

Abstract
The nonphosphorylated neurofilament marker SMI-32 stains motor neurons in spinal cord slices and stains a subset of cultured spinal neurons [“large SMI-32(+) neurons”], which have a morphology consistent with motor neurons identified in vitro: large cell body, long axon, and extensive dendritic arborization. They are found preferentially in ventral spinal cord cultures, providing further evidence that large SMI-32(+) neurons are indeed motor neurons, and SMI-32 staining often colocalizes with established motor neuron markers (including acetylcholine, calcitonin gene-related peptide, and peripherin). Additionally, choline acetyltransferase activity (a frequently used index of the motor neuron population) and peripherin(+) neurons share with large SMI-32(+) neurons an unusual vulnerability to AMPA/kainate receptor-mediated injury. Kainate-induced loss of these motor neuron markers is Ca2+-dependent, which supports a critical role of Ca2+ ions in this injury. Raising extracellular Ca2+exacerbates injury, whereas removal of extracellular Ca2+ is protective. A basis for this vulnerability is provided by the observation that most peripherin(+) neurons, like large SMI-32(+) neurons, are subject to kainate-stimulated Co2+ uptake, a histochemical stain that identifies neurons possessing Ca2+-permeable AMPA/kainate receptor-gated channels. Finally, of possibly greater relevance to the slow motor neuronal degeneration in diseases, both large SMI-32(+) neurons and peripherin(+) neurons are selectively damaged by prolonged (24 hr) low-level exposures to kainate (10 μm) or to the glutamate reuptake blockerl-trans-pyrrolidine-2,4-dicarboxylic acid (100 μm). During these low-level kainate exposures, large SMI-32(+) neurons showed higher intracellular Ca2+ concentrations than most spinal neurons, suggesting that Ca2+ ions are also important in this more slowly evolving injury.
Keywords: cell culture, AMPA, kainate, glutamate, motor neuron, calcium, SMI-32, peripherin, ChAT, neurotoxicity, cobalt, calcium imaging
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the progressive loss of upper (Betz cells) and lower (ventral horn) motor neurons. Although of unknown cause, findings of abnormalities in the uptake (Rothstein et al., 1992) or metabolism (Plaitakis and Caroscio, 1987; Hugon et al., 1989a; Rothstein et al., 1990) of glutamate and related excitatory amino acids suggest that excitotoxic injury may be a contributory factor.
Glutamate can kill neurons through effects at both NMDA and AMPA/kainate types of ionotropic receptors. Although NMDA receptors may mediate most acute neuronal injury, several factors suggest that AMPA/kainate receptors may be of greater importance to the slow neurodegeneration that occurs in ALS (Weiss and Choi, 1991). First, three syndromes with prominent motor system manifestations, i.e., lathyrism (Spencer et al., 1986), domoic acid toxicity (Teitelbaum et al., 1990), and BMAA toxicity (Spencer et al., 1987), are all linked to the consumption of environmental AMPA/kainate receptor agonists (Ross et al., 1987; Bridges et al., 1989; Debonnel et al., 1989; Richter and Mena, 1989; Weiss et al., 1989). Second, motor neurons are injured preferentially by intrathecal kainate (Hugon et al., 1989b), and selective AMPA/kainate receptor antagonists protect motor neurons against degeneration caused by chronic blockade of glutamate uptake in spinal cord slice (Rothstein et al., 1993).
Studies in culture have revealed that most central neurons are injured lethally by brief periods of NMDA receptor activation. This injury is Ca2+-dependent and likely reflects the high Ca2+ permeability of the NMDA receptor-gated channel (MacDermott et al., 1986). In contrast, AMPA/kainate receptors are generally Ca2+-impermeable and require longer periods (several hours) of activation for comparable widespread neuronal injury to occur (Choi, 1992). Certain populations of central neurons, however, possess Ca2+-permeable AMPA/kainate channels (Iino et al., 1990; Pruss et al., 1991; Brorson et al., 1992), a feature that may confer an enhanced intrinsic vulnerability to AMPA/kainate receptor-mediated injury (Koh and Choi, 1988;Brorson et al., 1994; Turetsky et al., 1994; Weiss et al., 1994a; Yin et al., 1994a,b).
SMI-32, a marker of nonphosphorylated neurofilaments, stains motor neurons in spinal cord slice (Gotow and Tanaka, 1994; Carriedo et al., 1995). In dissociated spinal cultures, we found SMI-32 to label a subset of neurons in vitro that had morphological characteristics of identified motor neurons [“large SMI-32(+) neurons”]), which were unusually vulnerable to rapidly triggered, Ca2+- dependent, AMPA/kainate receptor-mediated injury (Carriedo et al., 1995).
The present project extends these initial studies in several ways. First, we use anatomic considerations and labeling with other motor neuron markers to strengthen the hypothesis that large SMI-32(+) neurons are indeed motor neurons. Other experiments use choline acetyltransferase (ChAT) enzymatic activity and peripherin immunocytochemistry (both known indices of the motor neuronal population), in addition to SMI-32 staining, to examine further the role of Ca2+ ions in AMPA/kainate receptor-mediated injury to putative motor neurons in culture. Finally, we explored the vulnerability of large SMI-32(+) and peripherin(+) neurons to injury resulting from prolonged low-level exposures to kainate or the glutamate reuptake blockerl-trans-pyrrolidine-2,4-dicarboxylic acid (PDC) (Rothstein et al., 1993), a type of injury possibly more relevant to the chronic motor neuron degeneration seen in disease states such as ALS.
MATERIALS AND METHODS
Spinal cord cultures. Mixed spinal cord cultures were prepared, largely as described previously (Regan and Choi, 1991), from 13-d-old mouse embryos, except that the cultures were plated on a preestablished monolayer of cortical astrocytes. Recent studies have indicated that astrocytes (Eagleson et al., 1985; Schaffner et al., 1987; Martinou et al., 1989a; Ang et al., 1992) or mixed spinal cell conditioned media (Calof and Reichardt, 1984; Dohrmann et al., 1987) provide trophic support for and markedly enhance the survival of motor neurons in culture.
Both meninges and dorsal root ganglia were removed from spinal cords before the cords were plated in media consisting of Eagle’s minimal essential medium (MEM) (Earle’s salts, supplied glutamine-free serum; Gibco, Grand Island, NY) supplemented with 10% heat-inactivated horse serum (Gibco), 10% fetal bovine serum (Gibco), glutamine (2 mm) (Gibco), and glucose (total 25 mm) on an astrocyte monolayer in 15 mm Primaria-coated culture plates (Falcon, Franklin Lake, NJ) at a density of 3 “spinal cords” per 24-well plate (∼1–2 × 105 cells/cm2). Cultures were maintained at 37°C in a 5% CO2 incubator. After 4–6 d in vitro, non-neuronal cell division was halted by exposure to 10−5mcytosine arabinoside for 1–3 d. The cells were then shifted into a maintenance medium identical to the plating medium but lacking fetal serum. Subsequent media replacement occurred twice a week. Cultures were studied after 13–15 d in vitro.
Immunocytochemical labeling. For labeling motor neurons in spinal cord slices, spinal cords from adult mice were removed surgically and fixed in 4% paraformaldehyde overnight before paraffin embedding. Spinal cords were cut into 6 μm sections, deparaffinized, and quenched of endogenous peroxidase activity (by incubation in 0.3% H2O2 in absolute methanol). Slices were then incubated with primary antiserum (in PBS containing 10% serum from the species in which the secondary antibody was made, to minimize background staining) for 72 hr at 4°C: SMI-32 (1:1000 dilution; mouse monoclonal, Sternberger Monoclonals, Baltimore, MD), calcitonin gene-related peptide (CGRP) (1:2000 dilution; rabbit monoclonal, Amersham International, Arlington Heights, IL), peripherin (1:200 dilution; rabbit polyclonal, Chemicon International, Temecula, CA), ChAT (1:200 dilution; mouse monoclonal, Chemicon International), and acetylcholine (ACh) (1:2000 dilution; rabbit monoclonal, Biodesign International, Kennebunkport, ME). Appropriate biotinylated secondary antibody (Vector Laboratories, Burlingame, CA), avidin, and biotinylated horseradish peroxidase macromolecular complex (ABC solution, Vector), and diaminobenzidine (Sigma) were used to visualize stained cells.
