

Abstract
The effects of gonadal steroid hormones on dendritic spines were studied in hippocampal neurons that were dissociated and grown in culture for 2–3 weeks. Exposure to estradiol caused up to a twofold increase in dendritic spine density in these neurons. The effect of estradiol was stereospecific and blocked by the steroid antagonist tamoxifen. The estradiol-induced rise in spine density was blocked by the NMDA antagonist APV, but not by the AMPA/KA antagonist DNQX. The estradiol-induced rise in spine density was blocked by the serine/threonine kinase inhibitor H7, but not by the tyrosine kinase inhibitor genestein, and was partially mimicked by PMA, an activator of protein kinase C. Estradiol also caused an increase in the fluorescence intensity of synaptophysin-immunoreactive terminals, corresponding to presynaptic boutons. Finally, estradiol caused a rise in [Ca]i reactivity of the cultured neurons to topical application of glutamate. These studies are the first to examine receptor and second messenger regulation of dendritic spines, and they illustrate the viability of cultured neurons as a powerful test system to address issues related to the regulation of dendritic spine maturation.
Keywords: dendritic spines, estradiol, culture, hippocampal neurons, calcium, plasticity
Dendritic spines are the loci of synaptic interactions among central neurons. Their susceptibility to changes in afferent stimulation makes dendritic spines the prime candidates for serving the long-term morphological substrates of neuronal plasticity. In the hippocampus, which has been associated with learning and memory, spines may be essential for the induction, associativity, specificity, and endurance of long-term potentiation (Harris and Kater, 1994). Despite extensive interest in dendritic spines, progress toward understanding the mechanisms governing their generation and morphological plasticity has been rather slow. This is attributable partly to their minute size, which is at the limit of optical resolution, and their large density and heterogeneity in neurons of the intact brain.
Dissociated cultures of hippocampal neurons offer a viable and convenient means for studying the factors that regulate the development and functions of dendritic spines. The two-dimensional nature of cultured neurons, as well as the relative sparseness of dendritic spines in culture, which are approximately one third to one fourth the density of those neurons in vivo, makes their study technically achievable (Papa et al., 1995; Segal, 1995a,b).
It has been observed recently that the density of dendritic spines in the intact rat hippocampus undergoes marked variations during the estrous cycle. A higher density of dendritic spines coincides with high levels of estrogen (Woolley et al., 1990), and the addition of estradiol to ovariectomized rats causes a marked increase in spine density (Woolley and McEwen, 1994). Circulating estrogens are prevalent during neonatal development and so may be active in neonatal spine formation. In fact, estrogens may be regulating synaptic plasticity in adult brains as well. The hippocampus possesses estrogen receptors, and the brain is able to synthesize steroid hormones, independent of the gonads or adrenals (Singh et al., 1994). This would suggest that estrogens play a major role in neuronal activity, regulating not only reproductive behavior but also neuronal plasticity. In the present study, we measured the effects of steroid hormones on dendritic spine density in cultured hippocampal neurons and began a systematic analysis of the receptor types and second messenger mechanisms associated with the effects of estradiol on spine density.
MATERIALS AND METHODS
Culture of hippocampal neurons. Hippocampal cultures were prepared as described previously (Papa et al., 1995). Briefly, 19-d-old embryos were taken from anesthetized Wistar rats. Their brains were removed and placed in ice-cold (4°C) L15 medium supplemented with 0.6% glucose and 15 μg/ml gentamicin. The hippocampus was dissected out and mechanically disaggregated by gentle trituration using a Pasteur pipette. Dissociated cells (500,000 cells/well) were plated onto 12 mm glass coverslips that were coated with poly-l-lysine (15 μg/ml) and then sterilized by ultraviolet light. The plating medium was Eagle’s MEM containing 10% heat-inactivated horse serum, 5% fetal calf serum, 2 mm glutamine, 0.6% glucose, and 15 μg/ml gentamicin. Cells were incubated at 37°C with 8% CO2. The first change of medium, ∼4–6 d after plating, included 50 μg/ml uridine and 20 μg/ml deoxyuridine to prevent glial cell overgrowth; thereafter, the cultures were fed 1–2 times/week with Eagle’s MEM and 10% horse serum.
Cell dosing. Experimental solutions were prepared in hippocampal growth media on the day of dosing (14 d in vitro) and placed in the incubator to reach the appropriate temperature and pH. Half of the media was removed from wells and replaced with experimental media to give the appropriate final desired concentration. Unless indicated otherwise, 17-β-estradiol was used at the intermediate dose of 0.1 μg/ml. The estrogen receptor antagonist tamoxifen was used at 1 μg/ml to ensure saturation of binding sites. Other drugs that were used (all from Sigma, St. Louis, MO) included α-estradiol (1 μg/ml), progesterone (125 μg/ml), 2-APV (50 μm), DNQX (50 μm), tetrodotoxin (TTX) (1 μm), 1-(5-isoquinolinesulphonyl)−2-methylpiperazine hydrochloride (H7) (15 μm), phorbol 12-myristate 13-acetate (PMA) (150 nm), and genestein (20 μm). After 2–3 d in experimental solutions, cells were washed with PBS and fixed for 30 min at room temperature with 4% paraformaldehyde in PBS. In each of the experiments conducted in this study, a control and an estradiol-treated culture were always compared with the addition of the test drug in a 2 × 2 design.
Confocal imaging. Fixed cells were stained by pressure ejection of microdrops of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) dissolved in cod-liver oil (Hosokawa et al., 1992). Thirty to forty isolated, medium-sized (15–25 μm soma diameter) cells were labeled on each coverslip, and these were stored in PBS at 4°C for ∼6 hr. Cells were visualized with a Zeiss confocal laser scanning microscope (CLSM) (National Institute of Neurological Disorders and Stroke Imaging Facility, National Institutes of Health) using a 100×, 1.4 NA oil-immersion objective. A single, low-power scan was made of each individual cell soma, followed by four to eight serial, zoomed, optical sections of three to four regions of secondary dendrites around each cell. Individual dendrites were reconstructed three-dimensionally from the optical serial sections using National Institutes of Health Image software on a Power Macintosh. Individual spines were identified clearly as short, 1–3 μm protrusions at a right angle from the dendrite, some having a distinct spine head. These spines were different from filopodia in that the latter were longer and thinner at the limit of resolution. As seen before, filopodia were abundant in the younger cultures (Papa et al., 1995). Spines were counted in 50 μm segments of dendrites. In a typical experiment, ∼2000–4000 spines were counted on 200 dendritic segments in 50 neurons. Average spine densities per 50 μm dendritic segments were then calculated for each treatment group, along with the SE. Groups of spines were compared using the Student’s t test.
