

Abstract
Although the interleukin-1β converting enzyme (ICE)/CED-3 family of proteases has been implicated recently in neuronal cell deathin vitro and in ovo, the role of specific genes belonging to this family in cell death in the nervous system remains unknown. To address this question, we examined the in vivoexpression of one of these genes, Ice, after global forebrain ischemia in gerbils. Using RT-PCR and Western immunoblot techniques, we detected an increase in the mRNA and protein expression of ICE in hippocampus during a period of 4 d after ischemia. Chromatin condensation was observed in CA1 neurons within 2 d after ischemia. Internucleosomal DNA fragmentation and apoptotic bodies were observed between 3 and 4 d after ischemia, a period during which CA1 neuronal death is maximal. In nonischemic brains, ICE-like immunoreactivity was relatively low in CA1 pyramidal neurons but high in scattered hippocampal interneurons. After ischemia, ICE-like immunoreactivity was not altered in these neurons. ICE-like immunoreactivity, however, was observed in microglial cells in the regions adjacent to the CA1 layer as early as 2 d after ischemic insult. The increase in ICE-like immunoreactivity was robust at 4 d after ischemia, a period that correlates with the DNA fragmentation observed in hippocampal homogenates of ischemic brains. These results provide the first evidence for the localization and induction of ICE expression in vivo after ischemia and suggest an indirect role for ICE in ischemic damage through mediation of an inflammatory response.
Keywords: apoptosis, interleukin-1β converting enzyme, neuronal cell death, global forebrain ischemia, glia, hippocampus
The hypothesis that a genetic program induces cell death stems from studies demonstrating that programmed cell death is dependent on macromolecular synthesis (Tata, 1966; Martin et al., 1988;Johnson et al., 1989; Oppenheim et al., 1990; Johnson and Deckwerth, 1993). One of the genes required for somatic cell death duringCaenorhabditis elegans development, Ced-3 (Ellis and Horvitz, 1986), shares ∼28% amino acid identity with human interleukin-1β converting enzyme (ICE) (Yuan, 1993). Overexpression of ICE in mammalian cells is sufficient to cause apoptosis (Miura et al., 1993), a distinct morphological feature of programmed cell death. Although mice harboring a disrupted Ice gene are resistant to Fas-mediated apoptosis, they do not exhibit gross developmental abnormalities, which suggests that ICE may not be necessary for programmed cell death during development (Kuida et al., 1995; Li et al., 1995).
In the nervous system, the involvement of ICE, or homologous proteases, in apoptosis was deduced from the observation that microinjection into dorsal root ganglion cells of the cytokine response modifier A (crmA) gene product, a viral serpin-like inhibitor of ICE-like proteases (Komiyama et al., 1994), blocks apoptosis induced by NGF deprivation (Gagliardini et al., 1994). Furthermore, peptide inhibitors of ICE-like proteases prevent cell death of motor neurons that lack trophic support in vitro and in ovo(Milligan et al., 1995). Members of the Ice/Ced-3multigene family that potentially can regulate apoptosis areprIce (Lazebnik et al., 1994),Icerel-II/Ich-2/Tx(Faucheu et al., 1995; Munday et al., 1995),ICE-rel-III (Munday et al., 1995),Nedd-2/Ich-1 (Kumar et al., 1992; Wang et al., 1994), CPP32/Yama/Apopain(Fernandes-Alnemri et al., 1994; Nicholson et al., 1995; Tewari et al., 1995), Mch-2/Ice-Lap3 (Fernandes-Alnemri et al., 1995a; Duan et al., 1996), Mch3 (Fernandes-Alnemri et al., 1995b), and Cmh-1 (Lippke et al., 1996). The cellular localization of these gene products in vivo and their temporal expression during naturally occurring or induced apoptosis in the nervous system is unclear.
ICE and/or its homologs could also participate in cell death via indirect mechanisms such as inflammation. The preferred substrate for ICE is proIL-1β, a 31 kDa inactive precursor that is proteolytically cleaved to the 17.5 kDa mature form, a mediator of inflammation (Cerretti et al., 1992; Thornberry et al., 1992). The role of ICE in processing proIL-1β is demonstrated by the ability of ICE inhibitors to selectively inhibit production of mature IL-1β in macrophagesin vitro and in vivo (Thornberry et al., 1992;Uhl et al., 1993; Miller et al., 1995).
Global forebrain ischemia in gerbils is an attractive model for analyzing changes in the expression of cell death genes in vivo. The CA1 pyramidal neurons in gerbil hippocampus, which are selectively vulnerable to ischemic damage (Kirino and Sano, 1984a,b), undergo apoptotic cell death between 3 and 4 d after ischemia (Nitatori et al., 1995). In this report, we have evaluated the in vivo localization and temporal relationship of ICE expression to the apoptotic cell death observed after global forebrain ischemia.
MATERIALS AND METHODS
Animals. Male mongolian gerbils (65–75 gm; Harlan Sprague–Dawley) were housed six to a cage and given food and waterad libitum. All procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the Cephalon institutional animal care and use committee.
Surgical procedures and sample preparation. The common carotid arteries were exposed and occluded bilaterally for 7 min in gerbils anesthetized with 4% isofluorane volatilized by 30% O2 and 70% N2. Core temperature was regulated carefully at 38°C by a thermostatically controlled heat lamp. Reperfusion was instituted by the release of the arterial occlusion. The skin incision on the neck was closed with a wound clip, and the gerbils were allowed to recover from anesthesia in a temperature-controlled incubator for 1 hr, after which they were returned to home cages. Gerbils were killed at 1, 2, 3, and 4 d after ischemia, and brain tissue was prepared as described below. For RT-PCR, Western immunoblotting, and evaluating DNA fragmentation, brains were dissected rapidly, and the cortex was removed. The hippocampal sector containing the CA1 region was excised using a 3 mm hole punch, and the tissue was frozen rapidly in liquid nitrogen and stored at −70°C.