For staining, cultures were fixed for 40 min in 4% paraformaldehyde, washed three times with PBS, and incubated (30 min) with the appropriate blocking solution (10% serum in PBS) to minimize background staining. For SMI-32 and peripherin, the blocking solution included 0.2% Triton X-100. For CGRP staining, cultures were pretreated the day before with the axoplasmic transport inhibitor colchicine (10 μm), a maneuver that has been shown to increase staining intensity markedly (Juurlink et al., 1990). Primary antibody exposures were in “blocking solution” (with 10% serum) at the following concentrations: SMI-32 (1:6000), peripherin (1:2000), ACh (1:6000), CGRP (1:7000), and ChAT (1:2000). SMI-32 and peripherin exposures were carried out overnight at room temperature, whereas other antibody exposures were carried out for 48–72 hr at 4°C. Biotinylated secondary antibody (Vector Laboratories), ABC solution (Vector), and 3-amino-9-ethyl-carbazole (AEC) (Sigma) were used to visualize stained cells. For double staining, cultures were exposed simultaneously to SMI-32 antibody (1:1000 dilution) and antibody to either ACh, CGRP, or peripherin. SMI-32 staining was visualized under fluorescence microscopy (excitation, 510–560 nm; emission, >590 nm) after exposure to CY3-conjugated anti-mouse secondary antibody (1:100 dilution; Jackson Research, Westgrove, PA) for 1 hr. Visualization of the second stain was with biotinylated secondary antibody, ABC solution, and AEC (as described above). Controls omitting one or the other primary antibody revealed no cross-reactivity between the stains.
Co2+ labeling. Co2+labeling was carried out as described (Pruss et al., 1991; Turetsky et al., 1994), with minor modifications. Cultures were Co2+-loaded by exposure to kainate (100 μm) with Co2+ (2.5 mm) in uptake buffer (139 mm sucrose, 57.5 mm NaCl, 5 mm KCl, 2 mmMgCl2, 1 mmCaCl2, 12 mm glucose, 10 mm HEPES, pH 7.6) for 15 min. Cultures were then washed in uptake buffer with 3 mm EDTA to remove extracellular Co2+, incubated in 0.05% (NH4)2S for 5 min to precipitate intracellular Co2+, washed three times in uptake buffer, and subjected to fixation (4% paraformaldehyde, 30 min). For silver enhancement, cultures were washed three times in development buffer (292 mmsucrose, 15.5 mm hydroquinone, 42 mm citric acid) and incubated in 0.1% AgNO3 in development buffer at 50–55°C. This solution was changed at 15 min intervals, and enhancement was monitored periodically by microscopic observation. When enhancement was complete (usually after 30–50 min), the reaction was terminated by washing the cultures three times in warm development buffer.
Neurotoxicity experiments. Brief (15 min) toxic exposures were carried out in room air (25°C), using a HEPES-buffered salt solution (HSS) with the following composition (in mm): 130 Na+, 5.4 K+, 0.8 Mg2+, 1.8 Ca2+ (except where indicated), 130.6 Cl−, 20 HEPES, pH 7.4, at 25°C, and 15 glucose. Acute exposures (10–15 min) were terminated by replacing the exposure solution with MEM + glucose, along with 10 μm of the ionotropic glutamate receptor antagonists MK-801 (Research Biochemicals International, Natick, MA) and 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline (NBQX) (kindly provided by Novo Nordisk, Malov, Denmark), and returning the cultures to the 5% CO2 incubator at 37°C. Longer (24 hr) exposures to kainate or the glutamate reuptake blocker PDC (Tocris Cookson, Bristol, England) were in MEM + glucose in the 5% CO2 incubator at 37°C. MK-801 (10 μm) was added during all kainate exposures to prevent the activation of NMDA receptors from endogenously released glutamate and thereby to ensure a pure AMPA/kainate receptor-mediated injury mechanism.
With both acute and chronic exposures, overall neuronal injury was assessed 20–24 hr after the start of the exposure, by both morphological examination and quantitative measurement of lactate dehydrogenase (LDH) in the bathing medium, an index that is proportional to the total number of neurons damaged by excitotoxic exposure (Koh and Choi, 1987). LDH values were scaled to the near maximal mean value found in sister cultures exposed to 300 μm kainate for 24 hr (=100% cell loss).
Cultures were then fixed and stained for either SMI-32 or peripherin. Damage to labeled neurons from excitotoxic exposures was evaluated by direct counts of all stained neurons. Specific destruction of large SMI-32(+) or peripherin(+) neurons was assessed as the difference between the mean number of intact cells in neuronal cultures exposed to an excitotoxic agonist and the mean number in several sister cultures exposed to sham wash alone, expressed as a percentage of the latter.
ChAT activity assay. Overall neuronal injury was assessed by measurement of LDH release (as described above) before ChAT activity assays were performed. ChAT activity was assayed exactly as described previously (Weiss et al., 1994b), according to methods adopted fromFonnum (1975). [3H]acetyl-CoA (0.125 μCi/sample; DuPont NEN, Boston, MA) was used as reaction substrate, and counts revealed incorporation into [3H]ACh.
After subtracting background counts, specific loss of ChAT activity was assessed as the difference between the mean ChAT activities in sham-washed and kainate-exposed cultures and scaled to the near maximal loss of activity in sister cultures exposed to 300 μm kainate for 24 hr (=100% loss).
Intracellular free Ca2+([Ca2+]i) measurements. For Ca2+ imaging studies, cultures were prepared as described above except that cells were plated on poly-l-lysine-coated glass-bottomed plates (Plastek Cultureware, Ashland, MA). Cultures were Fura-2-loaded by exposure to 5 μm Fura-2 AM (Molecular Probes, Eugene, OR) with 0.1% pluronic acid in HSS in the dark (25°C for 30 min). Cultures were then washed and incubated an additional 30 min in the dark (for deesterification). Cultures were mounted in a microscope-stage adapter, and preselected fields were viewed with an inverted microscope (Nikon Diaphot equipped with xenon epifluorescence optics) at 200×. Cells were alternately illuminated with 340 and 380 nm light from a xenon source, and fluorescence (at 510 nm) was imaged by a Hamamatsu intensified CCD camera. Data were gathered on an 80486-based computer using fluor software from Universal Imaging (West Chester, PA). [Ca2+]i was determined by the equation [Ca2+]i = Kd(Fmin/Fmax){(R − Rmin)}/{(Rmax − R)}, using Kd = 224 nm. The system was recalibrated after any adjustments to the apparatus.
Imaging experiments were conducted at room temperature in a 2 ml static bath of HSS. After obtaining baseline values, kainate (10 μm) and MK-801 (10 μm) were added, and [Ca2+]ivalues were obtained every 30 sec for an additional 10 min. Because of the high affinity of Fura-2, peak [Ca2+]i values obtained immediately after the addition of kainate probably mildly underestimate true [Ca2+]i values. Cultures were then fixed and stained for SMI-32. In fields containing large SMI-32(+) neurons, all distinct neurons were analyzed for fluorescence. Only fields containing large SMI-32(+) neurons were analyzed.
Materials. All other chemicals and reagents were obtained from common commercial sources.
RESULTS
SMI-32 and other motor neuron markers label motor neurons in spinal cord slices
The nonphosphorylated neurofilament antibody SMI-32 strongly labels ventral horn motor neurons in spinal cord slices (Gotow and Tanaka, 1994; Carriedo et al., 1995). Initial studies compared SMI-32 labeling of motor neurons in slices with that of various other markers reported to stain motor neurons. Antibodies against the following markers were examined: the neurotransmitter ACh (Geffard et al., 1985), the ACh-synthesizing enzyme ChAT (Hietanen et al., 1990;Wang et al., 1990), peripherin (Parysek and Goldman, 1988; Escurat et al., 1990), a class III intermediate filament protein, and CGRP (Gibson et al., 1984; Kruger et al., 1988; Juurlink et al., 1990). Although each of these markers labeled motor neurons in the ventral horn, the degree of staining varied significantly (Fig. 1). SMI-32 and CGRP showed the most robust staining, although SMI-32 did show some labeling of small neurons outside the ventral horn. Both ChAT and ACh showed weaker staining and also labeled some small neurons in the dorsal horn, a finding that is consistent with previous studies of ChAT staining in spinal cord (Houser et al., 1983). Also consistent with previous studies, peripherin staining in spinal cord slice was faint but seemed to be selective for subsets of motor neurons in ventral horn.
Fig. 1.
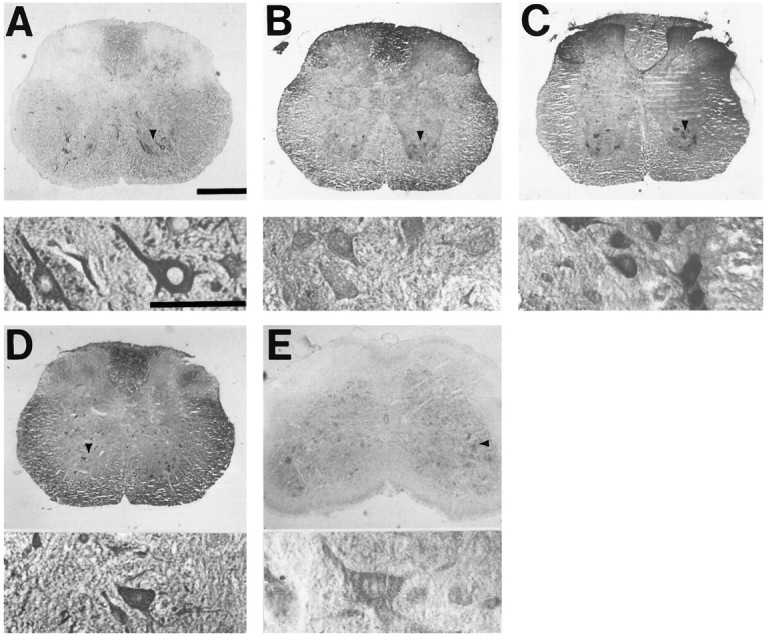
Motor neuron markers label large ventral horn neurons in spinal cord slices. Photomicrographs show slices of adult spinal cord stained (as described) with antibody to SMI-32 (A), peripherin (B), CGRP (C), ACh (D), or ChAT (E). For each marker, thebottom panel shows high-power (400×) detail of neurons indicated by arrowhead in top panel. Scale bar, 300 μm.