Synaptophysin (SF) immunoreactivity. Cultures were fixed in 4% paraformaldehyde at room temperature for 30 min. Cells were washed three times in PBS and subsequently blocked with 10% horse serum and 0.2% saponin in PBS for 20 min at room temperature. The blocking solution was shaken off, and the cells were incubated with rabbit anti-human SF (Dako, Glostrup, Denmark) overnight at 4°C. The following morning, cells were washed again in PBS and incubated with fluorescently labeled goat anti-rabbit IgG at room temperature for 60 min. Cells were washed in PBS and mounted in Vectashield (Vector Labs). Images of SF-immunoreactive nerve terminals were taken with the CLSM, at the same magnifications as for the DiI-stained cells, and stored for later analysis. The analysis involved automatic counting of SF-immunostained nerve terminals and measuring their size and fluorescence intensity, using National Institutes of Health image software. This analysis was conducted on 25 fields of controls and 27 fields of estradiol-treated cultures.
Calcium imaging. Cells were loaded with 5 μM Fura-2 AM, prepared from a stock DMSO solution of 1 mm(Molecular Probes), and 1 μl pluronic acid, prepared from a stock solution of 20 mg/ml DMSO. Dye loading was in a HEPES-buffered (10 mm) recording medium containing TTX (1 μm), Ca2+ (2 mm), Mg2+ (1 mm), NaCl (130 mm), KCl (4 mm), and glucose (10 mm). The medium had a pH of 7.4, and the osmolarity was adjusted to 320 mOsm with sucrose. The cultures were exposed to the dye for 45 min, followed by a 30 min wash. They were then used for imaging of glutamate-evoked [Ca]i changes. The culture coverslips were glued to the bottom of a small, 0.5 ml flow chamber placed in an inverted Zeiss microscope. The cells were imaged with a cooled CCD camera (Photometrics PXL) linked to an IBM computer. Images were taken at 5 sec intervals before and after exposure of the cells to pressure application of 1 mm glutamate applied through a micropipette with a tip diameter of 2–3 μm. The ratio of fluorescence emitted from the cells to excitation wavelengths of 340 and 380 nm were calculated before and after exposure to glutamate. The intensity of excitation with the two wavelengths was equated with neutral density filters to yield a ratio of ∼1 at resting [Ca]i. Groups of cells were compared using independent t tests and ANOVA wherever appropriate.
RESULTS
Effects of steroid hormones on dendritic spine density
Estradiol treatment for up to 3 d did not have an apparent effect on soma and primary dendritic morphology. The size of the somata was measured in 20 cells from each of the control and estradiol-treated cultures. The mean was about the same in the two groups: 334.5 ± 46.3 μm2 for control and 352.2 ± 72.8 μm2 for estradiol-treated cells. Likewise, dendritic diameters, measured randomly in the same regions where spines were counted, were not different between controls (1.12 ± 0.07 μm; mean ± SEM; n = 35 dendritic segments) and estradiol-treated cultures (0.97 ± 0.045 μm; n = 35;p > 0.05).
Spine density increased significantly after treatment of the cultures with estradiol; in control cells, spine density was 8.91 ± 0.57 (mean ± SEM) spines per 50 μm dendritic segment (Figs.1, 2), whereas in cells treated with 0.1 μg/ml 17-β estradiol, spine density was up to 20.44 ± 0.92 spines per 50 μm segment. This effect was stereoselective and was not seen after treatment of the cells with α-estradiol. Likewise, progesterone did not affect spine density in these cells, but it did block the action of estradiol (Fig. 2).
Fig. 1.
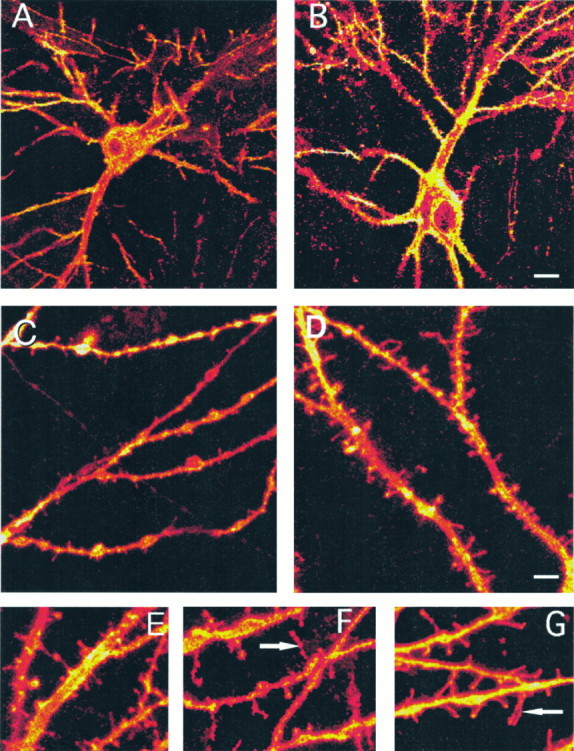
Three-dimensional, reconstructed images of DiI-stained cultured hippocampal neurons. A, B, Low-power images of control (A) and estradiol-treated (B) neurons. C, D, Zoomed images of dendrites of control (C) and estradiol-treated (D) cultured neurons. The counting of spines in these and similar dendrites requires going back and forth between the individual planes and the reconstructed image to verify that each and every spine is counted only once.E–G, Additional zoomed images of estradiol-treated (E), H7-treated (F), and combined H7/estradiol-treated cultures (G). Note the appearance of long filopodia-like spines (arrowheads) in the H7-treated cultures. Scale bar: A, B, 12 μm; C, D, 6 μm;E–G, 3 μm.
