

Abstract
Primary afferent neurons in mammalian dorsal root ganglia (DRGs) are anatomically isolated from one another and are not synaptically interconnected. As such, they are classically thought to function as independent sensory communication elements. However, it has recently been shown that most DRG neurons are transiently depolarized when axons of neighboring neurons of the same ganglion are stimulated repetitively. Here we further characterize this functional coupling. In electrophysiological recordings made from excised rat DRGs, we found that DRG “cross-depolarization” is excitatory in that it is accompanied by an increase in the probability of spiking in response to otherwise subthreshold test pulses delivered intracellularly. Cross-depolarization contributes to this mutual cross-excitation. However, at least as important a contribution comes from a net increase in the neurons’ input resistance (Rin) triggered by the stimulation of neighboring neurons. This change inRin occurs even when cross-depolarization is absent or is balanced out. The amplitude of cross-depolarization was found to be voltage-dependent, with a reversal potential at approximately −23 mV. Reversibility and the change inRin both indicate that activity of neighboring neurons causes a membrane conductance change that is chemically mediated. Thus, far from being isolated, most DRG neurons participate in ongoing mutual interactions in which neuronal excitability is continuously modulated by afferent spike activity. This intraganglionic dialog appears to be mediated, at least in part, by an activity-dependent diffusable substance(s) released from neuronal somata and/or adjacent axons, and detected by neighboring cell somata and/or axons.
Keywords: cross-excitation, cross talk, dorsal root ganglion, neuropathy, nonsynaptic neurotransmission, pain
Spike activity in primary afferent neurons in dorsal root ganglia (DRGs) in vivo excites adjacent passive neurons that share the same DRG (Devor and Wall, 1990). In cells with resting discharge, this is reflected in an acceleration of firing during and shortly after tetanic stimulation of neighboring afferents. Because synapses are virtually absent in DRGs and because adjacent neurons are isolated in individual satellite cell sheaths (Lieberman, 1976), cell-to-cell interactions within the DRG cannot involve synaptic or electrical junctions. The possibility that intraganglionic communication is based on a novel, nonconventional neural mechanism and that it has practical consequences for sensory conduction in health and disease motivates a deeper understanding of the underlying process.
Electrophysiological recordings in Devor and Wall’s (1990) in vivo study were made from axon microfilaments teased from the sciatic nerve at a distance from the DRG, and hence precluded observation of the underlying membrane potential. With this limitation in mind, Utzschneider et al. (1992) recorded intracellularly from rat DRG neurons in vitro using a setup that permitted selective tetanic stimulation of the axons of neighboring DRG neurons. They observed a transient “cross-depolarization” in ∼90% of the cells sampled. Because the time course of the depolarization resembled that of DRG cross-excitation in vivo, they inferred that the depolarization is responsible for DRG cross-excitation. However, the cells studied in vitro rarely had resting spike discharge, and they did not generate discharge on depolarization. It was therefore not clearly established whether DRG cross-excitation is a direct consequence of cross-depolarization. In the present study, we have combined intracellular subthreshold electrical stimulation of DRG neurons with repetitive stimulation of their neighbors to ask whether DRG cross-depolarization indeed increases the excitability of the affected cells and, if so, what is the underlying mechanism.
MATERIALS AND METHODS
Animals and preparation. The experiments were performed using 47 young rats (27 males, 20 females, 2–6 weeks old; 15–145 gm) of the Wistar-derived Sabra strain (Lutzky et al., 1984). The animals were deeply anesthetized with pentobarbital sodium (Nembutal, 60 mg/kg, i.p.) and euthanized by carotid exsanguination. DRGs L4 or L5 were excised with their dorsal roots (DRs), the spinal nerve, and a variable length of attached sciatic nerve. After ∼1 hr recovery in a modified Krebs’ solution containing (in mm): NaCl 124, NaHCO3 26, KCl 3, NaH2PO4 1.3, MgCl 2, dextrose 10, and saturated with 95% O2 and 5% CO2, pH 7.4 (290–300 mOsm, 20°C), the ganglia were mounted in a recording chamber and superfused with the Krebs’ solution (1–2 ml/min, 20 or 37°C) to which 2 mm CaCl2 was added. Sharp glass microelectrodes were used for intracellular recording and stimulation (20–40 MΩ filled with 3 m KCl or 2 m K-acetate). The electrodes were passed through the undissected DRG capsule, which is thin in young rats. All cells reported here had a stable resting membrane potential more negative than −40 mV and, after intracellular somatic or extracellular axonal stimulation, an overshooting spike. Data were recorded digitally on magnetic tape for off-line analysis.
Cross-depolarization and measurement of input resistance.Three alternative protocols were used to deliver conditioning stimulation trains (tetani) to the axons of neighboring DRG neurons while avoiding the axon of the impaled neuron. In the first, electrical pulses were delivered through an Ag/AgCl electrode pair placed across the sciatic nerve (SN in Fig.2A). Stimulus current was set just subthreshold for the impaled neuron. By adjusting the polarity, it was usually possible to activate a substantial proportion of the myelinated axons in the nerve, as judged from the size of the compound action potential (CAP) monitored through a suction electrode on the DR, without stimulating the axon of the impaled neuron itself. In the second protocol, pulses were delivered to the DR while the CAP was monitored from the sciatic nerve (SDR in Fig. 5A). Finally, in some experiments, the DR was split longitudinally and stimuli were delivered to each branch separately (Fig. 3A). The branch that when stimulated evoked a soma spike in the impaled neuron was designated S1. The other branch, which contained only axons of neighboring neurons and never evoked soma spikes, was designated S2. In all protocols, stimuli were monophasic 0.1–0.2 msec square pulses, ≤7 mA, delivered singly or in 10 sec tetani at 10–500 Hz (usually 50 and/or 100 Hz). Two to sixteen repeated trials were run for each set of stimulus parameters for a given cell. The stimulus parameters used were not sufficient to activate unmyelinated axons as judged both from the CAP and the failure to record evoked spikes from neurons with unmyelinated axons.
Fig. 2.

Cross-depolarization in DRG neurons. A, Sketch of one of the alternative experimental protocols (see Materials and Methods). B, Intracellular voltage (R&SIC) recording from an AINF neuron in response to sub- and suprathreshold pulses delivered through the intracellular micropipette (SIC, 35 msec pulse) and the sciatic nerve (SN, 0.1 msec pulse). The SN -evoked spike is small because it did not invade the soma (same calibration for both trials). C, Cross-depolarization in this cell to 10 sec SNtetani (dashed line) using stimuli subthreshold for the axon of the impaled neuron, and frequency as indicated.
Fig. 5.
