

Abstract
Lipopolysaccharide (LPS), an endotoxin, produces pain behavior, inflammation, and changes in immune function. Many of these effects are secondary to the production of cytokines. In the present study, we investigated the effect of LPS on the releasing function of afferent terminals as measured by calcitonin gene-related peptide (CGRP) release in ex vivo perfused rat trachea, and examined the possible role of the cytokines interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) as intermediaries in this effect. Systemic injection of LPS (0.75 mg/kg, i.p.) in adult rats induced an increase in body temperature followed by hypothermia, indicating ongoing infection. We observed that capsaicin-induced (0.1 μm) tracheal CGRP release was significantly enhanced in the LPS-treated animals after 5 hr. This enhancement of the peptide release by LPS was blocked by IL-1β tripeptide antagonist Lys-d-Pro-Thr (10 μm) and mimicked by IL-1β and TNF-α (10–100 pg/ml), suggesting that the potentiating effect of LPS on CGRP release is mediated by generation of IL-1β and TNF-α. IL-1β-induced augmentation of CGRP release was blocked by Lys-d-Pro-Thr. Additionally, the cyclooxygenase inhibitor ketorolac (10 μm) significantly attenuated the facilitatory effects of LPS and IL-1β, indicating involvement of prostanoids. These findings suggest that endotoxin treatment generated cytokines such as IL-1β and TNF-α that regulated the peripheral releasing function of primary sensory afferents by sensitizing the terminals and facilitating peptide release. This effect is prostanoid dependent.
Keywords: sensory nerves, calcitonin gene-related peptide, lipopolysaccharide, interleukin-1β, tumor necrosis factor-α, trachea
Gram-negative bacteria exert profound influences on the physiology of organisms through the actions of endotoxin. Lipopolysaccharide (LPS) is an endotoxin derived from the cell walls of these bacteria. Systemically delivered LPS thus mimics many effects of the bacterial infections including fever, pain (Watkins et al., 1994), inflammation (Vincent et al., 1991), and changes in immune functions (Morrison, 1987). The present work seeks to establish two points: (1) an important target of the action of LPS is the peripheral terminal of sensory afferents, and (2) this action may be secondary to the production of cytokines.
The terminals of unmyelinated sensory afferents serve two general functions. These terminals transduce thermal, mechanical, and chemical stimuli such that the rate of axon discharge is increased as a function of stimulus intensity. Exposure of the peripheral C-fiber terminals to inflammatory mediators such as bradykinin, serotonin, prostaglandins, leukotriences, and protons induces depolarization of the terminals as evidenced by the generation of action potentials and/or sensitization of the terminals (Basbaum, 1991). Such activation of afferent C fibers by the stimuli leads to evocation of nociceptive reflexes and pain behavior (Basbaum, 1991; Dray and Bevan, 1993). Aside from the generation of action potentials, the C-fiber terminal is a secretory system from which peptides such as calcitonin gene-related peptide (CGRP) and tachykinins are released (Holzer, 1988). The local action of these released peptides are known to increase tissue blood flow, capillary permeability, and inflammatory cell activities (Holzer, 1988). The airways, including the trachea, are innervated by vagal afferent fibers containing CGRP and tachykinins (Cadieux et al., 1986;Lundberg et al., 1987; Shimosegawa and Said, 1991). As discussed below, these peptides are released from afferent terminals in the airways by antidromic activity and local depolarization. It is now emphasized that local release from vagal C-fiber terminals in the airway may play a pervasive pathophysiological role in respiratory diseases such as airway hypersensitivity and asthma (Lundberg et al., 1987; Barnes, 1992).
The effects of LPS appear to be secondary to the production of cytokines (Morrison, 1987). Cytokines such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) can be generated by inflammatory and nonimmune cells in response to inflammation, tissue injury, microbial invasion, and immunological reactions (Dinarello, 1994). Given systemically or intradermally, these cytokines induce a hyperalgesic state, presumably by an action at the peripheral terminals, leading to decreased thresholds required for thermal and mechanical stimuli to activate C fibers (Ferreira et al., 1988; Cunha et al., 1992; Watkins et al., 1994). Consistent with this effect on peripheral terminals, it has been shown that IL-1β enhances capsaicin-induced vasodilatation in rat skin (Herbert and Holzer, 1994). All of these findings suggest the hypothesis that LPS exerts effects on peripheral terminals of sensory afferents via generation of cytokines that serve to stimulate and/or sensitize afferent nerves and regulate terminal release. We sought, in the present study, to directly examine this issue by studying the effect of LPS treatment on the releasing function of afferent terminals as measured by CGRP release inex vivo perfused rat trachea. Additionally, this work sought to establish the possible role of IL-1β and TNF-α as intermediaries in this effect.
MATERIALS AND METHODS
Animals and treatments. Male Harlan Sprague–Dawley rats (300–350 gm body weight) were studied in accordance with protocols approved by the Animal Care Committee, University of California, San Diego. LPS (0.75 mg/kg) was injected intraperitoneally in unanesthetized rats. In some animals, the body temperature was monitored periodically by insertion a thermistor probe (YSI) 3.5 cm into the rectum.
Tissue collection and extraction. Two or five hours after LPS administration (0.75 mg/kg, i.p.) or saline (same volume, i.p.), the animals were killed with sodium pentobarbital (70 mg/kg, i.p.). The whole trachea and cervical vagus nerve (1–1.5 cm) were dissected and frozen. The tissue specimens were placed in 1 ml of 0.1 N HCl in a boiling water bath. After 10 min of boiling, the tissues were homogenized using a Polytron device. The homogenates were subsequently centrifuged and the supernates collected, lyophilized, and subjected to radioimmunoassay (RIA) for analysis of CGRP content (see below).