Fig. 2.

Estradiol produces a stereoselective effect on dendritic spines. 17-β-est (17-b est.) (0.1 μg/ml) and not α-est (a-est.) (1 μg/ml) causes a larger than twofold increase in spine density. The effects of estradiol are not mimicked by progesterone (125 μg/ml), which blocks the effects of estradiol. In this and the following figures, ** indicates statistical difference from control using the Student’s t test atp < 0.001; * indicates statistical difference ofp < 0.05; n = 110, the total number of dendritic segments analyzed in this experiment.
The dose dependency of estradiol was tested first by using three different doses of the hormone: 0.01 μg/ml, 0.1 μg/ml, and 1 μg/ml. All three doses caused a significant, nearly twofold increase in spine density (data not shown). Thereafter, all other experiments were performed at the intermediate dose of 0.1 μg/ml.
The effect of 17-β estradiol is mediated by activation of an estrogen receptor. In the presence of the estrogen receptor antagonist tamoxifen, the effects of estradiol on spine density were completely blocked (Fig. 3), indicating that estradiol did not exert its action via some nonspecific route, but it acts on a genuine estrogen receptor. In fact, tamoxifen produced a slight but nonsignificant reduction in spine density in the control condition, indicating that the growth medium does not contain significant concentrations of estrogen.
Fig. 3.

Estrogen receptor antagonist tamoxifen (Tam.) blocks the effects of estradiol (Est.) on spine density. Tamoxifen (1 μg/ml) blocks the effects of 0.1 μg/ml estradiol (E+T), even when applied 12 or 24 hr after the beginning of exposure to estradiol. n = 259 dendritic segments.
The time course of the effect of estradiol was studied in two types of experiments. In the first, estradiol was kept in the culture medium for up to 4 d. At various time periods, cells were fixed, and the density of the spines was measured as before. In these experiments, it seems that estradiol began to have a significant enhancing effect on spine density within 48 hr of exposure to the drug (Fig.4). An additional rise in spine density was seen for up to 4 d after exposure to the drug. In the second series of experiments, the cultures were exposed first to estradiol, followed by their simultaneous exposure to tamoxifen for 12–24 hr (Fig. 3). With both time points analyzed, the addition of tamoxifen reduced significantly the action of estradiol. Thus, it seems that estradiol has to be present for at least 24 hr to exert its action in the hippocampus.
Fig. 4.

Time course of estradiol action on dendritic spine density. Estradiol (0.1 μg/ml) produces a significant effect on spine density within 48 hr of its application. n = 240 dendritic segments.
Dendritic spines are the sites of excitatory synaptic connections and are thus enriched in glutamate receptors. We therefore examined the role of glutamate receptors in the action of estradiol on dendritic spine density. For this we employed the NMDA receptor antagonist 2-APV and the AMPA/KA receptor antagonist DNQX. 2-APV completely reversed the action of estradiol on dendritic spine density (Fig. 5). By contrast, DNQX had only a negligible effect on estradiol-induced spine formation. These data indicate that the effects of estradiol are mediated by the activation of an NMDA receptor. The possibility that the activation of the NMDA receptor is an indirect effect resulting from an increase in spontaneous activity of the neurons in the culture was examined by blocking action-potential discharges using TTX. By itself, TTX had no effect on spine density in the culture, but it reduced the effects of estradiol by ∼35% (Fig. 5). This effect was smaller than that produced by 2-APV, but it indicates that an increase in spontaneous action-potential discharges may play a role in the effect of estradiol on spine formation.
Fig. 5.

The NMDA receptor antagonist APV blocks the effects of estradiol on spine density. Estradiol (0.1 μg/ml) produces a twofold increase in spine density. APV, which has no effect of its own at 50 μm, blocks the effect of estradiol. DNQX, an AMPA/KA antagonist, has only a partial but a significant effect on estradiol action. Likewise,TTX reduces significantly the effect of estradiol but does not eliminate it. n = 264 dendritic segments.
The involvement of second messenger systems in the action of estradiol on spine formation was examined by comparing the effects of a drug that blocks serine/threonine kinase (H7) with a drug that blocks tyrosine kinase (genestein). H7 by itself had no effect on spine density, but it caused the formation of new filopodia, defined as thin, long (>3 μm), headless appendages resembling headless spines in immature cultures (Fig. 1) (Papa et al., 1995). In the presence of H7, the effects of estradiol on spine density were blocked completely (Fig.6). In contrast, genestein did not affect spine density, and neither did it interact with the effects of estradiol. Thus, it seems that estradiol acts via a serine/threonine kinase, possibly protein kinase C (PKC), to enhance formation of new spines.
Fig. 6.
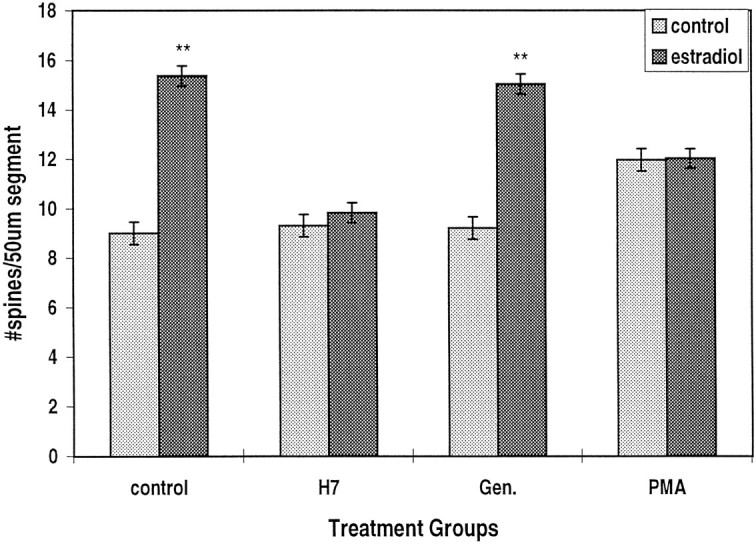
A serine/threonine kinase inhibitor, H7(15 μg/ml), but not a tyrosine kinase inhibitor, genestein (Gen.) (20 μg/ml), blocks the effect of estradiol on dendritic spine density. The phorbol ester PMA (150 nm) has a direct enhancing action on spine density but also reduces significantly the effect of estradiol.n = 300 dendritic segments.