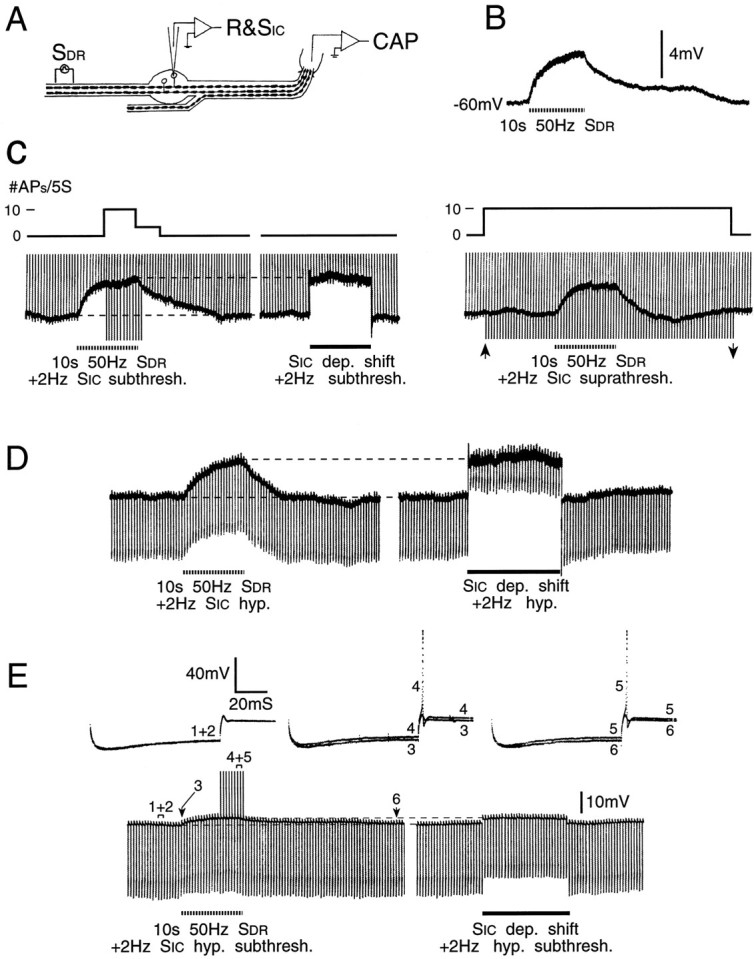
Increased firing probability during cross-depolarization is associated with increasedRin. A, B, A third experimental protocol, and an example of cross-depolarization evoked by DR conditioning (SDR) subthreshold for the axon of the impaled neuron. C, Left, Initially subthreshold test pulses were delivered at 2 Hz through the intracellular electrode (SIC, vertical voltage deviations). Firing probability increased during conditioning (note spikes during the peak of the cross-depolarization), but was unaffected when an equivalent depolarizing shift was imposed by passing current through the recording electrode. C, Right, Probing with just suprathreshold test pulses (betweenarrows) yielded no sign of excitability suppression. Upward deflections before and after this time are just subthreshold test pulses. D, Hyperpolarizing constant-current test pulses reveal slightly increased Rin during DR conditioning, but much decreased Rin during an equivalent imposed depolarizing shift. E, Conditioning also increased the probability of anodal break responses (using higher amplitude hyperpolarizing test pulses than in D). An imposed depolarizing shift of similar amplitude had no effect on firing probability. Note the different change inRin during cross-depolarization versus imposed depolarization. Sample voltage traces from time windows 1–6 are shown.
Fig. 3.

Firing probability is increased during cross-depolarization. A, A second experimental protocol. The DR was split, and conditioning tetani were delivered to the half not containing the axon of the impaled neuron (S2).B, (Initially) subthreshold test pulses delivered at 2 Hz (SIC) yielded firing probability of 0 action potentials/5 sec epoch (#APs/5S). During conditioning, firing probability increased, reaching the maximum possible of 10 APs/5 sec. C, Evoked responses during time windows 1–4 inB. Each trace shows three consecutive sweeps.
To test whether depolarization evoked by conditioning tetani is accompanied by a change in whole-cell membrane conductance, the amplifier bridge was carefully balanced, and constant current hyperpolarizing pulses (0.5–2 nA, 20–100 msec, usually ≥80 msec) were delivered intracellularly at 2 Hz. Input resistance (Rin) was calculated from the early voltage peak using Ohm’s law.
Measurement of change in cell excitability. Attempts to titrate changes in the actual current threshold for evoking spikes by intracellular stimulation failed because individual evoked spikes often caused prolonged changes in threshold. As an alternative, we monitored changes in the proportion of test pulses that evoked a spike during 10 sec periods of fixed intensity 1 or 2 Hz intracellular stimulation (“firing probability” = n responses/10 or 20 test stimuli). To check for excitation, stimulus intensity was initially set just below (∼15%) threshold. At this intensity, firing probability before conditioning was 0/10 or 0/20. An increase in this value to ≥3/10 or ≥5/20 during 10 sec of conditioning was considered statistically significant excitation (p < 0.05, one-tailed Fisher exact probabilities test). Suppression was checked using test pulses at just threshold intensity (the minimal current required to achieve a firing probability of 20/20) and asking whether the firing probability decreased during or after the conditioning tetanus (criterion for p < 0.05 was ≤15/20, one-tailed Fisher test). In some experiments, stimulus intensity was set to place firing probability between 0/20 and 20/20. In this way, excitation and suppression could be monitored in the same trial by obtaining an increase or a decrease in firing probability (two-tailed Fisher test). Finally, in a few experiments we examined the effect of conditioning tetani on the probability of evoking spikes at the termination of strong hyperpolarizing pulses (anodal break responses).
Stimulus pulses were applied to the impaled cell through the amplifier bridge circuit (5–40 msec depolarizing pulses, 0.1–7 nA, 1 or 2 Hz). Pulse duration was always much longer than the chronaxie for these cells (1.8 ± 0.4 msec; n = 25), and therefore changes in firing probability could not have reflected subtle, uncontrolled changes in effective pulse width.
Cell characterization. The preparation did not permit determination of receptive fields. Instead, we categorized neurons by axonal conduction velocity (CV) and the shape of the intracellularly recorded spike (Fig. 1). CV was calculated by dividing propagation distance by spike latency after single suprathreshold stimulus pulses to the sciatic nerve or the DR. For present purposes, if the CV was >1 m/sec, the neuron was categorized as having a myelinated axon (A-neuron) regardless of temperature or the animal’s age.
Fig. 1.

A0 and AINF spikes. A0(A) and AINF (B) spikes (direct and differentiated record) after axonal stimulation (*) just below and just above threshold (single sweeps). Note the inflection on the rising phase (downward pointing arrows) and on the falling phase (B only, upward pointing arrow). CV = 8.6 m/sec for A, 4.3 m/sec for B.