SMI-32 and other motor neuron markers label subsets of neurons in spinal cord cultures
Development of culture models to study motor neurons has been hindered by a paucity of well characterized markers that are both specific for motor neurons and show good morphological detail. Consistent with its staining of motor neurons in slice, SMI-32 labeled subsets of neurons in dissociated spinal cultures with the morphological characteristics of cultured motor neurons (Schaffner et al., 1987; Martinou et al., 1989a): a large (>20 μm) cell body, a prominent neuritic arborization, and generally a single long axon-like neurite, often extending over several millimeters (Fig.2). Furthermore, although SMI-32 labeled ∼3% of all neurons in the cultures, only a minority of labeled neurons (∼30%) had these characteristic features (“large SMI-32(+) neurons”) (Carriedo et al., 1995).
Fig. 2.
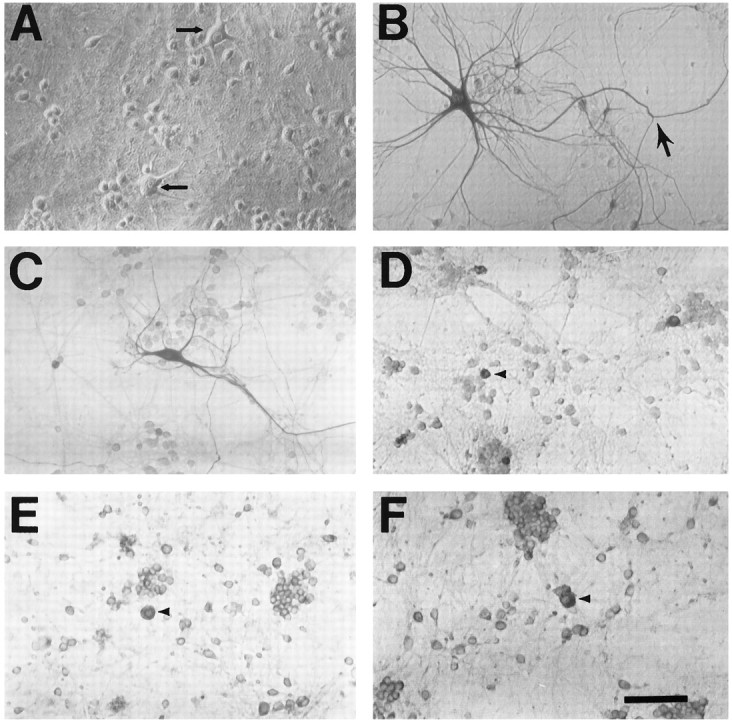
Motor neuron markers label neurons in spinal cord cultures. Photomicrographs show dissociated spinal cord cultures under phase-contrast microscopy (A, arrows show the typical appearance of putative motor neurons before staining) or after immunostaining (as described) with antibody to SMI-32 (B,arrow shows axonal branch), peripherin (C), CGRP (D), ACh (E), or ChAT (F). Note the extensive morphological detail provided by SMI-32 and peripherin staining (generally showing an extensive dendritic arborization as well as a single axon-like process that often extends for several millimeters). In comparison, relatively little morphological detail is provided by the other stains (D–F, arrowheadsindicate representative stained neurons). Scale bar, 100 μm.
Staining with the other motor neuron markers (ChAT, ACh, CGRP, and peripherin) was used to characterize better the motor neuronal population in our cultures (Richards et al., 1995) and to confirm further the presumed motor neuron identity of large SMI-32(+) neurons (Fig. 2). In contrast to the weak staining seen in slice, peripherin(+) neurons (∼0.5% of neurons in the cultures) were often strongly stained and demonstrated extensive morphological detail (often closely resembling large SMI-32(+) neurons). Approximately 1.5% of neurons in the cultures were CGRP(+). As reported previously (Juurlink et al., 1990), however, CGRP staining was obtained only after colchicine pretreatment (see Materials and Methods), a maneuver that increased staining intensity but generally caused a marked disruption of morphological features in our culture system. ChAT (∼0.25% of total neurons) and ACh (∼0.1% of total neurons) gave weaker and more variable staining in culture than in slice and revealed little neuritic morphology. Double immunocytochemistry revealed a majority of ACh(+) neurons (80%), peripherin(+) neurons (68%), and CGRP(+) neurons (60%) to be SMI-32(+) (≥50 labeled cells from ≥3 experiments counted for each marker; Fig. 3), supporting the motor neuron identity of large SMI-32(+) neurons.
Fig. 3.

Top left. Most peripherin(+) neurons are also SMI-32(+). Photomicrographs show a culture double-stained for peripherin and SMI-32 under visible light (A, peripherin staining) and under fluorescence microscopy (B, SMI-32 labeling). A majority of peripherin(+) neurons were found to express SMI-32 immunoreactivity. Other double-labeling studies showed most ACh- or CGRP-immunoreactive neurons to be SMI-32(+) (see Results). Scale bar, 50 μm.
To characterize large SMI-32(+) neurons further, we examined their distribution in spinal cord by preparing separate “ventral” and “dorsal” cultures (by splitting the cord longitudinally before plating). Large SMI-32(+) neurons were found preferentially in ventral cultures [they comprised 3.5% (283/8147) of neurons in ventral, but only 0.4% (14/3341) of neurons in dorsal spinal cord cultures], thus supporting a motor neuronal identity. Also, large SMI-32(+) neurons comprised 78% (283/364) of all SMI-32(+) neurons in ventral but only 17% (14/81) in dorsal spinal cord cultures (data compiled from three experiments).
Acute AMPA/kainate receptor-mediated injury to cultured motor neurons is Ca2+-dependent
We reported previously that removal of Ca2+from the media during brief kainate exposures lessened subsequent degeneration of large SMI-32(+) neurons, as assessed 20–24 hr later (Carriedo et al., 1995). These studies have now been extended. First, we find that raising extracellular Ca2+ (from 1.8 to 10 mm) during the exposure markedly enhances the damage to the SMI-32(+) neurons resulting from brief (10 min, 100 μm) kainate exposures and produces little overall neuronal injury (as assessed by measurement of LDH release) (Figs. 4, 5), further substantiating the critical role of Ca2+ entry in rapidly triggered injury to large SMI-32(+) neurons. In addition, because the motor neuron identity of large SMI-32(+) neurons has not been proven, we repeated Ca2+-dependence experiments with two other indices of the motor neuron population: peripherin immunocytochemistry (Parysek and Goldman, 1988; Escurat et al., 1990) and ChAT enzymatic activity (Schnaar and Schaffner, 1981; Schaffner et al., 1987; Martinou et al., 1989b; Rothstein et al., 1993). As with large SMI-32(+) neurons, both peripherin(+) neurons and ChAT activity were lost preferentially after brief kainate exposures. Furthermore, such rapidly triggered damage to each of these measures was Ca2+-dependent.
Fig. 4.

Kainate injury to peripherin(+) and large SMI-32(+) neurons is Ca2+-dependent: morphological appearance. Spinal cord cultures were exposed to kainate (100 μm for 10 min), in either the presence of 1.8 mm Ca2+ (A, C) or the absence of Ca2+ (B, D), and were stained 24 hr later for either peripherin (A, B) or SMI-32 (C, D). Although these submaximal exposures in the presence of Ca2+ caused severe damage to many (but not all) labeled neurons, removal of Ca2+during the exposure resulted in good preservation of most neurons. Scale bar, 100 μm.
Fig. 5.
