

Abstract
Seizures cause a persistent enhancement in dentate synaptic inhibition concurrent with, and possibly compensatory for, seizure-induced hippocampal hyperexcitability. To study this phenomenon, we evoked status epilepticus in rats with systemic kainic acid (KA), and 2 weeks later assessed granule cell inhibition with paired-pulse stimulation of the perforant path (PP) in vitro. Controls demonstrated three components of paired-pulse inhibition: early inhibition (10–30 msec), intermediate facilitation (30–120 msec), and late inhibition (120 msec to 120 sec). After seizures, inhibition in all components was enhanced significantly. The GABAA antagonist bicuculline blocked only early enhanced inhibition, demonstrating that both GABAA and GABABpostsynaptic receptors contribute to seizure-induced enhanced inhibition. In controls, the GABAB antagonist CGP 35348 increased both GABAA and GABAB responses in granule cells, suggesting that CGP 35348 acts presynaptically, blocking receptors that suppress GABA release. In contrast, slices from KA-treated rats were markedly less sensitive to CGP 35348. To test the hypothesis that GABAB receptors regulating GABA release are downregulated after seizures, we measured paired-pulse suppression of recurrent IPSPs, or disinhibition, using mossy fiber stimuli. Early disinhibition (< 200 msec) was reduced after seizures, whereas late disinhibition remained intact. CGP 35348 blocked the early component of disinhibition in controls and, to a lesser extent, reduced disinhibition in KA slices. However, paired monosynaptic IPSPs recorded intracellularly showed no difference in disinhibition between groups. Our findings indicate that seizure-induced enhancement in dentate inhibition is caused, at least in part, by reduced GABAB function in the polysynaptic recurrent inhibitory circuit, resulting in reduced disinhibition and heightened GABA release.
Keywords: hippocampus, dentate gyrus, paired-pulse inhibition, GABAB, disinhibition, presynaptic, autoreceptors, kainic acid, bicuculline, CGP 35348, epilepsy, seizures
Neuronal alterations produced by seizures have been implicated in promoting cellular hyperexcitability and seizure propagation. In animal models of epilepsy, including kainic acid (KA)-induced status epilepticus (Holmes and Thompson, 1988;Stafstrom et al., 1992; Lothman and Bertram, 1993) and kindling (Goddard et al., 1969; Wong and Moshé, 1987; Holmes et al., 1993), seizures alter the brain, increasing susceptibility to subsequent epileptogenic stimuli. Seizure-induced changes that contribute to this heightened excitability include enhanced NMDA receptor function (Mody et al., 1988; Martin et al., 1992; Kohr et al., 1993; Kohr and Mody, 1994), reduced inhibition in CA1 (King et al., 1985; Kapur et al., 1989) and CA3 (Zhao and Leung, 1992), death of hilar cells that normally excite recurrent inhibitory interneurons (Sloviter, 1987; Cavazos and Sutula, 1990), axonal sprouting, and formation of aberrant excitatory synapses (Sutula, 1988).
In contrast to these persistent changes that promote hyperexcitability, inhibition of dentate gyrus granule cells is lost only transiently during and immediately after seizures (Maru and Goddard, 1987; Milgram et al., 1991; Spiller and Racine, 1994). Within 24 hr, PP-evoked inhibition of granule neurons recovers to levels higher than those preceding KA administration (Milgram et al., 1991) or kindled stimulation (Tuff et al., 1983; King et al., 1985; Oliver and Miller, 1985; de Jonge and Racine, 1987; Maru and Goddard, 1987; Gilbert, 1991;Milgram et al., 1995), although this inhibition may be subject to more rapid fatigue (Sloviter, 1992; Buhl et al., 1996). Little is known about the underlying mechanisms or the functional role of this enhanced inhibition. The concurrent expression of increased dentate inhibition and hyperexcitability throughout the hippocampus (King et al., 1985;Cronin et al., 1992; Zhao and Leung, 1992; Bekenstein et al., 1993) suggests that enhanced inhibition may be a compensatory mechanism to suppress the spread of seizure activity.
Although studies have indicated an increase of late Cl−-independent synaptic potentials (Oliver and Miller, 1985), GABAA receptor number- (Otis et al., 1994) or both postsynaptic GABAA and GABAB receptor response- (Sperber, 1991; de Jonge and Racine, 1987; Gilbert, 1991) enhanced paired-pulse inhibition might not be mediated solely by postsynaptic mechanisms, because there is a complex presynaptic regulation of inhibitory transmission. Repeated stimuli produce a decrease in inhibitory postsynaptic potential (IPSP) amplitude, a phenomenon called disinhibition, produced in large part by activation of presynaptic GABAB receptors (Thompson, 1992; Lambert and Wilson, 1993; Pitler and Alger, 1994; Pearce, 1995). GABAB receptors that suppress neurotransmitter release have been found on presynaptic terminals of both GABAergic (Davies and Collingridge, 1993; Mott et al., 1993) and glutamatergic (Harrison et al., 1990; Scanziani et al., 1992; Davies et al., 1993) neurons in the hippocampus. Therefore, inhibition of granule neurons also could be enhanced either by raising the excitatory drive on inhibitory interneurons (Collins et al., 1982) or by reducing GABAB receptor-mediated suppression of GABA release from inhibitory terminals (Davies et al., 1990; Mott et al., 1993).
In the dentate gyrus of in vitro hippocampal slices, we have used selective antagonists to remove GABAA or GABAB inhibition to determine which receptor populations show seizure-induced alterations. In addition, extracellular and intracellular recordings from dentate granule neurons were used to examine frequency-dependent disinhibition of IPSPs in the recurrent inhibitory pathway. Our results demonstrate that although both postsynaptic GABAA and GABAB responses are enhanced by seizures, the underlying mechanism is a decreased GABABreceptor-mediated suppression of presynaptic GABA release. Portions of these results have been presented elsewhere (Sperber et al., 1991; Haas et al., 1994).
MATERIALS AND METHODS
KA status epilepticus. Seizures were induced in 60-d-old, male Sprague–Dawley rats (Taconic Farms, NY) by systemic KA injection (15 mg/kg, i.p.) (K 0250, Sigma, St. Louis, MO). Rats were monitored for seizure behavior for 24 hr after injection. Only rats displaying severe status epilepticus, defined as continuous tonic–clonic seizure behavior for at least 30 min, were used in this study.
Electrophysiology. Two weeks after exposure to KA status epilepticus, KA-treated rats (n = 28) and age-matched controls (n = 26) were decapitated under deep ether anesthesia and the brains rapidly removed. The hippocampus plus attached entorhinal cortex were dissected out, submerged in a reservoir containing chilled artificial CSF (ACSF), and sliced transversely with a vibratome (400 μm thick) (Ted Pella, Reading, CA). Slices were transferred to an interface perfusion chamber (Haas et al., 1979) and bathed continuously with ACSF containing (in mm): NaCl 126, KCl 5, CaCl22, MgCl2 2, NaH2PO4 1.25, NaHCO3 26, d-glucose 10, pH 7.2, 32–34°C. Slices were incubated for at least 30 min before recording. For extracellular recording of field potentials, glass microelectrodes (microfilament capillary 1.2 mm outer diameter; 5–10 MΩ) (A-M Systems) filled with NaCl (2 m) were placed in the granule cell body layer (stratum granulosum) of the suprapyramidal blade of the dentate gyrus. Bipolar, twisted tungsten stimulating electrodes (tip distance 50 μm) (Frederick Haer) were placed in the perforant path (PP) for orthodromic activation of granule cells and at the CA3–hilar border to stimulate mossy fibers antidromically, and stimuli were applied as DC square pulses (20–400 μA, 100 μsec duration). Extracellular population spikes were recorded with an Axoclamp-2A amplifier, and data were digitized and analyzed on an IBM PC-AT. To block GABA receptors, either the GABAA antagonist bicuculline (10 μm) (Tocris Cookson, St. Louis) or the GABAB antagonist 3-aminopropyl-diethoxymethyl-phosphinic acid, CGP 35348 (Olpe et al., 1990), 400 μm) (a generous gift from Ciba-Geigy, Basel, Switzerland) was bath applied for 1 hr before electrophysiological recording.
