;343:R24. [Google Scholar]
- 48.Oberhammer FA, Pavelka M, Sharma S, Tiefenbacher R, Purchio AF, Bursch W, Schulte-Hermann R. Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor β1. Proc Natl Acad Sci USA. 1992;89:5408–5412. doi: 10.1073/pnas.89.12.5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pasinetti GM, Nichols NR, Tocco G, Morgan T, Laping N, Finch CE. Transforming growth factor β1 and fibronectin messenger RNA in rat brain: responses to injury and cell-type localization. Neuroscience. 1993;54:893–907. doi: 10.1016/0306-4522(93)90583-2. [DOI] [PubMed] [Google Scholar]
- 50.Pearson H, Graham ME, Burgoyne RD. Relationship between intracellular free calcium concentration and NMDA-induced cerebellar granule cell survival in vitro. Eur J Neurosci. 1992;4:1369–1375. doi: 10.1111/j.1460-9568.1992.tb00162.x. [DOI] [PubMed] [Google Scholar]
- 51.Peng L, Zhang X, Hertz L. High extracellular potassium concentrations stimulate oxidative metabolism in a glutamatergic neuronal culture and glycolysis in cultured astrocytes but have no stimulatory effect in a GABAergic neuronal culture. Brain Res. 1994;663:168–172. doi: 10.1016/0006-8993(94)90475-8. [DOI] [PubMed] [Google Scholar]
- 52.Pfister H-W, Frei K, Ottnad B, Koedel U, Tomasz A, Fontana A. Transforming growth factor β2 inhibits cerebrovascular changes and brain edema formation in the tumor necrosis factor α-independent early phase of experimental pneumococcal meningitis. J Exp Med. 1992;176:265–268. doi: 10.1084/jem.176.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Piani D, Frei K, Quang Do K, Cuénod M, Fontana A. Murine brain macrophages induce NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neuroscience Lett. 1991;133:159–162. doi: 10.1016/0304-3940(91)90559-c. [DOI] [PubMed] [Google Scholar]
- 54.Poulsen KT, Armanini MP, Hynes MA, Phillips HS, Rosenthal A. TGF-β2 and TGF-β3 are potent survival factors for midbrain dopaminergic neurons. Neuron. 1994;13:1245–1252. doi: 10.1016/0896-6273(94)90062-0. [DOI] [PubMed] [Google Scholar]
- 55.Prehn JHM, Backhauss C, Krieglstein J. Transforming growth factor-β1 prevents glutamate neurotoxicity in rat neocortical cultures and protects mouse neocortex from ischemic injury in vivo. J Cereb Blood Flow Metab. 1993a;13:521–525. doi: 10.1038/jcbfm.1993.67. [DOI] [PubMed] [Google Scholar]
- 56.Prehn JHM, Peruche B, Unsicker K, Krieglstein J. Isoform-specific effects of transforming growth factors-β on degeneration of primary neuronal cultures induced by cytotoxic hypoxia or glutamate. J Neurochem. 1993b;60:1665–1672. doi: 10.1111/j.1471-4159.1993.tb13389.x. [DOI] [PubMed] [Google Scholar]
- 57.Prehn JHM, Bindokas VP, Marcuccilli CJ, Krajewski S, Reed JC, Miller RJ. Regulation of neuronal Bcl-2 expression and calcium homeostasis by transforming growth factor type β confers wide-ranging protection on rat hippocampal neurons. Proc Natl Acad Sci USA. 1994;91:12599–12603. doi: 10.1073/pnas.91.26.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- 59.Resink A, Hack N, Boer GJ, Balàsz R. Growth conditions differentially modulate the vulnerability of developing cerebellar granule cells to excitatory amino acids. Brain Res. 1994;655:222–232. doi: 10.1016/0006-8993(94)91617-9. [DOI] [PubMed] [Google Scholar]