Two criteria were set to identify that minority of neurons for which CV was not determined. The first was the presence of an inflection on the falling phase of the intracellularly recorded action potential (determined using an analog signal differentiator). Spikes were evoked by axonal stimulation or by 1 msec intracellular stimulus pulses. Neurons with CV <1 m/sec (C-neurons) consistently had an inflection, whereas neurons with CV >1 m/sec mostly had no inflection. A second criterion was spike width (measured at 1/2 peak-to-peak amplitude). C-neurons always had a spike width >2 msec; in A-neurons, spike width was always ≤2 msec. Thus, cells that showed no inflection and/or had a spike width ≤2 msec were categorized as A-neurons even if CV was not established. The observed relationship between CV, spike inflection, and spike width was consistent with previous findings in rat DRGs and sensory ganglia of other vertebrates (Gorke and Pierau, 1980; Harper and Lawson, 1985).
All means are given ±SD. Unless otherwise noted, statistical evaluations are based on Mann–Whitney U tests and significance of linear correlation coefficient (criterionp < 0.05).
RESULTS
Cell sample and spike characteristics
This report is based on data from 136 DRG A-neurons, 48 with an inflection on the spike falling phase (AINFneurons) and 88 with no inflection (A0 neurons). Some cells of both types also had an inflection on the spike rising phase (arrows in Fig. 1A,B; also see Ito, 1959). A0 neurons had narrower and lower amplitude spikes than AINF neurons, lower resting membrane potential, and faster axonal CV. In both cell types, spike amplitude and width were sensitive to bath temperature (20 or 37°C, Table 1). The A0 group represents predominantly Aβ afferents, most of which are low-threshold mechanoreceptors; the AINF group, containing mostly Aδ afferents, probably includes most of the myelinated nociceptors (Gorke and Pierau, 1980; Koerber and Mendell, 1992).
Table 1.
Action potential parameters (mean ± SD) for DRG neurons with (AINF) and without (A0) an inflection on the falling phase of the spike in experiments performed at 20 and 37°C
| Neuron type/ temperature | Cond. velocity (m/sec) | Resting potential (mV) | Spike amplitude (mV) | Spike duration (msec) |
|---|---|---|---|---|
| 20 and 37°C | ||||
| A0 | 7.0 ± 2.3 | −54.0 ± 6.6 | 67.0 ± 15.6 | 0.6 ± 0.3 |
| n = 47 | n = 88 | n = 88 | n = 88 | |
| AINF | 5.3 ± 2.7 | −57.6 ± 7.0 | 75.2 ± 14.0 | 1.3 ± 0.7 |
| n = 31 | n = 48 | n = 48 | n = 48 | |
| A0 vs AINF | p < 0.01 | p < 0.01 | p < 0.01 | p < 0.01 |
| A0 at 20°C | 6.0 ± 2.4 | −53.3 ± 5.8 | 60.5 ± 9.4 | 1.0 ± 0.3 |
| n = 13 | n = 21 | n = 21 | n = 21 | |
| AINF at 20°C | 4.9 ± 3.3 | −58.8 ± 5.5 | 66.2 ± 11.6 | 2.7 ± 1.1 |
| n = 4 | n = 6 | n = 6 | n = 6 | |
| A0 at 37°C | 7.3 ± 2.2 | −54.2 ± 6.8 | 69.0 ± 16.6 | 0.5 ± 0.1 |
| n = 34 | n = 67 | n = 67 | n = 67 | |
| AINF at 37°C | 5.3 ± 2.7 | −57.4 ± 7.2 | 76.5 ± 14 | 1.1 ± 0.4 |
| n = 27 | n = 42 | n = 42 | n = 42 | |
| Both at 20°C | 5.7 ± 2.6 | −54.5 ± 6.1 | 61.8 ± 10 | 1.4 ± 0.9 |
| n = 17 | n = 27 | n = 27 | n = 27 | |
| Both at 37°C | 6.4 ± 2.6 | −55.4 ± 7.1 | 71.9 ± 16 | 0.7 ± 0.4 |
| n = 61 | n = 109 | n = 109 | n = 109 | |
| 20°C vs 37°C | p > 0.2 | p > 0.2 | p < 0.01 | p < 0.001 |
p values are based on two-tailed Mann–WhitneyU tests.
Cross-depolarization
We challenged 95 DRG neurons (54 A0; 41 AINF) with 10 sec, 50 or 100 Hz conditioning tetani to the axons of neighboring neurons. Nearly all (90 of 95, or 95%) showed cross-depolarization (Fig. 2C); only five were unaffected. Depolarization began within 500 msec of tetanus onset, increased monotonically at a gradually decreasing rate [rising phase time constant τ = 2.6 ± 0.7 sec (mean ± SD)], and at the end of stimulation returned to baseline over 1.5–57 sec (recovery τ = 5.9 ± 4.6 sec). Peak depolarization amplitude ranged from 0.5 to 25 mV. No such response occurred with the microelectrode positioned extracellularly.
Responses were larger and more reliable using 100 Hz than 50 Hz tetani (4.5 ± 4.0 mV vs 3.2 ± 3.1, n = 30, p < 0.01; 47 of 47 vs 74 of 82 responded,p = 0.05). Still larger responses were obtained using 200 and 500 Hz tetani (Fig. 2C). Stimulation of whole DRs at an intensity subthreshold for the axon of the impaled neuron yielded somewhat larger and more reliable responses than stimulation of longitudinally split DRs or stimulation of the sciatic nerve (SN). All three sites, however, were effective (Table 2). Responses were smoothly graded as stimulus intensity was increased, with a direct correlation between cross-depolarization amplitude and the size of the evoked CAP until saturation (n = 4). Thus, the reason for the larger responses using the whole DR protocol presumably was that a larger number of adjacent neurons were stimulated. Neither the probability of evoking responses nor response amplitude depended on cell type (A0 vs AINF), axonal CV, or bath temperature (p > 0.2 in each case). None of the responding cells fired action potentials during the course of cross-depolarization, although we have seen such crossed afterdischarge on occasion in vitro. Other parameters of crossed depolarization are provided by Utzschneider et al. (1992).
Table 2.
Comparison of three sites for delivering conditioning stimulation (all tetani were 50 Hz for 10 sec)
| Site of conditioning stimulation | Prevalence of crossdepolarization* (n responses/n tested, %) | Cross-depolarization amplitude** (mV) |
|---|---|---|
| Sciatic nerve (SN) | 9/11 (82%) | 2.9 ± 2.4 |
| Dorsal root (DR) | p > 0.2 51/55 (93%) | p > 0.2 3.4 ± 3.0 |
| Split DR | 14/16 (88%) | 1.9 ± 1.4 |
*, Two-tailed Fisher test.