Intracellular recordings from granule cells were made with glass electrodes filled with 3 mK+ acetate (120–170 MΩ). Stimulating electrodes were placed in the stratum granulosum proximal to the recording electrode to activate interneurons directly and evoke monosynaptic IPSPs. Excitatory amino acid neurotransmission was blocked with the AMPA receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione [CNXQ (10 μm)] plus the NMDA receptor antagonist d-APV (40 μm), bath applied for 30 min before recording. Intracellular data were filtered at 3 kHz and sampled at 20 kHz. CNQX andd-APV were purchased from Tocris Neuramin (Bristol, England).
Stimulation paradigms. The amplitude and time course of feedforward and feedback inhibition of dentate granule cells were assessed with paired-pulse stimulation of the PP. PP stimulus evoked EPSPs, and action potentials (population spikes) were recorded extracellularly in the stratum granulosum (see Fig.2A, Control PS1, 10 msec). Two identical stimuli were delivered with interstimulus intervals (ISIs) ranging from 10 msec to 9 sec. Stimulus intensities were chosen such that the first evoked population spike was ∼75% of maximum. Paired-pulse inhibition was measured as the ratio of the second, test, population spike (PS2) amplitude to the amplitude of the first, conditioning, population spike (PS1). A decrease in the test spike amplitude compared with the conditioning spike indicated paired-pulse inhibition (Fig. 2A, Control, 10 msec), whereas a relative increase represented facilitation (Fig.2A, Control, 50 msec). No paired-pulse effects were observed with intervals longer than 10 sec, so the time between pairs of stimuli was maintained at 20 sec to avoid interference between trials.
Fig. 2.

Enhanced paired-pulse inhibition in hippocampal slices from KA-treated rats 2 weeks after seizures.A, Field population spikes (PS) recorded in the stratum granulosum elicited by pairs of PP stimulation. The three ISIs shown demonstrate paired-pulse early inhibition (10 msec), intermediate facilitation (50 msec), and late inhibition (500 msec) in control slices. Enhanced inhibition after KA status is apparent as a marked decrease in amplitude of the second response (PS2), compared with the first (PS1), and a complete loss of paired-pulse facilitation. (Closed circles denote truncated stimulus artifacts.) B, Paired-pulse profile from control (open circles) and KA-treated rats 2 weeks after seizures (closed circles). The percent change in the second population spike (PS2) amplitude compared with the first (PS1) (mean ± SEM) is presented at each interstimulus interval tested. The dotted line marks no change in the amplitude of the second response compared with the first (PS2 = PS1). Points above the dotted line represent facilitation, whereas values below indicate inhibition. Control slices (n = 29) showed early (10–30 msec) and late (120–6000 msec) paired-pulse inhibition, separated by paired-pulse facilitation (30–120 msec). In contrast, hippocampal slices from KA-treated rats (n = 21) showed enhanced paired-pulse inhibition at intervals from 10 to 500 msec (asterisks, one-way ANOVA, p < 0.05, compared with controls).
Disinhibition of recurrent inhibition, the decrease in IPSP amplitude with repeated stimuli, was measured using paired-pulse stimulation of mossy fibers. Stimulation of mossy fibers at the hilar–CA3 border evoked a negative-going antidromic population action potential without an EPSP (see Fig. 5A) and activated feedback inhibition. Because pure IPSPs cannot be directly recorded extracellularly (Brunner and Misgeld, 1993), IPSP strength was inferred from the decrease in amplitude that an antidromic mossy fiber stimulus produced on a subsequent orthodromic PP-evoked population spike (see Fig.5A, compare amplitude of the PP response alone,left, to the PP response in the MF–PPpair, right). Mossy fiber and PP stimulus intensities were chosen such that the mossy fiber-evoked recurrent IPSP inhibited the orthodromic population spike by 50% at an interpulse interval of 5 msec. To produce disinhibition of recurrent inhibition, paired mossy fiber stimuli were delivered with ISIs ranging from 10 msec to 9 sec, with the second mossy fiber stimulus followed 5 msec later by a test orthodromic stimulus to the PP (see Fig. 5B). The decrease in the second mossy fiber-evoked IPSP was apparent as an increase in amplitude of the test PP-evoked population spike (Fig. 5B) compared with inhibition produced by a single MF stimulation (Fig.5A, MF–PP).
Fig. 5.

Recurrent inhibition on granule cells was reduced by paired mossy fiber stimulation. Disinhibition of recurrent inhibition was measured as a reduction in the inhibitory effect on a PP-evoked population spike. A, Field potentials evoked by orthodromic stimulation of the PP (PP) alone and an identical PP stimulation preceded by a mossy fiber stimulus (MF) to illustrate mossy fiber-evoked inhibition of the population spike. The interval between the MF and PP stimuli was 5 msec, which produced a 50% reduction in the population spike amplitude. Stimulus artifacts have been truncated. B, Disinhibition of recurrent inhibition was produced with pairs of MF stimuli. Here, MF stimuli are separated by 200 msec, which reduces inhibition on the test population spike. Compare amplitude of the test PP-evoked population spike with that in the MF–PP pair inA. Stimulus artifacts have been truncated. C, The time course of disinhibition of recurrent inhibition in slices from controls (open circles, n = 17) versus KA-treated rats (closed circles, n = 20). Plot is of the mean ratios ± SEM of the amplitudes of the population spike in the MF–PP pair preceded by an initial MF stimulus (as in B) to the population spike in a MF–PP pair alone (as in A). Slices from KA-treated rats showed significantly reduced disinhibition at ISIs ranging from 30 to 120 msec (asterisks, p < 0.05, one-way ANOVA, compared with control slices).
In intracellular recordings from granule neurons, pure monosynaptic IPSPs were evoked by proximal stimulation of the stratum granulosum in the presence of CNQX (10 μm) andd-APV (40 μm). Disinhibition of monosynaptic IPSPs was examined using paired-pulse stimulations in the stratum granulosum. Intervals between stimulations ranged from 30 to 900 msec. Intracellular recordings were only made from cells with resting membrane potentials more hyperpolarized than −60 mV, input resistances >30 MΩ, and overshooting action potentials.
Data from paired-pulse experiments were compared using a one-way ANOVA. Statistical significance of intracellular measurements were determined with the Student’s t test for unpaired data.
Histology. Histological examination for cell loss and synaptic reorganization were performed on a subset of the rats used for electrophysiological studies (n = 5 KA treated; 5 controls). Brains were removed and bisected, with half used for paired-pulse electrophysiology as described above. The other half was prepared for histological examination using the Timm silver sulfide stain for mossy fiber terminals and thionin stain for neuronal loss.
The hemisections were immersed in a 0.4% sodium sulfide solution for 20 min followed by overnight fixation in a 1% paraformaldehyde and 1.25% glutaraldehyde solution (Tauck and Nadler, 1985), followed by 24 hr in fixation solution with 30% sucrose. Brains were frozen rapidly with methylbutane (−35°C) and cut horizontally in a cryostat (30 μm sections). The sections were developed in the dark for 45–60 min in a solution of 20% (w/v) gum arabic, 5.6% (w/v) hydroquinone, and citric acid/sodium citrate buffer with a solution of 17% silver nitrate. After staining, the sections were dehydrated in alcohol. Alternate sections were hydrated and stained with thionin. The sections then were dehydrated in alcohol and examined microscopically.
RESULTS
KA-induced seizures
60-d-old rats received a single, systemic, intraperitoneal injection of KA (15 mg/kg), which produced severe tonic–clonic seizures and a high mortality rate (40%). Intermittent seizure behavior and mouth and forelimb clonus developed within 30–60 min after injection, and then progressed to continuous forelimb clonus, with rearing and falling lasting from 30 min to >1 hr. After termination of status, rats continued to express sporadic, short-duration bouts of seizure behavior for 24 hr. All rats examined histologically 2 weeks after seizures (n = 5) showed dense supragranular Timm staining (Fig.1A, KA,arrows), indicative of mossy fiber sprouting and aberrant synaptogenesis (Tauck and Nadler, 1985; Sperber et al., 1991), and loss of CA3 pyramidal cells with thionin staining (Fig. 1B). Alterations in Timm staining and cell loss were consistent in all rats examined, and seizure severity among all KA-treated rats was similar.
Fig. 1.