Antibodies. A rabbit antiserum specific for ICE (Ab122) was prepared using the synthetic peptide EHKTSDST conjugated through a C-terminal alanine-cysteine to keyhole limpet hemocyanin. The peptide corresponds to a region of the mouse ICE proenzyme and p20 subunit (residues 223–230) that is completely conserved in human ICE but has one or more nonconservative differences with all other known ICE family members. The reactivity of Ab122 with ICE was confirmed by immunoblotting with recombinant baculovirus-derived human ICE (S. Meyer, unpublished observation). ICE-like immunoreactivity was also determined using a commercially available polyclonal antibody, AbM-20 (Santa Cruz Biotech, Santa Cruz, CA), which detects pro-ICE of mouse origin. Astrocytes were visualized with rabbit polyclonal antisera directed against glial fibrillary acidic protein (GFAP) (Sigma, St. Louis, MO) at a dilution of 1:2000.
Gel electrophoresis of fragmented DNA. Genomic DNA was extracted from hippocampal tissue of nonischemic control and ischemic gerbils (1–4 d after ischemia) using an Easy DNA extraction kit (Invitrogen, San Diego, CA). Ten micrograms of DNA were electrophoresed on a 3% agarose gel. Fragmented DNA was visualized by ethidium bromide under an ultraviolet light source.
Isolation of RNA and RT-PCR analysis. Total RNA was isolated from hippocampal tissue (0–4 d after ischemia) using RNAzol B (Cinna Biotecx Labs, Houston, TX). RNA integrity was determined by electrophoresis on a 1% agarose gel followed by visualization of intact 18S and 28S ribosomal RNA bands. RNA purity was measured using the A260/A280ratio (average ratio was ≥ 1.85). To ensure that the optical density at A260 was in the linear range, various concentrations of RNA were plotted against absorbance.
cDNA was synthesized from 1 μg of mRNA by reverse transcription using 200 U of Superscript II reverse transcriptase (Life Technologies, Gaithersburg, MD) and oligo (dT) as primers in a 15 μl reaction containing 1× Superscript buffer (Life Technologies), 500 μm of each dNTP, 0.1 mDTT, and 40 U of RNAsin (Promega). After incubation for 1 hr at 42°C, the reaction was terminated by denaturing the enzyme for 5 min at 95°C. Reverse transcription was followed by amplification using individual primer sets for Ice or GAPDH. The following oligonucleotides derived from the corresponding nucleotide sequence were used as primers:
hIce:
5′ primer: 5′-CCAGAGCACAAGACCTCTGAC-3′ (hIce exon 6; 661–681).
3′ primer: 5′-GCTTTCTGCTCTTCCACACCA-3′ (hIce exon 7; 978–998).
GAPDH: 0.45 kb (Clontech Labs, Palo Alto, CA)
5′ primer: 5′-ACCACAGTCCATGCCATCAC-3′.
3′ primer: 5′-TCCACCACCCTGTTGCTGTA-3′.
For PCR amplification, 1 μl of cDNA was incubated in a 40 μl reaction mix containing 0.5 μm of each primer, 1 U of Taq polymerase (Promega, Madison, WI), 1×Taq buffer, 150 μm of each NTP, 1.5 μm MgCl2, and 400 nCi [32P]dCTP. PCR was performed in a DNA Thermal Cycler (Perkin-Elmer Cetus, Norwalk, CT) using the following conditions: 1 min at 95°C, 1 min at 58°C, and 2 min at 72°C. After amplification, samples (10 μl of each reaction) were subjected to 5% PAGE. Gels were quantified by PhosphorImager (Molecular Dynamics, Sunnyvale, CA) analysis.
In initial experiments, the optimal numbers of PCR cycles forIce and GAPDH were determined to be 35 and 25, respectively. The volume of each amplified product was integrated and plotted graphically against the number of PCR cycles to determine whether the increase in intensity of the amplified product was linear with the number of PCR cycles. In addition, experiments were conducted to determine the relationship between the amount of mRNA used as starting material and the corresponding amplified product intensity.
Western immunoblot analysis. Samples from the CA1 hippocampal sector (0–4 d after ischemia) were dissected as described above and homogenized in buffer containing 10 mmTris HCl, 1 mm EGTA, 1 mmEDTA, 0.02% sodium azide, and 0.1% PMSF. The mixture was then added to 3× Laemmli sample buffer containing 2% SDS, 125 mm Tris-HCl, pH 6.8, 10% w/v glycerol, and 5% β-mercaptoethanol in a 2:1 ratio, sonicated briefly, and boiled for 1 min. Protein concentration was determined in each sample before the addition of SDS. Proteins (20 μg) were separated on a 4–20% gradient gel in Tris-glycine buffer (Bio-Rad, Melville, NY) and then transferred to nitrocellulose membranes. Filters were probed with the ICE antibodies at a concentration of 1:800. Detection was carried out by use of a secondary horseradish peroxidase-linked anti-rabbit antibody (1:2000) (Bio-Rad) and developed using the chemiluminescence procedure (Amersham, Buckinghamshire, UK).
Immunohistochemistry. Gerbils were anesthetized (0–4 d after is- chemia) with 50 mg/kg nembutol, and brains were perfusion-fixed with 4% paraformaldehyde. Brains were postfixed for 30 min and cryoprotected in 30% sucrose at 4°C, overnight. Brains were then frozen rapidly using isopentane (kept at −30°C) and stored at −70°C. Fifty micrometer sections were cut on a sliding microtome, and immunohistochemistry was performed on free-floating sections using the Vectastain ABC Elite kit (Vector Laboratories, Burlingame, CA). Primary antibodies to ICE (Santa Cruz Biotech) were used at a dilution of 1:800 in 5% blotto containing 0.25% Triton X-100 and 2% normal goat serum. Incubations were performed for 2 nights at 4°C.
Histopathology. Gerbils were perfusion-fixed with 4% paraformaldehyde and 0.25% glutaraldehyde, 0–4 d after ischemia. The hippocampi were dissected and sliced into 2 mm cross-sectional slabs. These were dehydrated in 95% ethanol and embedded in glycol methacrylate (Historesin; Leica, Heidelberg, Germany). Sections were cut at 2 μm and stained with cresyl violet.
RESULTS
Apoptotic cell death in hippocampal CA1 sector after ischemia
Cell death in the hippocampus was characterized in the CA1 sector after global forebrain ischemia in gerbils to provide a framework for evaluating changes in the expression of ICE. Gerbils were killed at 1, 2, 3, and 4 d after ischemia. Histological examination of sections stained with cresyl violet indicated that loss of CA1 pyramidal neurons was minimal up to 3 d after ischemia; however, the vast majority of these cells was lost between 3 and 4 d after ischemia (Fig.1A).
Fig. 1.