- 60.Saad B, Constam DB, Ortmann R, Moos M, Fontana A, Schachner M. Astrocyte-derived TGF-β2 and NGF differentially regulate neural recognition molecule expression by cultured astrocytes. J Cell Biol. 1991;115:473–484. doi: 10.1083/jcb.115.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tymianski M, Wang LY, MacDonald JF. Alteration of neuronal calcium homeostasis and excitotoxic vulnerability by chronic depolarization. Brain Res. 1994;648:291–295. doi: 10.1016/0006-8993(94)91129-0. [DOI] [PubMed] [Google Scholar]
- 62.Unsicker K, Flanders KC, Cissel DS, Lafyatis R, Sporn MB. Transforming growth factor beta isoforms in the adult rat central and peripheral nervous system. Neuroscience. 1991;44:613–625. doi: 10.1016/0306-4522(91)90082-y. [DOI] [PubMed] [Google Scholar]
- 63.van der Wal EA, Gómez-Pinilla F, Cotman CW. Transforming growth factor-β1 is in plaques in Alzheimer and Down pathologies. NeuroReport. 1993;4:69–72. doi: 10.1097/00001756-199301000-00018. [DOI] [PubMed] [Google Scholar]
- 64.Weller M, Marini AM, Paul SM. Niacinamide blocks 3-acetylpyridine toxicity of cerebellar granule cells in vitro. Brain Res. 1992;594:160–164. doi: 10.1016/0006-8993(92)91043-e. [DOI] [PubMed] [Google Scholar]
- 65.Weller M, Constam DB, Malipiero U, Fontana A. Transforming growth factor-β2 induces apoptosis of murine T cell clones without down-regulating bcl-2 mRNA expression. Eur J Immunol. 1994a;24:1293–1300. doi: 10.1002/eji.1830240608. [DOI] [PubMed] [Google Scholar]
- 66.Weller M, Frei K, Groscurth P, Krammer PH, Yonekawa Y, Fontana A. Anti-Fas/APO-1 antibody-mediated apoptosis of cultured human glioma cells. Induction and modulation of sensitivity by cytokines. J Clin Invest. 1994b;94:954–964. doi: 10.1172/JCI117462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weller M, Malipiero U, Aguzzi A, Reed JC, Fontana A. Protooncogene bcl-2 gene transfer abrogates Fas/APO-1 antibody- mediated apoptosis of human malignant glioma cells and confers resistance to chemotherapeutic drugs and therapeutic irradiation. J Clin Invest. 1995a;95:2633–2643. doi: 10.1172/JCI117965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weller M, Malipiero U, Rensing-Ehl A, Barr PJ, Fontana A. Fas/APO-1 gene transfer for human malignant glioma. Cancer Res. 1995b;55:2936–2944. [PubMed] [Google Scholar]
- 69.Weller M, Fontana A (1996) The failure of current immunotherapy for malignant glioma. Tumor-derived TGF-β, T cell apoptosis and the immune privilege of the brain. Brain Res Rev, in press. [DOI] [PubMed]
- 70.Wood KA, Dipasquale B, Youle RJ. In situ labeling of granule cells for apoptosis-associated DNA fragmentation reveals different mechanisms of cell loss in developing cerebellum. Neuron. 1993;11:621–632. doi: 10.1016/0896-6273(93)90074-2. [DOI] [PubMed] [Google Scholar]
- 71.Wrann M, Bodmer S, de Martin R, Siepl C, Hofer-Warbinek R, Frei K, Hofer E, Fontana A. T cell suppressor factor from human glioblastoma cells is a 12.5 kDa protein closely related to transforming growth factor-beta. EMBO J. 1987;6:1633–1636. doi: 10.1002/j.1460-2075.1987.tb02411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yan G-M, Ni B, Weller M, Wood KA, Paul SM. Depolarization or glutamate receptor activation blocks apoptotic cell death of cultured cerebellar granule neurons. Brain Res. 1994;656:43–51. doi: 10.1016/0006-8993(94)91364-1. [DOI] [PubMed] [Google Scholar]
- 73.Yanagihara K, Tsumuraya M. Transforming growth factor β1 induces apoptotic cell death in cultured human gastric carcinoma cells. Cancer Res. 1992;52:4042–4045. [PubMed] [Google Scholar]