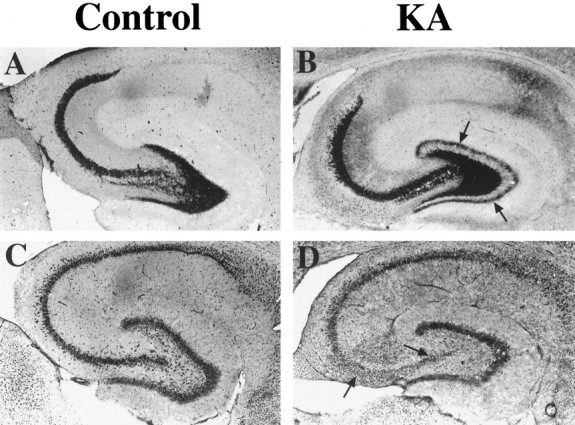
Synaptic reorganization and cell loss 2 weeks after KA-induced status epilepticus (KA).A, B, Timm silver sulfide stain for mossy fiber axonal terminals. In controls (A), positive Timm staining (dark granular stain) demonstrates granule cell axonal termination in the hilus and proximal dendrites of CA3 pyramidal cells. Two weeks after KA-induced status epilepticus (B), aberrant Timm staining (arrows) is evident in supragranular layers throughout the dentate crests. C, D, Thionin staining shows cell bodies of dentate granule cells and pyramidal neurons of the hippocampus (C). KA-treated rats (D) have marked cell loss in area CA3 and the hilus 2 weeks after seizures (arrows).
KA status epilepticus produces a persistent increase in perforant path inhibition
PP stimulation evoked a positive-going EPSP and a superimposed negative-going population action potential, or spike, in field recordings from the stratum granulosum (Fig.2A). PP stimulation also activated inhibitory interneurons mediating feedforward inhibition, whereas granule cell discharge evoked recurrent inhibition. Once activated, both feedforward and feedback inhibition suppressed granule cell firing, producing paired-pulse inhibition.
Paired-pulse stimulation of the PP in hippocampal slices from control rats showed a triphasic profile of granule cell inhibition (Fig.2B, open circles, each point = mean ± SEM). A rapidly activating, short-latency inhibition was present at ISIs ranging from 10 to 30 msec (Fig. 2B,Control, 10 msec). ISIs between 30 and 120 msec caused facilitation of the second population spike (Fig.2A, Control, 50 msec), whereas longer ISIs (120 msec to 6 sec) produced a late-activating, long-lasting inhibition (Fig. 2A, Control, 200 msec). The time courses of early and late paired-pulse inhibition are consistent with those of GABAA receptor- and GABAB receptor-mediated IPSPs, respectively.
Two weeks after KA-induced seizures, slices from these rats showed a significant increase in paired-pulse inhibition over ISIs ranging from 10 to 500 msec, a duration that encompasses both early and late inhibition, as well as facilitation (Fig. 2B,closed circles, asterisks, p < 0.05, one-way ANOVA compared with controls). This apparent enhancement of inhibition completely eliminated paired-pulse facilitation in slices from KA-treated rats (Fig. 2A,KA, 50 msec). Electrophysiological responses from KA-treated rats otherwise appeared normal; in particular, multiple population spikes were not evoked either by PP or mossy fiber stimulation.
Role of GABAA and GABAB receptors in seizure-enhanced inhibition
The GABAA receptor antagonist bicuculline (10 μm) completely blocked early inhibition in slices from both control and KA-treated rats (Fig. 3). Blockade of GABAAreceptor-mediated inhibition by bicuculline produced multiple spiking in response to PP stimulation and unmasked paired-pulse facilitation at short ISIs in both groups. In bicuculline, there was no longer any significant difference in paired-pulse inhibition in control and KA slices at short ISIs (Fig. 3) (ISI < 70 msec). In contrast, late GABAB receptor-mediated inhibition was unaltered by bicuculline (compare ISIs > 90 msec in Figs. 3and 2B), and the significant difference between slices from control and KA rats was still present at longer ISIs, 70–400 msec (Fig. 3, asterisks, p < 0.05). These results indicate that enhanced early inhibition is caused by increased GABAA receptor activation, whereas KA-induced enhancement of late inhibition is mediated through enhanced GABAB receptor function. Therefore, enhanced inhibition after KA status is not attributable to the selective enhancement of activation of only one subtype of postsynaptic GABA receptor.
Fig. 3.

The GABAA receptor antagonist bicuculline (10 μm) blocked early paired-pulse inhibition in both control slices (open circles, n = 20) and slices from KA-treated rats (closed circles, n = 30) (mean ± SEM PS2/PS1. The blockade of early inhibition unmasked equal paired-pulse facilitation in both groups, but late, GABAB receptor-mediated inhibition in slices from KA-treated rats still was enhanced significantly compared with controls (asterisks, p < 0.05, one-way ANOVA; points with no error bars had SEMs smaller than symbols).
The GABAB receptor antagonist CGP 35348 acts as a selective blocker of presynaptic GABAB receptors
To determine the contribution of GABABreceptors to control paired-pulse inhibition and enhanced inhibition in KA-treated rats, the selective GABAB antagonist CGP 35348 was bath applied to slices. CGP 35348 (400 μm) produced a significant increase in control-slice paired-pulse inhibition at ISIs ranging from 10 to 600 msec (Fig. 4A, closed circles, asterisks, p < 0.05 compared with controls without CGP 35348, open circles). The late paired-pulse inhibitory component was not decreased by CGP 35348, as would be expected from a postsynapticGABAB receptor antagonist. Thus, the drug-induced enhancement of both GABAA and GABAB postsynaptic responses is consistent with selective blockade of presynaptic GABABreceptors, which normally act to suppress GABA release.
Fig. 4.
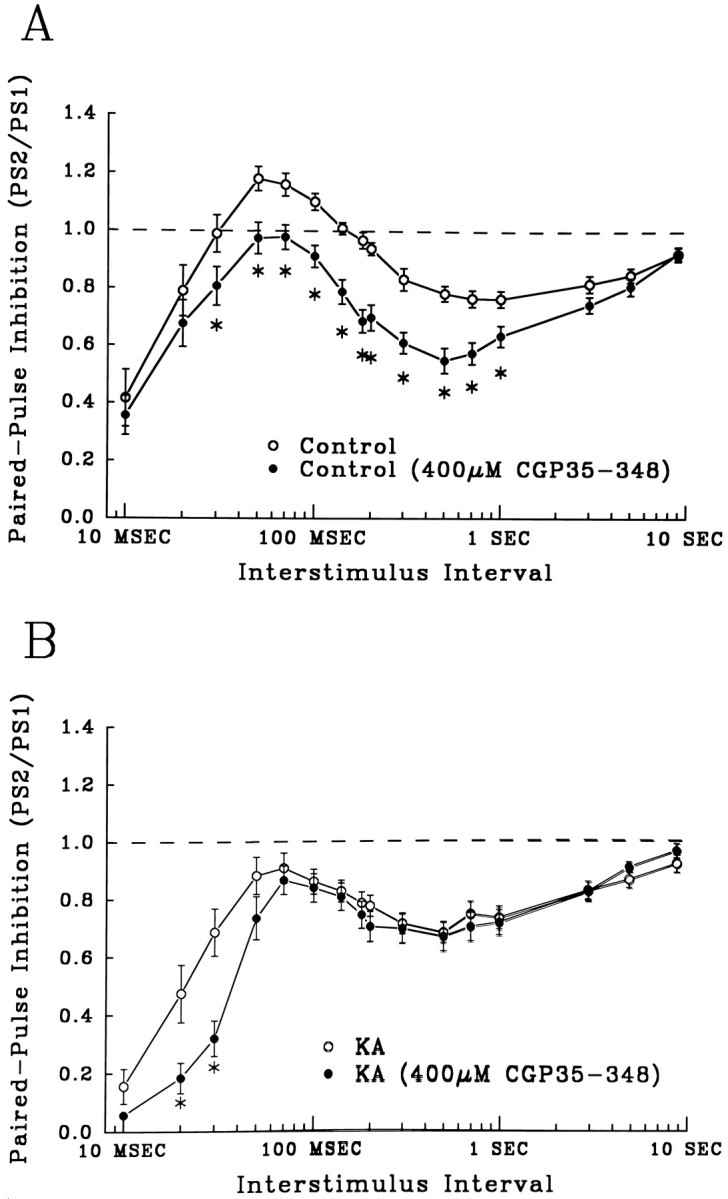
The GABAB receptor antagonist CGP 35348 acted as a blocker of presynaptic GABAB autoreceptors in control slices, but was largely ineffective in slices from KA-treated rats.A, CGP 35348 (400 μm) increased early and late paired-pulse inhibition significantly in control slices at ISIs of 30 msec to 1 sec. Open circles graph control paired-pulse profiles (n = 29), and closed circles are from control slices perfused with CGP 35348 (400 μm) (n = 26, asterisks, p < 0.05, compared with untreated controls). B, In slices from KA-treated rats, CGP 35348 enhanced only early inhibition, ISIs of 20–30 msec (closed circles = CGP 35348 treated, n = 25; open circles = untreated, n = 21;asterisks, p < 0.05, compared with controls). In contrast to control slices, CGP 35348 had no significant effect on late paired-pulse inhibition after KA-induced seizures.
