

Abstract
We conducted single cell metabolomics studies of live cancer cells through online single cell mass spectrometry (SCMS) experiments combined with a generalized comprehensive data analysis workflow. The SCMS experiments were carried out using the Single-probe device coupled with a mass spectrometer to measure molecular profiles of cells in response to two mitotic inhibitors, taxol and vinblastine, under a series of treatment conditions. SCMS metabolomic data were analyzed using a comprehensive approach, including data pre-treatment, visualization, statistical analysis, machine learning, and pathway enrichment analysis. For comparative studies, traditional liquid chromatography-MS (LC-MS) experiments were conducted using lysates prepared from bulk cell samples. Metabolomic profiles of single cells were visualized through Partial Least Square-Discriminant Analysis (PLS-DA), and the phenotypic biomarkers associated with emerging phenotypes induced by drug treatment were discovered and compared through a series of rigorous statistical analysis. Species of interest were further identified at both the single cell and population levels. In addition, four biological pathways potentially involved in the drug treatment were determined through pathway enrichment analysis. Our work demonstrated the capability of comprehensive pipeline studies of single cell metabolomics. This method can be potentially applied to broader types of SCMS datasets for future pharmaceutical and chemotherapeutic research.
Keywords: Single cell metabolomics, Mass spectrometry, Single-probe, Phenotype, Biomarker
Graphical Abstract
1. Introduction
Cell, as a fundamental component of living organisms, regulates cellular metabolic activities through a variety of biological pathways.[1, 2] In recent years, a tremendous number of metabolites that participate in rapid and subtle biological and physiological activities[3, 4] were intensively investigated to gain a profound perspective towards the dynamic nature of the cell.[5, 6] Metabolomics, serving as a bridge between cellular metabolism and phenotypes,[7, 8] becomes an increasingly intriguing research field where modern instrumentation and methodologies are involved.[9, 10] Among all techniques for metabolomics studies, mass spectrometry (MS) based approaches possess considerable advantages over others for providing a large amount of molecular information from complex samples. MS methods are widely used in metabolomic studies due to their high sensitivity to detect low-abundance cellular metabolites,[11] high mass resolution to resolve isobaric species,[12] flexible capabilities to be coupled to versatile chromatographic separations to enhance metabolite coverage,[13, 14] and wide selections of orthogonal yet compatible analytical methods to discriminant isomers.[15, 16] To date, the majority of current MS based cell metabolomic studies are carried out by analyzing cell lysates prepared from a large cohort of cells, and consequently, leading to an accumulative result of populations analyzed.[17] However, each cell is an individually functional unit that is encoded with heterogeneous genomic information, and presents diverse biological status in different microenvironment.[18] Single cell MS (SCMS), as an emerging field of study, appreciates such cell-to-cell heterogeneity masked by conventional liquid chromatography-mass spectrometry (LC-MS) methods through interrogating cellular contents of individual cells. A number of SCMS techniques have been dedicatedly developed, and they are roughly classified as ion-beam based,[19] laser based,[20–23] probe based,[24–28] and other techniques.[29–32] Typically, they have been employed for distinguishing cellular fingerprints, identifying intracellular metabolites, and discovering new biological mechanisms through single cell metabolomic analysis (i.e., single cell metabolomics).[33, 34] However, to the best of our knowledge, the majority of reported single cell metabolomic studies rely on the non-specialized software, which is either vendor-specific (MassLynx,[35, 36] Compass Data Analysis,[29, 37] etc.) or derived from conventional LC-MS analysis (e.g., Decon2LS),[38] to process the datasets. Therefore, further efforts are needed to establish the standardized data analysis procedure for the single cell metabolomic analysis of data obtained from broader types of MS instruments. On the other hand, cells are sensitive to their surrounding microenvironment, and cellular metabolites have rapid turnover rate upon subtle changes,[39] which adds another layer of complexity to single cell metabolomics in native status. Facing those challenges, it is imperative to develop a comprehensive single cell metabolomics approach consisting of SCMS experiments and a generalized pipeline for SCMS metabolomic data analysis. Ultimately, a fully developed single cell metabolomics method can be used to capture metabolomic signatures of individual cells, identify metabolic phenotypes, and disclose underlying biological principles of live single cells.