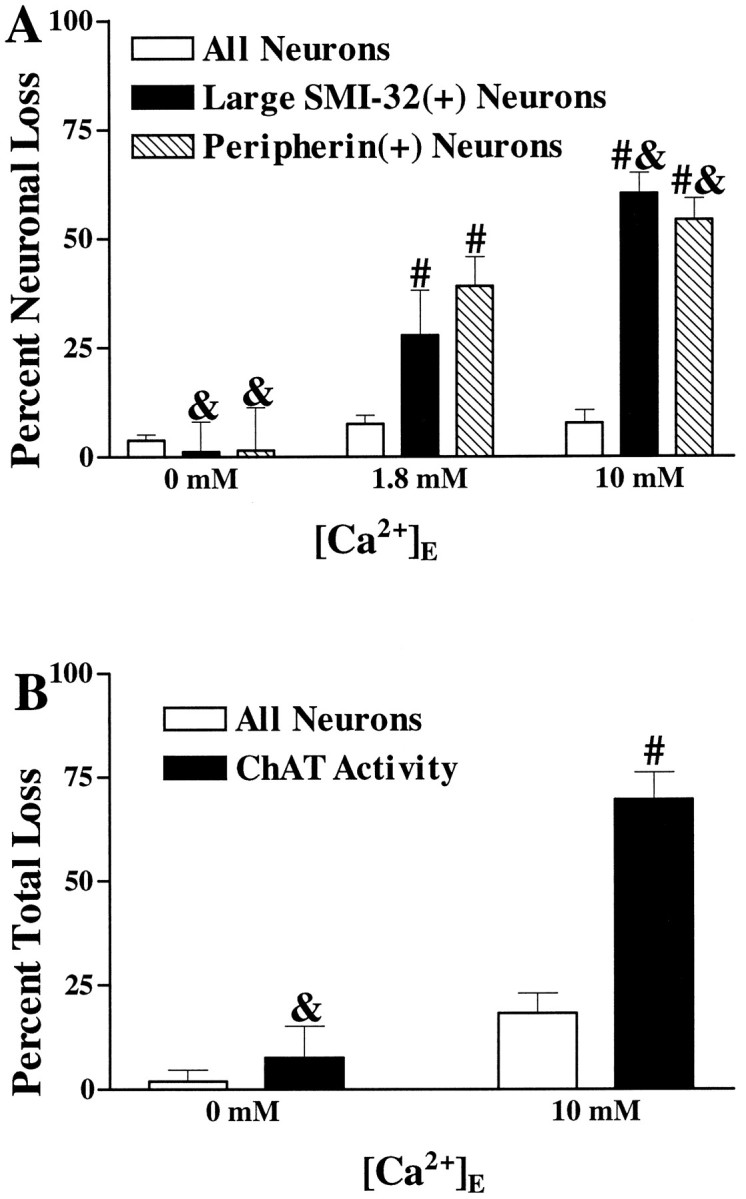
Kainate injury to the spinal motor neuronal population is Ca2+-dependent. A, Kainate injury to large SMI-32(+) and peripherin(+) neurons is Ca2+-dependent. Cultures were exposed to kainate (100 μm for 10 min) in the presence of the indicated Ca2+ concentration. Overall neuronal loss and loss of large SMI-32(+) or peripherin(+) neurons were evaluated 20–24 hr later (as described in Materials and Methods). Values represent mean ± SEM compiled from four experiments;n = 9–15 cultures per condition. & indicates labeled neuronal loss significantly different from labeled neuronal loss seen in the 1.8 mmCa2+ condition (p < 0.05 by two-tailed t test). # indicates labeled neuronal loss significantly different from total neuronal loss after the same exposure (p < 0.01 by two-tailedt test). B, Kainate-induced loss of spinal cord ChAT activity is Ca2+-dependent. Cultures were exposed to kainate (100 μm for 15 min) in the presence of the indicated Ca2+ concentration. Overall neuronal loss and loss of ChAT activity were assessed 20–24 hr later (as described). Values represent mean ± SEM compiled from 10 experiments; n = 27–36 cultures for each condition.& indicates ChAT activity loss significantly different from that seen in the 10 mm Ca2+condition (p < 0.01 by two-tailed ttest). # indicates ChAT activity loss significantly different from overall neuronal injury after the same exposure (p < 0.01 by two-tailed t test).
Motor neurons possess Ca2+-permeable AMPA/kainate channels
Neurons possessing Ca2+-permeable AMPA/kainate channels can be identified by a histochemical technique based on kainate-stimulated uptake of Co2+ ions (“Co2+(+) neurons”) (Pruss et al., 1991). The specificity of this stain for Ca2+-permeable AMPA/kainate channels is indicated by the inability of NMDA or high K+ to substitute for kainate in triggering Co2+ entry. We reported previously that a substantial subpopulation (∼40%) of all neurons in dissociated spinal cultures were Co2+(+) (Yin et al., 1995). Furthermore, consistent with studies of Co2+(+) neurons in other regions of the central nervous system (Brorson et al., 1994;Turetsky et al., 1994), these spinal Co2+(+) neurons were more vulnerable than other spinal neurons to AMPA/kainate receptor-mediated injury (Yin et al., 1995).
In our previous brief report, we found that a majority (∼80%) of large SMI-32(+) neurons were Co2+(+) (Carriedo et al., 1995). Our additional finding that a large majority of peripherin(+) neurons (84%; 42/50 from four experiments) are also Co2+(+) (Fig. 6) supports the hypothesis that possession of Ca2+-permeable AMPA/kainate channels is one factor that contributes to the high vulnerability of motor neurons to AMPA/kainate receptor-mediated injury. Preliminary observations suggest the possibility that these putative motor neurons are somewhat more vulnerable than some other spinal Co2+(+) neurons to kainate toxicity. An interesting topic of future studies thus will be the search for additional factors that may contribute to their high vulnerability.
Fig. 6.
Bottom. Peripherin(+) neurons and large SMI-32(+) neurons are Co2+(+). Photomicrographs show a peripherin(+) neuron (A, B) and a large SMI-32(+) neuron (C, D) before (A, C) and after (B, D) development of the Co2+stain. Co2+ stains were made light to preserve clarity of pictures. In many neurons, Co2+staining is evident only through nuclear darkening. Arrows(A, C) indicate axonal projections. Scale bar, 100 μm.
Motor neurons are selectively injured by prolonged low-level kainate exposures
Although the rapid, intense kainate exposures used in the above experiments may be directly relevant to the pathophysiology of acute spinal cord injury that occurs in trauma or ischemia, their relevance to potential excitotoxic contributions to the chronic motor neuron degeneration seen in ALS is less clear. We thus examined the vulnerability of large SMI-32(+) and peripherin(+) neurons to injury resulting from more prolonged low-level kainate exposures. Spinal cord cultures were exposed to kainate at concentrations ranging from 5 to 20 μm for 24 hr, followed by evaluation of both overall neuronal injury and labeled neuronal injury. Although a 10 μm kainate exposure caused relatively little overall neuronal injury, this exposure caused substantial damage to both the large SMI-32(+) and the peripherin(+) neuronal populations (Fig. 7A).
Fig. 7.
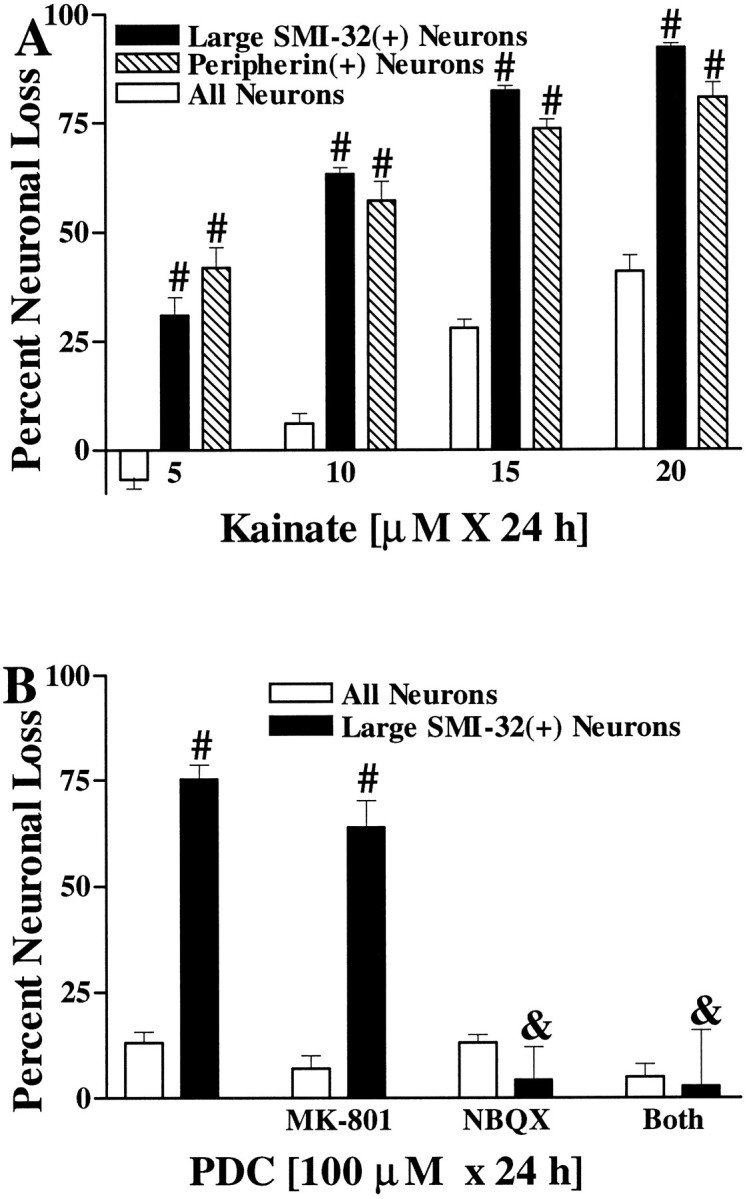
Motor neurons are selectively vulnerable to slow, excitotoxic injury. A, Large SMI-32(+) neurons and peripherin(+) neurons are selectively damaged by chronic kainate exposures. Cultures were exposed to the indicated kainate concentration for 20–24 hr, followed by evaluation of injury to the overall neuronal population and to the labeled neuronal population. Values represent mean ± SEM compiled from three to four representative experiments;n = 10–12 cultures per condition. # indicates labeled neuronal loss significantly different from total neuronal loss after the same exposure (p < 0.01 by two-tailedt test). B, Large SMI-32(+) neurons are selectively damaged by chronic exposure to the glutamate reuptake blocker PDC. Cultures were exposed for 24 hr to PDC (100 μm) alone or with the addition of glutamate receptor antagonists as indicated (each at 10 μm), followed by evaluation of damage to the overall neuronal population and to large SMI-32(+) neurons. Values represent mean ± SEM compiled from three to four representative experiments; n = 9–12 cultures per condition.# indicates large SMI-32(+) neuronal loss significantly different from total neuronal loss after the same exposure (p < 0.01 by two-tailed t test).& indicates large SMI-32(+) neuronal loss significantly different from that obtained in the 100 μm PDC condition (p < 0.01 by two-tailed ttest).