The magnitude and time course of CGP 35348-induced increases in inhibition in control slices were very similar to changes produced by KA seizures (compare Fig. 2B, closed circles with Fig. 4A, closed circles). Furthermore, in marked contrast to controls, CGP 35348 was largely ineffective on slices from KA-treated rats (Fig.4B). The GABAB antagonist produced an increase in inhibition only at short ISIs (10–30 msec) (Fig.4B, asterisks, p < 0.05). These data suggest that CGP 35348- and KA status-mediated enhancement of inhibition might be produced through the same mechanism, a functional downregulation of presynaptic GABABreceptors.
Measuring recurrent disinhibition in control and KA-treated rats
Extracellular field potential measurements of paired-pulse depression of recurrent IPSPs were carried out to assess directly presynaptic GABAB receptor function. Disinhibition, the decrease in IPSP strength with repeated stimuli, has been shown to be mediated, in large part, by the activation of presynaptic GABAB receptors (Deisz and Prince, 1989; Thompson and Gahwiler, 1989; Brucato et al., 1992; Mott et al., 1993). Figure 5C illustrates the profile of disinhibition produced by paired mossy fiber stimuli in control hippocampal slices. Pairs of mossy fiber stimuli, which evoked recurrent inhibition of granule cells, increased or decreased the amount of inhibition induced by the second stimulus of the pair, depending on the ISI. This distribution can be measured by the amount of inhibition produced by the pair on a third, PP-evoked response. In control rats, at short ISIs (10–20 msec), the amount of recurrent inhibition was increased, probably because of the summation of GABAA IPSPs (Fig. 5C, open circles). ISIs between 50 msec and 3 sec produced a decrease in recurrent inhibition, evident by an increase in orthodromic population spike (Fig. 5B). This disinhibition is thought to be attributable, in part, to activation of GABABautoreceptors, which limit GABA release.
In comparison to controls, disinhibition was decreased significantly in slices from KA-treated rats (Fig. 5C, closed circles, asterisks, p < 0.05, one-way ANOVA). ISIs between 30 and 180 msec failed to evoke a decrease in recurrent inhibition. Disinhibition was evoked by longer ISIs, 300 msec to 3 sec, and in contrast to shorter intervals, the profiles of late disinhibition at these ISIs were not different in KA versus control slices. The reduction in disinhibition produced by KA seizures results in enhanced GABA release during paired stimuli and may explain enhanced PP paired-pulse inhibition.
GABAB receptors contribute to recurrent disinhibition
To assess the role of presynaptic GABABreceptors in paired-pulse IPSP disinhibition, we bath applied CGP 35348. Figure 6A shows that in control slices, CGP 35348 (400 μm) blocked disinhibition only at ISIs shorter than 200 msec (closed circles, asterisks, p < 0.05). Disinhibition at longer intervals was unaltered by the GABAB antagonist, suggesting that there are at least two components of disinhibition mediated by separate mechanisms. Similar to its effect on the orthodromic paired-pulse profile in controls (Fig. 4A), the blockade by CGP 35348 of control disinhibition looked very similar to the decrease in disinhibition seen in the absence of drug in slices from KA-treated rats. CGP 35348 still produced some additional depression of disinhibition in slices from KA-treated rats (Fig. 6B, asterisks, p < 0.05). The percent decrease was markedly less than that seen in CGP 35348-treated controls, suggesting that KA seizures already had caused a functional downregulation of the contribution of presynaptic GABAB receptors to paired-pulse IPSP disinhibition.
Fig. 6.
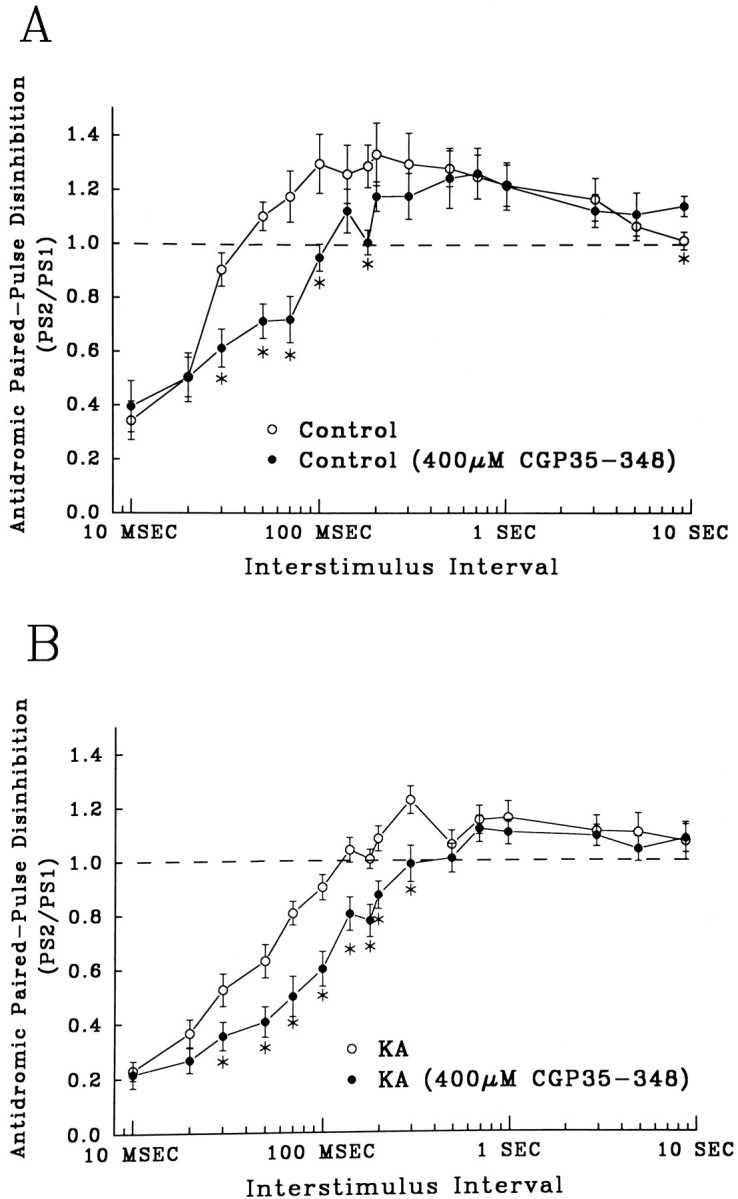
The GABAB receptor antagonist CGP 35348 (400 μm) reduced an early component of disinhibition. A, Control recurrent disinhibition (open circles, n = 17) and disinhibition in slices perfused in CGP 35348 (closed circles, n = 12). An early component of disinhibition in control slices (30–120 msec) was blocked by CGP 35348 (asterisks, p < 0.05). B, Disinhibition of recurrent inhibition in slices from KA-treated rats (open circles, n = 20) and disinhibition in slices from KA-treated rats in the presence of CGP 35348 (closed circles, n = 18). Slices from KA-treated rats also show a significant reduction in disinhibition at intervals of 30–120 msec (asterisks,p < 0.05).
Disinhibition of monosynaptic IPSPs recorded intracellularly is not reduced by KA treatment
Given the extracellular data described above, we recorded intracellularly in granule cells to examine directly disinhibition of monosynaptic IPSPs. Intracellular parameters including resting membrane potential (mean ± SEM, control −77.5 ± 1.7 mV, KA −79.6 ± 3.2 mV) and input resistance (control 78.3 ± 8.4 MΩ, KA 87.5 ± 6.8 MΩ) were not significantly different (Student’s t test for unpaired measures). Monosynaptic IPSPs were readily elicited by stimulation of the stratum granulosum close to an intracellularly impaled granule cell (Fig.7) in the presence of CNXQ (10 μm) and D-APV (40 μm). Monosynaptic IPSPs were composed of an early and a late component. The early component of these IPSPs reversed at −71.2 ± 1.4 mV (controls) and −69.7 ± 2.8 mV (KA), and was blocked completely by the GABAA receptor antagonist bicuculline (10 μm).