In our single cell metabolomics approach, we used a miniaturized multifunctional sampling device, the Single-probe,[27, 40–45] coupled to MS to analyze live single cells in ambient conditions, followed by multivariate and univariate data analysis. We selected human cervical cancer cell line, HeLa, as our model system, to demonstrate changes of metabolomic profile of each cell upon exposure to external stimuli (i.e., anticancer drugs). Specifically, two types of mitotic inhibitors, paclitaxel (taxol) and vinblastine, were selected for a series of time- and concentration-dependent treatments. Both taxol and vinblastine inhibit cell mitotic process in G2/M phase by either stabilizing (taxol) or destabilizing (vinblastine) microtubules, and ultimately induce cell apoptosis.[46, 47] Although both drug compounds share similarities such as the binding target (microtubules) and IC50 values,[48] their influence on the cellular metabolism needs to be further understood at the single cell level.
Similar to the untargeted LC-MS metabolomics data handling procedures, our SCMS metabolomic data processing aims to discriminate metabolic phenotypes, discover phenotypic biomarkers (i.e., characteristic species closely related to specific phenotypes), and unveil related biological pathways. However, due to the nature of cell heterogeneity, each cell may have a different response to drug treatment resulting in varied metabolomic profiles. Therefore, it is impractical to directly apply the conventional metabolomic data analysis procedure to SCMS datasets, as the underlying assumption (i.e., homogeneity of variance) of a variety of statistical tests are challenged. Here, we developed a comprehensive approach to SCMS metabolomics studies by performing data pre-treatment, visualization, statistical analysis, machine learning, and pathway enrichment analysis (Figure 1).
Figure 1.
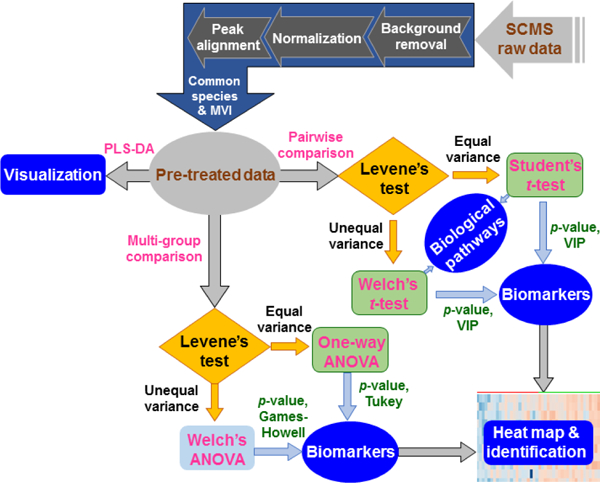
Workflow of SCMS data analysis consisting of data pre-treatment, multivariate analysis, and univariate analysis. This generalized procedure can be coupled to raw datasets obtained from broader types of SCMS platforms for single cell metabolomic analysis.
2. Experimental
2.1. SCMS data acquisition
Detailed fabrication procedure and working mechanisms of the Single-probe device are provided in our previous publications [27, 40] and briefly summarized in the Supporting Information (see “Fabrication and Working Mechanisms of the Single-probe” in the Supporting Information). To conduct the SCMS experiment, the Single-probe device is coupled to a Thermo LTQ Orbitrap XL mass spectrometer. The tip (size < 10 µm) of the Single-probe is inserted into a target cell to extract cellular contents through a liquid junction at the probe tip, and then the extracted mixture is driven towards the nano-ESI emitter for immediate ionization and MS detection (Figure 2B). Cell selection and penetration are precisely controlled by our in-house built XYZ-translational stage system (Figure 2A), and these processes are visualized using a stereo microscope (Figure 2C). The experimental MS parameters are listed as follows: ionization voltage +4.5 kV, mass range 150–1500 m/z (mass-to-charge ratio), mass resolution 60,000 at m/z 400, 1 microscan, and 100 ms max injection time and automatic gain control (AGC) on.
Figure 2.

Experimental setup of the Single-probe SCMS system. (A) Key components of the in-house developed Single-probe SCMS platform. (B) A zoomed-in photo of the Single-probe and illustration of its working mechanism. (C) The insertion of the Single-probe tip into a single cell monitored using a high-resolution digital stereo microscope during a SCMS experiment.