Additional experiments sought to examine the vulnerability of large SMI-32(+) neurons to injury from prolonged exposures to increased levels of endogenous glutamate by exposing the cultures for 24 hr to the glutamate reuptake blocker PDC (100 μm). Such exposures caused substantial degeneration of the large SMI-32(+) neurons but little damage to the overall spinal neuronal population. Consistent with previous studies in spinal cord slice culture models (Rothstein et al., 1993), this injury seemed to be mediated primarily through activation of AMPA/kainate-type glutamate receptors. The selective AMPA/kainate receptor antagonist NBQX (10 μm) was fully protective, whereas the NMDA receptor antagonist MK-801 (10 μm) had no effect (Fig. 7B).
Unlike acute excitotoxicity protocols, in which the role of Ca2+ ions in kainate injury to large SMI-32(+) or peripherin(+) neurons is readily demonstrable, the role of Ca2+ ion entry in these prolonged toxic exposures is harder to demonstrate, because removal of Ca2+from the media for extended periods is in itself injurious. We therefore used Fura-2 imaging to compare intracellular free Ca2+ levels ([Ca2+]i) in large SMI-32(+) neurons with [Ca2+]i in other spinal neurons during low level (10 μm) kainate exposures. Resting [Ca2+]i levels differed little between neurons. When kainate was added, however, [Ca2+]i levels increased rapidly in most cells before gradually declining to sustained [Ca2+]i values close to baseline. Large SMI-32(+) neurons showed sustained [Ca2+]i levels that were significantly higher than average (Figs. 8,9), thus supporting a role of Ca2+ions in their vulnerability to low levels of kainate.
Fig. 8.
Top right. Large SMI-32(+) neurons show substantial [Ca2+]ielevations during low-level kainate exposures. A, Pseudocolor image of a culture field showing [Ca2+]i levels in individual neurons 8 min after the addition of kainate (10 μm). B, Photomicrograph of the same field after development of the SMI-32 stain. Note that the large SMI-32(+) neuron had one of the highest [Ca2+]i levels. Scale bar, 100 μm.
Fig. 9.
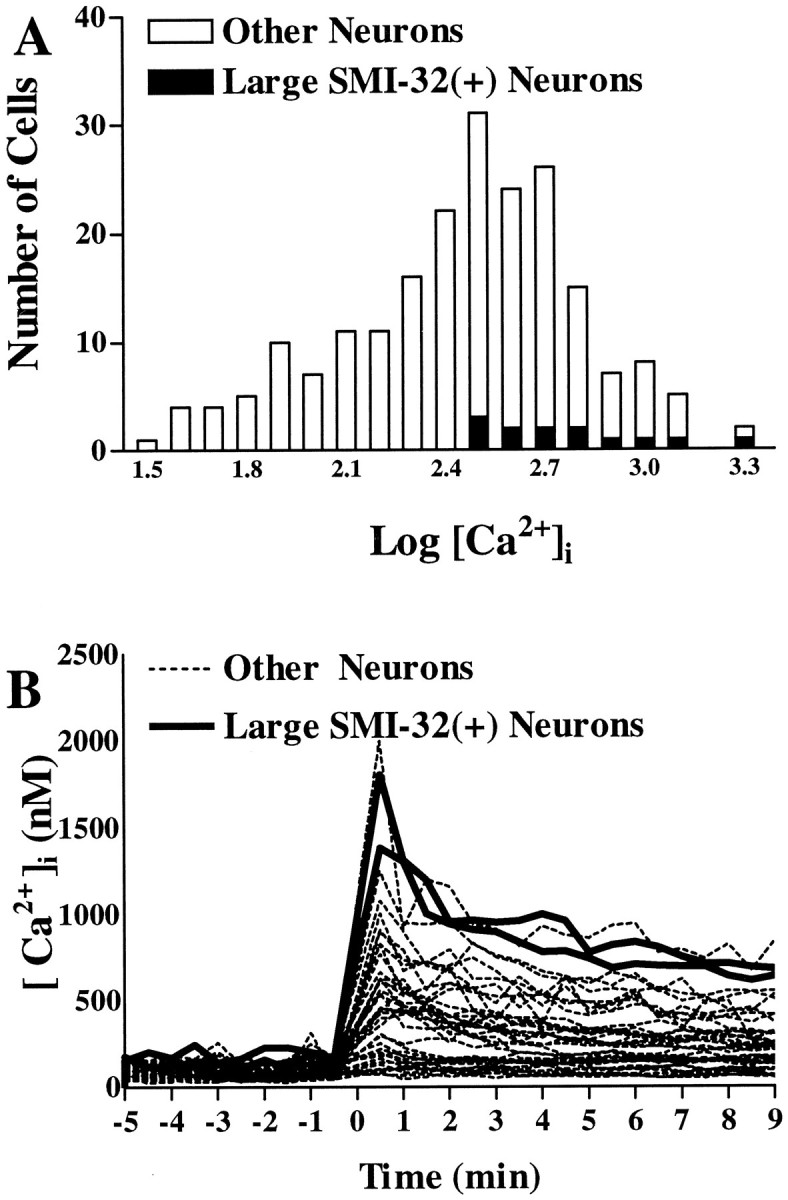
Large SMI-32(+) neurons show substantial [Ca2+]i elevations during low-level kainate exposures. A, Distribution of [Ca2+]i values in spinal neurons (528 neurons from eight experiments) 8–10 min after the addition of kainate (10 μm). All large SMI-32(+) neurons have higher than average [Ca2+]i values.B, Time course of [Ca2+]i changes. [Ca2+]i levels are plotted in individual neurons before and for 9 min after addition of kainate (10 μm). Of the 44 neurons shown, the 2 neurons represented by solid lines are large SMI-32(+) neurons.
DISCUSSION
A dissociated culture model for studies of motor neurons
For studies examining mechanisms of selective neuronal vulnerability, culture systems offer certain advantages over in vivo systems: they permit excellent control over the extracellular environment and monitoring of responses in individual neurons. Recent studies have examined the excitotoxic vulnerability of motor neurons using spinal cord slice cultures (Rothstein et al., 1993; Rothstein and Kuncl, 1995), which allow good environmental control while maintaining more physiological patterns of connectivity than dissociated cultures. For the present study, we have opted to use a dissociated culture model, which provides more precise environmental control and instantaneous monitoring of responses in individual neurons. Such models of motor neuron injury have been used infrequently, however, in large part because of difficulty in maintaining, identifying, and visualizing morphological characteristics of the motor neurons.
For instance, transmitter-specific markers such as ChAT decrease after several days in culture (Lombard-Golly et al., 1990), often give poor staining (Schaffner et al., 1987), and are not specific for motor neurons (Houser et al., 1983). Motor neuron enrichment techniques such as density gradients (sorting by size: Schnaar and Schaffner, 1981;Dohrmann et al., 1986; Martinou et al., 1989a), retrograde labeling (Calof and Reichardt, 1984; O’Brian and Fischbach, 1986a; Schaffner et al., 1987), and immunopanning (Camu and Henderson, 1992; Mettling et al., 1995) have been used often, but generally give limited yield or lack complete specificity for motor neurons. In addition, enriching for motor neurons does not in itself provide the morphological detail that can be revealed with direct labeling, and removal of other spinal cord neurons with which motor neurons may interact may markedly alter their survival or phenotype (O’Brian and Fischbach, 1986b).