**, Two-tailed Mann–Whitney U test.
Changes in excitability during conditioning-evoked cross-depolarization
Depolarization does not necessarily yield excitation. For example, activation of a Cl− conductance may depolarize neurons, but it shunts membrane current, hence usuallysuppressing cell excitability (Dudel and Kuffler, 1961). We therefore checked the effect of conditioning tetani on cell excitability directly using subthreshold intracellular test pulses. During conditioning, most cells (53 of 57, or 93%) showed an increase in their probability of being fired by these test pulses (p < 0.001; Fig. 3). Indeed, the change was large enough to reach our statistical criterion individually in 41 of the 53 responding cells (77%). Both 50 and 100 Hz tetani were effective. Some cells fired occasional bursts of 2–3 spikes in response to test pulses. In these, the probability of bursting increased, and the interval between spikes within a burst decreased during cross-depolarization (Fig.4A). Finally, seven cells were tested with hyperpolarizing test pulses. In each there was an increase in anodal break spiking, with firing probability rising from 0/20 to ≥5/20 (p < 0.05) in six of the seven cells (Fig.5E). All of these observations indicate that conditioning tetani evoke cross-excitation in vitro as they do in vivo.
Fig. 4.

Cross-excitation. A, Increased firing probability during cross-depolarization in a cell that fired bursts in response to intracellular stimulus pulses (10 msec, 2 Hz). The experimental protocol was as in Figure 5A. Conditioning, with resulting cross-depolarization, caused an increase in firing probability (bursts/5 sec). Spike bursts during time windows 1–4 (3 or 4 superimposed traces each) are shown below. Note that spikes in A and in subsequent figures are truncated.B, Conditioning tetani may produce cross-excitation even without triggering cross-depolarization. The experimental protocol was as in Figure 3. Conditioning did not trigger cross-depolarization in this AINF neuron (top), but nonetheless produced cross-excitation (bottom). Firing probability, initially set at 5/20, increased to 13/20 during the conditioning tetanus (p < 0.05). Downward deflections are shock artifacts, and upward deflections are spikes (note the prolonged postspike AHPs).
Spike discharge in a small proportion (∼2%) of DRG neurons studiedin vivo was suppressed during periods of tetanic stimulation of their neighbors (Devor and Wall, 1990). To test for such cross-suppression in vitro, we applied intracellular test pulses that were near or just suprathreshold. As expected, in most cells cross-depolarization caused firing probability either to increase (7 of 8 cells with initial firing probability set <20/20) or to remain unchanged (15 of 16 cells with initial firing probability = 20/20; Figs. 4B, 5C). However, two cells were exceptional. In one, firing probability fell slightly during the conditioning tetanus (from 5/20 to 2/20, p > 0.2), and in the other it fell significantly (from 20/20 to 10/20,p < 0.01). Thus, some cells appear to show cross-suppression.
Relation between cross-depolarization and firing probability
Although increased firing probability coincided with cross-depolarization, there was no significant correlation between their magnitudes (r = 0.1, p > 0.2). For example, firing probability sometimes increased even when there was no cross-depolarization (Fig. 4B). This suggests that depolarization per se is not the only key parameter controlling firing probability. To check this directly, we used two alternative strategies. In the first, we monitored firing probability while imposing 10 sec depolarizing current steps through the microelectrode. For most of the cells sampled (9 of 14), test pulses were set so that during the imposed depolarization, firing probability remained 0/20. Then, in a separate trial, conditioning tetani were given yielding the same level of depolarization. In all 14 cells, firing probability increased, reaching ≥5/20 in 12 (p < 0.005). Thus, overall, cross-depolarization evoked a significantly larger increase than did imposed depolarization of equal amplitude (Fig.5C, Table 3). Cross-depolarization also increased the probability of obtaining anodal break spikes more than equivalent imposed depolarizing steps did (n = 5; Fig.5E).
Table 3.
Effect of cross-depolarization evoked by a conditioning tetanus (10 sec, 50 or 100 Hz) and of an equivalent depolarization induced by intracellular current injection onRin (ΔRinconditioning and ΔRin depolarization, respectively) and on firing probability
| Neuron type | Rin(MΩ) | Crossdepolarization (mV) | ΔRin conditioning (%) | ΔRin depolarization (%) | ΔRin# difference (%) | Δ Firing probability | |
|---|---|---|---|---|---|---|---|
| Conditioning (n/20) | Depolarization (n/20) | ||||||
| A0 mean ± SD | 11.3 ± 5.8 | 3.9 ± 3.4 | 1.3 ± 9.5 | −16.8 ± 13.9 | 18.1** ± 16.2 | 9.9 ± 4.7 | 2.9* ± 6.2 |
| (ncells) | (23) | (25) | (23) | (23) | (23) | (10) | (10) |
| AINF mean ± SD | 13.7 ± 9.6 | 4.6 ± 4.9 | −5.9 ± 8.5 | −22.1 ± 18.6 | 16.2** ± 15.6 | 8.0 ± 2.4 | 5.5 ± 4.4 |
| (ncells) | (19) | (19) | (19) | (19) | (19) | (4) | (4) |
| A0 + AINF mean ± SD | 12.4 ± 7.8 | 4.5 ± 4.2 | −2.0 ± 9.6 | −19.2 ± 16.2 | 17.2** ± 15.7 | 9.4 ± 4.1 | 4.0* ± 6.4 |
| (ncells) | (42) | (44) | (42) | (42) | (42) | (14) | (14) |
*p < 0.05, **p < 0.001 using two-tailed Mann–Whitney tests comparing conditioning versus an equivalent depolarizing pulse.
#ΔRin difference = ΔRin conditioning − ΔRin depolarization.
The second strategy was to balance out cross-depolarization with hyperpolarizing current applied through the microelectrode during conditioning. All five cells tested in this way continued to show increased firing probability despite the absence of depolarization (Fig. 6C). These results prompted us to ask whether firing probability is affected by a change in the conductance of the cell membrane as well as by membrane depolarization.
Fig. 6.