To study changes of metabolomic profiles of cancer cells induced by taxol and vinblastine, we cultured HeLa cells under normal condition (control), and treated them using a series of drug treatment conditions (Table 1). Individual cells in both control and treatments groups were randomly selected for analysis using the Single-probe SCMS technique (see “Sample Preparation” and “Single-probe SCMS” in the Supporting Information). We carefully designed our treatment conditions allowing for sufficient cellular metabolomic changes to be detected, while minimizing other factors (environmental perturbations, mutations, etc.) that could potentially interfere with phenotypic identification. In our experiments, 22–28 cells were sampled from the control group and each of those drug treatment groups.
Table 1.
HeLa cells in the control and treatment groups for SCMS experiments.
| Condition | Drug | Concentration (µM) | Time (h) | Group Name | Number of Cells |
|---|---|---|---|---|---|
| Control | N/A | N/A | N/A | Control | 23 |
|
Treatment |
Taxol | 0.1 | 2 | TaxA | 25 |
| 0.1 | 6 | TaxB | 28 | ||
| 1.0 | 2 | TaxC | 22 | ||
| Vinblastine | 0.1 | 2 | VinA | 28 | |
| 0.1 | 6 | VinB | 23 | ||
| 1.0 | 2 | VinC | 24 | ||
2.2. SCMS data pre-treatment
Following online data acquisition, we performed a generalized comprehensive SCMS metabolomics data analysis, including multivariate and univariate analysis, to gain biological insights into raw data matrices (Figure 1). Particularly, to preserve metabolomic information of endogenous species from single cells while avoiding interference with other species (exogenous species from the sampling environment, detection noise, etc.), we conducted data pre-treatment that can be generally divided into three consecutive steps.
2.2.1. Generation of metabolomic peak list
The acquired raw data files (.raw) from our SCMS experiments were accessed using Xcalibur 3.0 (Thermo Fisher Scientific). A common cellular species with relatively high ion intensity, PC (34:1),[27] was selected as an indicator of successful MS detection of cellular contents from individual cells (Figure S1). We exported an averaged MS spectrum from each cell containing all detected peaks (i.e., m/z values) along with their corresponding ion intensities as the metabolic peak list. Similar lists of metabolites can be generated from all other major types of MS platforms, including quadrupole time-of-flight (qTOF), fourier transform-ion cyclotron resonance (FT-ICR), and Orbitraps, using vendor-specific software.
2.2.2. MS background removal
Under our experimental conditions, a raw file typically consists of more than 6,000 distinct peaks, which can be attributed to endogenous species (i.e., cellular metabolites), exogeneous species (i.e., from surrounding matrix such as cell culture medium and sampling solvent), and instrument noise. Based on the data obtained from 10 randomly selected cells, we estimated the total ion current (TIC) of exogenous species and noise are ~11 fold higher than that of endogenous species (Figure S2). This result could be attributed to both the extremely limited amount of analytes within a single cell (in picoliter range)[39, 49] and the reduced detection sensitivity due to the matrix effect.[50] Because only the relative abundances of cellular metabolites from single cells were used in the downstream analysis, we excluded interfering ion as described below. First, we removed ion signals of exogenous species (i.e., background ion signals), which were detected from cell culture medium and the sampling solvent used in SCMS experiments. Second, we filtered out instrument noise, which may result in false positive discovery and unnecessary computational burden to data analysis. Instrument noise accounts for ~20–40% of the total number of peaks detected in SCMS experiments, and it was removed by eliminating ions with evidently lower ion intensities (< 103). Removing background and noise greatly reduced the dimensionality of SCMS data matrices and preserved the molecular information of endogenous cellular metabolites. Lastly, we normalized the ion intensity of each metabolite to TIC prior to the following data processing steps. It is worth noting that our background removal method is similar to those used in prevalent LC-MS metabolomics data analysis software (e.g., MZmine 2). However, discriminating instrument noise from low-abundance MS peaks of cellular metabolites is challenging. Other advanced noise removal algorithms, such as repetition rate filtering (RRF) that has been demonstrated effective in shotgun lipidomics,[51] can be incorporated in future studies.