Fig. 7.

Intracellular recordings of GABAA IPSPs from dentate granule cells.A, Intracellularly recorded monosynaptic IPSPs at different holding potentials induced by proximal stimulation of the stratum granulosum in the presence CNQX (10 μm) and D-APV (40 μm). The early component reversed at −75 mV and was blocked completely by bicuculline (10 μm). B, The amplitude of GABAA IPSPs at various holding potentials from two representative cells, control (open circles) and after KA-induced status epilepticus (closed circles). Although current clamp recording precluded direct comparison, IPSP amplitudes were consistently similar between groups.
Pairs of stratum granulosum stimulations proximal to the recording electrode evoked paired-pulse inhibition, or disinhibition, of the second monosynaptic IPSP (Fig. 8). Disinhibition was observed in slices from both control and KA status rats, with no significant difference between groups, indicating that the changes in inhibition induced by KA are not attributable to a functional decrease in GABAB receptors on interneuron terminals that mediate recurrent inhibition directly on granule neurons.
Fig. 8.
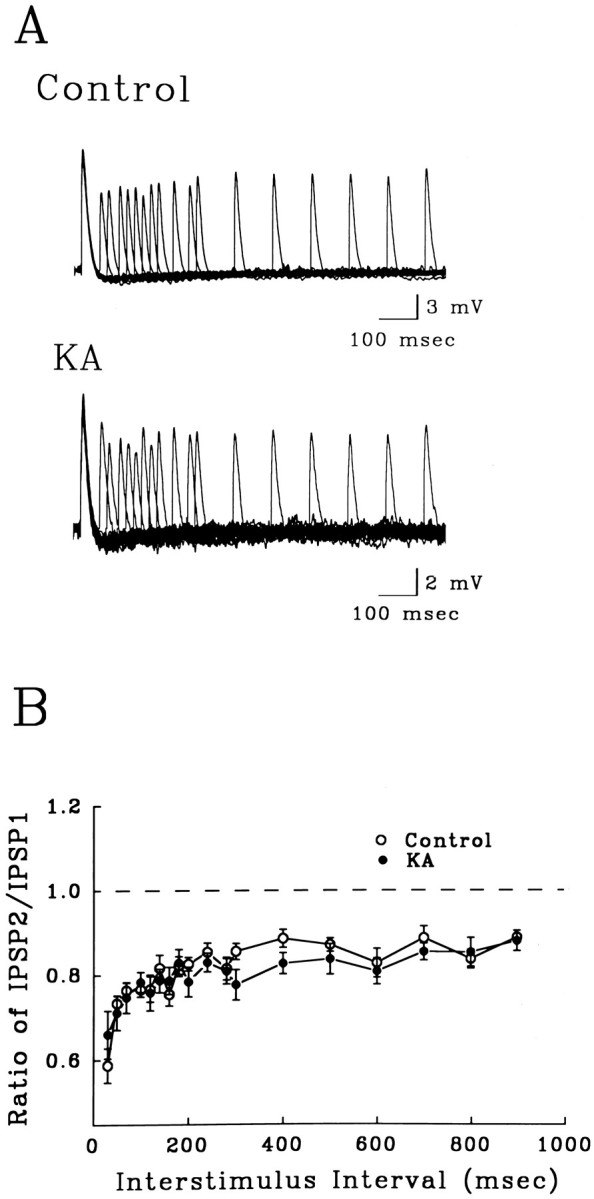
Disinhibition of monosynaptic IPSPs in dentate granule cells (holding potential = −100 mV). A, Overlaid traces from 17 paired-pulse trials with ISIs ranging from 50 to 900 msec. The amplitude of the second IPSP was depressed compared with the first at all ISIs in slices from control and KA-treated rats. Seven-point smoothing was done to limit noise for overlaying traces. B, Paired-pulse profile of suppression of monosynaptic IPSPs at ISIs ranging from 30 msec to 9 sec. There was no significant difference in the monosynaptic disinhibition between controls (open circles,n = 9) and slices from KA-treated rats (closed circles, n = 13).
DISCUSSION
Although others have described an increase in paired-pulse inhibition after seizures (Tuff et al., 1983; King et al., 1985; Oliver and Miller, 1985; de Jonge and Racine, 1987; Maru and Goddard, 1987;Milgram et al., 1991; Sperber et al., 1991), the locus for this change has not been determined. Up to now, it has not been clear whether enhanced granule cell inhibition is produced by a heightened postsynaptic response to GABA or an increase in inhibitory synaptic transmission impinging on granule cells. Although extensive mossy fiber sprouting into supragranular layers is also present 2 weeks after seizures (Sperber et al., 1991), the possibility that these new collaterals drive inhibitory interneurons to produce enhanced inhibition is not supported by the time courses of these alterations. Enhanced paired-pulse inhibition arises within 24 hr after seizure termination, plateaus rapidly, and returns to preseizure levels after 4–5 weeks (de Jonge and Racine, 1987; Maru and Goddard, 1987; Gilbert, 1991; Milgram et al., 1991; Spiller and Racine, 1994). In contrast, aberrant supragranular Timm staining requires at least 4 d to develop, increases in intensity over several weeks, and appears to be permanent (Cavazos et al., 1991; Mello et al., 1993; Okazaki et al., 1995). The new mossy fiber collaterals appear to terminate on granule neurons, not inhibitory basket cells (Represa et al., 1993; Okazaki et al., 1995), forming new excitatory feedback inputs to granule cells (Tauck and Nadler, 1985; Cronin et al., 1992). Indeed, enhanced granule cell inhibition might function to counter this new excitation directly.Cronin et al. (1992) have observed that dentate responses in slices from KA status rats appear normal in control solutions, but exhibited abnormal hyperactivity when GABAergic inhibition was blocked. The rapid development of enhanced inhibition suggests that it may be independent of extensive anatomic alteration.
Results differ about whether seizures enhance early [GABAA receptor-mediated inhibition (Milgram et al., 1991; Otis et al., 1994; Spiller and Racine, 1994)], late [GABAB receptor-mediated inhibition (Oliver and Miller, 1985)], or both (Tuff et al., 1983; de Jonge and Racine, 1987;Gilbert, 1991; Sperber, 1991). Oliver and Miller (1985) found that kindling selectively enhances a Cl−-independent late component of paired-pulse inhibition, consistent with GABAB receptor enhancement, whereas Otis and Mody (1994) described a kindling-induced enhancement of postsynaptic GABAA receptor activation, possibly attributable to an increase in GABAA receptor number. In the present studies, we show that KA seizure-induced enhanced inhibition encompasses both early and late components of paired-pulse inhibition, suggesting that there is an increased postsynaptic activation of both GABAA and GABAB receptors.
We assessed the relative involvement of GABAA and GABAB inhibition and paired-pulse facilitation in enhanced inhibition by the selective removal of the early inhibitory component. Blockade of GABAA inhibition with bicuculline unmasked paired-pulse facilitation (Steffensen and Henriksen, 1991) in both controls and KA-treated rats. Because paired-pulse modulation represents the summation of inhibition and facilitation, the apparent enhancement of inhibition also could be attributable to a decrease of paired-pulse facilitation. Facilitation, produced by accumulation of Ca2+ in presynaptic terminals (Zucker, 1993) or NMDA receptor activation (Joy and Albertson, 1993), has a time course consistent with the apparent enhancement of inhibition after seizures. However, our data show that paired-pulse facilitation was not different after KA seizures and, therefore, did not contribute to enhanced inhibition. Because bicuculline blocked only the early component of seizure-enhanced inhibition, while not affecting the late phase, we conclude that enhanced inhibition after KA seizures consists of increases in postsynaptic responses of both GABAA and GABAB receptors, and not a decrease in facilitation or granule cell excitability.
The differences in GABAA and GABAB receptor structure (Barnard et al., 1992;Kuriyama et al., 1993; Stephenson, 1995) and signal transduction mechanisms (Alger and Nicoll, 1982; Schofield et al., 1987; Dutar and Nicoll, 1988) make it less likely that the enhancement of both responses is attributable to a single postsynaptic modification. A simpler hypothesis is that inhibitory output of GABAergic interneurons is increased after KA seizures, producing enhanced levels of synaptic GABA release that act indiscriminately on both GABAA and GABAB receptors. This hypothesis is supported by our data using the GABAB antagonist CGP 35348. Although a blocker of postsynaptic GABAB receptors on granule cells would be expected to remove late paired-pulse inhibition selectively, CGP 35348 increased equally both early and late inhibition. These results demonstrate that CGP 35348 blocks GABABreceptors that normally suppress GABA release. The similarity in time course of CGP 35348- and KA seizure-induced enhancement of inhibition, as well as the relative ineffectiveness of CGP 35348 to enhance inhibition further in slices from KA-treated rats, suggests that both treatments alter inhibition through the same mechanism, i.e., a reduction in functional activation of GABAB receptors presynaptic to granule neurons.