Release experiment. Tracheas (from larynx to carina) were dissected out and connected to glass pipette holders to allow intraluminal perfusion (Hua and Yaksh, 1992). The cannulated trachea then was placed in a perfusion bath containing oxygenated Krebs/bicarbonate solution (see below) and maintained at 37°C. Intraluminal tracheal perfusion was performed with oxygenated Krebs’ solution at a perfusion flow rate of 0.2 ml/min. After a 30 min equilibration period, perfusates (2 ml) were collected at 10 min intervals in test tubes containing acetic acid (final concentration of 2 m). All fractions were then frozen and lyophilized, and CGRP concentrations were measured by RIA.
CGRP RIA. In the current study, we used CGRP antibody G 2027 with [I125]Tyr0CGRP as tracer. G 2027, a polyclonal antibody raised against rat CGRP, was developed in our laboratory (Hastings and Hua, 1995). The working dilution of G 2027 was 1:21,000, and the absolute sensitivity of G 2027 detection was 3 fmol/assay tube. The antisera G 2027 cross-reacted 100% with human CGRP-α and human CGRP-β but did not cross-react to substance P (SP), neurokinin A (NKA), or cholecystokinin-8 (<1%), although there was some cross-reactivities (18–30%) with amylin, calcitonin, and CGRP29–37 at high concentration (1 μg/ml). In a previous study, we demonstrated that the immunoreactivity detected by antibody G 2027 was eluted as a single peak in the same fraction as synthetic rat CGRP-β (Hastings and Hua, 1995). The tracer [I125]Tyr0CGRP was iodinated by the chloramine-T method and purified by elution from a reverse-phase HPLC column using an acetonitrile gradient. All the assays were carried out in duplicate. Nonspecific binding and blanks were also assessed with the background blanks examined in normal buffer and buffer containing the examined substances. Under no condition did the buffer interfere with the CGRP assay.
Solution and drugs. The perfusion media consisted of a Krebs’ buffer solution containing (in mm): NaCl 118.3, KCl 4.7, CaCl2 2.5, MgSO4 1.2, NaHCO3 25, KH2PO4 1.2, and glucose 11. Bacitracin (0.3%) was added to reduce possible peptide degradation. Bubbling of the solution with 95% O2/5% CO2 resulted in a pH between 7.35 and 7.45.
LPS, capsaicin, indomethacin, recombinant mouse IL-1β, and bacitracin were obtained from Sigma (St. Louis, MO), and interleukin 1β 193–195 analog (Lys-d-Pro-Thr) was obtained from Peninsula (Belmont, CA). Recombinant murine TNF-α was obtained from Research and Development Systems (Minneapolis, MN) and ketorolac tromethamine from Syntex (Palo Alto, CA). Capsaicin and indomethacin were initially dissolved in ethanol and subsequently diluted in Krebs’ buffer with a final ethanol concentration of 0.6–1.0%. This vehicle did not influence the basal release of CGRP (Hua and Yaksh, 1992). The other agents were dissolved in Krebs’ buffer. To study evoked release, agents were added to the perfusate during the appropriate stimulation interval. The effect of Lys-d-Pro-Thr, ketorolac, and indomethacin were studied by applying the drug to the trachea 20 min before and during the challenge with the examined agents. IL-1β and TNF-α were added 10 min before and during application of capsaicin.
Data analysis. All the data are present as mean ± SEM. Statistical significance was calculated using unpaired ttest or one-way ANOVA followed by a Fisher test. Differences at the level of p < 0.05 were considered significant.
RESULTS
Behavioral observation
LPS injection (0.75 mg/kg, i.p.) induced an initial transient increase in body temperature (1–1.5°C) by 15 min, which was followed by a pronounced decrease in temperature (2–2.5°C) with a maximal effect at ∼2 hr. The temperature returned to baseline level by ∼5 hr (Fig. 1A). Some animals also had diarrhea and reduced locomotive activity after LPS application. All the LPS-treated rats had normal pinna and cornea reflexes, and none showed motor weakness or airway distress.
Fig. 1.
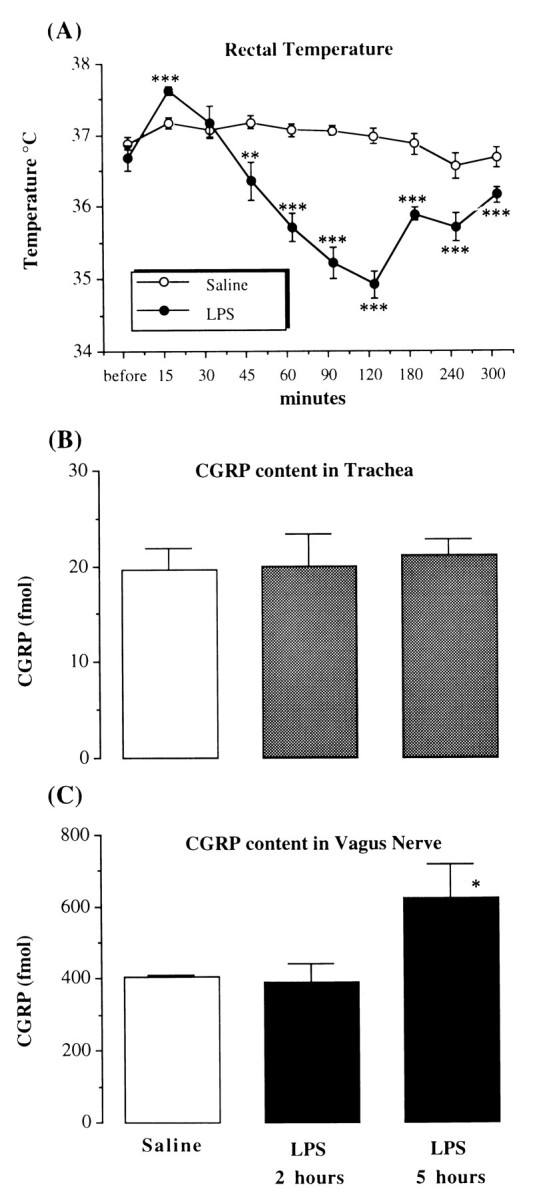
The effect of LPS on rectal temperature and CGRP content in tissues. A, The time course of rat rectal temperature before and after intraperitoneal injection of saline or LPS (0.75 mg/kg). The data are presented as mean ± SEM of five to six animals in each group. **p < 0.01, ***p < 0.001, unpaired t test; LPS group versus saline group in the same time point. B, The concentration of CGRP (fmol/mg tissue) in the rat trachea;C, the CGRP concentration (fmol/mg protein) in vagus nerve 2 and 5 hr after intraperitoneal injection of LPS (0.75 mg/kg) or 5 hr after intraperitoneal injection of saline. *p < 0.05, one-way ANOVA; LPS 5 hr versus saline group.