2.2.3. Peak alignment and common species determination
The SCMS datasets obtained from the previous step were submitted to Geena 2[52] for MS peak alignment. We then utilized MetaboAnalyst[53, 54] to determine the common species, which are defined as cellular species that can be frequently detected from measured cells in each group. Here, we referred to the standard 80% rule (i.e., excluding species with > 20% missing values from all measured cells), a broadly accepted rule for feature selection in untargeted LC-MS metabolomics research,[55] as the criterion to determine common species. In addition, a missing value imputation (MVI) algorithm, K-nearest neighbor (KNN),[56] was employed to eliminate missing values and reduce false positive results in our analysis. Using the above data pre-treatment procedures, we promptly reduced the size of our datasets while retaining the essential metabolomic information from individual cells. However, this 80% rule may eliminate rare cells, which can be critical for a variety of biological mechanisms present in a large population of cells.[57, 58] To include more measured cells for SCMS data analysis, a lenient missing value threshold can be employed. However, applying a loose missing value threshold may reduce the statistical power, introduce bias, and increase computing demand.[59]
2.3. SCMS data visualization.
To evaluate the differences of metabolomic profiles of single cells among all groups, we conducted the dimensionality reduction of pre-treated SCMS datasets, which facilitates the visualization of high-dimensional data matrices in a low-dimensional space through multivariate analysis. Here, we employed Partial Least Square-Discriminant Analysis (PLS-DA), a supervised method, to achieve phenotypic separation when the within-group variation (i.e., variation of cellular metabolite abundance within the control and each of the treatment groups) is pronounced.[60] To evaluate the quality of PLS-DA models and avoid data overfitting, the explained variation (R2) and the predictive ability (Q2) were calculated through a 10-fold cross validation procedure.[61] PLS-DA models with Q2 > 0.5 were considered to be robust,[62] and they were further analyzed using permutation tests[63] to identify significantly separated phenotypes. We performed 2,000 permutation tests for each model, and a small statistic p-value (< 0.05) indicated a significant phenotypic discrimination.
2.4. Discovery of phenotypic biomarkers
To study changes of metabolomic profiles of single cells induced by microenvironmental alternation (i.e., drug treatment) and discover phenotypic biomarkers, we utilized statistical methods to process SCMS metabolomic datasets after data pre-treatment.
2.4.1. Biomarkers from pairwise group comparison
To discover phenotypic biomarkers corresponding to a particular treatment condition, we utilized the pre-treated SCMS datasets from the control group and that treatment group for the PLS-DA. We then calculated Variable Importance in Projection (VIP) scores for all cellular species, and selected those with VIP scores >1.2, representing major contributions to group discrimination,[64] as biomarker candidates. These biomarker candidates were subsequently subjected to unpaired two-sample t-test for the comparison of abundances. Due to cell heterogeneity, cellular response to the drug treatment is different. Therefore, Levene’s test was conducted prior to t-test to evaluate the homogeneity of variance of each metabolite, allowing us to determine which type of t-test to be performed. Depending on the results from Levene’s test, we performed Student’s t-test (data with equal within-group variance) or Welch’s t-test (data with unequal within-group variance). Cellular species with both VIP scores > 1.2 and p-value (from t-test) < 0.05 were marked as potential biomarkers related to the examined phenotypes. More technical details can be found in “Terminology” section in the Supporting Information.
2.4.2. Biomarkers from multi-group comparison
To obtain common biomarkers reflecting the influence of drug molecules on cellular metabolism, we compared the ion abundance of detected species in the control and all treatment groups for each drug compound. First, similar to the pairwise group comparison in the previous step, we conducted Levene’s test to evaluate the homogeneity of variance for each metabolite among multiple examined groups. Second, to determine if there are statistically significant differences of metabolites among all groups, we used one-way (data with equal within-group variance) or Welch’s (data with unequal within-group variance) Analysis of Variance (ANOVA). Third, to rigorously select biomarkers (i.e., metabolites with significant abundance change), we then performed two types of post hoc tests for metabolites with p-value < 0.05 (from ANOVA): Tukey’s HSD (Honestly Significant Difference) and Games-Howell tests for one-way ANOVA and Welch’s ANOVA tests, respectively. Cellular species with p-values < 0.05 (from both the ANOVA and the corresponding post hoc tests) among all examined groups were highlighted as biomarkers. The above procedures have been previously used in LC-MS metabolomics studies to discover biomarkers corresponding to dyslipidemia progression.[65] All above statistical analyses were performed in R environment with functions available in Metabox, a toolbox for metabolomic studies.[66] More technical details can be found in “Terminology” and “Source Code Availability” in the Supporting Information.