Presynaptic GABAB receptors have been shown to be responsible, in part, for the suppression of IPSPs during repeated stimulation, a phenomenon called disinhibition (Deisz and Prince, 1989;Thompson and Gahwiler, 1989; Brucato et al., 1992; Mott et al., 1993;Lambert and Wilson, 1994; Olpe et al., 1994). The involvement of a population of GABAB receptors that regulates GABA release is supported further by our finding that a CGP 35348-sensitive component of disinhibition of recurrent inhibition is decreased after KA status. Disinhibition in slices from KA status rats and control slices treated with CGP 35348 was decreased significantly at ISIs from 30 to 180 msec, whereas disinhibition at longer intervals was unaltered. The selective loss of an early component of disinhibition by the downregulation of mechanisms that suppress GABA release would lead to greater than normal GABA output and enhanced postsynaptic inhibition. The ability of CGP 35348 to further reduce disinhibition in slices from KA-treated rats, but to a lesser extent than controls, demonstrates that although GABAB autoreceptor function may be downregulated, it was not completely absent. The effects of KA seizures and CGP 35348 support the theory that there are at least two components to disinhibition, one seizure- and CGP 35348-sensitive, at ISIs shorter than 200 msec, and one seizure- and CGP 35348-insensitive, at intervals longer than 200 msec (Lambert and Wilson, 1994; Olpe et al., 1994). The lack of CGP 35348- or KA-induced reduction in disinhibition at ISIs of 10–20 msec may suggest that an alternative mechanism underlies enhanced inhibition at short intervals.
There are three GABAB receptor populations presynaptic to granule neurons in the polysynaptic feedback inhibitory circuit that could control disinhibition and for which downregulation would enhance inhibition: (1) autoreceptors on GABAergic terminals synapsing directly on granule cells (Davies et al., 1990; Mott et al., 1993), (2) presynaptic receptors on glutamatergic terminals, which synapse on inhibitory interneurons (Collins et al., 1982), and (3) somatic or dendritic receptors directly on inhibitory interneurons (Misgeld et al., 1989). In the hippocampus, the release of both GABA (Davies et al., 1990; Mott et al., 1993) and glutamate (Harrison et al., 1990) has been shown to be suppressed by the activation of GABAB receptors on presynaptic terminals. Our finding that paired monosynaptic IPSPs did not show a decrease in disinhibition after KA status discounts a role for autoreceptors on the presynaptic GABAergic terminals synapsing directly on granule neurons. In contrast, Buhl et al. (1996) have reported recently that kindling did produce a decrease in disinhibition of monosynaptic GABAB-mediated IPSCs in granule cells, suggesting that altered GABAB autoreceptor function may contribute to enhanced inhibition in kindled seizures.
Although GABAB autoreceptors directly presynaptic to granule cells have been shown to be involved in disinhibition, other GABAB receptor populations in the polysynaptic inhibitory pathway also mediate frequency-dependent suppression of inhibition. For example, the GABAB agonist baclofen depresses polysynaptic IPSPs to a greater extent than monosynaptically evoked IPSPs (Mott et al., 1993). GABA release from interneurons synapsing on granule cells can be modulated by GABAB receptors at excitatory and inhibitory inputs to these interneurons. In the polysynaptic recurrent inhibitory circuit, mossy fiber collaterals either directly activate inhibitory interneurons or activate glutamatergic mossy cells, which then drive the interneurons. Activation of presynaptic GABABreceptors on mossy fiber terminals or axonal terminals of mossy cells would suppress glutamate release and, therefore, decrease excitation of inhibitory interneurons (Collins et al., 1982). A decrease of these presynaptic GABAB receptors on glutamate terminals would allow more glutamate release and heightened activation of interneurons. Such an increase in excitatory activation of inhibitory interneurons has also been demonstrated after kindling (Buhl et al., 1996). However, Misgeld et al. (1989) found that the GABAB agonist baclofen did not effect mossy fiber-evoked EPSPs in hilar inhibitory interneurons, suggesting an absence of GABAB receptors on these glutamatergic synapses. Interneuron excitability also is controlled by GABAergic synapses from other inhibitory neurons (Scharfman et al., 1990). GABAB agonists hyperpolarize hilar interneurons, which mediate recurrent inhibition in granule cells (Misgeld et al., 1989). Thus, it is possible that downregulation of postsynaptic GABAB receptors on interneurons mediates the KA-induced loss of disinhibition and enhanced paired-pulse inhibition.
In conclusion, our data indicate that the mechanism for seizure-induced enhancement of dentate inhibition is, at least in part, through downregulation of GABAB receptors in the polysynaptic recurrent inhibitory circuit. These GABAB receptors normally cause suppression of GABAergic transmission, or disinhibition, with repetitive stimulation. Downregulation of a single receptor population could be induced rapidly, consistent with the time course of the appearance of enhanced inhibition, and would not require slower mechanisms such as sprouting and synaptogenesis. Such a mechanism would be well suited to enhance both early and late inhibition during trains of inputs, as might be produced in a hyperexcitable, epileptic circuit. Because enhanced inhibition develops while other seizure-induced alterations promote excitability elsewhere in the hippocampus, this may be an important compensatory mechanism in limiting seizure spread.
Footnotes
This work was supported in part by National Institutes of Health Training Grant T32DK07513 (K.Z.H.), the Klingenstein Foundation, the Office of Naval Research (P.K.S.), and National Institute of Neurological Diseases and Stroke Research Grants NS-20253 (S.L.M.) and NS-30387 (E.F.S.). Data in this paper are from a thesis to be submitted in partial fulfillment of the requirements for the degree of Doctorate of Philosophy in the Sue Golding Graduate Division of Medical Sciences, Albert Einstein College of Medicine, Yeshiva University.
Correspondence should be addressed to Kurt Z. Haas, Albert Einstein College of Medicine, Laboratory of Developmental Epilepsy, 316 Kennedy Center, 1410 Pelham Parkway South, Bronx, NY 10461-1602.