To examine this hypothesis further, we exposed cells to the phorbol ester PMA, an activator of PKC. After 30 hr of exposure to PMA, there was a modest but significant increase in mean spine density from 9.0 ± 0.31 to 11.96 ± 0.42 spines per 50 μm segment (Fig. 6). In the same experiment, estradiol caused a rise of spine density to 15.4 ± 0.46 spines per 50 μm dendrite. Interestingly, there was no additive effect of the two drugs on spine density. In fact, the presence of PMA significantly reduced the effect of estradiol to 12.0 ± 0.43 spines per 50 μm segment.
Effects of estradiol on synapse formation
Estradiol may cause formation of new spines by converting existing shaft synapses into spine synapses or by producing new synapses. Alternatively, the new spines may be simple, nonfunctional extrusions from the dendrites, having no presynaptic terminals attached to them. To examine these possibilities, we stained control and estradiol-treated cultures with SF antibodies. Analysis of bouton staining in 25 and 27 random fields taken from control and estradiol-treated cultures, respectively, revealed no difference in the size of the terminals (32.1 ± 0.3 and 32.9 ± 0.3 pixels, where each pixel is 0.125 μm × 0.125 μm, corresponding to an approximate area of 0.6 μm2). There was, however, a significant difference in the staining intensity per terminal: the controls were 76.5 ± 0.78 intensity units, compared with 102.7 ± 0.82 units in the estradiol-treated cultures (p < 0.001) (Fig.7). This may indicate an increased density of SF protein, suggesting that the new spines may represent an enhanced density of synaptic connections. It is still unclear whether all of the newly formed spines contain functional synapses, or whether the SF-immunoreactive boutons form multiple contacts with one or several spines. This remains to be determined by additional experimentation with high-power electron microscopy.
Fig. 7.
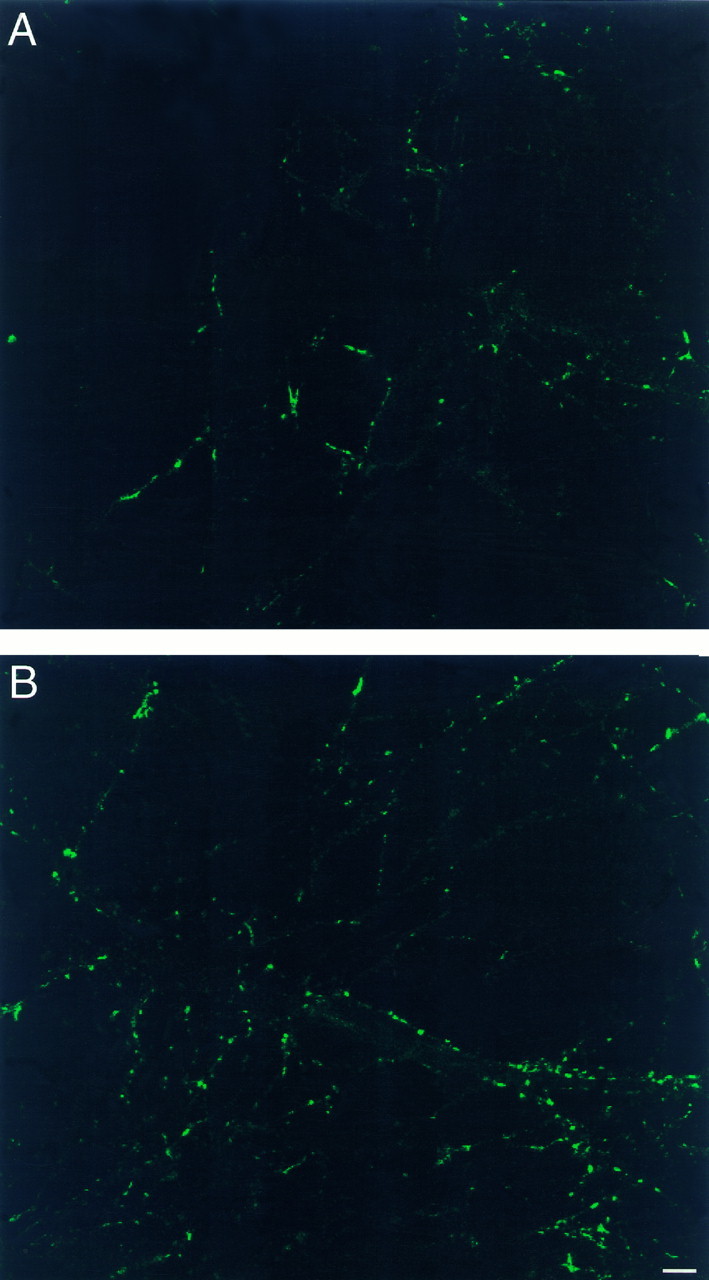
SF-containing boutons are different between control (A) and estradiol-treated (B) cultures. The intensity of staining is higher in the estradiol-treated cultures. Consult text for details. Scale bar, 10 μm.