2.4.3. Tentative assignment and identification of biomarkers
To identify discovered biomarkers, their accurate m/z values were compared with those registered at online metabolome database, METLIN[67] and HMDB[68]. Moreover, we performed online MS/MS analysis of biomarkers with relatively higher abundance at the single cell level, whereas conventional LC-MS/MS experiments were also carried out as a complimentary approach to molecular identification at the population level (see “Complimentary LC-MS Analysis and LC-MS/MS Identification” in the Supporting Information).
2.5. Potential biological pathways
Mummichog, a program for data analysis in untargeted metabolomics studies,[69] was used in the current work to address potential biological pathways involved in drug treatment at the single cell level. Unlike many other available programs, Mummichog only utilizes the information of accurate m/z values, rather than identified metabolites, to perform pathway enrichment analysis.[70] Required inputs, such as m/z values, t-test p-values, and fold change of all cellular species, were fulfilled based on results from the data analysis as described in the previous steps, and Mummichog was operated using default settings.
3. Results and discussion
3.1. Metabolic response to drug treatment
To study cellular metabolic response and visualize phenotypic separation induced by drug treatment, we constructed PLS-DA models for SCMS datasets collected from the control and each of those drug treatment groups. As shown in Figure 3, a data point represents the metabolomic profile of a single cell, and the cell-to-cell heterogeneity can be reflected by the distribution of data points within a group.[71, 72] The phenotypic separation can be visualized by the distance of data points between two groups. Generally, the first PLS-DA component explains more than 25% of variance (i.e., Component 1 > 25%) in all score plots, and significant phenotypic discrimination (p < 0.0035) between two groups is further demonstrated through permutation tests (Figure S3). However, the overlapped regions can still be observed between the control and a “shorter” treatment time condition (i.e., TaxA, VinA, TaxC, or VinC) for both drugs. In contrast, complete group separation (no overlapped region) can be observed between the control and a “longer” treatment time condition (TaxB or VinB). This trend is also visually reflected on PLS-DA score plots containing multiple groups (Figure S4), in which a complete phenotypic separation is only observed between the control and “longer” treatment time condition. From a biological perspective, though cellular xenobiotic activity was reported to be both time- and concentration-dependent,[73] our SCMS results demonstrate that treatment time has a more significant influence on cells’ metabolomic profiles, at least at early treatment stage (e.g., treatment time < 6 h). To validate our SCMS results, we prepared lysates using cells, which were treated under the same conditions as those in the SCMS experiments, for LC-MS analysis (see “Complimentary LC-MS Analysis and LC-MS/MS Identification” in the Supporting Information). We further conducted principal component analysis (PCA) of the LC-MS results (Figure S5), and obtained similar trends observed in the SCMS studies: longer treatment time resulted in more evident changes of cellular metabolomic profiles. In addition, we compared the number of metabolites detected using the LC-MS and SCMS approaches. As shown in the Venn diagram (Figure S6), 230 cellular metabolites can only be detected in the SCMS datasets; these metabolites are likely to have rapid turnover rates, and therefore could be potentially lost during the lengthy LC-MS sample preparation process. On the other hand, due to significantly larger amounts of cellular species contained in the cell lysate and chromatographic separation (i.e., minimized matrix effect) in the LC-MS measurement, more metabolites were detected in the LCMS (1612) than SCMS measurements (340). Thus, traditional LC-MS measurements can provide complementary information to our novel SCMS studies.
Figure 3.
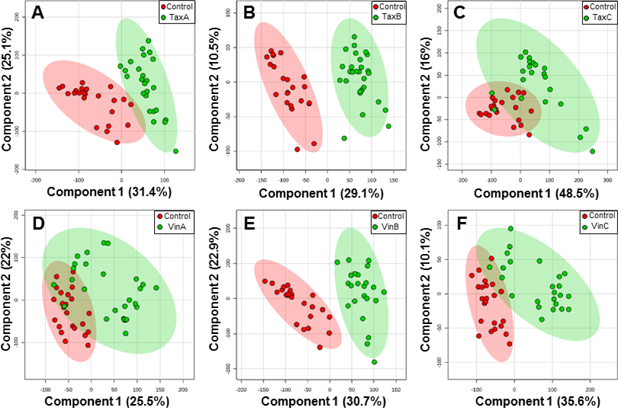
PLS-DA score plots in 2D space for phenotypic discrimination between the control and a drug treatment group, including (A) TaxA, (B) TaxB, (C) TaxC, (D) VinA, (E) VinB, and (F) VinC. Each data point represents the metabolomic profile of an individual cell, and the ellipse highlights the 95% confidence region.