REFERENCES
- 1.Alger BE, Nicoll RA. Feed-forward dendritic inhibition in rat hippocampal pyramidal cells studied in vitro. J Physiol (Lond) 1982;328:105–123. doi: 10.1113/jphysiol.1982.sp014255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnard EA, Bateson AN, Darlison MG, Glencorse TA, Harvey RJ, Hicks AA, Lasham A, Shingai R, Usherwood PN, Vreugdenhil El El. Genes for the GABAA receptor subunit types and their expression. Adv Biochem Psychopharmacol. 1992;47:17–27. [PubMed] [Google Scholar]
- 3.Bekenstein J, Rempe D, Lothman E. Decreased heterosynaptic and homosynaptic paired pulse inhibition in the rat hippocampus as a chronic sequela to limbic status epilepticus. Brain Res. 1993;601:111–120. doi: 10.1016/0006-8993(93)91701-s. [DOI] [PubMed] [Google Scholar]
- 4.Buhl EH, Otis TS, Mody I. Zinc-induced collapse of augmented inhibition by GABA in a temporal lobe epilepsy model. Science. 1996;271:369–373. doi: 10.1126/science.271.5247.369. [DOI] [PubMed] [Google Scholar]
- 5.Brucato FH, Morrisett RA, Wilson WA, Swartzwelder HS. The GABAB receptor antagonist, CGP-35348, inhibits paired-pulse disinhibition in the rat dentate gyrus in vivo . Brain Res. 1992;588:150–153. doi: 10.1016/0006-8993(92)91355-i. [DOI] [PubMed] [Google Scholar]
- 6.Brunner H, Misgeld U. Synaptic activation in guinea-pig dentate area: dependence on the stimulation site. Pflügers Arch. 1993;423:497–503. doi: 10.1007/BF00374947. [DOI] [PubMed] [Google Scholar]
- 7.Cavazos JE, Golarai G, Sutula TP. Mossy fiber synaptic reorganization induced by kindling: time course of development, progression, and permanence. J Neurosci. 1991;11:2795–2803. doi: 10.1523/JNEUROSCI.11-09-02795.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavazos JE, Sutula TP. Progressive neuronal loss induced by kindling: a possible mechanism for mossy fiber synaptic reorganization and hippocampal sclerosis. Brain Res. 1990;527:1–6. doi: 10.1016/0006-8993(90)91054-k. [DOI] [PubMed] [Google Scholar]
- 9.Collins GG, Anson J, Kelly EP. Baclofen: effects on evoked field potentials and amino acid neurotransmitter release in the rat olfactory cortex slice. Brain Res. 1982;238:371–383. doi: 10.1016/0006-8993(82)90111-1. [DOI] [PubMed] [Google Scholar]
- 10.Cronin J, Obenaus A, Houser CR, Dudek FE. Electrophysiology of dentate granule cells after kainate-induced synaptic reorganization of the mossy fibers. Brain Res. 1992;573:305–310. doi: 10.1016/0006-8993(92)90777-7. [DOI] [PubMed] [Google Scholar]
- 11.Davies CH, Collingridge GL. The physiological regulation of synaptic inhibition by GABABautoreceptors in rat hippocampus. J Physiol (Lond) 1993;472:245–265. doi: 10.1113/jphysiol.1993.sp019945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies CH, Davies SN, Collingridge GL. Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. J Physiol (Lond) 1990;424:513–531. doi: 10.1113/jphysiol.1990.sp018080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies CH, Pozza MF, Collingridge GL. CGP 55845A: a potent antagonist of GABAB receptors in the CA1 region of rat hippocampus. Neuropharmacology. 1993;32:1071–1073. doi: 10.1016/0028-3908(93)90073-c. [DOI] [PubMed] [Google Scholar]
- 14.de Jonge M, Racine RJ. The development and decay of kindling-induced increases in paired-pulse depression in the dentate gyrus. Brain Res. 1987;412:318–328. doi: 10.1016/0006-8993(87)91139-5. [DOI] [PubMed] [Google Scholar]
- 15.Deisz RA, Prince DA. Frequency-dependent depression of inhibition in guinea-pig neocortex in vitro by GABAB receptor feed-back on GABA release. J Physiol (Lond) 1989;412:513–541. doi: 10.1113/jphysiol.1989.sp017629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dutar P, Nicoll RA. A physiological role for GABAB receptors in the central nervous system. Nature. 1988;332:156–158. doi: 10.1038/332156a0. [DOI] [PubMed] [Google Scholar]
- 17.Gilbert ME. Potentiation of inhibition with perforant path kindling: an NMDA-receptor dependent process. Brain Res. 1991;564:109–116. doi: 10.1016/0006-8993(91)91359-9. [DOI] [PubMed] [Google Scholar]
- 18.Goddard GV, McIntyre DC, Leech CK. A permanent change in brain function resulting from daily electrical stimulation. Exp Neurol. 1969;25:295–330. doi: 10.1016/0014-4886(69)90128-9. [DOI] [PubMed] [Google Scholar]
- 19.Haas HL, Schaerer B, Vosmansky H. A simple perfusion chamber for the study of nervous tissue slices in vitro. J Neurosci Methods. 1979;1:323–325. doi: 10.1016/0165-0270(79)90021-9. [DOI] [PubMed] [Google Scholar]
- 20.Haas KZ, Stanton PK, Moshé SL. Contribution of GABAA and GABAB receptors to enhanced dentate paired-pulse inhibition following kainic acid seizures. Soc Neurosci Abstr. 1994;20:408. [Google Scholar]
- 21.Harrison NL, Lovinge DM, Lambert NA, Teyler TJ, Prager R, Ong J, Kerr DI. The actions of 2-hydroxy-saclofen at presynaptic GABAB receptors in the rat hippocampus. Neurosci Lett. 1990;119:272–276. doi: 10.1016/0304-3940(90)90851-y. [DOI] [PubMed] [Google Scholar]
- 22.Holmes GL, Thompson JL. Effects of kainic acid on seizure susceptibility in the developing brain. Brain Res. 1988;467:51–59. doi: 10.1016/0165-3806(88)90066-1. [DOI] [PubMed] [Google Scholar]
- 23.Holmes GL, Chronopoulos A, Stafstrom CE, Mikati MA, Thurber SJ, Hyde PA, Thompson JL. Effects of kindling on subsequent learning, memory, behavior, and seizure susceptibility. Brain Res Dev Brain Res. 1993;73:71–77. doi: 10.1016/0165-3806(93)90047-e. [DOI] [PubMed] [Google Scholar]
- 24.Joy RM, Albertson TE. NMDA receptors have a dominant role in population spike-paired pulse facilitation in the dentate gyrus of urethane-anesthetized rats. Brain Res. 1993;604:273–282. doi: 10.1016/0006-8993(93)90379-2. [DOI] [PubMed] [Google Scholar]
- 25.Kapur J, Michelson HB, Buterbaugh GG, Lothman EW. Evidence for a chronic loss of inhibition in the hippocampus after kindling: electrophysiological studies. Epilepsy Res. 1989;4:90–99. doi: 10.1016/0920-1211(89)90013-2. [DOI] [PubMed] [Google Scholar]
- 26.King GL, Dingledine R, Giacchino JL, McNamara JO. Abnormal neuronal excitability in hippocampal slices from kindled rats. J Neurophysiol. 1985;54:1295–1304. doi: 10.1152/jn.1985.54.5.1295. [DOI] [PubMed] [Google Scholar]
- 27.Kohr G, De Koninck Y, Mody I. Properties of NMDA receptor channels in neurons acutely isolated from epileptic (kindled) rats. J Neurosci. 1993;13:3612–3627. doi: 10.1523/JNEUROSCI.13-08-03612.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kohr G, Mody I. Kindling increases N -methyl-d-aspartate potency at single N -methyl-d-aspartate channels in dentate gyrus granule cells. Neuroscience. 1994;62:975–981. doi: 10.1016/0306-4522(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 29.Kuriyama K, Hirouchi M, Nakayasu H. Structure and function of cerebral GABAA and GABAB receptors. Neurosci Res. 1993;17:91–99. doi: 10.1016/0168-0102(93)90087-7. [DOI] [PubMed] [Google Scholar]
- 30.Lambert NA, Wilson WA. Discrimination of post- and presynaptic GABAB receptor-mediated responses by tetrahydroaminoacridine in area CA3 of the rat hippocampus. J Neurophysiol. 1993;69:630–635. doi: 10.1152/jn.1993.69.2.630. [DOI] [PubMed] [Google Scholar]
- 31.Lambert NA, Wilson WA. Temporally distinct mechanisms of use-dependent depression at inhibitory synapses in the rat hippocampus in vitro. J Neurophysiol. 1994;72:121–130. doi: 10.1152/jn.1994.72.1.121. [DOI] [PubMed] [Google Scholar]
- 32.Lothman EW, Bertram EH., III Epileptogenic effects of status epilepticus. Epilepsia. 1993;34:S59–S70. doi: 10.1111/j.1528-1157.1993.tb05907.x. [DOI] [PubMed] [Google Scholar]
- 33.Martin D, McNamara JO, Nadler JV. Kindling enhances sensitivity of CA3 hippocampal pyramidal cells to NMDA. J Neurosci. 1992;12:1928–1935. doi: 10.1523/JNEUROSCI.12-05-01928.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maru E, Goddard GV. Alteration in dentate neuronal activities associated with perforant path kindling. III. Enhancement of synaptic inhibition. Exp Neurol. 1987;96:46–60. doi: 10.1016/0014-4886(87)90167-1. [DOI] [PubMed] [Google Scholar]