We thus have opted to grow cultures in the simplest way that permits survival under mixed culture conditions: plating total spinal cord neurons on a feeder layer of astrocytes, a maneuver that provides a more physiological environment than artificial substrates do and markedly enhances survival of cultured motor neurons (Eagleson et al., 1985; Schaffner et al., 1987; Martinou et al., 1989a; Ang et al., 1992). Motor neurons in these mixed cultures were studied through direct labeling with several putative motor neuronal markers.
Of the markers studied, SMI-32 seems to be particularly useful. Large SMI-32(+) neurons have appropriate morphological features (Martinou et al., 1989a; Schaffner et al., 1987) and are found preferentially in ventral but not dorsal spinal cord cultures, which suggests that they are indeed motor neurons. In addition, neurons labeled by other motor neuron markers frequently express SMI-32 immunoreactivity and show excitotoxic vulnerability similar to that of large SMI-32(+) neurons. A particular advantage of using SMI-32 for studies of motor neuron injury, however, is the consistency and robustness of the staining and the extensive morphological detail that is revealed. Except for peripherin, the other motor neuron markers that were examined gave weak staining with little morphological detail and often showed high levels of background staining.
In addition to staining large SMI-32(+) neurons, SMI-32 also stains a group of smaller spinal neurons in culture with less evident resemblance to motor neurons. Although the identity of these neurons is uncertain, subpopulations of small (<13 μm) motor neurons have been identified in other culture studies (Mettling et al., 1995), and some small as well as large peripherin- and CGRP-stained neurons were SMI-32(+), suggesting that at least some of the small SMI-32(+) neurons may be motor neurons.
Excitotoxic vulnerability of cultured motor neurons
Present results suggesting that cultured motor neurons are highly vulnerable to rapidly triggered AMPA/kainate receptor-mediated injury are in general agreement with previous studies of the excitotoxic vulnerability of motor neurons carried out in slice cultures (Rothstein et al., 1993; Rothstein and Kuncl, 1995) or animal models (Hugon et al., 1989b). Thus, motor neurons seem to maintain critical features pertinent to their excitotoxic vulnerability when transplanted into a dissociated culture system, suggesting that this simplified system may be useful for studying their vulnerability under controlled conditions. Indeed, we have been able to elucidate a feature that may contribute critically to their unusual excitotoxic vulnerability. Possession of AMPA/kainate receptors gating channels with direct Ca2+ permeability may permit receptor activation to cause unusually high Ca2+influx rates (Carriedo et al., 1995; Lu et al., 1995).
Additional studies will be necessary to determine the precise molecular basis of this Ca2+ permeability. Although expression studies in non-neural cells as well as studies of native neurons suggest that in heteromeric AMPA channels an absence of the GluR2 subunit may confer Ca2+ permeability (Hollman et al., 1991; Verdoon et al., 1991; Bochet et al., 1994; Jonas et al., 1994; Yin et al., 1994a), motor neurons have been found to express GluR2 mRNA (Tölle et al., 1993). It is possible that this mRNA is not translated into protein with high efficiency or that heterogeneous populations of receptors may be expressed on the neurons, only some of which may lack the GluR2 subunit (Geiger et al., 1995;Goldstein et al., 1995). Alternatively, motor neurons could express AMPA/kainate channels composed of kainate-preferring subunits that gate Ca2+ permeable channels (Sommer et al., 1991; Kohler et al., 1993; Vickers et al., 1993; Paternain et al., 1995).
One way in which Ca2+ entry can injure neurons is by triggering excess production of free radicals (Dykens, 1994), which results in oxidative injury. Recent studies indicate that NMDA receptor-mediated intracellular Ca2+ loads trigger a burst of injurious free radicals, likely emanating from the mitochondria (Lafon-Cazal et al., 1993; Dugan et al., 1995; Reynolds and Hastings, 1995). Possession of AMPA/kainate receptors gating channels with direct Ca2+ permeability might result in similar excess free-radical production in motor neurons during AMPA/kainate receptor activation. If so, free-radical mediated oxidative injury to motor neurons could be an injury mechanism common to excitotoxic models of ALS (Plaitakis and Caroscio, 1987;Rothstein et al., 1990, 1992) as well as to recently described familial forms of the disease linked to mutations in the free-radical metabolizing enzyme superoxide dismutase (Rosen et al., 1993).
Cultured motor neurons are vulnerable to slow AMPA/kainate receptor-mediated injury
The present culture system has been useful for studying the role of Ca2+ in triggering motor neuron degeneration after brief intense periods of AMPA/kainate receptor activation. Such acute insults, however, although possibly of direct relevance to acute motor neuron degeneration in conditions such as ischemia or trauma, may have less direct bearing on the very slow neurodegeneration that occurs in chronic conditions such as ALS. For instance, slow excitotoxic exposures might trigger low rates of Ca2+ entry that do not rapidly overwhelm the homeostatic abilities of the cells, possibly allowing different injury mechanisms to become activated. Thus, for comparative purposes, experiments that used more chronic (24 hr) exposures to lower levels of kainate or to the glutamate reuptake blocker PDC were undertaken. Not surprisingly, in light of previous studies in spinal cord slice using prolonged exposures (Rothstein et al., 1993; Rothstein and Kuncl, 1995), present experiments also show that prolonged periods of exposure to kainate or endogenous glutamate produce preferential degeneration of motor neurons through activation of AMPA/kainate receptors. In addition, although additional studies will be needed to elucidate the principal injury mechanisms downstream from Ca2+ion entry, present [Ca2+]i imaging experiments, which showed unusually high [Ca2+]i responses in large SMI-32(+) neurons during low-level kainate exposures, suggest that Ca2+ ions play a central role in slow as well as rapidly triggered AMPA/kainate receptor-mediated motor neuron injury.
Conclusions
In summary, we have developed a dissociated cell culture system for studying motor neurons. The nonphosphorylated neurofilament antibody SMI-32 seems to be particularly useful for identifying and examining morphological features of motor neurons in these cultures. We find, consistent with previous in vivo and spinal cord slice studies, that motor neurons seem to be highly vulnerable to both rapidly triggered and more chronic AMPA/kainate receptor-mediated injury. We also find that this vulnerability likely reflects, at least in part, possession of Ca2+-permeable AMPA/kainate receptor-gated channels. If the present findings in cultured mouse neurons pertain to motor neurons in mature humans, it is possible that possession of these channels may be one factor that helps predispose motor neurons to degenerate in diseases such as ALS. The present model system may be useful for examining how multiple factors may interact in producing degeneration of motor neurons and may allow for rapid screening of protective interventions.
Footnotes
This work was supported by National Institutes of Health Grant NS 30884 (J.H.W.), by a grant from the Pew Scholars Program in the Biomedical Sciences (J.H.W.), and by an American Psychological Association Fellowship (S.G.C.). We thank Juna Chiang for expert cell culture assistance.
Correspondence should be addressed to John H. Weiss, Department of Psychobiology, University of California-Irvine, Irvine, CA 92717-4290.