Imaging of calcium variations in estradiol-treated cultures
The association of estradiol effects on spine formation with activation of the NMDA receptor led us to examine the effects of estradiol on the reactivity of cultured hippocampal neurons to glutamate. Two types of experiments were conducted. In the first, we studied the acute effects of estradiol on reactivity to glutamate in drug-naive cultures; in the second, we compared control and estradiol-treated cultures for reactivity to glutamate acting on NMDA and non-NMDA receptors. If indeed estradiol interacts directly with the NMDA receptor, one can expect that exposure to estradiol will modify reactivity to glutamate. The magnitude of the response and the time course of the recovery from calcium load after exposure to glutamate were compared before and after superfusion of cells with 1 μm estradiol. Exposure to glutamate caused a transient rise of [Ca]i in all cells examined to a level of ∼300 nm, as estimated from the ratio of emitted fluorescence to 340/380 nm illumination. This response peaked within 5 sec after the onset of exposure to glutamate and recovered to near basal levels within the following 10 sec. Exposure to estradiol caused an initial small and insignificant decline in reactivity to glutamate (Fig. 8B). No further decrease in reactivity to glutamate was seen with extended exposure to estradiol.
Fig. 8.
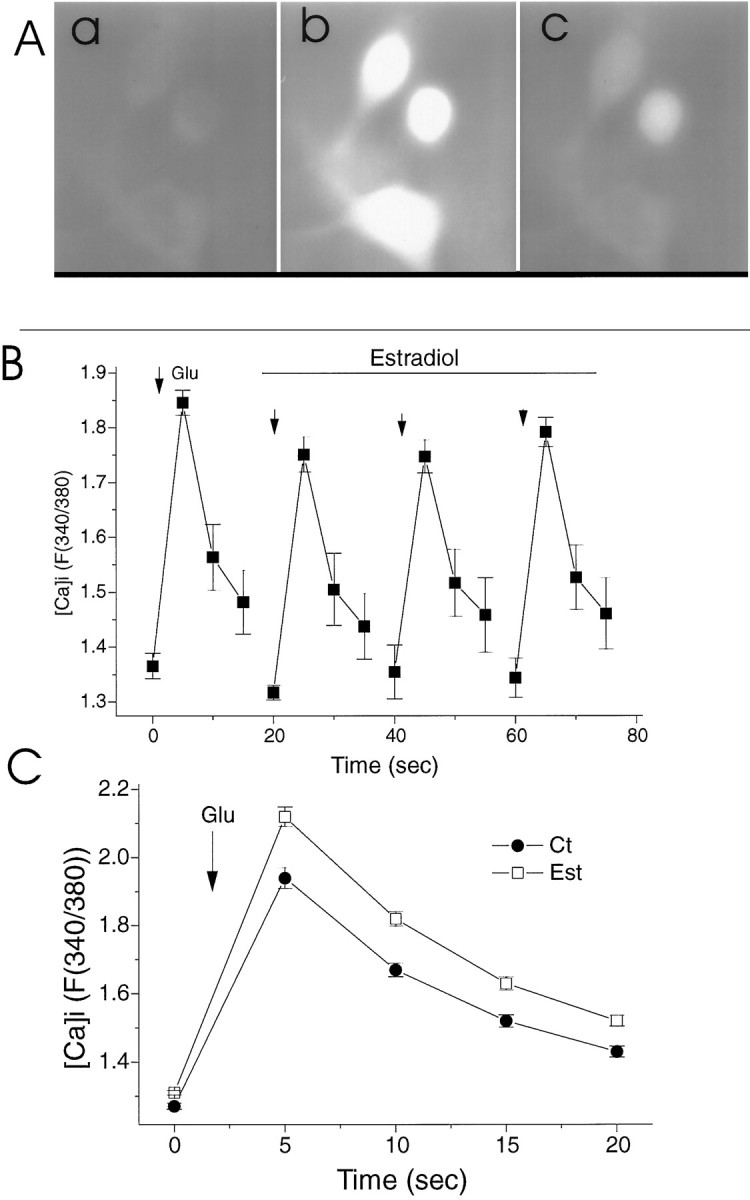
Intracellular calcium ([Ca]i) changes after exposure to pulse application of glutamate. A, An illustration of a ratioed image of a Fura-2-loaded cell at rest (left) and after exposure to glutamate (right). The measurements in this and the following figure were made of the region in the center of the cell, ∼15 μm in diameter, which includes the nucleus. B, Lack of acute effects of estradiol (0.1 μm) on reactivity to successive application of glutamate (Glu). For each sequence, a baseline control value was measured, followed by puff application of glutamate (arrowhead) followed by three measurements of fluorescence, taken at 5 sec intervals. Five to ten minutes elapsed between successive applications of glutamate. Note that at the peak response to glutamate, the heterogeneity (i.e., the SEM) is smaller than during recovery, indicating that the rate of recovery is widely distributed among different cells. C, A comparison of responses to glutamate of control (Ct) and estradiol-treated (Est) cultured neurons. A significant difference between the two groups in both the peak and the recovery of rest [Ca]i is evident.
In the other series of experiments, reactivity to glutamate was compared between the estradiol-treated and control cultures; 82 estradiol-treated cells exhibited a significantly larger (by 23%) and more persistent response to glutamate than 48 control cells did (Fig.8C).
In an attempt to dissociate between NMDA- and non-NMDA- (primarily voltage-gated calcium channels activated by glutamate-induced depolarization) mediated calcium changes, we repeated this study using conditions that favor activation of one of the two types of receptors (Fig. 9). As seen in the previous experiment, estradiol-treated cells (n = 24) exhibited a significantly larger (by 33%) response to glutamate than control cells did (n = 49). The cells were then washed with normal medium containing 0 mm Mg2+, glycine (10 μm), and a Ca2+ channel blocker, verapamil (20 μm). Under these conditions, which favor activation of NMDA receptors, estradiol-treated (n = 34) cells exhibited an even larger (by 95%) response to glutamate than control (n = 19) cells did. The cultures were then washed with normal medium containing 50 μm 2-APV. Under these conditions, the responses to glutamate, which now activates only non-NMDA receptors, was larger by 80% in estradiol-treated cells (n = 31) than in control cells (n = 31). These experiments indicate that estradiol causes an increase in reactivity of chronically exposed neurons to glutamate. Whether this is caused by an increase in the density of glutamate receptors, by a change in receptor affinity to glutamate, or by an increase in some second messenger systems associated with the glutamate receptors remains to be determined. A correlation seems to exist between the effects of estradiol on cellular morphology (i.e., spine formation) and function (glutamate responses).