A, Loss of CA1 pyramidal neurons 4 d after global forebrain ischemia. Sections from nonischemic control (CON) gerbil hippocampus and hippocampus obtained from gerbils 4 d (4d) after ischemia were stained with cresyl violet. Low-power photomicrographs show the cell loss in the CA1 region of the hippocampus (arrowhead) 4 d after global forebrain ischemia. dg, Dentate gyrus. B, Time course of DNA fragmentation in hippocampal tissue after ischemia. Genomic DNA was extracted from ischemic and nonischemic hippocampi and subjected to gel electrophoresis. Laddering of fragmented DNA is observed between 3 and 4 d after ischemia.
To characterize the DNA fragmentation that occurred in this model, genomic DNA from hippocampi (0–4 d after ischemia) was extracted and electrophoresed on a 3% agarose gel stained with ethidium bromide. Biochemically, cells undergoing apoptosis are associated with cleavage of genomic DNA into nucleosomal fragments differing in length by multiples of 200 bp, which give rise to a characteristic DNA ladder when subjected to agarose gel electrophoresis. Consistent with a previous report (Nitatori et al., 1995), DNA fragmentation was observed between 3 and 4 d after ischemia (Fig. 1B), which suggests that the delayed cell death is apoptotic.
DNA degradation by endonucleases and subsequent laddering is not the sole criterion for apoptosis (Cohen et al., 1992; Schulze-Osthoff et al., 1994). Therefore, to characterize further the delayed cell death of CA1 pyramidal neurons after ischemia, 2 μm plastic-embedded hippocampal sections were stained with cresyl violet. In comparison with nonischemic neurons (Fig. 2A), neurons in the CA1 region seemed to undergo the classic morphological changes associated with apoptosis (Clarke, 1990) after ischemia. As shown in Figure 2B, within 2 d after ischemia, CA1 pyramidal neurons began to show nuclear condensation and in some cases cytoplasmic shrinkage. Dense chromatin masses appeared in the nucleus, which was followed by margination of condensed chromatin along the nuclear envelope. Occasionally, a microglial cell was associated with a neuron. By 3 d after ischemia, most of the neurons exhibited late stages of apoptosis. The membranes appeared convoluted, clumping of chromatin was more evident, and cytoplasmic organelles were no longer visible (Fig. 2C). These changes culminated in the formation of apoptotic bodies 4 d after ischemia (Fig. 2D). As is true in apoptosis induced in vitro, neurons did not seem to undergo apoptosis in a synchronous fashion, as is evident in Figure2B,D. Although most of the neurons exhibited signs of apoptosis, some of the neurons seemed necrotic (Fig. 2D,inset), because in these neurons blebbing of membranes was clearly evident in the absence of pyknosis. The time course of the late stages of apoptosis observed histologically was consistent with the time course of DNA fragmentation.
Fig. 2.

Morphological evidence for a- poptosis in CA1 neurons after ischemia. A, Normal CA1 pyramidal neurons from nonischemic gerbil. B, Margination of condensed chromatin along the nuclear envelope of CA1 neurons 2 d after ischemia. Some neurons exhibit nuclear shrinkage (inset). Occasionally microglia are seen attached to a neuronal cell body (arrowhead). C, Most CA1 neurons have shrunken nuclei by 3 d after ischemia. Clumping of chromatin and late stages of apoptotic morphology are evident. Numerous microglia are found in the CA1 region. D, End-stage apoptosis is evident by 4 d after ischemia (arrow). Some neurons exhibit morphological features of necrosis (inset).
Time course of Ice mRNA levels in hippocampus after ischemia
To determine whether the expression of Ice mRNA is altered by ischemia, RT-PCR was performed on mRNA isolated from hippocampal tissue 0–4 d after the ischemic insult. A single amplified product of the predicted fragment size was detected. As shown in Figure3A, a robust increase in the expression ofIce was observed in the hippocampus after ischemia. This increase in Ice expression was observed in six gerbils at each time point after ischemia. Sham-operated gerbils did not show any change in levels of Ice expression. The integrated volume of the amplified product (as a percentage of control) was plotted against the time post-ischemia to visualize the average changes (n = 6) that occurred in Ice mRNA expression (Fig. 3B). An increase in Ice mRNA levels was evident as early as 1 d and continued to increase at 3–4 d after ischemic insult. As a control, the expression of the housekeeping gene (GAPDH) was examined. No changes in the mRNA levels ofGAPDH were observed after ischemia.
Fig. 3.

RT-PCR analysis of the time course ofIce mRNA after ischemia. A, The time course of expression of Ice mRNA levels in sham-operated and in ischemic gerbil hippocampi shows that low levels of Ice mRNA are detected in sham-operated and nonischemic gerbil hippocampus. After ischemia, an increase in Ice mRNA levels is observed as early as 24 hr and continues to increase during the 4 d after ischemia. mRNA isolated from the same tissues was subjected to RT-PCR using primers for the GAPDH gene. No significant alteration in mRNA expression is observed for this gene. B, Representation of amplified products (volumetric quantitation) as a percentage of nonischemic control at various post-ischemic intervals.
Temporal expression of ICE protein after ischemia
To determine whether a corresponding change in ICE protein could be observed, Western immunoblotting was performed using homogenates from hippocampal tissue. No detectable ICE immunoreactivity was observed in immunoblots of hippocampal tissue from nonischemic gerbils. When hippocampal homogenates from ischemic brain (4 d after ischemia) were used, however, an immunoreactive species corresponding to ∼45 kDa, which is the size of pro-ICE, was detected (Fig.4a). This indicated that the expression of ICE in nonischemic hippocampi was low but increased after ischemia. This increase in ICE protein expression after ischemia was observed using two ICE antibodies (AbM-20 and Ab122). The addition of a 100-fold excess of a blocking peptide to Ab122 resulted in the reduction of the 45 kDa immunoreactivity, demonstrating the specificity of the antibody for pro-ICE. The appearance of either the p20 or p10 catalytic subunits of ICE was not detected after ischemia. The time course of hippocampal pro-ICE expression (Fig. 4b) shows a gradual increase during the 4 d post-ischemic interval.
Fig. 4.