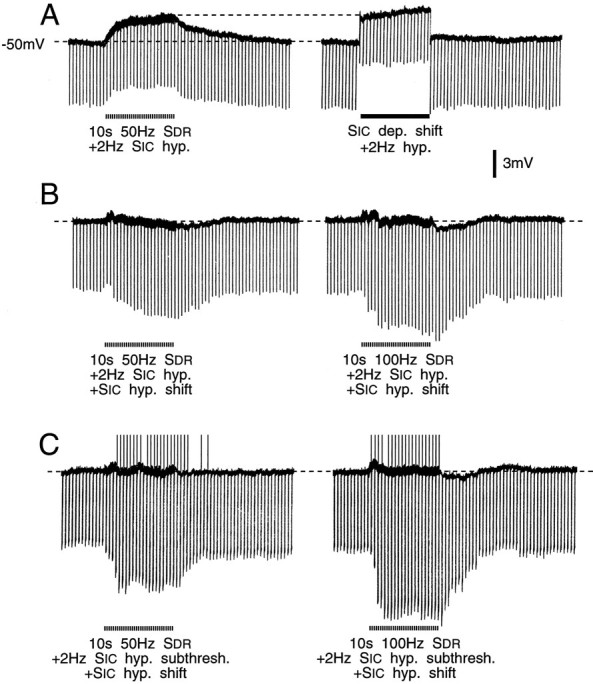
Balancing out cross-depolarization did not abolish cross-excitation. A, Hyperpolarizing test pulses (1 nA, 20 msec, 2 Hz) were applied during cross-depolarization (left trace) and indicated ∼10% increase inRin from a control level of 7 MΩ. Identical test pulses revealed a 25% decrease inRin when depolarization of similar amplitude was imposed intracellularly (right trace).B, Cross-depolarization was balanced out with intracellular hyperpolarizing current. This revealed a 35% increase inRin during conditioning at 50 Hz (left) and a 45% increase during conditioning at 100 Hz (right). C, Intracellular hyperpolarizing test pulses initially 25% subthreshold for anodal break spikes (1.3 nA, 85 msec, 2 Hz) evoked such spikes during 50 Hz (left) and 100 Hz (right) conditioning tetani (upward deflections, spikes truncated). Cross-depolarization was quenched during these trials.Rin increased ∼55% and 75%, respectively.
Conductance change associated with conditioning tetani
Constant current hyperpolarizing pulses were used to monitor cell Rin (Figs. 5, 6). In some of the cells, there was a voltage sag after an initial early peak (Fig.5E). This sag represents inward rectification. Measurements were made at the voltage peak to avoid possible effects of conditioning on the process responsible for the sag. However, we confirmed that had measurements been made at the end of the hyperpolarizing pulse, the basic conclusions would not have been materially altered. Inward rectification in DRG cells has been described by others and will not be commented on further here (Gorke and Pierau, 1980; Belmonte and Gallego, 1983; Harper and Lawson, 1985). We examined 52 neurons, all showing cross-depolarization (mean 4.5 ± 3.7 mV, range 0.5–18 mV). Results were variable. In 19 cells (37%), conditioning tetani had no effect on Rin despite cross-depolarization of as much as 4.5 mV. In 23 cells (44%), Rindecreased (by a maximum of 10.3 ± 7.5%, range 2–28%) and in 10 (19%) it increased (by 8.8 ± 6.4, range 1–22%). The time course paralleled that of the cross-depolarization.
To evaluate how much of the change in Rinwas attributable to membrane depolarization and how much to other effects of conditioning, we imposed equivalent 10 sec depolarizing steps. These almost always yielded a decrease inRin (outward rectification) even when cross-depolarization yielded an increase. Subtracting this depolarization-evoked change in Rin from the change produced by conditioning tetani nearly always yielded a netincrease in Rin (38 of 42 cells, Figs. 5D,E and 6A, ΔRin difference in Table 3). In these cells, there was a significant positive correlation between the magnitude of the Rin increase and the magnitude of cross-depolarization (ΔRindifference vs cross-depolarization, r = 0.37, p < 0.05, Table 3). There also was a significant correlation between the magnitude of theRin increase and the degree of cross-excitation (ΔRin difference vs Δ firing probability, r = 0.77, p < 0.05, Table 3). Two cells showed a net decrease inRin and two showed no change.
We also tried balancing out cross-depolarization with hyperpolarizing current. Here, conditioning almost always yielded increasedRin (15 of 18 cells tested, mean 26.9 ± 21.9%, Fig. 6B). In the three exceptional cells,Rin decreased (by 9, 12, and 14%). Likewise, in four cells in which conditioning increased the probability of anodal break spikes despite quenching of cross-depolarization, there was an increase in Rin (Fig.6C). Changes in Rin were the same for A0 and AINFneurons (p > 0.2, Table 3). There was no significant correlation between the change inRin and either CV (r = −0.14, p > 0.2) or resting membrane potential (r = −0.08, p > 0.2).
Reversal of cross-depolarization
The observation that conditioning tetani increasedRin even when membrane potential was held constant implies that they affect ion channels via a chemical mediator(s). This was confirmed by measuring the amplitude of cross depolarization while systematically varying the membrane potential. Hyperpolarization usually increased cross-depolarization amplitude (15/18). Unfortunately, significant depolarization generally triggered membrane instability and/or failure of recovery to rest. However, in nine neurons part of the trajectory was revealed, including four neurons in which the response to conditioning reversed and became hyperpolarizatory (by linear interpolation at mean = −19.9 mV). In two of these, closely spaced data points were obtained near the reversal potential, which was at −23 mV in both cases (Fig.7).
Fig. 7.

Reversal of cross-depolarization.Right, Peak amplitude of voltage shift evoked by 10 sec, 50 Hz conditioning tetani (Vtet., ordinate) as a function of membrane holding potential (Vm). Symbols identify five neurons, two of which reversed. Traces on theleft are from cell (○). Resting potentials = −55 mV (○), −54 mV (▴), −61 mV(+), −53 mV (✖), −56mV (▪).
Spike shape during conditioning tetani
In most cells, the action potential evoked by axonal or intracellular stimulation was followed by an afterhyperpolarization (AHP), usually brief (<20 msec), but sometimes prolonged (up to several seconds). During conditioning tetani, in the presence of cross-depolarization or in its absence, the brief AHP amplitude decreased (34.8 ± 10.3%, n = 11), following a time course that paralleled that of cross-depolarization. There was also a small delay in the falling phase of the spike (by 13.0 ± 6.7%, n = 11, Fig. 8). Both changes imply a reduction in K+ current. There was no consistent change in spike amplitude, and there was no noticeable effect on the prolonged AHP when present.
Fig. 8.

Spike AHP decreases during conditioning tetani (10 sec, 100 Hz), with kinetics resembling those of cross-depolarization. In the record shown, cross-depolarization was quenched. Traces inB show spike shape before (pre) andduring conditioning. Note delay in spike falling phase during conditioning. The horizontal dashed line indicates the resting membrane potential.
DISCUSSION
Despite the fact that primary sensory neurons in mammalian DRGs are anatomically isolated from one another with no synaptic interconnections, our data indicate that they are nearly all coupled in an activity-dependent manner. Recording from excised rat DRG A-neuronsin vitro, we confirmed the finding of Utzschneider et al. (1992) that impulse activity evokes cross-depolarization of passive neighbors. The depolarization proved to be excitatory as evidenced by the appearance of spike responses to previously subthreshold test pulses. DRG cross-excitation is associated with an increase inRin. Presumably, both the cross-depolarization and the increase inRin contribute to cross-excitation.