CGRP content in tissues
CGRP content in the trachea from saline-treated animals was 18 ± 1 fmol/mg tissue (n = 11). There were no significant changes in the first 2 hr (20 ± 3 fmol/mg tissue, n = 6) or at 5 hr (20 ± 2 fmol/mg tissue, n = 6) after LPS treatment (see Fig. 1B). CGRP level in the vagi from saline-treated animals was 385 ± 17 fmol/mg protein (n = 9). This was not altered after LPS injection at 2 hr (392 ± 49 fmol/mg protein, n = 5), but by 5 hr, the mean CGRP content in vagi was significantly enhanced (623 ± 94 fmol/mg protein,n = 5, p < 0.05, Fig.1C).
LPS on tracheal CGRP release
Basal CGRP levels in nonstimulated fractions were 30–50 fmol/10 min fraction. Application of capsaicin (0.1 μm) to an intraluminally perfused in vitro rat trachea induced an increase in CGRP outflow in saline-treated animals (CGRP level in capsaicin-stimulated fraction: 178 ± 25 fmol/fraction,n = 10) (Fig. 2A). The capsaicin-evoked CGRP release was not significantly changed in the tracheas from LPS-treated rats (0.75 mg/kg) at 2 hr (225 ± 22 fmol/fraction, n = 4) (Fig. 2B), but it was enhanced 5 hr after treatment (348 ± 64 fmol/fraction,n = 8, p < 0.05) (Fig. 2C). LPS (10 μg/ml) added directly to tracheal perfusates did not evoke an increase in CGRP release (38 ± 4 fmol/fraction, n = 4) (Fig. 2D). Basal release of CGRP from the trachea was not changed by LPS treatment at either 2 or 5 hr.
Fig. 2.

The effect of LPS on tracheal CGRP release. CGRP concentration (fmol/10 min fraction) in the perfusates from intraluminally perfused rat trachea by capsaicin (Caps, 0.1 μm) in control group (A), in LPS-treated rats (0.75 mg/kg, i.p.) 2 hr after drug administration (B), and LPS-treated animals (0.75 mg/kg, i.p.) 5 hr after drug administration (C), and LPS (10 μg/ml) was applied directly to the tracheas in untreated rats (D).Basal, Perfused with Krebs’ buffer; n = 8–10/group. *p < 0.05, one-way ANOVA; LPS 5 hr versus saline group.
Antagonism of LPS-induced facilitation
Enhancement of capsaicin-evoked CGRP release by LPS (ΔCGRP 294 ± 59 fmol/fraction, basal level subtracted, n = 8) was inhibited by IL-1β antagonist Lys-d-Pro-Thr at concentrations of 1 μm (ΔCGRP 192 ± 39 fmol/fraction, 62% reduction of enhanced release, n = 10) (Fig. 3) and 10 μm (ΔCGRP 153 ± 17 fmol/fraction, 86% reduction, p < 0.01, n = 8) (Fig. 3). Lys-d-Pro-Thr at 10 μm did not antagonize capsaicin-evoked release in untreated animals (ΔCGRP 170 ± 43 fmol/fraction,n = 6). Ketorolac (10 μm) did not alter the facilitatory effect of LPS (ΔCGRP 297 ± 43 fmol/fraction, no reduction, n = 4) (Fig. 3), but at the higher concentration of 100 μmsignificantly blocked the effect (ΔCGRP 124 ± 4 fmol/fraction, 100% reduction, p < 0.05, n = 4) (Fig. 3). Indomethacin (10 μm) attenuated the LPS-induced enhancement (ΔCGRP 191 ± 28 fmol/fraction, 63% reduction,n = 6) (Fig. 3), although it did not reach a statistical significance.
Fig. 3.

Antagonism of the potentiating effect of LPS. Effects of IL-1β antagonist: Lys-d-Pro-Thr or cyclooxygenase inhibitors ketorolac (Keto) or indomethacin (Indo) on capsaicin-evoked (0.1 μm) CGRP release (Δfmol/10 min fraction, basal levels were subtracted) in the tracheas of LPS-treated or saline-treated (No LPS) animals. No Ant, No antagonist; n = 4–10/group; *p < 0.05, **p < 0.01, one-way ANOVA.
Effects of IL-1β and TNF-α
The basal release of CGRP was not changed by application of IL-1β (1 pg/ml to 10 ng/ml) to tracheal perfusates; however, capsaicin-induced release was significantly elevated by IL-1β at a concentration of 10 pg/ml (ΔCGRP: 319 ± 54 fmol/fraction, 235% increase, p < 0.05, n = 7) (Fig.4). This enhancement declined when the concentration of IL-1β added to the perfusates was increased (from 100 pg/ml to 10 ng/ml); IL-1β at a concentration of 10 ng/ml did not demonstrate any potentiating activity (Fig. 4). TNF-α displayed an activity on tracheal CGRP release similar to that of IL-1β. TNF-α (10 pg/ml to 10 ng/ml) did not cause an increase in basal CGRP release, but augmented capsaicin-induced release (Fig. 5). TNF-α at 100 pg/ml significantly elevated capsaicin-evoked CGRP release (ΔCGRP: 254 ± 45 fmol/fraction, 187% increase, p < 0.05, n = 4) (Fig. 5).