Western immunoblot analysis of ICE expression.a, Twenty micrograms of protein from hippocampal homogenates from control (C) and 4 d post-ischemic brains were subjected to gel electrophoresis, and the blots were probed with polyclonal antibodies to ICE (AbM-20 and Ab122). Both antibodies detect an increase in the 45 kDa immunoreactive species (arrowhead) 4 d after ischemia. Preadsorption with 100-fold excess peptide prepared against Ab122 significantly reduced the intensity of this band. b, Time course of ICE protein expression detected by ABM-20 in hippocampal homogenates in nonischemic control (C) and 1–4 d after ischemia.
Cellular localization of ICE-like immunoreactivity
Although the RT-PCR and Western immunoblot analyses demonstrated an increase in ICE expression after ischemia, these analyses do not describe the localization of ICE or the changes associated within a particular cell type. To facilitate the cellular localization of this protein, we examined ICE-like immunoreactivity in gerbil hippocampal sections after ischemia. The Ab122 and AbM-20 gave comparable staining in gerbil hippocampus. In nonischemic hippocampal sections, the stratum pyramidale of CA1 and the granule cell layer of the dentate gyrus were moderately immunoreactive when examined under low power (Fig.5a). Examination of this material under high power (Fig. 6a), however, showed that most of the immunoreactivity was associated with interneurons within the CA1. Twenty-four hours after ischemia, ICE-like immunoreactivity increased in the stratum lacunosum-moleculare of CA1, a region occupied by the apical and innervated by projections from the midline thalamic nuclei and entorhinal cortex (data not shown). In this layer, ICE-like immunoreactive material was dispersed evenly and was not localized to any particular cell type. Furthermore, ICE-like immunoreactivity was not altered in CA1 pyramidal neurons. In addition to the staining in stratum lacunosum-moleculare, ICE-like immunoreactivity appeared as a faint band in the outer two thirds of the stratum moleculare of the dentate gyrus 2 d after ischemia (Fig. 5b). At this time, cells in the region of the hippocampal fissure were lightly immunoreactive for ICE. These cells were small, with elongated cell bodies, and had one or more thick processes and numerous short secondary branches along their length, characteristic of a microglial cell (Fig. 6f, inset). By 3 d after ischemia, ICE-like immunoreactivity disappeared from the stratum moleculare of the dentate gyrus but persisted in the stratum lacunosum-moleculare of CA1 (Fig. 5c). In addition, ICE-like immunoreactivity appeared in microglia in the stratum radiatum. Some of these microglia seemed to be attached to the apical dendrites of the CA1 pyramidal neurons. ICE-like immunoreactivity was relatively high in microglia that seemed to be infiltrating from the areas adjacent to the ventricle. By 4 d after ischemia, ICE-like immunoreactivity was robust in the activated microglial cells in all layers of the hippocampus, including the alveus, stratum oriens, stratum radiatum, and stratum lacunosum-moleculare (Fig. 5d). During the period after ischemia, ICE-like immunoreactivity was not altered in either the CA1 pyramidal neurons that undergo apoptotic cell death or the interneurons within this region.
Fig. 5.
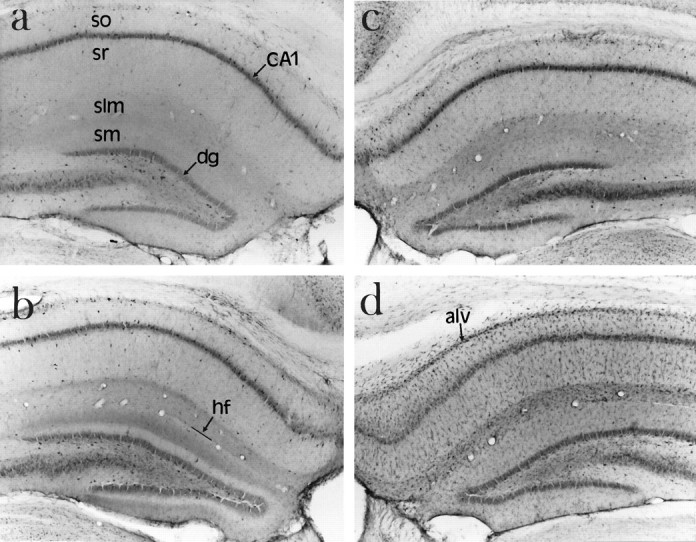
Increased expression of ICE-like immunoreactivity in hippocampus after ischemia. Gerbils were perfusion-fixed and 50 μm frozen sections were stained free-floating for ICE immunoreactivity using AbM-20. Low-power photomicrographs show (a) ICE-like immunoreactivity in control (nonischemic) brain section (b) 2 d after ischemia, (c) 3 d after ischemia, and (d) 4 d after ischemia. so, Stratum oriens;sr, stratum radiatum; slm, stratum lacunosum-moleculare; sm, stratum moleculare; hf, hippocampal fissure; alv, alveus; dg, dentate gyrus.
Fig. 6.
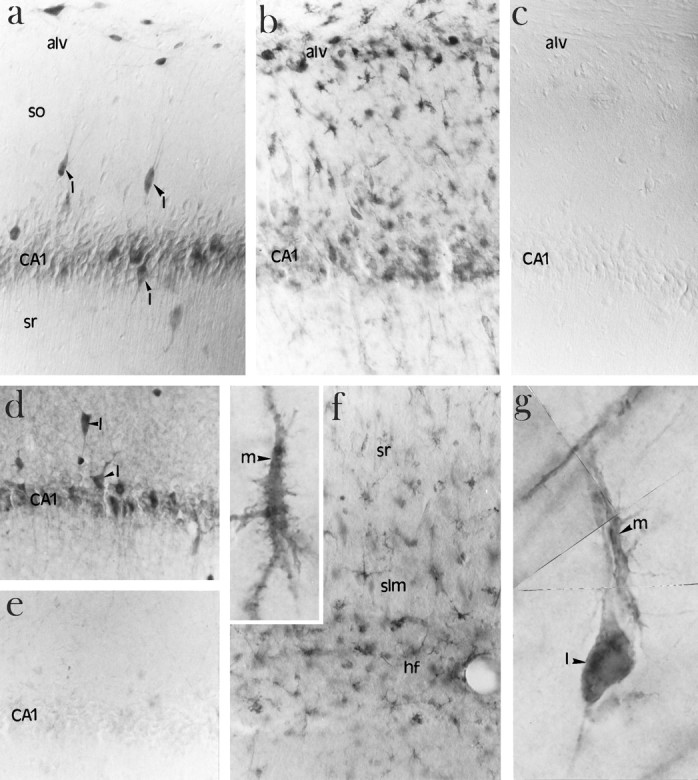
Immunolocalization of ICE in microglia after ischemia. a, High-power photomicrograph of ICE-like immunoreactivity localized to interneurons (arrowheads) in the CA1 region of the hippocampus of a nonischemic brain using AbM-20.I, Interneurons; so, stratum oriens;sr, stratum radiatum; alv, alveus. b, High-power photomicrograph of ICE-like immunoreactivity in the CA1 region of the hippocampus of a 4 d ischemic brain showing an increase in ICE-like immunoreactivity in microglia. alv, Alveus. c, Lack of immunostaining in the absence of primary antibody. d, A second antibody (Ab122) showing a similar pattern of immunostaining in nonischemic CA1 sector of hippocampus.I, interneurons. e, Reduction in staining with the Ab-122 in the presence of 100-fold excess blocking peptide in the nonischemic CA1 sector of hippocampus. f, Increased ICE-like immunoreactivity in the region of the hippocampal fissure (hf) 4 d after ischemia. High-magnification photomicrograph showing ICE-like immunoreactivity in microglia (m, inset). sr, Stratum radiatum;slm, stratum lacunosum-moleculare. g, High-magnification photomicrograph showing a composite of an ICE-positive microglial cell (m) in the process of attaching to an ICE-positive interneuron (I).