Relation of cross-excitation to cross-depolarization and increasedRin
Cross-depolarization is expected to be excitatory because it brings the cell closer to its firing threshold. However, there were several indications that this is only one of the mechanisms underlying cross-excitation. First, cells showed a smaller increase in firing probability when they were depolarized by an intracellular voltage step than when cross-depolarization of the same amplitude was evoked by conditioning tetani. Second, there was no correlation between the magnitude of the cross-depolarization and the increase in firing probability. We documented increased firing probability in cells, the membrane potential of which did not change in response to tetanic stimulation of neighbors, or in which cross-depolarization was quenched with applied hyperpolarizing current. In a few cases, conditioning increased firing probability during periods when the cell was actually hyperpolarized as a result of spike-evoked AHP (5/41 cells). The apparent reason for these anomalies is that even without a change in membrane potential, conditioning stimuli increaseRin, rendering test pulses more effective. Indeed, there was a significant positive correlation between the net increase in Rin and cell excitability. We conclude that, typically, both the cross-depolarization and the increase in Rin triggered by conditioning tetani contribute to mutual cross-excitation within DRGs.
Mechanism of cross-depolarization
Synaptic contacts are vanishingly rare in DRGs, if they exist at all (Lieberman, 1976; Kayahara et al., 1981; Pannese, 1981). Furthermore, with only infrequent exceptions (Pannese et al., 1991), each neuron in the DRG is wrapped in an individual satellite cell sheath (Lieberman, 1976; Pannese, 1981). This prevents close apposition between adjacent neurons in the ganglion, restricting the possibility of electrical contact through gap junctions and other ephaptic interactions. There remain two obvious candidate communication mechanisms: (1) activity-evoked elevation of extracellular K+ concentration ([K+]o) and (2) nonsynaptic release of a chemical mediator(s). By nonsynaptic chemical communication, we envisage the release of mediator molecules from cell somata and/or intraganglionic axons (or axon varicosities), in the absence of a closely apposed postsynaptic membrane, and their diffusion from the release site to a relatively distant receptive membrane. A priori, both of these candidate mechanisms are possible, and they are not mutually exclusive.
With respect to K+ coupling, Utzschneider et al. (1992) demonstrated doubling of the baseline 3 mm[K+]o in rat DRGs following conditioning tetani of the sort used here (also see Deschenes and Feltz, 1976). An increase in [K+]o in the bulk extracellular medium is expected to equilibrate through the seam that separates adjacent ensheathing satellite cells (Shinder and Devor, 1994) and through the thin satellite cell sheath itself, and hence to present a similar concentration change to the soma surface. Dye and electrical coupling has been demonstrated among satellite cells in autonomic ganglia (Maudlej and Hanani, 1992). If this also occurs in DRGs, then ionic equilibration through this glial syncitium (Karwosky et al., 1989; Brunet and Jirounek, 1994) could also contribute to the spread of K+ depolarization. Applying the Goldman (constant field) equations, we calculate that an increase in [K+]o from 3 to 6 mm is expected to evoke a depolarization of ∼8.5 mV (Hille, 1992). This is well within the range of cross-depolarization amplitudes observed in our DRG cells.
Conditions within DRGs also permit nonsynaptic chemically mediated coupling. Release of a number of candidate mediators from DRGs has been documented. These include a range of neuropeptides, excitatory amino acids, eicosanoids, and nitric oxide (references in Shinder and Devor, 1994). In some instances, neurotransmitter/peptide-containing axonal “baskets” (“glomeruli,” “pericellular nests”) surround the perineuronal satellite cell sheath in a manner that should favor nonsynaptic chemical interactions (Matsuda and Uehara, 1981; Kuramoto et al., 1990; Devor et al., 1995). As for reception of the chemical signal, DRG neurons are known to be invested with a wealth of receptors for the above and other neurotransmitters (references in Shinder and Devor, 1994). Small molecules in the bulk extracellular space have access to the soma membrane, especially in the axon hillock region by diffusion through the seam that separates adjacent ensheathing satellite cells (Shinder and Devor, 1994). Thus, in principle, molecules of a chemical mediator released during the conditioning tetanus could diffuse into the bulk extracellular medium in the DRG and from there gain access to receptors on neighboring DRG cells.
The observed changes in spike shape during conditioning are consistent with both the [K+]o and the neurotransmitter hypotheses. The falling phase of spikes and the brief AHP reflect the opening of delayed rectifier K+ channels. Thus, the increase in spike duration and the reduction in AHP amplitude during conditioning could result from a reduction in K+ driving force because of increased [K+]o. Likewise, the changes could be attributable to a transmitter-mediated decrease in K+ conductance.
How can we chose between these two coupling options? One major distinction is the predicted effect on membrane conductance as measured by Rin. Neurotransmitters open (or close) ion channels and hence (usually) change Rin(Hille, 1992). Nernstian depolarization by elevated [K+]o, on the other hand, is not expected to yield a conductance change. As noted, we consistently observed a change in Rin, favoring the neurotransmitter hypothesis. It is true that K+-evoked depolarization may secondarily change (decrease) Rin by activating outward rectifying K+ channels. However, we found that the net effect of conditioning was to increaseRin. Only 2 cells out of 42 showed a net decrease. A second piece of evidence favoring a chemical mediator is the observation that cross-depolarization can be reversed by shifting the membrane potential far enough in the positive direction. Classically, mediators affect the opening of ion channels, but the direction of ion flow through the channels is determined by the electrochemical gradient present. Hence, flow direction is reversible if ion channels are involved.
We know of only one report in which K+ ions were seen to affect membrane conductance directly. Rudomin et al. (1979)showed in molluscan neurons that depolarization evoked by increased [K+]o can trigger a small decrease in Rin over and above that produced by the depolarization-evoked membrane rectification. The current could not be reversed, and its mechanism was not determined. Because cross-depolarization is associated with increasedRin and does reverse, we are clearly not dealing with the same phenomenon. Taken together, all of these observations argue in favor of chemically mediated coupling, although they do not rule out a [K+]o component as well. At present, we are unable to apportion the amplitude of cross-excitation between the effects of increased [K+]o and those of the putative chemical mediator(s).
Candidate chemical mediators
Our data constrain the possible range of candidate mediators of DRG cross-excitation, although identification will have to await completion of the appropriate pharmacological manipulations under voltage clamp. First, the mediator probably depolarizes the membrane and attenuates K+ current, although this is uncertain because of the unknown contribution of [K+]o. Second, it triggers increased Rin and elevated excitability. A priori, the most likely cause for this constellation of changes is closure of K+ channels (Hille, 1992). However, because the response reverses at −23 mV, well positive of the K+ battery, another ionic conductance(s) must be involved (Brown et al., 1971). The Cl− battery in DRG neurons, on the other hand, is not far from the reversal potential, and GABAA receptors, which couple to Cl− channels, are known to be expressed by DRG neurons (Deschenes et al., 1976; Gallagher et al., 1978). However, GABAA receptor activation would be expected to decrease, not increase, Rin. No other single ionic conductance meets all of the known constraints. Possible ways out of this dilemma are if more than one ion passes through a single channel (Callahan and Korn, 1994) or if the mediator affects more than one channel type. Indeed, there may be more than one mediator involved.