Fig. 4.
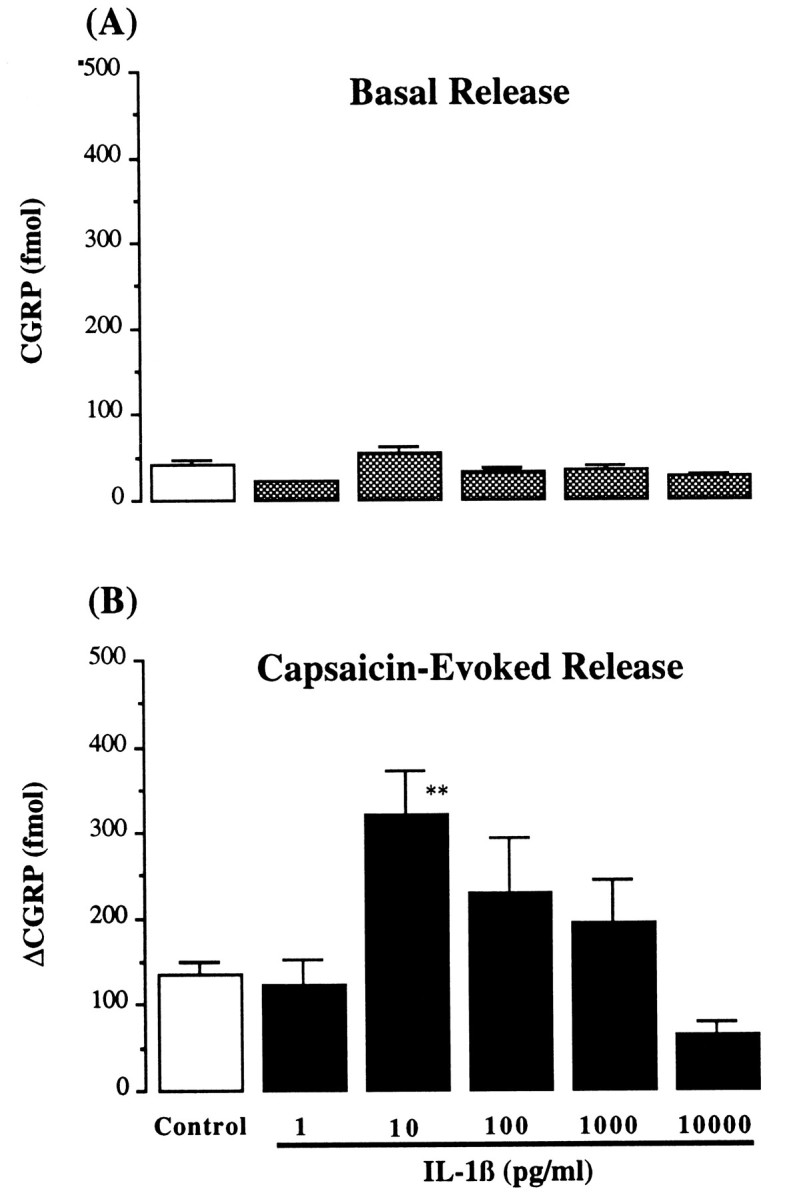
Basal and capsaicin-evoked release of CGRP by IL-1β. Basal CGRP release (A) (fmol/10 min fraction) and capsaicin-evoked (0.1 μm) CGRP release (B) (Δfmol/10 min fraction) from intraluminally perfused rat trachea in presence of IL-1β (1–10,000 pg/ml). Control, Absence of IL-1β; n = 4–12/each group; **p < 0.01, one-way ANOVA, IL-1β versus control.
Fig. 5.
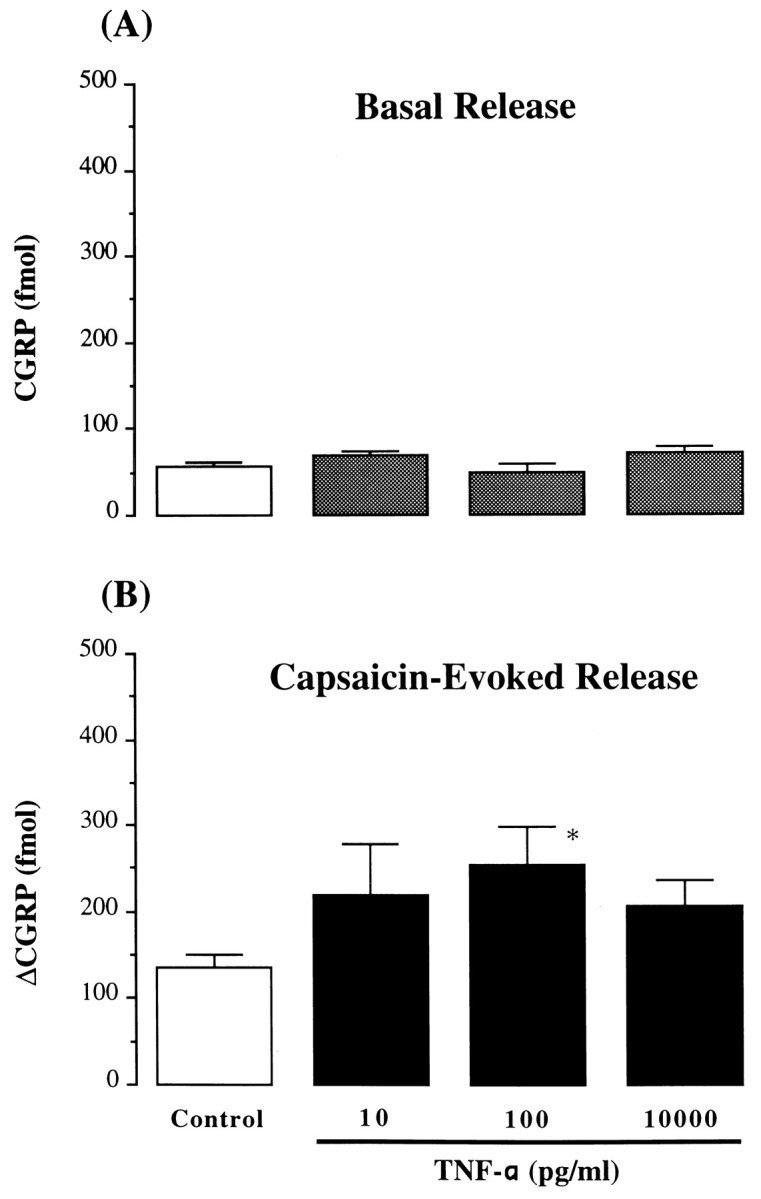
Basal and capsaicin-evoked release of CGRP by TNF-α. Basal CGRP release (A) (fmol/10 min fraction) and capsaicin-evoked (0.1 μm) CGRP release (B) (Δfmol/10 min fraction) from intraluminally perfused rat trachea in presence of TNF-α (10–10,000 pg/ml). Control, Absence of TNF-α; n = 4–16/each group; *p < 0.05, one-way ANOVA, TNF-α versus control.