To assess the astroglial response to ischemia, adjacent brain sections were also processed for GFAP immunoreactivity (data not shown). Unlike ICE-like immunoreactivity, GFAP staining was increased within 1 d and was maximal at 2 d after ischemia in the hippocampus, after which a gradual decline in GFAP immunoreactivity was observed. The time course of the increase in GFAP immunoreactivity precedes the ICE-like immunoreactivity in microglia after ischemia.
ICE-like immunoreactivity in nonischemic and ischemic brains at high-power magnification is shown in Figure 6. In nonischemic brains, low levels of ICE-like immunoreactivity were detected in the CA1 pyramidal neurons relative to adjacent stratum radiatum and stratum oriens. High levels of ICE-like immunoreactivity were detected in cells whose distribution pattern is consistent with interneurons of the CA1 layer (Fig. 6a). The distribution pattern is consistent with interneurons on the basis of the pattern of γ-aminobutyric acid decarboxylase (GAD) immunoreactivity (which stains GABAergic interneurons in the hippocampus) on adjacent sections (data not shown). A second ICE antibody, Ab122, also showed a similar pattern of immunostaining in nonischemic sections (Fig. 6d) and after ischemia (data not shown). The addition of a 100-fold excess peptide to block the antibody resulted in the disappearance of specific staining (Fig. 6e). No detectable staining was observed in the absence of primary antibody (Fig. 6c). Figure 6bshows a robust increase in ICE-like immunoreactivity in ameboid microglia of the alveus and stratum pyramidale of the CA1 region of the hippocampus 4 d after ischemia. An increase in ICE-like immunoreactivity was not observed in the CA1 pyramidal neurons. ICE-like immunoreactivity in microglia is also observed in the hippocampal fissure (Fig. 6f). These microglia seem to be adjacent to the distal apical dendrites of the CA1 neurons (Fig.6f, inset). A high-magnification composite of an ICE-positive microglial cell apposed to an ICE-positive interneuron is shown in Figure 6g. ICE-like immunoreactivity was detectable but relatively low in cortical neurons and was not altered after ischemia. ICE-like immunoreactivity was not detectable in cortical microglia during the 4 d post-ischemic period (data not shown).
DISCUSSION
This study provides the first demonstration of an increase in thein vivo expression of ICE and its cellular localization after global forebrain ischemia. The results indicate that ICE-like immunoreactivity was not altered in CA1 neurons that undergo apoptosis after ischemia. Rather, the increase in ICE-like immunoreactivity was associated with microglia.
In nonischemic hippocampal tissue, low levels of ICE expression were observed. Although ICE-like immunoreactivity was high in scattered hippocampal interneurons, it was relatively low in CA1 pyramidal neurons. Within 24 hr after ischemia, an increase in IcemRNA was detected by RT-PCR. During this period, a dramatic increase in GFAP-positive astrocytes was observed in the hippocampus. Most of the CA1 pyramidal neurons seemed to be intact, but a few neurons exhibited shrunken nuclei. Neither immunoblot analysis of hippocampal homogenates nor immunohistochemistry performed on sections showed any detectable increase in ICE protein expression at 24 hr after ischemia. Within 2 d after ischemia, ICE-like immunoreactivity increased in a few microglial cells in the hippocampal region. Consistent with this observation, a small increase in 45 kDa pro-ICE was detected in Western immunoblots. Also at 2 d, CA1 pyramidal neurons displayed condensation and segregation of chromatin toward the nuclear envelope. By 3 d after ischemia, a decrease in GFAP immunostaining in astrocytes was observed; however, ameboid microglia were abundant near the hippocampal fissure and areas adjacent to the ventricle and were highly immunoreactive for ICE. It is not clear whether the resident microglia were activated or whether these cells were infiltrating from the vasculature. By the fourth day after ischemia, the majority of the CA1 neurons exhibited late stages of apoptosis, including DNA fragmentation. At this time, a further increase in ICE mRNA and protein expression was observed, and immunolocalization of ICE revealed that this increase seems to be accounted for largely by the increase in ICE-like immunoreactivity in microglia. During the 4 d after ischemia, no changes in ICE-like immunoreactivity were observed in the CA1 neurons or the hippocampal interneurons.
Despite the observation that apoptotic cell death occurs in CA1 pyramidal neurons, ICE expression was not altered in these neurons. Instead, ICE-like immunoreactivity increased in microglia after ischemia. It is well known that microglia play an important role in inflammation and repair by acting as scavengers of cellular debris (Killackey, 1984; Giulian, 1987). Several reports also suggest that microglia produce and release neurotoxic substances, such as glutamate (Giulian and Vaca, 1993; Giulian et al., 1993) and free radicals (Colton and Gilbert, 1987; Thery et al., 1991), and contribute to tissue damage after cerebral ischemia (Gehrmann et al., 1995).