Despite significant phenotypic separation demonstrated by permutation tests (Figure S3) for each PLD-DA analysis, certain types of uncertainties are regarded as “noise”,[74] including cell heterogeneity and technical variation (e.g., the sampling process, ionization stability, and instrument condition in SCMS experiments), and they may interfere with phenotypic separation in the SCMS data. To evaluate the influence of such “noise” on our data analysis, we employed a well-established machine learning algorithm in metabolomics studies, random forest,[75] to perform phenotypic classification. Each pair of pre-treated SCMS datasets were subjected to the classification, and results are summarized in the confusion matrices (Tables S1–S6) and pie plots (Figure 4). Among them, cells in the control and a “shorter” time treatment condition yielded a low misclassification rate (6–11%, Figures 4A, 4C–4D, and 4F), implying a minor interference of such “noise” on the phenotypic separation. More interestingly, the misclassification rate is even lower (0 and 2%, Figures 4B and4E) in the pair of datasets from the control and a “longer” treatment time condition, agreeing with the complete separation observed in the PLS-DA score plots (Figure 3). To our best knowledge, this is the first report of employing random forest as an alternative approach to evaluate the influence of the “noise”[74] on phenotypic separation in single cell metabolomics studies.
Figure 4.
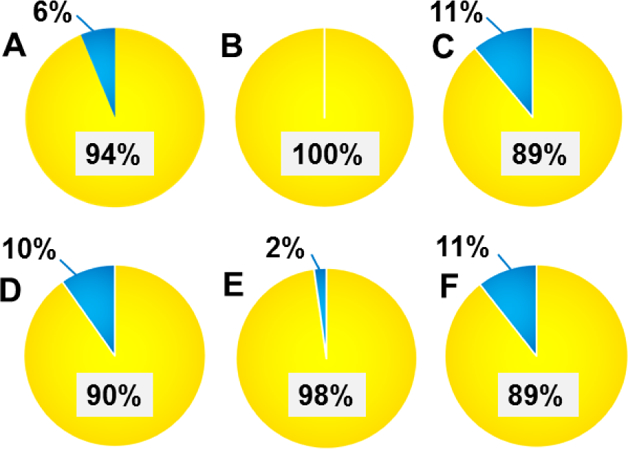
Cellular species correctly classified (yellow) and misclassified (blue) by random forest classification between the control and a drug treatment group, including (A) TaxA, (B) TaxB, (C) TaxC, (D) VinA, (E) VinB, and (F) VinC, determined from confusion matrix (Table S1–S6).
3.2. Study of phenotypic biomarkers
As previously reported, the magnitude of the abundance change of cellular metabolites represents the degree of difference between phenotypes.[76] Therefore, metabolites with significant abundances change after drug treatment are suitable candidates for phenotypic biomarkers, and they may arise biological interest and further suggest related cellular xenobiotic activities. Through our biomarker selection criteria as described above, we discovered a variety of phenotypic biomarkers corresponding to multiple treatment conditions (Tables S7–S15 in the Supporting Information), followed by tentative assignment of those biomarkers based on accurate mass. To further confirm the chemical identities of tentatively labeled biomarkers, we performed MS/MS analysis at the single cell level for those with relatively higher ion abundances. Six phenotypic biomarkers, i.e., [PC(16:0) + Na]+ (m/z 518.3194), [PC(18:0) + Na]+ (m/z 546.3506), [SM(34:1) + Na]+ (m/z 725.5539), [PC(32:1) + Na]+ (m/z 754.5345), [PC(34:1) + Na]+ (m/z 782.5660), and [PC(36:2) + Na]+ (m/z 808.5813), were identified from single cells (Figure S7). As a complimentary approach to enhance the biomarker identification, LC-MS/MS was also utilized to analyze cell lysates. In addition to the above six identified biomarkers, three more identifications, i.e., [PC(34:1) + H]+ (m/z 760.5860), [PC(34:2) + Na]+ (m/z 780.5561), and [PC(36:3) + Na]+ (m/z 806.5643), were obtained (Figure S8). The majority of the identified biomarkers are phospholipids, which are related to the regulation of cell signal transduction in response to external stimuli.[77] Heat maps were constructed to intuitively visualize the relative abundances of the discovered biomarkers of cells in each treatment group (Figures S9 and S10). The overall color clusters (i.e., red and blue) matched well with cell attributes, although slight color variations can be observed for each biomarker among multiple cells likely due to cell heterogeneity.