IL-1β-induced enhancement (10 pg/ml) was significantly inhibited by Lys-d-Pro-Thr (50 μm) (ΔCGRP: 198 ± 47 fmol/fraction, 66% reduction of enhanced release,p < 0.05, n = 6) and by ketorolac at 10 μm (ΔCGRP: 194 ± 39 fmol/fraction, 67% reduction of enhanced release, p < 0.05, n = 6) (Fig. 6). Lys-d-Pro-Thr (10 μm) did not antagonize the potentiating effect of 100 pg/ml of TNF-α (ΔCGRP: 249 ± 40 fmol/fraction, n = 4).
Fig. 6.

Antagonism of the potentiating effect of IL-1β. Effects of IL-1β antagonist Lys-d-Pro-Thr (50 μm) or cyclooxygenase inhibitor ketorolac (Keto, 10 μm) on IL-1β-potentiated (10 pg/ml) CGRP release by capsaicin (0.1 μm) (Δfmol/10 min fraction, basal level were subtracted);n = 6–16/group; *p < 0.05, one-way ANOVA.
DISCUSSION
In the present study, we found that endotoxin treatment influences the peripheral activity of primary sensory afferent fibers by sensitizing the terminals and facilitating release of neuropeptides such as CGRP. This effect is apparently mediated by production of endogenous cytokines such as IL-1β and TNF-α.
LPS activity
It is well known that LPS stimulates the in vitro andin vivo expression of a large number of cytokines that orchestrate inflammation, the immune response, and some acute phase responses such as fever. A brief increase in body temperature after LPS injection was observed in the present study. IL-1β is believed to be one of the important mediators for LPS-induced fever (Long et al., 1990). The subsequent hypothermia could be attributable to release of other cytokines such as TNF-α, which acts as an endogenous antipyretic agent (Long et al., 1990). It has been demonstrated that IL-1α/β mRNA expression in the rat airway is induced by LPS injection (Ulich et al., 1991). Specific binding sites of IL-1 are located on the vagus (Watkins et al., 1995), and IL-1 is able to increase afferent activity in rat vagus nerve (Niijima, 1992). We found that LPS treatment enhanced capsaicin-induced tracheal CGRP release, and this enhancement was mimicked by IL-1β and TNF-α, but blocked by the tripeptide IL-1β antagonist Lys-d-Pro-Thr, which has been shown to inhibit IL-1β-induced effects such as fever (Marceau et al., 1991) and hyperalgesia (Ferreira et al., 1988). These data suggest that endotoxin-induced facilitation of tracheal CGRP release is mediated by generation of cytokines, such as IL-1β, and perhaps TNF-α. The observation of an increased CGRP level in vagus nerve after LPS treatment implies an enhancement of the peptide synthesis. An important biological activity of IL-1 is to upregulate gene expression and initiate and/or facilitate protein and peptide synthesis (Dinarello, 1994). However, the fact that CGRP content in trachea tissue wasnot elevated by LPS treatment suggests that the enhancement of release is likely attributable to an action on the terminals, although facilitation of peptide synthesis could exist as well.
Role of IL-1β and TNF-α
A study on inflammation in rabbit knee joint shows that intra-articular injection of IL-1α increases SP level in the synovial fluid (O’Byrne et al., 1990). Neither IL-1β nor TNF-α, however, induced a direct stimulating effect on tracheal peptide release in this study. Our result is consistent with observations in rat skin, in which IL-1β had no effect on blood flow alone, but potentiated capsaicin-induced neurogenic vasodilatation (Herbert and Holzer, 1994). In the rat trachea, both IL-1β and TNF-α displayed a potent potentiating action on capsaicin-evoked CGRP release. It suggests that the cytokines may exert a modulatory, rather than a direct stimulatory, effect on regulation of sensory nerve activity. The present finding further supports the hypothesis that the facilitatory effect of IL-1β on vasodilatation is attributable to an enhanced release of neuropeptides such as CGRP with potent vasodilatory activity from sensory afferent terminals (Brain et al., 1985). IL-1β does not alter CGRP-induced hyperemia in skin (Herbert and Holzer, 1994).