The lack of change in ICE-like immunoreactivity in neurons, however, does not exclude an increase in ICE proteolytic activity in neurons. Active ICE is an oligomeric enzyme composed of two subunits, p20 and p10, both of which are required for catalytic activity (Thornberry et al., 1992; Wilson et al., 1994). The subunits are cleaved from a single proenzyme, p45, which itself is an ICE substrate, suggesting that ICE is autocatalytic (Thornberry et al., 1992). The lack of detection of the p10 and p20 subunits after ischemia could be attributable to the lack of adequate detection sensitivity or to a rapid turnover of the products. This observation is consistent with a previous report, which indicates that ICE predominantly exists in the precursor 45 kDa form in the cytoplasm of both stimulated and unstimulated monocytes (Ayala et al., 1994). In that study, neither the p20 nor the p10 subunits of ICE were detected in cells, even after a strong apoptotic stimulus such as lipopolysaccharide.
Pro-IL-1β, a highly preferred substrate of ICE (Howard et al., 1991), is synthesized as an inactive 31–33 kDa precursor (Dinarello, 1991,1993) that is proteolytically cleaved by ICE to the mature form of IL-1β, a proinflammatory cytokine. Mature IL-1β is secreted by polymorphonuclear leukocytes and activated microglia (Plantais and Vogelzang, 1990; Lee et al., 1993), induces synthesis of other cytokines, and participates in leukocyte adhesion, tissue destruction, and edema formation (Dinarello, 1991; Yamasaki et al., 1992). IL-1β disrupts the blood-brain barrier and serves as a chemoattractant for the recruitment of neutrophils (Quagliarello et al., 1991). After transient forebrain ischemia, IL-1β mRNA is increased in rat brain (Minami et al., 1992). This observation coupled with our result demonstrating an increase in ICE after ischemia possibly accounts for an increase in microglial IL-1β secretion. IL-1β released by microglia after ischemia could recruit additional microglia locally, thereby contributing to increased cell killing of healthy and/or compromised neurons. Thus ICE could indirectly play a role in contributing to tissue damage after ischemia. Support for this theory is demonstrated in a recent report indicating that intracerebroventricular administration of IL-1 receptor antagonists significantly reduces neuronal damage that occurs in focal models of ischemia as well as after excitotoxic lesions (Realton and Rothwell, 1992).
Despite these findings demonstrating the adverse influence of IL-1β on neuronal survival, IL-1β has been considered to be neuroprotective because it increases inhibitory GABAergic action in neurons (Miller at al., 1990) and could potentially counteract the increased excitotoxicity observed after ischemia. In our study, ICE-like immunoreactivity is high in GABAergic interneurons of the hippocampus in nonischemic gerbils. Although ICE expression in these interneurons does not seem to change after ischemia, the constitutive expression of ICE itself suggests that it could play a neuromodulatory role in these cells.
Previous studies have demonstrated that administration of the protein synthesis inhibitor cycloheximide decreases the ischemia-induced delayed cell death in the hippocampus (Goto et al., 1990), suggesting that a genetic program could contribute to the pathogenesis after ischemia. Overexpression of ICE is a sufficient but not a necessary cause of apoptosis in mammalian cells (Miura et al., 1993), becauseIce knock-out mice are resistant to Fas-mediated apoptosis but do not exhibit gross developmental abnormalities (Kuida et al., 1995; Li et al., 1995). In fact, direct evidence that regulation ofIce at the transcriptional or translational level causes apoptosis in vivo is lacking. Although we have shown an increase in Ice mRNA levels in the hippocampus after ischemia, increased ICE-like immunoreactivity was localized to microglia in this region. Accordingly, the increase in hippocampalIce mRNA may simply reflect an increase in the population of microglial cells. Our study supports an indirect role for ICE in neuronal damage after ischemia but does not rule out a direct role caused by modulation of ICE proteolytic activity in neurons. Alternatively, the possibility that apoptotic death of CA1 neurons after ischemia is mediated by other proteases belonging to the ICE/CED-3 family must be considered. It is unlikely that any single protease could be solely responsible for apoptotic cell death (Martin and Green, 1995); hence, understanding the role of each of these proteases in cell death is critical.
In summary, we have provided evidence for an increase in ICE mRNA and protein levels in hippocampus after ischemia. Furthermore, we have found that the increase in ICE-like immunoreactivity was localized selectively in microglia. Given the observation that microglia secrete IL-1β and that IL-1 receptor antagonists are neuroprotective in ischemia, we suggest an indirect role for ICE in ischemic damage through mediation of inflammation in neuronal tissue.
Footnotes
We thank Drs. A. Maroney, M. Ator, and S. Carswell for helpful discussions, and S. Trusko and Y.-G. Lin for technical assistance.
Correspondence should be addressed to Dr. Ratan Bhat, Cephalon, Inc., 145 Brandywine Parkway, West Chester, PA 19380.