3.3. Potential biological pathways
As biomarkers are tightly related to biological pathways regulating cellular metabolism,[78] we used the biomarkers discovered from our SCMS studies to unveil potential biological pathways related to metabolomic response to the drug treatment (Figure S11). We found two pathways, biopterin metabolism (p-value = 0.025) and glycerophospholipid metabolism (p-value = 0.041), were significantly enriched by taxol treatment. Other two pathways, bile acid biosynthesis (p-value = 0.021) and de novo fatty acid biosynthesis (p-value = 0.043), were significantly enriched by vinblastine treatment. The altered biopterin metabolism may be attributed to enzymatic activities related to guanosine triphosphate (GTP) cyclohydrolase I, which regulates biopterin metabolism[79] and is sensitive to drug treatment.[80] Glycerophospholipid metabolism involves a variety of phospholipids, which are responsible for cellular signal transduction[77] sensitive to surrounding microenvironment.[81] Bile acids are cell signaling molecules that are closely related to the regulation of energy and metabolic homeostasis,[82] and our SCMS results achieved good agreement with previous publications reporting a suppressed bile acid metabolism following vinblastine treatment.[83] De novo fatty acid biosynthesis has been reported to be suppressed upon drug treatment.[84] Our results, from the perspective of single cell metabolomics, suggest that those biological pathways may be significantly influenced by the treatment of mitotic inhibitors. It is worth noting that MS experimental conditions, such as the solvent composition[85], ionization polarity[86], and instrument type and tuning, can affect the detection sensitivity of different classes of species. Because our SCMS measurements were conducted under the same condition (i.e., using acidified acetonitrile sampling solvent, positive ion mode, and only one model of mass spectrometer), metabolites detected in the current study are likely within a limited coverage range. Therefore, more comprehensive experimental conditions can be used in future studies for broader coverage of metabolites, and ultimately enhance the statistical power in pathway enrichment analysis.
4. Conclusions
We performed live single cell metabolomics studies using the Single-probe SCMS experiments in combination with a generalized comprehensive data analysis procedure. Cellular response to two mitotic inhibitors, taxol and vinblastine, were investigated and compared under multiple treatment conditions. Through the visualization using PLS-DA and the following permutation tests, our SCMS metabolomics results showed a rapid emergence of new phenotypes upon drug treatment. Similar trends were observed from traditional LC-MS experiments utilizing lysates prepared from population cells treated under the same conditions. Phenotypic biomarkers corresponding to two or multiple treatment conditions were discovered through statistical tests, with some of those further identified at both single cell and population levels. Based on the information of discovered biomarkers, potential biological pathways related to drug treatment were unveiled using the pathway enrichment analysis. Our methodology holds a promising potential to be readily coupled to other SCMS datasets produced from broader types of MS based analytical approaches to implement metabolomics at the single cell level, and ultimately gain insights into biological principles that regulate cellular metabolism.
Supplementary Material
Highlights.
Live single cell metabolomics was performed using MS and comprehensive data analysis.
Cells’ metabolomic response to anticancer drugs was investigated.
Phenotypic biomarkers reflecting drug treatment were discovered and identified.
Biological pathways related to drug treatment were revealed at the single cell level.
Acknowledgement
We acknowledge Drs. Steven B. Foster, Vincent Bonifay, Anthony A.W.G. Burgett, Yihan Shao, and Laura Isobel-McCall (University of Oklahoma) for their generous help on experimental design and SCMS data processing. We thank Dr. Paolo Romano (Ospedale Policlinico San Martino) and Dr. Shuzhao Li (Emory University, School of Medicine) for their valuable suggestions on data analysis. This research project is partially supported by grants from National Institutes of Health (R01GM116116 and R21CA204706) and National Science Foundation Major Research Instrumentation Program (NSF-MRI 1626372).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interests
The authors declare no conflicts of interests.