In the pg/ml concentration range, IL-1β and TNF-α induced facilitation of CGRP release. This is comparable to the high potency of these cytokines in augmenting neurogenic vasodilatation (Herbert and Holzer, 1994) and in sensitizing the rat hindpaw to noxious stimuli (Ferreira et al., 1988; Follenfant et al., 1989). The narrow effective dose range (10–100 pg/ml) of both cytokines may indicate that other inhibitory mediators may be released by higher concentration of the agents. A similar phenomenon was also observed in an electrophysiological study performed on single fibers of rat sciatic nerves, in which TNF-α induced background firing of A-δ and C fibers from 5–100 pg/ml, but at higher concentrations, the effect was diminished (Dr. L. Sorkin, University of California, San Diego, personal communication). One of the possible mechanisms underlying the sensitization of peripheral peptide release by the cytokines observed in the present study could be attributable to generation of prostanoids, because the effects of LPS and IL-1β were antagonized by the cyclooxygenase inhibitor ketorolac. Prostanoids are well-documented to sensitize the nociceptors and augment the excitability of sensory afferents (Martin et al., 1987; Basbaum, 1991) (see below). Other factors, however, may also contribute to this facilitation. LPS, as well as the cytokines IL-1β and/or TNF-α, causes induction of excitatory receptors, including bradykinin B1 receptors (Regoli et al., 1981; Davis and Perkins, 1994) and production of inducible nitric oxide synthase (Liu et al., 1993; Robbins et al., 1994). These factors are known to be involved in hyperalgesia (Dray and Perkins, 1993) and airway hypersensitivity (Barnes and Belvisi, 1993). Thus, we hypothesize that similar mechanisms may underlie the phenomenon of terminal release facilitation seen in this study.
Role of prostanoids
The present observation that LPS- and IL-1β-induced enhancement of CGRP release was blocked by the cyclooxygenase inhibitor ketorolac suggests that the facilitatory action could be attributable to generation of prostanoids. Both LPS and IL-1β are able to stimulate the arachidonic acid cascade resulting in prostanoid production (Morrison, 1987). The increase in synthesis of cyclooxygenase produced by LPS in the present study is likely attributable to the effect of the subsequently generated cytokines, because IL-1β-induced augmentation of CGRP release was also blocked by ketorolac. It has been demonstrated that prostaglandins display a potent sensitizing activity on nociceptive neurons (Martin et al., 1987; Basbaum, 1991). Prostaglandins facilitate CGRP release from rat dorsal root ganglia cells through activation of the cAMP transduction cascade (Vasko et al., 1994; Hingtgen et al., 1995). IL-1β-induced hyperalgesia and neurogenic inflammation are mediated by prostanoids (Follenfant et al., 1989; Herbert and Holzer, 1994). We have found in a previous study that several prostaglandins such as PGE1, PGE2, PGF2α, and PGI2 display excitatory actions on tracheal CGRP release. None of them, however, have showed sensitizing effects (Hua et al., 1994). Prostanoid-mediated facilitatory action on terminal release was observed previously in the same model. Serotonin-enhanced CGRP release by capsaicin was indomethacin-sensitive (Hua and Yaksh, 1993). Instability of thromboxane A2 inhibits the investigation of the biological activity of this agent. Additional work using selective competitive receptor antagonists and stable agonists for the several prostanoids is required to further elucidate the role of prostanoids on facilitated release.
Significance of interaction between cytokines and afferent terminals
Evidence from several lines of investigation has suggested that cytokines play an important role in airway inflammation and disease, such as airway hyper-responsiveness and chronic asthma (Barnes, 1994). IL-1β and TNF-α are secreted predominantly from macrophases and monocytes, but are also produced from several other cell types, including mast cells (Gordon et al., 1990) and Schwann cells (Wagner and Myers, 1996). Increased production of IL-1 has been found in asthmatic airways (Mattoli et al., 1991). IL-1 has broad proinflammatory effects on airways and can activate T lymphocytes to express IL-2, stimulate epithelial cells to produce IL-8 and other cytokines, initiate neutrophil infiltration, and increase airway responsiveness to other inflammatory mediators (Barnes, 1994). Activation of peripheral terminals of sensory nerves and subsequent release of CGRP and tachykinins have been considered an important component in neurogenic inflammation in various visceral organs including airways (Lundberg et al., 1987; Holzer, 1988; Barnes, 1992). Thus, given that cytokines interact with sensory nerves and facilitate peripheral release of neuropeptides, these potent intermediaries may contribute significantly to infection-related airway inflammation and hypersensitivity. Moreover, interaction of cytokines with sensory afferent nerves is also known to exist in other pathophysiological situation. Elevated plasma and synovial levels of IL-1β have been observed in arthritic patients (Eastgate et al., 1988), and intra-articular injection of IL-1α causes an increase in SP efflux in synovial fluid (O’Byrne et al., 1990). As noted above, IL-1β and TNF-α have been implicated in inflammation- and nerve injury-induced hyperalgesia (Ferreira et al., 1988; Cunha et al., 1992; Sommer and Myers, 1994).
In summary, the present study has provided evidence that endotoxin treatment induces generation of cytokines such as IL-1β and TNF-α, which may target primary afferent fibers and regulate their peripheral releasing function by sensitizing the terminals. Amplification of the local secretion of neuropeptides such as CGRP with well-defined effects in neurogenic inflammation suggests that the action of cytokines on sensory afferent nerves serves a complex role not only to provide the afferent information necessary to generating centrally mediating responses, but also to contribute to the reinforcement of local inflammatory process.
Footnotes
This work was supported by National Institutes of Health Grants R29 HL 50403 (X.-Y.H.) and NS 18715 (R.R.M.), and Wellcome Travel Fund (X.-Y.H.). We gratefully recognize the expert technical assistance of Fran Simonet-Magnuson and Alan Moore, and helpful discussion from Dr. Tony L. Yaksh.
Correspondence should be addressed to Xiao-Ying Hua, Anesthesia Research Laboratory, 0818, Department of Anesthesiology, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0818.