REFERENCES
- 1.Ayala JM, Yamin T-T, Egger LA, Chin J, Kostura MJ, Miller DK. IL-1β-converting enzyme is present in monocytic cells as an inactive 45 kDa precursor. J Immunol. 1994;153:2592–2599. [PubMed] [Google Scholar]
- 2.Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Ness KV, Greenstreet TA, March CJ, Kronheim SR, Druck T, Cannizzaro LA, Huebner K, Black RA. Molecular cloning of the interleukin-1β converting enzyme. Science. 1992;256:97–100. doi: 10.1126/science.1373520. [DOI] [PubMed] [Google Scholar]
- 3.Clarke PGH. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 4.Cohen GM, Sun X-M, Snowden RT, Dinsdale D, Skilter DN. Key morphological features of apoptosis may occur in the absence of internucleosomal DNA fragmentation. Biochem J. 1992;286:331–334. doi: 10.1042/bj2860331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colton CA, Gilbert DL. Production of the superoxide anion by a CNS macrophage, the microglia. FEBS Lett. 1987;223:284–288. doi: 10.1016/0014-5793(87)80305-8. [DOI] [PubMed] [Google Scholar]
- 6.Dinarello CA. Interleukin-1 and interleukin-1 antagonism. Blood. 1991;77:1627–1645. [PubMed] [Google Scholar]
- 7.Dinarello CA. Modalities for reducing interleukin-1 activity in disease. Trends Pharmacol Sci. 1993;14:155–159. doi: 10.1016/0165-6147(93)90200-4. [DOI] [PubMed] [Google Scholar]
- 8.Duan H, Chinnaiyan AM, Hudson PL, Wing JP, He W-W, Dixit VM. ICE-LAP3, a novel mammalian homologue of the Caenorhabditis elegans cell death protein ced-3 is activated during fas- and tumor necrosis factor-induced apoptosis. J Biol Chem. 1996;271:1621–1625. doi: 10.1074/jbc.271.3.1621. [DOI] [PubMed] [Google Scholar]
- 9.Ellis RE, Horvitz HR. Genetic control of programmed cell death in the nematode Caenorhabditis elegans . Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- 10.Faucheu C, Diu A, Chan AWE, Blanchet A-M, Miossec C, Hervé F, Collard-Dutilleul V, Gu Y, Aldape RA, Lippke JA, Rocher C, Su MS-S, Livingston DJ, Hercend T, Lalanne J-L. A novel human protease similar to interleukin-1β converting enzyme induces apoptosis in transfected cells. EMBO J. 1995;14:1914–1922. doi: 10.1002/j.1460-2075.1995.tb07183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernandes-Alnemri T, Litwack G, Alnemri ES. CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein ced-3 and mammalian interleukin-1β converting enzyme. J Biol Chem. 1994;269:30761–30764. [PubMed] [Google Scholar]
- 12.Fernandes-Alnemri T, Litwack G, Alnemri ES. Mch2, a new member of the apoptotic Ced-3/ICE cysteine protease gene family. Cancer Res. 1995a;55:2737–2742. [PubMed] [Google Scholar]
- 13.Fernandes-Alnemri T, Takahashi A, Armstrong R, Krebs J, Fritz L, Tomaselli KJ, Wang L, Croce CM, Salveson G, Earnshaw WC, Litwack G, Alnemri ES. Mch3, a novel human apoptotic cysteine protease highly related to CPP32. Cancer Res. 1995b;55:6045–6052. [PubMed] [Google Scholar]
- 14.Gagliardini V, Fernandez PA, Lee RKK, Drexler HCA, Rotello RJ, Fishman MC, Yuan J-Y. Prevention of vertebrate neuronal death by the crmA gene. Science. 1994;263:816–828. doi: 10.1126/science.8303301. [DOI] [PubMed] [Google Scholar]
- 15.Gehrmann J, Banati RB, Wiessner C, Hossmann KA, Kreutzberg GW. Reactive microglia in cerebral ischemia: an early mediator of tissue damage? Neuropathol Appl Neurobiol. 1995;21:277–289. doi: 10.1111/j.1365-2990.1995.tb01062.x. [DOI] [PubMed] [Google Scholar]
- 16.Goto K, Ishige A, Sekiguchi K, Iizuka S, Sugimoto A, Yuzurihara M, Aburada M, Hosoya E, Kogure K. Effects of cycloheximide on delayed neuronal death in rat hippocampus. Brain Res. 1990;534:299–302. doi: 10.1016/0006-8993(90)90144-z. [DOI] [PubMed] [Google Scholar]
- 17.Giulian D. Amoeboid microglia as effectors of inflammation in the central nervous system. J Neurosci Res. 1987;18:155–171. doi: 10.1002/jnr.490180123. [DOI] [PubMed] [Google Scholar]
- 18.Giulian D, Vaca K (1993) Inflammatory glia mediate delayed neuronal damage after ischemia in the central nervous system. Stroke 24[Suppl I]:184–190. [PubMed]
- 19.Giulian D, Vaca K, Corpuz M. Brain glia release factors with opposing actions upon neuronal survival. J Neurosci. 1993;13:29–37. doi: 10.1523/JNEUROSCI.13-01-00029.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howard AD, Kostura MJ, Thornberry N, Ding GJ-F, Limjuco G, Weidner J, Salley JP, Hogquist KA, Chaplin DD, Mumford RA, Schmidt JA, Tocci MJ. IL-1β-converting enzyme requires aspartic acid residues for processing of the IL-1β precursor at two distinct sites and does not cleave 31 kDa IL-1α. J Immunol. 1991;147:2964–2969. [PubMed] [Google Scholar]
- 21.Johnson EM, Chang JY, Koike T, Martin DP. Why do neurons die when deprived of trophic factor? Neurobiol Aging. 1989;10:549–552. doi: 10.1016/0197-4580(89)90127-9. [DOI] [PubMed] [Google Scholar]
- 22.Johnson EM, Deckwerth TL. Molecular mechanisms of developmental neuronal death. Annu Rev Neurosci. 1993;16:31–46. doi: 10.1146/annurev.ne.16.030193.000335. [DOI] [PubMed] [Google Scholar]
- 23.Killackey HP. Glia and the elimination of transient cortical projections. Trends Neurosci. 1984;7:225–226. [Google Scholar]
- 24.Kirino T, Sano K. Selective vulnerability in the gerbil hippocampus following transient ischemia. Acta Neuropathol (Berl) 1984a;62:201–208. doi: 10.1007/BF00691853. [DOI] [PubMed] [Google Scholar]
- 25.Kirino T, Sano K. Fine structural nature of delayed neuronal death following ischemia in the gerbil hippocampus. Acta Neuropathol (Berl) 1984b;62:209–218. doi: 10.1007/BF00691854. [DOI] [PubMed] [Google Scholar]
- 26.Komiyama T, Ray CA, Pickup DJ, Howard AD, Thornberry NA, Peterson EP, Salvesen G. Inhibition of interleukin-1β converting enzyme by the cowpox virus serpin CrmA. J Biol Chem. 1994;269:19331–19337. [PubMed] [Google Scholar]
- 27.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS-S, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1β converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 28.Kumar S, Tomooka Y, Noda M. Identification of genes with developmentally down-regulated expression in the mouse brain. Biochem Biophys Res Commun. 1992;185:1155–1161. doi: 10.1016/0006-291x(92)91747-e. [DOI] [PubMed] [Google Scholar]