Fig. 9.
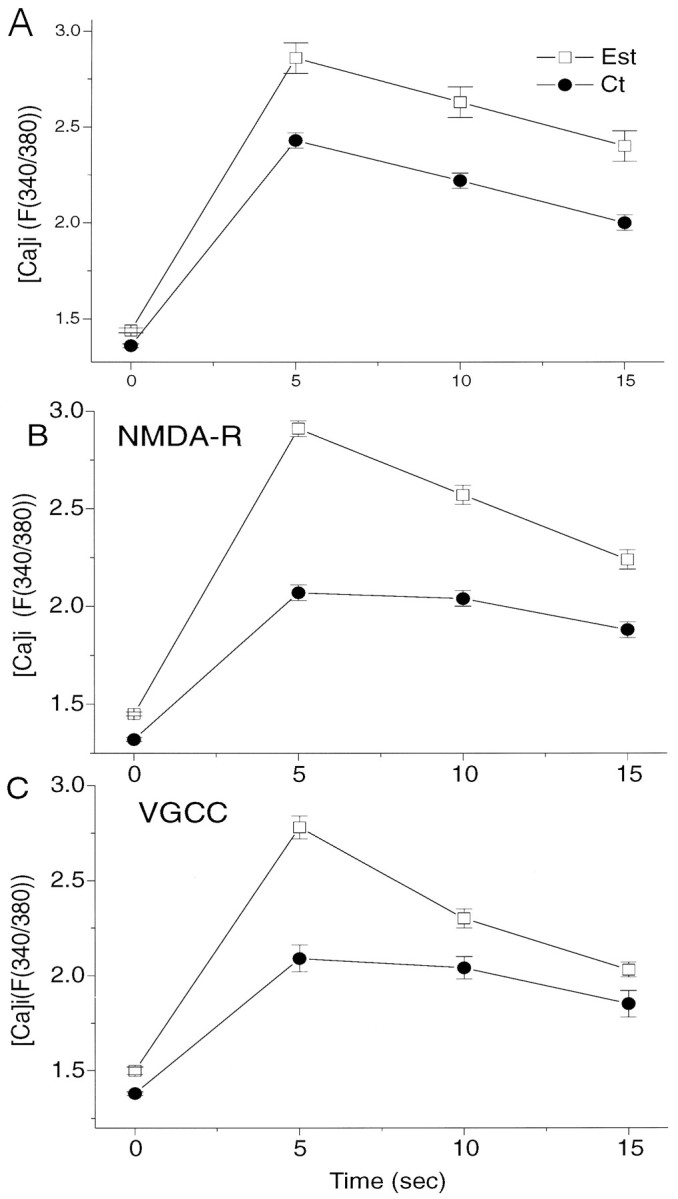
Effects of estradiol (Est) on [Ca]i reactivity of neurons to topical application of glutamate in conditions that favor activation of NMDA- and non-NMDA-linked [Ca]i changes.A, The total response to glutamate under normal conditions, as in Figure 8C, are seen. B, The culture medium contains nominally zero Mg2+, glycine, and a voltage-gated calcium Ca2+ blocker, verapamil (see Results for further details), to allow activation of NMDA receptor type. C, The cultures were treated with normal medium containing 1 mm Mg2+, to which 50 μm APV was added to allow detection of non-NMDA receptor/channels. In both B and C, a marked difference between normal and estradiol-treated cultures is seen.
DISCUSSION
Throughout the past decade, studies have shown the involvement of steroid hormones in the CNS (for review, see McEwen et al., 1993).In vivo models have explored the effects of estradiol in the hippocampus. Ovariectomized adult rats display a dramatic decrease in the density of dendritic spines, which can be reversed by the replacement of estradiol (Gould et al., 1990), and supplemental estradiol treatment can increase the density of dendritic spines. It has also been shown that normal fluctuations of estradiol during the 5-d estrous cycle in rats causes fluctuations in dendritic spines (Woolley et al., 1990) as well as in the density of synapses in the hippocampus (Woolley and McEwen, 1993). These effects seem to be mediated by an NMDA receptor-dependent mechanism (Woolley and McEwen, 1994). Behavioral studies have demonstrated that ovariectomized adult rats displayed impaired learning and memory, which was reversible with estradiol replacement (Singh et al., 1994). Clearly, estradiol has a marked effect on the modulation of spines in vivo and may be related to the effect of estradiol on hippocampal functions in the intact brain.
In the present study, we have successfully reproduced the in vivo estradiol response in a tissue culture model of hippocampal neurons. Our results are similar to those seen in vivo: 17-β-estradiol significantly increased the density of spines on hippocampal neurons. This effect is seen 48 hr, not 24 hr, after the onset of estradiol treatment, which is the same time course seenin vivo (Woolley and McEwen, 1993).
The cells that we analyzed are most likely medium-size CA1 pyramidal neurons, as the larger CA3 pyramids do not survive well in our culture conditions, and we were unable to detect the large mossy fiber-related dendritic spines typical of these neurons. Also the smaller, granular cells of the dentate gyrus have a distinctly different morphology and tend to produce clumps of cells, which were not studied herein. CA1 cells were those studied in vivo. One major difference between the in vivo and in vitro CA1 cells is the density of their dendritic spines, which are up to 8–10 times higherin vivo than in vitro (Harris et al., 1992). This difference can be caused by the relatively small number of afferents impinging on the dendrites of the in vitro cell, by the growth conditions, and by the possible lack of intrinsic growth factors. Additionally, the cultures are studied in a juvenile state, 2–3 weeks in vitro, corresponding to 10–17 d in neonatal rats. Despite the diminished number of spines, it is easier and more reliable to study dendritic spine density in the isolated culture dish, where each spine can be counted and the spine density analysis is fast and unequivocal. The initial low density of spines in the culture may underlie the larger effect of estradiol seen here by comparison with the in vivo case, where the reported changes are 30–50%.