Several neuropeptides, including substance P, CCK, and CGRP, evoke depolarization with increased Rin when applied to mammalian myenteric ganglion cells (Nemeth et al., 1985;Palmer et al., 1986; Hanani et al., 1988), but these are found mostly in small-diameter neurons, most of which are C-neurons (Lawson, 1992). In the present study, the mediator is almost certainly associated with A-neurons, as the conditioning tetani used to evoke cross-excitation drove few, if any, C-neurons. A more likely possibility is serotonin/5HT. This has the appropriate membrane effects (Christian et al., 1989; Todorovic and Anderson, 1990) and is detectable histologically in rat DRG, albeit only with special enhancement procedures and mostly in small neurons (Lawson, 1992). Another possibility is ATP, which is abundant in rat DRG A-cells. External application of ATP normally induces depolarization with decreasedRin (Krishtal et al., 1988; Bean et al., 1990). However, Tokimasa et al. (1993, their Fig. 3) reported recently that in bullfrog DRG A-cells, ATP application blocks a K+ conductance yielding increasedRin.
Functional implications of DRG cross-excitation in health and disease
Our results indicate that, far from being isolated as classically believed, most DRG neurons participate in ongoing mutual interactions in which neuronal excitability is continuously modulated by afferent spike activity in neighboring neurons. This dialog appears to be mediated, at least in part, by an activity-dependent diffusable substance(s) released from neuronal somata and/or adjacent axons, and detected by neighboring cell somata and/or axons. In vitroin DRGs taken from nerve-intact animals, the mutual interaction is reflected in subthreshold cross-depolarization and increased firing probability. In vivo, however, particularly in the presence of nerve injury, it may occasion accelerated spontaneous firing and, in some cells, recruitment of activity in silent neurons (Devor and Wall, 1990).
Extrapolating from these observations, one can imagine how, under conditions of pathological afferent hyperexcitability, excitatory coupling among DRG neurons could enter a positive feedback mode. For example, an afferent volley from the skin might evoke cross-excitation in the DRG. The resulting increase in intrinsic DRG activity would then cross-excite still more DRG cells. In principle, reiteration of this process could bring about an excitatory “explosion,” involving a large proportion of the ganglion. The expected result is the sort of paroxysmal, electric shock-like sensation characteristic of certain neuropathic pain conditions (White and Sweet, 1969; Rappaport and Devor, 1994). If cross-excitation occurs in other primary sensory ganglia, it could contribute to corresponding functional pathologies (e.g., tinitus, syncope, etc.).
It is not obvious to us what role subthreshold DRG cross-excitation might play in the normal economy of sensory systems. Its prevalence in DRGs in nerve-intact animals, however, suggests that it might play some yet-to-be-defined role, perhaps of a metabolic nature, in communication or coordination among sensory neurons sharing a particular ganglion. For example, cross-excitation might provide each neuron in the ganglion with information about the net amount of spike activity being generated in its dermatome. Such information might be used to help regulate the synthesis and transport of transducer molecules, and hence to stabilize, over time, the sensitivity of the peripheral sensory ending.
Footnotes
This work was supported by grants from the Israel Ministry of Science and Arts, the United States-Israel Binational Science Foundation, and the German-Israeli Foundation for Research and Development. We thank J. Kocsis, P. D. Wall, and Y. Yarom for useful comments on this manuscript.
Correspondence should be addressed to Marshall Devor, Department of Cell and Animal Biology, Life Sciences Institute, Hebrew University of Jerusalem, Jerusalem 91904, Israel.
REFERENCES
- 1.Bean BP, Williams CA, Ceelen PW. ATP-activated channels in rat and bullfrog sensory neurons: current-voltage relation and single-channel behavior. J Neurosci. 1990;10:11–19. doi: 10.1523/JNEUROSCI.10-01-00011.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belmonte C, Gallego R. Membrane properties of cat sensory neurones with chemoreceptor and baroreceptor endings. J Physiol (Lond) 1983;342:603–614. doi: 10.1113/jphysiol.1983.sp014871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown JE, Muller KJ, Murray G. Reversal potential for an electrophysiological event generated by conductance changes: mathematical analysis. Science. 1971;174:318. doi: 10.1126/science.174.4006.318. [DOI] [PubMed] [Google Scholar]
- 4.Brunet PC, Jirounek P. Long-range intercellular signalling in glial cells of the peripheral nerve. NeuroReport. 1994;5:635–638. doi: 10.1097/00001756-199401000-00026. [DOI] [PubMed] [Google Scholar]
- 5.Callahan MJ, Korn SJ. Permeation of Na+ through a delayed rectifier K+ channel in chick dorsal root ganglion neurons. J Gen Physiol. 1994;104:747–772. doi: 10.1085/jgp.104.4.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christian EP, Taylor GE, Weinreich D. Serotonin increases excitability of rabbit C-fiber neurons by two distinct mechanisms. J Appl Physiol. 1989;67:584–591. doi: 10.1152/jappl.1989.67.2.584. [DOI] [PubMed] [Google Scholar]
- 7.Deschenes M, Feltz P. GABA-induced rise of extracellular potassium in rat dorsal root ganglia: an electrophysiological study in vivo. Brain Res. 1976;118:494–499. doi: 10.1016/0006-8993(76)90319-x. [DOI] [PubMed] [Google Scholar]
- 8.Deschenes M, Feltz P, Lamour Y. A model for an estimate of the ionic basis of presynaptic inhibition: an intracellular analysis of the GABA-induced depolarization in rat dorsal root ganglia. Brain Res. 1976;118:486–493. doi: 10.1016/0006-8993(76)90318-8. [DOI] [PubMed] [Google Scholar]