REFERENCES
- 1.Ang LC, Bhaumich B, Munoz DG, Sass J, Juurlink BHJ. Effects of astrocytes, insulin and insulin-like growth factor 1 on the survival of motor neurons in vitro . J Neurol Sci. 1992;109:168–172. doi: 10.1016/0022-510x(92)90164-g. [DOI] [PubMed] [Google Scholar]
- 2.Bochet P, Audinet E, Lambolez B, Crepel F, Rossier J, Iino M, Tsuzuki K, Ozawa S. Subunit composition at the single-cell level explains functional properties of a glutamate-gated channel. Neuron. 1994;12:383–388. doi: 10.1016/0896-6273(94)90279-8. [DOI] [PubMed] [Google Scholar]
- 3.Bridges RJ, Stevens DR, Kahle JS, Nunn PB, Kadri M, Cotman CW. Structure-function studies on N -oxaly-diamino-dicarboxylic acids and excitatory amino acid receptors: evidence that β-l-ODAP is a selective non-NMDA agonist. J Neurosci. 1989;9:2073–2079. doi: 10.1523/JNEUROSCI.09-06-02073.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brorson JR, Bleakman D, Chard PS, Miller RJ. Calcium directly permeates kainate/alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid receptors in cultured cerebellar Purkinje neurons. Mol Pharmacol. 1992;41:603–608. [PubMed] [Google Scholar]
- 5.Brorson JR, Manzolillo PA, Miller RJ. Ca2+ Entry via AMPA/KA receptors and excitotoxicity in cultured cerebellar Purkinje cells. J Neurosci. 1994;14:187–197. doi: 10.1523/JNEUROSCI.14-01-00187.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calof AL, Reichardt LF. Motoneurons purified by cell sorting respond to two distinct activities in myotube-conditioned medium. Dev Biol. 1984;106:194–210. doi: 10.1016/0012-1606(84)90075-7. [DOI] [PubMed] [Google Scholar]
- 7.Camu W, Henderson CE. Purification of embryonic rat motoneurons by panning on a monoclonal antibody to the low-affinity NGF receptor. J Neurosci Methods. 1992;44:59–70. doi: 10.1016/0165-0270(92)90114-s. [DOI] [PubMed] [Google Scholar]
- 8.Carriedo SG, Yin H-Z, Lamberta R, Weiss JH. In vitro kainate injury to large SMI-32(+) spinal neurons is Ca2+ dependent. NeuroReport. 1995;6:945–948. doi: 10.1097/00001756-199504190-00030. [DOI] [PubMed] [Google Scholar]
- 9.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 10.Debonnel G, Beauchesne L, de Montigny C. Domoic acid, the alleged “mussel toxin” might produce its neurotoxic effect through kainate receptor activation: an electrophysiological study in dorsal hippocampus. Can J Physiol Pharmacol. 1989;67:29–33. doi: 10.1139/y89-005. [DOI] [PubMed] [Google Scholar]
- 11.Dohrmann U, Edgar D, Sendtner M, Thoenen H. Muscle-derived factors that support survival and promote fiber outgrowth from embryonic chick spinal motor neurons in culture. Dev Biol. 1986;118:209–221. doi: 10.1016/0012-1606(86)90089-8. [DOI] [PubMed] [Google Scholar]
- 12.Dohrmann U, Edgar D, Thoenen H. Distinct neurotrophic factors from skeletal muscle and the central nervous system interact synergistically to support the survival of cultured embryonic spinal motor neurons. Dev Biol. 1987;124:145–152. doi: 10.1016/0012-1606(87)90467-2. [DOI] [PubMed] [Google Scholar]
- 13.Dugan LL, Sensi SL, Canzoniero LMT, Handran SD, Rothman SM, Lin T-S, Goldberg MP, Choi DW. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N -methyl-d-aspartate. J Neurosci. 1995;15:6377–6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dykens JA. Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated Ca2+ and Na+: implications for neurodegeneration. J Neurochem. 1994;63:584–591. doi: 10.1046/j.1471-4159.1994.63020584.x. [DOI] [PubMed] [Google Scholar]
- 15.Eagleson KL, Raju TR, Bennett MR. Motoneurone survival is induced by immature astrocytes from developing spinal cord. Dev Brain Res. 1985;17:95–104. doi: 10.1016/0165-3806(85)90135-x. [DOI] [PubMed] [Google Scholar]
- 16.Escurat M, Djabali K, Gumpel M, Gros F, Portier MM. Differential expression of two neuronal intermediate-filament proteins, peripherin and the low-molecular-mass neurofilament protein (NF-L), during the development of the rat. J Neurosci. 1990;10:764–784. doi: 10.1523/JNEUROSCI.10-03-00764.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fonnum F. A rapid radiochemical method for the determination of choline acetyltransferase. J Neurochem. 1975;24:407–409. doi: 10.1111/j.1471-4159.1975.tb11895.x. [DOI] [PubMed] [Google Scholar]
- 18.Geffard M, McRae-Degueurce A, Souan ML. Immunocytochemical detection of acetylcholine in the rat central nervous system. Science. 1985;229:77–79. doi: 10.1126/science.3892687. [DOI] [PubMed] [Google Scholar]
- 19.Geiger JRP, Melcher T, Koh D-S, Sakmann B, Seeburg PH, Jonas P, Monyer H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 20.Gibson SJ, Polak JM, Bloom SR, Sabate IM, Mulderry PM, Ghatel MA, McGregor GP, Morrison JFB, Kelly JS, Evans RM, Rosenfeld MG. Calcitonin gene-related peptide immunoreactivity in the spinal cord of man and of eight other species. J Neurosci. 1984;4:3101–3111. doi: 10.1523/JNEUROSCI.04-12-03101.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldstein PA, Justin Lee C, MacDermott AB. Variable distributions of Ca2+-permeable and Ca2+-impermeable AMPA receptors on embryonic rat dorsal horn neurons. J Neurophysiol. 1995;73:2522–2534. doi: 10.1152/jn.1995.73.6.2522. [DOI] [PubMed] [Google Scholar]
- 22.Gotow T, Tanaka J. Phosphorylation of neurofilament H subunit as related to arrangement of neurofilaments. J Neurosci Res. 1994;37:691–713. doi: 10.1002/jnr.490370604. [DOI] [PubMed] [Google Scholar]
- 23.Hietanen M, Pelto-Huikko M, Rechardt L. Immunocytochemical study of the relations of acetylcholinesterase, enkephalin-, substance P-, choline acetyltransferase- and calcitonin gene-related peptide-immunoreactive structures in the ventral horn of rat spinal cord. Histochemistry. 1990;93:473–477. doi: 10.1007/BF00266403. [DOI] [PubMed] [Google Scholar]
- 24.Hollman M, Hartley M, Heinemann S. Ca2+ permeability of KA/AMPA-gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 25.Houser CR, Crawford GD, Barber RP, Salvaterra PM, Vaughn JE. Organization and morphological characteristics of cholinergic neurons: an immunocytochemical study with a monoclonal antibody to choline acetyltransferase. Brain Res. 1983;266:97–119. doi: 10.1016/0006-8993(83)91312-4. [DOI] [PubMed] [Google Scholar]
- 26.Hugon J, Tabaraud F, Rigaud M, Vallat JM, Dumas M. Glutamate dehydrogenase and aspartate aminotransferase in leukocytes of patients with motor neuron disease. Neurology. 1989a;39:956–958. doi: 10.1212/wnl.39.7.956. [DOI] [PubMed] [Google Scholar]
- 27.Hugon J, Vallat JM, Spencer PS, Leboutet MJ, Barthe D. Kainic acid induces early and late delayed degenerative neuronal changes in rat spinal cord. Neurosci Lett. 1989b;104:258–262. doi: 10.1016/0304-3940(89)90585-5. [DOI] [PubMed] [Google Scholar]
- 28.Iino M, Ozawa S, Tsuzuki K. Permeation of calcium through excitatory amino acid receptor channels in cultured rat hippocampal neurons. J Physiol (Lond) 1990;424:151–165. doi: 10.1113/jphysiol.1990.sp018060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jonas P, Racca C, Sakmann B, Seeburg PH, Monyer H. Differences in Ca2+permeability of AMPA-type glutamate receptor channels in neocortical neurons caused by differential GluR-B subunit expression. Neuron. 1994;12:1281–1289. doi: 10.1016/0896-6273(94)90444-8. [DOI] [PubMed] [Google Scholar]
- 30.Juurlink BHJ, Munoz DG, Devon RM. Calcitonin gene-related peptide identifies spinal motoneurons in vitro . J Neurosci Res. 1990;26:238–241. doi: 10.1002/jnr.490260214. [DOI] [PubMed] [Google Scholar]
- 31.Koh J, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 32.Koh J, Choi DW. Vulnerability of cultured cortical neurons to damage by excitotoxins: differential susceptibility of neurons containing NADPH-diaphorase. J Neurosci. 1988;8:2153–2163. doi: 10.1523/JNEUROSCI.08-06-02153.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kohler M, Burnashev N, Sakmann B, Seeburg PH. Determinants of Ca2+permeability in both TM1 and TM2 of high affinity kainate receptor channels: diversity by RNA editing. Neuron. 1993;10:491–500. doi: 10.1016/0896-6273(93)90336-p. [DOI] [PubMed] [Google Scholar]
- 34.Kruger L, Mantyh PW, Sternini C, Brecha NC, Mantyh CR. Calcitonin gene-related peptide (CGRP) in the rat central nervous system: patterns of immunoreactivity and receptor binding sites. Brain Res. 1988;463:223–244. doi: 10.1016/0006-8993(88)90395-2. [DOI] [PubMed] [Google Scholar]
- 35.Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 36.Lombard-Golly D, Wong V, Kessler JA. Regulation of cholinergic expression in cultured spinal cord neurons. Dev Biol. 1990;139:396–406. doi: 10.1016/0012-1606(90)90308-6. [DOI] [PubMed] [Google Scholar]
- 37.Lu YM, Yin H-Z, Weiss JH. Ca2+permeable AMPA/kainate channels permit rapid injurious Ca2+ entry. NeuroReport. 1995;6:1089–1092. doi: 10.1097/00001756-199505300-00004. [DOI] [PubMed] [Google Scholar]