The in vitro controlled tissue culture conditions allow us to study the mechanisms of estradiol action in relative isolation and to begin analysis of the factors involved in dendritic spine formation. The fact that the estradiol response can be blocked by tamoxifen and that α-estradiol does not mimic the effect of β-estradiol suggest that β-estradiol binds specifically to the estrogen receptor. The mechanism of action of estradiol may involve activation of PKC, as seen elsewhere. Estradiol upregulates PKC in ovaries and pituitary of several mammalian species, and this upregulation is inhibited by tamoxifen (Hahnel and Gschwendt, 1995). If estradiol activates PKC in hippocampal neurons, this may indicate that spine formation is initiated at the molecular level by this second messenger system. Indeed, the effect of estradiol is blocked by the broad-band serine/threonine kinase inhibitor H7, indicating that a kinase is involved in this action of estradiol. We chose H7 rather than a more selective PKC or other kinase antagonist to maximize our ability to observe some blocking effects on the action of estradiol. This should not be taken as direct evidence that PKC is the specific kinase associated with the action of estradiol and that more specific antagonists should be used in the context. The present results encourage such a search, in that they suggest that a serine/threonine but not a tyrosine kinase is activated by estradiol to increase dendritic spine density.
The role of the NMDA receptor in the action of estradiol is not entirely clear. Activation of the NMDA receptor is crucial for the induction of long-term potentiation in some synapses in the hippocampus and elsewhere (Bliss and Collingridge, 1993). If spine formation is associated with long-term plasticity in the brain, it will be intuitively associated with activation of the NMDA receptor. Is stimulation of kinases by estradiol causing the activation of the NMDA receptor, which forms new spines, or does estradiol affect NMDA receptors directly, which stimulates kinases to form new spines? Also, how is the rather gradual (48 hr) response to estradiol related to the fast action of glutamate at the NMDA receptor? These and related questions await further experimentation. Estradiol is also likely to increase spine density by affecting spontaneous synaptic activity of cells in culture. Blockade of action-potential discharges with TTX caused a partial blockade of the effects of estradiol on spine density. This indicates that estradiol may cause the formation of new synapses by increasing network activity, probably by increasing NMDA-mediated synaptic potentials.
The estrogen-initiated large fluctuations in dendritic spine density in the hippocampus, a structure traditionally associated with cognitive processes in the brain, have important theoretical implications with respect to the roles of dendritic spines in synaptic integration. If the spine is the site of memory storage, i.e., modification in spine shape/density represents the means by which a long-term change in synaptic efficacy is maintained, how can these large but transient changes in spine density be related to long-term memory storage? In fact, the estradiol effects may indicate just the opposite: that spines may also serve transient roles related to changes in synaptic strength occurring, for example, during the estrous cycle. The more enduring changes associated with long-term memory may not be seen in the hippocampus, or they may be associated with other morphological or nonmorphological changes in neurons. These may include receptor clustering, change in synaptic contact area, or spine form or shape (Fifkova and Van Harreveld, 1977; Lee et al., 1980; Chang and Greenough, 1984). The idea that spine density is not a vehicle for long-term memory storage is not consistent with other recent observations (Moser et al., 1994), and it is possible that spines have multiple functions in the brain that encompass long-term and short-term plasticity.
If not solely memory devices, what other functions can spines serve? It has been suggested recently that intracellular calcium concentrations can be regulated independent of the parent dendrites (Andrews et al., 1988; Wickens, 1988; Guthrie et al., 1991; Gold and Bear, 1994; Segal, 1995a). Thus, dendritic spines may prevent calcium surges from spreading into the parent dendrite and in that respect can be considered neuroprotectants (Harris and Kater, 1994; Segal, 1995b). Dendritic spines proliferate in the presence of excessive synaptic activity (Annis et al., 1994; Bundman et al., 1994; Papa and Segal, 1996), although some reports claim the opposite (Muller et al., 1993;Rocha and Sur, 1995). It is thought that most spines have one excitatory synapse on the head (Harris et al., 1992) and that most, if not all, excitatory synapses are on spines. Thus the doubling of spines in response to estradiol represents a doubling of excitatory synaptic inputs on each single spiny neuron. These dendritic spines can be absorbed back to their parent dendrites when the need to deal with this excessive synaptic activity is reduced. Our present observations are congruent with this hypothesis.
When imaging calcium transients in response to glutamate stimulation, we found that there was virtually no difference between the control responses and those seen during acute exposure to estradiol. Cells that had been treated with estradiol chronically (several days in culture), however, displayed intracellular calcium transients that were larger and longer in duration than those in controls. These findings would imply that the morphological change in neurons, i.e., the increase in spine density, may be responsible for the enhanced reactivity to glutamate. We cannot rule out the alternative possibility of a genomic action of estradiol on the glutamate receptors unrelated to morphological changes. Further analysis is needed to determine the causality between spine density and glutamate-evoked [Ca]i.
The present results illustrate the viability of the culture as a model system for the analysis of factors affecting the production of dendritic spines in central neurons. The ability to control the microenvironment of the cells and to expose the cells to drugs that may have adverse effects in vivo, as well as the relative simplicity of the cultured neurons, allows rapid progress in the study of the mechanisms associated with dendritic spines.
Footnotes
We thank Drs. T. S. Reese and S. B. Andrews for their support and Dr. C. Collin for help with the calcium imaging system.
Correspondence should be addressed to M. Segal, Department of Neurobiology, The Weizmann Institute, Rehovot 76100, Israel.