REFERENCES
- 1.Barnes P. Neurogenic inflammation and asthma. J Asthma. 1992;29:165–180. doi: 10.3109/02770909209099025. [DOI] [PubMed] [Google Scholar]
- 2.Barnes P. Cytokines as mediators of chronic asthma. Am J Respir Crit Care Med. 1994;150:42–49. doi: 10.1164/ajrccm/150.5_Pt_2.S42. [DOI] [PubMed] [Google Scholar]
- 3.Barnes PJ, Belvisi MG. Nitric oxide and lung disease. Thorax. 1993;48:1034–1043. doi: 10.1136/thx.48.10.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basbaum A. Peripheral mechanisms of sensitization and hyperalgesia. In: Stanley T, Ashburn M, Fine P, editors. Anesthesiology and pain management. Kluwer Academic Publishers; Norwell, MA: 1991. pp. 31–37. [Google Scholar]
- 5.Brain SD, Williams TJ, Tippins JR, Morris HR, MacIntyre I. Calcitonin gene-related peptide is a potent vasodilator. Nature. 1985;313:54–56. doi: 10.1038/313054a0. [DOI] [PubMed] [Google Scholar]
- 6.Cadieux A, Springall DR, Mulderry PK, Rodrigo J, Chatei MA, Terenghi G, Bloom SR, Polak JM. Occurrence, distribution and ontogeny of CGRP immunoreactivity in the rat lower respiratory tract: effect of capsaicin treatment and surgical denervation. Neuroscience. 1986;19:605–627. doi: 10.1016/0306-4522(86)90285-x. [DOI] [PubMed] [Google Scholar]
- 7.Cunha F, Poole S, Lorenzetti B, Ferreira S. The pivotal role of tumour necrosis factor alpha in the development of inflammatory hyperalgesia. Br J Pharmacol. 1992;107:660–664. doi: 10.1111/j.1476-5381.1992.tb14503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis AJ, Perkins MN. The involvement of bradykinin B1 and B2 receptor mechanisms in cytokine-induced mechanical hyperalgesia in the rat. Br J Pharmacol. 1994;113:63–68. doi: 10.1111/j.1476-5381.1994.tb16174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dinarello CA. The interleukin-1 family: 10 years of discovery. FASEB J. 1994;8:1314–1325. [PubMed] [Google Scholar]
- 10.Dray A, Bevan S. Inflammation and hyperalgesia: highlighting the team effort. Trends Pharmacol. 1993;14:287–290. doi: 10.1016/0165-6147(93)90041-H. [DOI] [PubMed] [Google Scholar]
- 11.Dray A, Perkins M. Bradykinin and inflammatory pain. Trends Neurosci. 1993;16:99–104. doi: 10.1016/0166-2236(93)90133-7. [DOI] [PubMed] [Google Scholar]
- 12.Eastgate JA, Symons JA, Wood NC, Grinlinton FM, di Giovine FS, Duff GW. Correlation of plasma interleukin 1 levels with disease activity in rheumatoid arthritis. Lancet. 1988;2:706–709. doi: 10.1016/s0140-6736(88)90185-7. [DOI] [PubMed] [Google Scholar]
- 13.Ferreira SH, Lorenzetti BB, Bristow AF, Poole S. Interleukin-1β as a potent hyperalgesic agent antagonized by a tripeptide analogue. Nature. 1988;334:698–700. doi: 10.1038/334698a0. [DOI] [PubMed] [Google Scholar]
- 14.Follenfant RL, Nakamura-Craig M, Henderson B, Higgs GA. Inhibition by neuropeptides of interleukin-1β-induced, prostaglandin-independent hyperalgesia. Br J Pharmacol. 1989;98:41–43. doi: 10.1111/j.1476-5381.1989.tb16860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gordon JR, Burd PR, Galli SJ. Mast cells as a source of multifunctional cytokines. Immunol Today. 1990;12:458–464. doi: 10.1016/0167-5699(90)90176-a. [DOI] [PubMed] [Google Scholar]
- 16.Hastings RH, Hua X-Y. Expression of calcitonin gene-related peptide by cultured rat alveolar type II cells. Am J Respir Cell Mol Biol. 1995;13:563–569. doi: 10.1165/ajrcmb.13.5.7576692. [DOI] [PubMed] [Google Scholar]
- 17.Herbert MK, Holzer P. Interleukin-1β enhances capsaicin-induced neurogenic vasodilatation in the rat skin. Br J Pharmacol. 1994;111:681–686. doi: 10.1111/j.1476-5381.1994.tb14791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hingtgen CM, Waite KJ, Vasko MR. Prostaglandins facilitate peptide release from rat sensory neurons by activating the adenosine 3′,5′-cyclic monophosphate transduction cascade. J Neurosci. 1995;15:5411–5419. doi: 10.1523/JNEUROSCI.15-07-05411.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holzer P. Local effector functions of capsaicin-sensitive sensory nerve endings: involvement of tachykinins, calcitonin gene-related peptide and other neuropeptides. Neuroscience. 1988;24:739–768. doi: 10.1016/0306-4522(88)90064-4. [DOI] [PubMed] [Google Scholar]
- 20.Hua X-Y, Jinno S, Back SM, Tam EK, Yaksh TL. Multiple mechanisms for the effects of capsaicin, bradykinin and nicotine on CGRP release from tracheal afferent nerves: role of prostaglandins, sympathetic nerves and mast cells. Neuropharmacology. 1994;33:1147–1154. doi: 10.1016/s0028-3908(05)80004-8. [DOI] [PubMed] [Google Scholar]
- 21.Hua X-Y, Yaksh TL. Release of calcitonin gene-related peptide and tachykinins from the rat trachea. Peptides. 1992;13:113–120. doi: 10.1016/0196-9781(92)90148-v. [DOI] [PubMed] [Google Scholar]