- 38.MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurons. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- 39.Martinou J-C, Bierer F, Thai ALV, Weber MJ. Influence of the culture substratum on the expression of choline acetyltransferase activity in purified motoneurons from rat embryos. Dev Brain Res. 1989a;47:251–262. doi: 10.1016/0165-3806(89)90180-6. [DOI] [PubMed] [Google Scholar]
- 40.Martinou JC, Thai ALV, Cassar G, Roubinet F, Weber MJ. Characterization of two factors enhancing choline acetyltransferase activity in cultures of purified rat motoneurons. J Neurosci. 1989b;9:3645–3656. doi: 10.1523/JNEUROSCI.09-10-03645.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mettling C, Gouin A, Robinson M, El M’Hamdi H, Camu W, Bloch-Gallego E, Buisson B, Tanaka H, Davies AM, Henderson CE. Survival of newly postmitotic motoneurons is transiently independent of exogenous trophic support. J Neurosci. 1995;15:3128–3137. doi: 10.1523/JNEUROSCI.15-04-03128.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Brian RJ, Fischbach GD. Isolation of embryonic chick motoneurons and their survival in vitro . J Neurosci. 1986a;6:3265–3274. doi: 10.1523/JNEUROSCI.06-11-03265.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Brian RJ, Fischbach GD. Modulation of embryonic chick motoneuron glutamate sensitivity by interneurons and agonists. J Neurosci. 1986b;6:3290–3296. doi: 10.1523/JNEUROSCI.06-11-03290.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parysek LM, Goldman RD. Distribution of a novel 57 kDa intermediate filament (IF) protein in the nervous system. J Neurosci. 1988;8:555–563. doi: 10.1523/JNEUROSCI.08-02-00555.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paternain AV, Morales M, Lerma J. Selective antagonism of AMPA receptors unmasks kainate receptor-mediated responses in hippocampal neurons. Neuron. 1995;14:185–189. doi: 10.1016/0896-6273(95)90253-8. [DOI] [PubMed] [Google Scholar]
- 46.Plaitakis A, Caroscio JT. Abnormal glutamate metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1987;22:575–579. doi: 10.1002/ana.410220503. [DOI] [PubMed] [Google Scholar]
- 47.Pruss RM, Akeson RL, Racke MM, Wilburn JL. Agonist-activated cobalt uptake identifies divalent cation-permeable kainate receptors on neurons and glia. Neuron. 1991;7:509–519. doi: 10.1016/0896-6273(91)90302-g. [DOI] [PubMed] [Google Scholar]
- 48.Regan RF, Choi DW. Glutamate neurotoxicity in spinal cord cell culture. Neuroscience. 1991;43:585–591. doi: 10.1016/0306-4522(91)90317-h. [DOI] [PubMed] [Google Scholar]
- 49.Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richards LJ, Murphy M, Dutton R, Kilpatrick TJ, Puche AC, Key B, Tan S-S, Talman PS, Bartlett PF. Lineage specification of neuronal precursors in the mouse spinal cord. Proc Natl Acad Sci USA. 1995;92:10079–10083. doi: 10.1073/pnas.92.22.10079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richter KE, Mena EE. l-beta-methylaminoalanine inhibits [3H] glutamate binding in the presence of bicarbonate ions. Brain Res. 1989;492:385–388. doi: 10.1016/0006-8993(89)90925-6. [DOI] [PubMed] [Google Scholar]
- 52.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tunzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH., Jr Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 53.Ross SM, Seelig M, Spencer PS. Specific antagonism of excitotoxic action of “uncommon” amino acids assayed in organotypic mouse cortical cultures. Brain Res. 1987;425:120–127. doi: 10.1016/0006-8993(87)90490-2. [DOI] [PubMed] [Google Scholar]
- 54.Rothstein JD, Kuncl RW. Neuroprotective strategies in a model of chronic glutamate-mediated motor neuron toxicity. J Neurochem. 1995;65:643–651. doi: 10.1046/j.1471-4159.1995.65020643.x. [DOI] [PubMed] [Google Scholar]
- 55.Rothstein JD, Jin L, Dykes-Hoberg M, Kuncl RW. Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc Natl Acad Sci USA. 1993;90:6591–6595. doi: 10.1073/pnas.90.14.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- 57.Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, Pestronk A, Stauch BL, Coyle JT. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1990;28:18–25. doi: 10.1002/ana.410280106. [DOI] [PubMed] [Google Scholar]
- 58.Schaffner AE, St John PA, Barker JL. Fluorescence-activated cell sorting of embryonic mouse and rat motoneurons and their long-term survival in vitro . J Neurosci. 1987;7:3088–3104. doi: 10.1523/JNEUROSCI.07-10-03088.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schnaar RL, Schaffner AE. Separation of cell types from embryonic chicken and rat spinal cord: characterization of motoneuron-enriched fractions. J Neurosci. 1981;1:204–217. doi: 10.1523/JNEUROSCI.01-02-00204.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sommer B, Kohler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67:11–19. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- 61.Spencer PS, Hugon J, Ludolph A, Nunn PB, Ross SM, Roy DN, Schaumburg HH. Discovery and partial characterization of primate motor-system toxins. Ciba Found Symp. 1987;126:221–238. doi: 10.1002/9780470513422.ch14. [DOI] [PubMed] [Google Scholar]
- 62.Spencer PS, Ludolph A, Dwivedi MP, Roy DN, Hugon J, Schaumberg HH. Lathyrism: evidence for the role of the neuroexcitatory amino acid BOAA. Lancet. 1986;2:1066–1067. doi: 10.1016/s0140-6736(86)90468-x. [DOI] [PubMed] [Google Scholar]
- 63.Teitelbaum JS, Zatorre RJ, Carpenter S, Gendron D, Evans AC, Gjedde A, Cashman NR. Neurologica sequelae of domoic acid intoxication due to the ingestion of contaminated mussels. N Engl J Med. 1990;322:1781–1787. doi: 10.1056/NEJM199006213222505. [DOI] [PubMed] [Google Scholar]
- 64.Tölle TR, Berthele A, Zieglgänsberger W, Seeburg PH, Wisden W. The differential expression of 16 NMDA and non-NMDA receptor subunits in the rat spinal cord and in periaqueductal gray. J Neurosci. 1993;13:5009–5028. doi: 10.1523/JNEUROSCI.13-12-05009.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Turetsky DM, Canzoniero LMT, Sensi SL, Weiss JH, Goldberg MP, Choi DW. Cortical neurones exhibiting kainate-activated Co2+ uptake are selectively vulnerable to AMPA/kainate receptor-mediated injury. Neurobiol Dis. 1994;1:101–110. doi: 10.1006/nbdi.1994.0013. [DOI] [PubMed] [Google Scholar]
- 66.Verdoon TA, Burnashev N, Monyer H, Seeburg PH, Sakmann B. Structural determinants of ion flow through recombinant glutamate receptor channels. Science. 1991;252:1715–1718. doi: 10.1126/science.1710829. [DOI] [PubMed] [Google Scholar]
- 67.Vickers JC, Huntley GW, Edwards AM, Moran T, Rogers SW, Heinemann SF, Morrison JH. Quantitative localization of AMPA/kainate and kainate glutamate receptor subunit immunoreactivity in neurochemically identified subpopulations of neurons in the prefrontal cortex of the macaque monkey. J Neurosci. 1993;13:2982–2992. doi: 10.1523/JNEUROSCI.13-07-02982.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang FZ, Nelson PG, Fitzgerald SC, Hersh LB, Neale EA. Cholinergic function in cultures of mouse spinal cord neurons. J Neurosci Res. 1990;25:312–323. doi: 10.1002/jnr.490250307. [DOI] [PubMed] [Google Scholar]
- 69.Weiss JH, Choi DW. Slow non-NMDA receptor mediated neurotoxicity and amyotrophic lateral sclerosis. Adv Neurol. 1991;56:311–318. [PubMed] [Google Scholar]
- 70.Weiss JH, Koh JY, Choi DW. Neurotoxicity of beta-N -methylamino-l-alanine (BMAA) and beta- N -oxalylamino-l-alanine (BOAA) on cultured cortical neurons. Brain Res. 1989;497:64–71. doi: 10.1016/0006-8993(89)90970-0. [DOI] [PubMed] [Google Scholar]
- 71.Weiss JH, Turetsky D, Wilke G, Choi DW. AMPA/kainate receptor-mediated damage to NADPH-diaphorase-containing neurons is Ca2+dependent. Neurosci Lett. 1994a;167:93–96. doi: 10.1016/0304-3940(94)91035-9. [DOI] [PubMed] [Google Scholar]
- 72.Weiss JH, Yin HZ, Choi DW. Basal forebrain cholinergic neurons are selectively vulnerable to AMPA/kainate receptor-mediated neurotoxicity. Neuroscience. 1994b;60:659–664. doi: 10.1016/0306-4522(94)90494-4. [DOI] [PubMed] [Google Scholar]
- 73.Yin HZ, Turetsky D, Choi DW, Weiss JH. Cortical neurons with Ca2+ permeable AMPA/kainate channels display distinct receptor immunoreactivity and are GABAergic. Neurobiol Dis. 1994a;1:43–49. doi: 10.1006/nbdi.1994.0006. [DOI] [PubMed] [Google Scholar]
- 74.Yin HZ, Lindsay AD, Weiss JH. Kainate injury to cultured basal forebrain cholinergic neurons is Ca2+ dependent. NeuroReport. 1994b;5:1477–1480. doi: 10.1097/00001756-199407000-00017. [DOI] [PubMed] [Google Scholar]
- 75.Yin HZ, Park DD, Lindsay AD, Weiss JH. Spinal cord neurons are vulnerable to rapidly triggered kainate neurotoxicity in vitro. Brain Res. 1995;689:265–270. doi: 10.1016/0006-8993(95)00532-u. [DOI] [PubMed] [Google Scholar]