REFERENCES
- 1.Andrews SB, Leapman RD, Landis DMD, Reese TS. Activity dependent accumulation of calcium in Purkinje cell dendritic spines. Proc Natl Acad Sci USA. 1988;85:1682–1685. doi: 10.1073/pnas.85.5.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Annis CM, O’Dowd DK, Robertson RT. Activity-dependent regulation of dendritic spine density on cortical pyramidal neurons in organotypic slice culture. J Neurobiol. 1994;25:1483–1493. doi: 10.1002/neu.480251202. [DOI] [PubMed] [Google Scholar]
- 3.Bliss TVP, Collingridge GL. A synaptic model of memory: long term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 4.Bundman MC, Pico RM, Gall CM. Ultrastructural plasticity of the dentate gyrus granule cells following recurrent limbic seizures: increase in somatic spines. Hippocampus. 1994;4:601–610. doi: 10.1002/hipo.450040510. [DOI] [PubMed] [Google Scholar]
- 5.Chang F, Greenough WT. Transient and enduring morphological correlates of synaptic activity and efficacy in the rat hippocampal slice. Brain Res. 1984;309:35–46. doi: 10.1016/0006-8993(84)91008-4. [DOI] [PubMed] [Google Scholar]
- 6.Fifkova E, Van Harreveld A. Long lasting morphological changes in dendritic spines of dentate granular cells following stimulation of the entorhinal area. J Neurocytol. 1977;6:211–230. doi: 10.1007/BF01261506. [DOI] [PubMed] [Google Scholar]
- 7.Gold JI, Bear MF. A model of dendritic spine Ca2+ concentration exploring possible bases for a sliding synaptic modification threshold. Proc Natl Acad Sci USA. 1994;91:3941–3945. doi: 10.1073/pnas.91.9.3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gould E, Woolley C, Frankfurt M, McEwen B (1990) Gonadal steroids regulate dendritic spine density in hippocampal pyramidal cells in adulthood. J Neurosci 10:1286–1291. [DOI] [PMC free article] [PubMed]
- 9.Guthrie PB, Segal M, Kater SB. Independent regulation of calcium revealed by imaging dendritic spines. Nature. 1991;354:76–80. doi: 10.1038/354076a0. [DOI] [PubMed] [Google Scholar]
- 10.Hahnel R, Gschwendt M. Interaction between protein kinase C (PKC) and estrogens. Int J Oncol. 1995;7:11–16. doi: 10.3892/ijo.7.1.11. [DOI] [PubMed] [Google Scholar]
- 11.Harris KM, Kater SB. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci. 1994;17:341–371. doi: 10.1146/annurev.ne.17.030194.002013. [DOI] [PubMed] [Google Scholar]
- 12.Harris KM, Jensen FE, Tsao BH. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12:2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hosokawa T, Bliss TVP, Fine A. Persistence of individual dendritic spines in living brain slices. NeuroReport. 1992;3:477–480. doi: 10.1097/00001756-199206000-00005. [DOI] [PubMed] [Google Scholar]
- 14.Lee KS, Schottler F, Oliver M, Lynch G. Brief bursts of high-frequency stimulation produce two types of structural change in the rat hippocampus. J Neurophysiol. 1980;44:247–258. doi: 10.1152/jn.1980.44.2.247. [DOI] [PubMed] [Google Scholar]
- 15.McEwen BS, Cameron H, Chao H, Gould E, Magarinos A, Watanabe Y, Woolley C. Adrenal steroids and plasticity of hippocampal neurons: toward an understanding of underlying cellular and molecular mechanisms. Cell Mol Neurobiol. 1993;13:457–482. doi: 10.1007/BF00711583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moser MB, Trommald M, Andersen P. An increase in dendritic spine density on hippocampal CA1 cells following spatial-learning in adult rats suggests the formation of new synapses. Proc Natl Acad Sci USA. 1994;91:12673–12675. doi: 10.1073/pnas.91.26.12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muller M, Gahwiler BH, Rietschin L, Thompson SM. Reversible loss of dendritic spines and altered excitability after chronic epilepsy in hippocampal slice cultures. Proc Natl Acad Sci USA. 1993;90:257–261. doi: 10.1073/pnas.90.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papa M, Segal M (1996) Morphological plasticity in dendritic spines of cultured hippocampal neurons. Neuroscience, 71:1005–1011. [DOI] [PubMed]
- 19.Papa M, Bundman MC, Greenberger V, Segal M. Morphological analysis of dendritic spine development in primary cultures of hippocampal neurons. J Neurosci. 1995;15:1–11. doi: 10.1523/JNEUROSCI.15-01-00001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rocha M, Sur M. Rapid acquisition of dendritic spines by visual thalamic neurons after blockade of N -methyl-d-aspartate receptors. Proc Natl Acad Sci USA. 1995;92:8026–8030. doi: 10.1073/pnas.92.17.8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Segal M. Imaging of calcium variations in living dendritic spines of cultured rat hippocampal neurons. J Physiol (Lond) 1995a;486:285–296. doi: 10.1113/jphysiol.1995.sp020811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Segal M. Spines for neuroprotection: a hypothesis. Trends Neurosci. 1995b;11:468–471. doi: 10.1016/0166-2236(95)92765-i. [DOI] [PubMed] [Google Scholar]
- 23.Singh M, Meyer E, Millard W, Simpkins J. Ovarian steroid deprivation results in a reversible learning impairment and compromised cholinergic function in female Sprague–Dawley rats. Brain Res. 1994;644:305–312. doi: 10.1016/0006-8993(94)91694-2. [DOI] [PubMed] [Google Scholar]
- 24.Wickens J. Electrically coupled but chemically isolated synapses: dendritic spines and calcium in a rule for synaptic modification. Prog Neurobiol. 1988;31:507–528. doi: 10.1016/0301-0082(88)90013-5. [DOI] [PubMed] [Google Scholar]
- 25.Woolley C, McEwen B. Roles of estradiol and progesterone in regulation of hippocampal dendritic spine density during the estrous cycle in the rat. J Comp Neurol. 1993;336:293–306. doi: 10.1002/cne.903360210. [DOI] [PubMed] [Google Scholar]
- 26.Woolley C, McEwen B. Estradiol regulates hippocampal dendritic spine density via an N -methyl-d-aspartate receptor dependent mechanism. J Neurosci. 1994;14:7680–7687. doi: 10.1523/JNEUROSCI.14-12-07680.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woolley CS, Gould E, Frankfurt M, McEwen BS. Naturally occurring fluctuation in dendritic spine density on adult hippocampal pyramidal neurons. J Neurosci. 1990;10:4035–4039. doi: 10.1523/JNEUROSCI.10-12-04035.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]