- 35.Mello LE, Cavalheiro EA, Tan AM, Kupfer WR, Pretorius JK, Babb TL, Finch DM. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: cell loss and mossy fiber sprouting. Epilepsia. 1993;34:985–995. doi: 10.1111/j.1528-1157.1993.tb02123.x. [DOI] [PubMed] [Google Scholar]
- 36.Milgram NW, Yearwood T, Khurgel M, Ivy GO, Racine R. Changes in inhibitory processes in the hippocampus following recurrent seizures induced by systemic administration of kainic acid. Brain Res. 1991;551:236–246. doi: 10.1016/0006-8993(91)90938-r. [DOI] [PubMed] [Google Scholar]
- 37.Milgram NW, Michael M, Cammisuli S, Head E, Ferbinteanu J, Reid C, Murphy MP, Racine R. Development of spontaneous seizures over extended electrical kindling. II. Persistence of dentate inhibitory suppression. Brain Res. 1995;670:112–120. doi: 10.1016/0006-8993(94)01277-o. [DOI] [PubMed] [Google Scholar]
- 38.Misgeld U, Muller W, Brunner H. Effects of (−)baclofen on inhibitory neurons in the guinea pig hippocampal slice. Pflügers Arch. 1989;414:139–144. doi: 10.1007/BF00580955. [DOI] [PubMed] [Google Scholar]
- 39.Mody I, Stanton PK, Heinemann U. Activation of N -methyl-d-aspartate receptors parallels changes in cellular and synaptic properties of dentate gyrus granule cells after kindling. J Neurophysiol. 1988;59:1033–1054. doi: 10.1152/jn.1988.59.3.1033. [DOI] [PubMed] [Google Scholar]
- 40.Mott DD, Xie CW, Wilson WA, Swartzwelder HS, Lewis DV. GABAB autoreceptors mediate activity-dependent disinhibition and enhance signal transmission in the dentate gyrus. J Neurophysiol. 1993;69:674–691. doi: 10.1152/jn.1993.69.3.674. [DOI] [PubMed] [Google Scholar]
- 41.Okazaki MM, Evenson DA, Nadler JV. Hippocampal mossy fiber sprouting and synapse formation after status epilepticus in rats: visualization after retrograde transport of biocytin. J Comp Neurol. 1995;352:515–534. doi: 10.1002/cne.903520404. [DOI] [PubMed] [Google Scholar]
- 42.Oliver MW, Miller JJ. Alterations of inhibitory processes in the dentate gyrus following kindling-induced epilepsy. Exp Brain Res. 1985;57:443–447. doi: 10.1007/BF00237830. [DOI] [PubMed] [Google Scholar]
- 43.Olpe HR, Karlsson G, Pozza MF, Brugger F, Steinmann M, Van Riezen H, Fagg G, Hall RG, Froestl W, Bittiger H. CGP 35348: a centrally active blocker of GABAB receptors. Eur J Pharmacol. 1990;187:27–38. doi: 10.1016/0014-2999(90)90337-6. [DOI] [PubMed] [Google Scholar]
- 44.Olpe HR, Steinmann MW, Greiner K, Pozza MF. Contribution of presynaptic GABA-B receptors to paired-pulse depression of GABA-responses in the hippocampus. Naunyn Schmiedebergs Arch Pharmacol. 1994;349:473–477. doi: 10.1007/BF00169135. [DOI] [PubMed] [Google Scholar]
- 45.Otis TS, De Koninck Y, Mody I. Lasting potentiation of inhibition is associated with an increased number of gamma-aminobutyric acid type A receptors activated during miniature inhibitory postsynaptic currents. Proc Natl Acad Sci USA. 1994;91:7698–7702. doi: 10.1073/pnas.91.16.7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pearce RA, Grunder SD, Faucher LD. Different mechanisms for use-dependent depression of two GABAA-mediated IPSCs in rat hippocampus. J Physiol (Lond) 1995;484:425–435. doi: 10.1113/jphysiol.1995.sp020675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pitler TA, Alger BE. Differences between presynaptic and postsynaptic GABAB mechanisms in rat hippocampal pyramidal cells. J Neurophysiol. 1994;72:2317–2327. doi: 10.1152/jn.1994.72.5.2317. [DOI] [PubMed] [Google Scholar]
- 48.Represa A, Jorquera I, Le Gal La Salle G, Ben-Ari Y. Epilepsy induced collateral sprouting of hippocampal mossy fibers: does it induce the development of ectopic synapses with granule cell dendrites? Hippocampus. 1993;3:257–268. doi: 10.1002/hipo.450030303. [DOI] [PubMed] [Google Scholar]
- 49.Scanziani M, Capogna M, Gahwiler BH, Thompson SM. Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron. 1992;9:919–927. doi: 10.1016/0896-6273(92)90244-8. [DOI] [PubMed] [Google Scholar]
- 50.Scharfman HE, Kunkel DD, Schwartzkroin PA. Synaptic connections of dentate granule cells and hilar neurons: results of paired intracellular recordings and intracellular horseradish peroxidase injections. Neuroscience. 1990;37:693–707. doi: 10.1016/0306-4522(90)90100-i. [DOI] [PubMed] [Google Scholar]
- 51.Schofield PR, Darlison MG, Fujita N, Burt DR, Stephenson FA, Rodriguez H, Rhee LM, Ramachandran J, Reale V, Glencorse TA. Sequence and functional expression of the GABA A receptor shows a ligand-gated receptor super-family. Nature. 1987;328:221–227. doi: 10.1038/328221a0. [DOI] [PubMed] [Google Scholar]
- 52.Sloviter RS. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science. 1987;235:73–76. doi: 10.1126/science.2879352. [DOI] [PubMed] [Google Scholar]
- 53.Sloviter RS. Possible functional consequences of synaptic reorganization in the dentate gyrus of kainate-treated rats. Neurosci Lett. 1992;137:91–96. doi: 10.1016/0304-3940(92)90306-r. [DOI] [PubMed] [Google Scholar]
- 54.Sperber EF, Haas KZ, Stanton PK, Moshé SL. Resistance of the immature hippocampus to seizure-induced synaptic reorganization. Brain Res Devel Brain Res. 1991;60:88–93. doi: 10.1016/0165-3806(91)90158-f. [DOI] [PubMed] [Google Scholar]
- 55.Spiller AE, Racine RJ. The effect of kindling beyond the “stage 5” criterion on paired-pulse depression and hilar cell counts in the dentate gyrus. Brain Res. 1994;635:139–147. doi: 10.1016/0006-8993(94)91433-8. [DOI] [PubMed] [Google Scholar]
- 56.Stafstrom CE, Thompson JL, Holmes GL. Kainic acid seizures in the developing brain: status epilepticus and spontaneous recurrent seizures. Brain Res Devel Brain Res. 1992;65:227–236. doi: 10.1016/0165-3806(92)90184-x. [DOI] [PubMed] [Google Scholar]
- 57.Steffensen SC, Henriksen SJ. Effects of baclofen and bicuculline on inhibition in the fascia dentata and hippocampus region superior. Brain Res. 1991;538:46–53. doi: 10.1016/0006-8993(91)90374-5. [DOI] [PubMed] [Google Scholar]
- 58.Stephenson FA. The GABAAreceptors. Biochem J. 1995;310:1–9. doi: 10.1042/bj3100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sutula T, He XX, Cavazos J, Scott G. Synaptic reorganization in the hippocampus induced by abnormal functional activity. Science. 1988;239:1147–1150. doi: 10.1126/science.2449733. [DOI] [PubMed] [Google Scholar]
- 60.Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thompson SM, Gahwiler BH. Activity-dependent disinhibition. III. Desensitization and GABABreceptor-mediated presynaptic inhibition in the hippocampus in vitro. J Neurophysiol. 1989;61:524–533. doi: 10.1152/jn.1989.61.3.524. [DOI] [PubMed] [Google Scholar]
- 62.Thompson SM, Gahwiler BH. Comparison of the actions of baclofen at pre- and postsynaptic receptors in the rat hippocampus in vitro. J Physiol (Lond) 1992;451:329–345. doi: 10.1113/jphysiol.1992.sp019167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tuff LP, Racine RJ, Adamec R. The effects of kindling on GABA-mediated inhibition in the dentate gyrus of the rat. I. Paired-pulse depression. Brain Res. 1983;277:79–90. doi: 10.1016/0006-8993(83)90909-5. [DOI] [PubMed] [Google Scholar]
- 64.Wong BY, Moshé SL. Mutual interactions between repeated flurothyl convulsions and electrical kindling. Epilepsy Res. 1987;1:159–164. doi: 10.1016/0920-1211(87)90036-2. [DOI] [PubMed] [Google Scholar]
- 65.Zhao D, Leung LS. Hippocampal kindling induced paired-pulse depression in the dentate gyrus and paired-pulse facilitation in CA3. Brain Res. 1992;582:163–167. doi: 10.1016/0006-8993(92)90333-5. [DOI] [PubMed] [Google Scholar]
- 66.Zucker RS. Calcium and transmitter release. J Physiol (Paris) 1993;87:25–36. doi: 10.1016/0928-4257(93)90021-k. [DOI] [PubMed] [Google Scholar]