- 29.Lazebnik YA, Kaufman SH, Desnoyers S, Poirier GG, Earnshaw WC. Clevage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 30.Lee SC, Liu W, Dickson DW, Bronson CF, Berman JW. Cytokine production by human fetal microglia and astrocytes. J Immunol. 1993;150:2659–2667. [PubMed] [Google Scholar]
- 31.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, Towine E, Tracey D, Wardwell S, Wei F-Y, Wong W, Kamen R, Sheshadri T. Mice deficient in IL-1β-converting enzyme are defective in the production of mature IL-1β and resistant to endotoxic shock. Cell. 1995;80:401–412. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 32.Lippke JA, Gu Y, Sarnecki C, Caron PR, Su MS-S. Identification and characterization of CPP32/Mch2 Homolog 1, a novel cysteine protease similar to CPP32. J Biol Chem. 1996;271:1825–1828. doi: 10.1074/jbc.271.4.1825. [DOI] [PubMed] [Google Scholar]
- 33.Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin SJ, Green DR. Protease activation during apoptosis: death by a thousand cuts? Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 35.Miller BE, Krasney PA, Gauvin DM, Holbrook KB, Koonz DJ, Abruzzese RV, Miller RE, Pagani KA, Dolle RE, Ator MA, Gilman SC. Inhibition of mature IL-1β production in murine macrophages and a murine model of inflammation by WIN 67694, an inhibitor of IL-1β converting enzyme. J Immunol. 1995;154:1331–1338. [PubMed] [Google Scholar]
- 36.Miller LG, Galpern WR, Dunlap K, Dinarello CA, Turner TJ. Interleukin-1 augments γ-aminobutyric acidA receptor function in brain. Mol Pharmacol. 1990;39:105–108. [PubMed] [Google Scholar]
- 37.Milligan CE, Prevette HY, Homma S, Cardwell C, Fritz LC, Tomaselli KJ, Oppenheim RW, Schwartz LM. Peptide inhibitors of the ICE protease family arrest programmed cell death of motoneurons in vivo and in vitro. Neuron. 1995;15:385–393. doi: 10.1016/0896-6273(95)90042-x. [DOI] [PubMed] [Google Scholar]
- 38.Minami M, Kuraishi Y, Yabuuchi K, Yamazaki A, Satoh M. Induction of interleukin-1β mRNA in rat brain after transient forebrain ischemia. J Neurochem. 1992;58:390–392. doi: 10.1111/j.1471-4159.1992.tb09324.x. [DOI] [PubMed] [Google Scholar]
- 39.Miura M, Zhu H, Rotello R, Hartwig EA, Yuan J. Induction of apoptosis in fibroblasts by IL-β converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- 40.Munday NA, Vailancourt JP, Ali A, Casano FJ, Miller DK, Molineaux SM, Yamin T-T, Yu VL, Nicholson DW. Molecular cloning and pro-apoptotic activity of ICErelII and ICErelIII, members of the ICE/CED-3 family of cysteine proteases. J Biol Chem. 1995;270:15870–15876. doi: 10.1074/jbc.270.26.15870. [DOI] [PubMed] [Google Scholar]
- 41.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Raju SM, Smulson ME, Yamin T-T, Yu VL, Miller DK. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 42.Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oppenheim RW, Prevette D, Tytell M, Homma S. Naturally occurring and induced neuronal death in the chick embryo in vivo requires protein and RNA synthesis: evidence for the role of cell death genes. Dev Biol. 1990;138:104–113. doi: 10.1016/0012-1606(90)90180-q. [DOI] [PubMed] [Google Scholar]
- 44.Plantais LC, Vogelzang NJ. Interleukin-1: biology, pathophysiology, and clinical prospects. Am J Med. 1990;89:621–629. doi: 10.1016/0002-9343(90)90181-c. [DOI] [PubMed] [Google Scholar]
- 45.Quagliarello VJ, Wispelway B, Long WJ, Scheld WM. Recombinant human interleukin-1 induces meningitis and blood-brain barrier injury in rat. J Clin Invest. 1991;87:1360–1366. doi: 10.1172/JCI115140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Realton JK, Rothwell NJ. Interleukin-1 receptor antagonist inhibits ischaemic and excitotoxic neuronal damage in the rat. Brain Res Bull. 1992;29:243–246. doi: 10.1016/0361-9230(92)90033-t. [DOI] [PubMed] [Google Scholar]
- 47.Schulze-Osthoff K, Walczak H, Droge W, Krammer PH. Cell nucleus and DNA fragmentation are not required for apoptosis. J Cell Biol. 1994;127:15–20. doi: 10.1083/jcb.127.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tata JR. Requirement for RNA and protein synthesis for induced regression of the tadpole tail in organ culture. Dev Biol. 1966;13:77–94. doi: 10.1016/0012-1606(66)90050-9. [DOI] [PubMed] [Google Scholar]
- 49.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32β, a mammalian homolog of ced-3 is a crmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 50.Thery C, Chamak B, Mallat M. Free radical killing of neurons. Eur J Neurosci. 1991;3:1155–1164. doi: 10.1111/j.1460-9568.1991.tb00050.x. [DOI] [PubMed] [Google Scholar]
- 51.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, Elliston KO, Ayala JM, Casano FJ, Chin J, Ding GJ-F, Egger LA, Gaffney EP, Limjuco G, Palyha OC, Raju SM, Rolando AM, Salley JP, Yamin TT, Lee TD, Shively JE, MacCross M, Mumford RA, Schmidt JA, Tocci MJ. A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 52.Uhl J, Brophy L, Stevenson J, Dolle R, Helaszek C, Miller R, Ator M (1993) Secretion of human monocyte mature IL-1β: optimization of culture conditions and inhibition by ICE inhibitors. Mol Biol Cell 4[Suppl]:341a. [DOI] [PubMed]
- 53.Wang L, Miura M, Bergeron L, Zhu H, Yuan J. Ich-1 an ICE/ced-3 related gene encodes both positive and negative regulators of programmed cell death. Cell. 1994;78:739–750. doi: 10.1016/s0092-8674(94)90422-7. [DOI] [PubMed] [Google Scholar]
- 54.Wilson KP, Black J-AF, Thomson JA, Kim EE, Griffith JP, Navia MA, Murcko MA, Chambers SP, Aldape RA, Raybuck SA, Livingston DJ. Structure and mechanism of interleukin-1β converting enzyme. Nature. 1994;370:270–275. doi: 10.1038/370270a0. [DOI] [PubMed] [Google Scholar]
- 55.Yamasaki Y, Susuki T, Yamaya H, Mastsuura N, Onodera H, Kogure K. Possible involvement of interleukin-1 in ischemic brain edema formation. Neurosci Lett. 1992;142:45–47. doi: 10.1016/0304-3940(92)90616-f. [DOI] [PubMed] [Google Scholar]
- 56.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1β converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]