- 22.Hua X-Y, Yaksh TL. Pharmacology of the effects of bradykinin, serotonin, and histamine on the release of calcitonin gene-related peptide from C-fiber terminals in the rat trachea. J Neurosci. 1993;13:1947–1953. doi: 10.1523/JNEUROSCI.13-05-01947.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu S, Adcock IM, Old RW, Barnes PJ, Evans TW. Lipopolysaccharide treatment in vivo induces widespread tissue expression of inducible nitric oxide synthase mRNA. Biochem Biophys Res Commun. 1993;196:1208–1213. doi: 10.1006/bbrc.1993.2380. [DOI] [PubMed] [Google Scholar]
- 24.Long NC, Otterness I, Kunkel SL, Vander AJ, Kluger J. Roles of interleukin 1β and tumor necrosis factor in lipopolysaccharide fever in rats. Am J Physiol. 1990;28:R724–R728. doi: 10.1152/ajpregu.1990.259.4.R724. [DOI] [PubMed] [Google Scholar]
- 25.Lundberg JM, Lundblad L, Martling C-R, Saria A, Stjarne P, Anggard A (1987) Coexistance of multiple peptides and classic transmitters in the airway neurons: functional and pathophysiologic aspects. Am Rev Respir Dis 136:s16–s22. [DOI] [PubMed]
- 26.Marceau F, Petitclerc E, DeBlois D, Pradelles P, Poubelle PE. Human interleukin-1 induces a rapid relaxation of the rabbit isolated mesenteric artery. Br J Pharmacol. 1991;103:1367–1372. doi: 10.1111/j.1476-5381.1991.tb09795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin HA, Basbaum AI, Kwiat GC, Goetzl EJ, Levine JD. Leukotriene and prostaglandin sensitization of cutaneous high-threshold C- and Ad-mechanonociceptors in the hairy skin of rat hindlimbs. Neuroscience. 1987;22:651–659. doi: 10.1016/0306-4522(87)90360-5. [DOI] [PubMed] [Google Scholar]
- 28.Mattoli S, Mattoso VL, Soloperto M, Allegra L, Fasoli A. Cellular and biochemical characteristics of bronchoalveolar lavage fluid in symptomatic non allergic asthma. J Allergy Clin Immunol. 1991;84:794–802. doi: 10.1016/0091-6749(91)90125-8. [DOI] [PubMed] [Google Scholar]
- 29.Morrison DC. Endotoxin and disease mechanisms. Annu Rev Med. 1987;38:417–432. doi: 10.1146/annurev.me.38.020187.002221. [DOI] [PubMed] [Google Scholar]
- 30.Niijima A (1992) The afferent discharges from sensors for interleukin-1β in the hepato-portal system in the anaesthetized rat. J Physiol (Lond) 446:236p.
- 31.O’Byrne EM, Blancuzzi V, Wilson DE, Wong M, Jeng AY. Elevated substance P and accelerated cartilage degradation in rabbit knees injected with interleukin-1 and tumor necrosis factor. Arthritis Rheum. 1990;33:1023–1028. doi: 10.1002/art.1780330715. [DOI] [PubMed] [Google Scholar]
- 32.Regoli D, Marceau F, Lavigne J. Induction of the B1-receptors for kinins in the rabbit by lipopolysaccharide. Eur J Pharmacol. 1981;71:105–115. doi: 10.1016/0014-2999(81)90391-5. [DOI] [PubMed] [Google Scholar]
- 33.Robbins RA, Springall DR, Warren JB, Kwon OJ, Buttery LDK, Wilson AJ, Adock IM, Riveros-Moreno V, Moncada S, Polak J, Barnes PJ. Inducible nitric oxide synthase is increased in murine lung epithelial cells by cytokine stimulation. Biochem Biophys Res Commun. 1994;198:835–843. doi: 10.1006/bbrc.1994.1119. [DOI] [PubMed] [Google Scholar]
- 34.Shimosegawa T, Said SI. Pulmonary calcitonin gene-related pep- tide immunoreactivity: nerve-endocrine cell interrelationships. Am J Respir Cell Mol Biol. 1991;4:126–134. doi: 10.1165/ajrcmb/4.2.126. [DOI] [PubMed] [Google Scholar]
- 35.Sommer C, Myers RR. Thalidomide inhibition of TNF reduces hyperalgesia in neuropathic rats. Reg Anesth. 1994;19:1. [Google Scholar]
- 36.Ulich TR, Yin S, Guo K, Eisenberg SP, Thompson RC. The intratracheal administration of endotoxin and cytokines. III. The IL-1 receptor antagonist inhibits endotoxin- and IL-1-induced acute inflammation. Am J Pathol. 1991;138:521–524. [PMC free article] [PubMed] [Google Scholar]
- 37.Vasko MR, Campbell WB, Waite KJ. Prostaglandin E2 enhances bradykinin-stimulated release of neuropeptides from rat sensory neurons in culture. J Neurosci. 1994;14:4987–4997. doi: 10.1523/JNEUROSCI.14-08-04987.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vincent D, Lefort J, Bureau M, Dry J, Vazgaftig BB. Dissociation between LPS-Induced bronchial hyperreactivity and airway edema in the guinea-pig. Agents Actions. 1991;34:203–204. doi: 10.1007/BF01993279. [DOI] [PubMed] [Google Scholar]
- 39.Wagner R, Myers RR. Schwann cells produce TNF-alpha: expression in injured and non-injured nerves. Neuroscience. 1996;73:625–629. doi: 10.1016/0306-4522(96)00127-3. [DOI] [PubMed] [Google Scholar]
- 40.Watkins LR, Wiertelak EP, Goelher LE, Smith KP, Martin D, Maier SF. Characterization of cytokine-induced hyperalgesia. Brain Res. 1994;654:15–26. doi: 10.1016/0006-8993(94)91566-0. [DOI] [PubMed] [Google Scholar]
- 41.Watkins LR, Maier SF, Goehler LE. Cytokine-to-brain brain communication: a review and analysis of alternative mechanism. Life Sci. 1995;57:1011–1026. doi: 10.1016/0024-3205(95)02047-m. [DOI] [PubMed] [Google Scholar]