- 9.Devor M, Wall PD. Cross-excitation in dorsal root ganglia of nerve-injured and intact rats. J Neurophysiol. 1990;64:1733–1746. doi: 10.1152/jn.1990.64.6.1733. [DOI] [PubMed] [Google Scholar]
- 10.Devor M, Shinder V, Govrin-Lippmann R. Sympathetic sprouting in axotomized rat DRG: ultrastructure. Soc Neurosci Abstr. 1995;21:894. [Google Scholar]
- 11.Dudel J, Kuffler SW. The presynaptic inhibition at the crayfish neuromuscular junction. J Physiol (Lond) 1961;155:543–562. doi: 10.1113/jphysiol.1961.sp006646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallagher JP, Higashi H, Nishi S. Characterization and ionic basis of GABA-induced depolarizations recorded in vitro from cat primary afferent neurones. J Physiol (Lond) 1978;275:263–282. doi: 10.1113/jphysiol.1978.sp012189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorke K, Pierau FK. Spike potentials and membrane properties of dorsal root ganglion cells in pigeons. Pflügers Arch. 1980;386:21–28. doi: 10.1007/BF00584182. [DOI] [PubMed] [Google Scholar]
- 14.Hanani M, Chorev M, Gilon C, Selinger Z. The actions of receptor selective substance-P analogs on myenteric neurons: an electrophysiological investigation. Eur J Pharmacol. 1988;153:247–253. doi: 10.1016/0014-2999(88)90612-7. [DOI] [PubMed] [Google Scholar]
- 15.Harper AA, Lawson SN. Electrical properties of rat dorsal root ganglion neurones with different peripheral nerve conduction velocities. J Physiol (Lond) 1985;359:47–63. doi: 10.1113/jphysiol.1985.sp015574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hille B. Sinauer; Sunderland, MA: 1992. Ionic channels of excitable membranes. 2nd Ed. [Google Scholar]
- 17.Ito M. An analysis of potentials recorded intracellularly from the spinal ganglion cell. Jpn J Physiol. 1959;9:20–32. doi: 10.2170/jjphysiol.9.20. [DOI] [PubMed] [Google Scholar]
- 18.Karwosky CJ, Lu HK, Newman EA. Spatial buffering of light-evoked potassium increases by retinal Muller (glial) cells. Science. 1989;244:578–580. doi: 10.1126/science.2785716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kayahara T, Takimoto T, Sakashita S. Synaptic junctions in the cat spinal ganglion. Brain Res. 1981;216:277–290. doi: 10.1016/0006-8993(81)90130-x. [DOI] [PubMed] [Google Scholar]
- 20.Koerber HR, Mendell LM. Functional heterogeneity of dorsal root ganglion cells. In: Scott SA, editor. Sensory neurons. Oxford UP; New York: 1992. pp. 77–96. [Google Scholar]
- 21.Krishtal OA, Marchenko SM, Obukhov AG. Cationic channels activated by extracellular ATP in rat sensory neurons. Neuroscience. 1988;27:995–1000. doi: 10.1016/0306-4522(88)90203-5. [DOI] [PubMed] [Google Scholar]
- 22.Kuramoto H, Furness JB, Gibbins IL. Calbindin immunoreactivity in sensory and autonomic ganglia in the guinea pig. Neurosci Lett. 1990;115:68–73. doi: 10.1016/0304-3940(90)90519-f. [DOI] [PubMed] [Google Scholar]
- 23.Lawson SN. Morphological and biochemical cell types of sensory neurons. In: Scott SA, editor. Sensory neurons. Oxford UP; New York: 1992. pp. 27–59. [Google Scholar]
- 24.Lieberman AR. Sensory ganglia. In: Landon DN, editor. The peripheral nerve. Chapman and Hall; London: 1976. pp. 188–278. [Google Scholar]
- 25.Lutzky I, Aizer F, Mor N. The “Sabra” rat: definition of a laboratory animal. Isr J Med Sci. 1984;20:603–612. [PubMed] [Google Scholar]
- 26.Matsuda S, Uehara Y. Cytoarchitecture of the rat dorsal root ganglia as revealed by scanning electron microscopy. J Electron Microsc. 1981;30:136–140. [PubMed] [Google Scholar]
- 27.Maudlej N, Hanani M. Modulation of dye coupling among glial cells in the myenteric and submucosal plexuses of the guinea pig. Brain Res. 1992;578:94–98. doi: 10.1016/0006-8993(92)90234-z. [DOI] [PubMed] [Google Scholar]
- 28.Nemeth PR, Zafirov DH, Wood JD. Effects of cholecystokinin, caerulein and pentagastrin on electrical behaviour of myenteric neurons. Eur J Pharmacol. 1985;116:263. doi: 10.1016/0014-2999(85)90161-x. [DOI] [PubMed] [Google Scholar]
- 29.Palmer JM, Schemann M, Tamura K, Wood JD. Calcitonin gene-related peptide excites myenteric neurons. Eur J Pharmacol. 1986;132:163. doi: 10.1016/0014-2999(86)90601-1. [DOI] [PubMed] [Google Scholar]
- 30.Pannese E. The satellite cells of the sensory ganglia. Adv Anat Embryol Cell Biol. 1981;47:1–97. doi: 10.1007/978-3-642-67750-2. [DOI] [PubMed] [Google Scholar]
- 31.Pannese E, Ledda M, Arcidiano G, Rigamonti I. Clusters of nerve cell bodies enclosed within a common connective tissue envelope in the spinal ganglia of the lizard and rat. Cell Tissue Res. 1991;264:209–214. doi: 10.1007/BF00313957. [DOI] [PubMed] [Google Scholar]
- 32.Rappaport ZH, Devor M. Trigeminal neuralgia: the role of self-sustaining discharge in the trigeminal ganglion (TRG). Pain. 1994;56:127–138. doi: 10.1016/0304-3959(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 33.Rudomin P, Stefani E, Werman R. Voltage sensitivity of small, focal transient potassium depolarizations in snail neurons: relevance for diagnosis of chemical synaptic activity. J Physiol (Lond) 1979;42:912–924. doi: 10.1152/jn.1979.42.3.912. [DOI] [PubMed] [Google Scholar]
- 34.Shinder V, Devor M. Structural basis of neuron-to-neuron cross-excitation in dorsal root ganglia. J Neurocytol. 1994;23:515–531. doi: 10.1007/BF01262054. [DOI] [PubMed] [Google Scholar]
- 35.Todorovic S, Anderson EG. 5-HT2and 5-HT3 receptors mediate two distinct depolarizing responses in rat dorsal root ganglion neurons. Brain Res. 1990;511:71–79. doi: 10.1016/0006-8993(90)90226-2. [DOI] [PubMed] [Google Scholar]
- 36.Tokimasa T, Tsurusaki M, Akasu T. Chemosensitivity of C-cells in bullfrog dorsal root ganglia to substance P and adenosine 5′-triphosphate. Neurosci Lett. 1993;163:169–172. doi: 10.1016/0304-3940(93)90374-t. [DOI] [PubMed] [Google Scholar]
- 37.Utzschneider D, Kocsis J, Devor M. Mutual excitation among dorsal root ganglion neurons in the rat. Neurosci Lett. 1992;146:53–56. doi: 10.1016/0304-3940(92)90170-c. [DOI] [PubMed] [Google Scholar]
- 38.White JC, Sweet WH (1969) Pain and the neurosurgeon. Springfield, MA: Thomas.