Declaration of interests
☒ The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Reference
- [1].McKnight SL, On Getting There from Here, Science, 330 (2010) 1338. [DOI] [PubMed] [Google Scholar]
- [2].Metallo CM, Heiden MGV, Understanding metabolic regulation and its influence on cell physiology, Mol Cell, 49 (2013) 388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Stopka SA, Mansour TR, Shrestha B, Maréchal É, Falconet D, Vertes A, Turnover rates in microorganisms by laser ablation electrospray ionization mass spectrometry and pulse-chase analysis, Anal Chim Acta, 902 (2016) 1–7. [DOI] [PubMed] [Google Scholar]
- [4].Knolhoff AM, Nautiyal KM, Nemes P, Kalachikov S, Morozova I, Silver R, Sweedler JV, Combining Small-Volume Metabolomic and Transcriptomic Approaches for Assessing Brain Chemistry, Anal Chem, 85 (2013) 3136–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chen WW, Freinkman E, Wang T, Birsoy K, Sabatini DM, Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism, Cell, 166 (2016) 1324–1337.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].You M, Litke JL, Jaffrey SR, Imaging metabolite dynamics in living cells using a Spinach-based riboswitch, Proc Natl Acad Sci U S A, 112 (2015) E2756–E2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Newgard CB, Metabolomics and Metabolic Diseases: Where Do We Stand?, Cell Metab, 25 (2017) 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Patti GJ, Yanes O, Siuzdak G, Innovation: Metabolomics: the apogee of the omics trilogy, Nat Rev Mol Cell Biol, 13 (2012) 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang A, Sun H, Wang P, Han Y, Wang X, Modern analytical techniques in metabolomics analysis, Analyst, 137 (2012) 293–300. [DOI] [PubMed] [Google Scholar]
- [10].Nicholson JK, Lindon JC, Systems biology: Metabonomics, Nature, 455 (2008) 1054–1056. [DOI] [PubMed] [Google Scholar]
- [11].Si X, Xiong X, Zhang S, Fang X, Zhang X, Detecting Low-Abundance Molecules at Single-Cell Level by Repeated Ion Accumulation in Ion Trap Mass Spectrometer, Anal Chem, 89 (2017) 2275–2281. [DOI] [PubMed] [Google Scholar]
- [12].Ghaste M, Mistrik R, Shulaev V, Applications of Fourier Transform Ion Cyclotron Resonance (FT-ICR) and Orbitrap Based High Resolution Mass Spectrometry in Metabolomics and Lipidomics, Int J Mol Sci, 17 (2016) 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Madji Hounoum B, Blasco H, Nadal-Desbarats L, Diémé B, Montigny F, Andres CR, Emond P, Mavel S, Analytical methodology for metabolomics study of adherent mammalian cells using NMR, GC-MS and LC-HRMS, Anal Bioanal Chem, 407 (2015) 8861–8872. [DOI] [PubMed] [Google Scholar]
- [14].Cuykx M, Negreira N, Beirnaert C, Van den Eede N, Rodrigues R, Vanhaecke T, Laukens K, Covaci A, Tailored liquid chromatography–mass spectrometry analysis improves the coverage of the intracellular metabolome of HepaRG cells, J Chromatogr A, 1487 (2017) 168–178. [DOI] [PubMed] [Google Scholar]
- [15].Zhang W, Hankemeier T, Ramautar R, Next-generation capillary electrophoresis–mass spectrometry approaches in metabolomics, Curr Opin Biotech, 43 (2017) 1–7. [DOI] [PubMed] [Google Scholar]
- [16].Mairinger T, Causon TJ, Hann S, The potential of ion mobility–mass spectrometry for non-targeted metabolomics, Curr Opin Chem Biol, 42 (2018) 9–15. [DOI] [PubMed] [Google Scholar]
- [17].Halama A, Metabolomics in cell culture—A strategy to study crucial metabolic pathways in cancer development and the response to treatment, Arch Biochem Biophys, 564 (2014) 100–109. [DOI] [PubMed] [Google Scholar]
- [18].Walker BN, Antonakos C, Retterer ST, Vertes A, Metabolic Differences in Microbial Cell Populations Revealed by Nanophotonic Ionization, Angew Chem Int Ed, 52 (2013) 3650–3653. [DOI] [PubMed] [Google Scholar]
- [19].Waki M, Ide Y, Ishizaki I, Nagata Y, Masaki N, Sugiyama E, Kurabe N, Nicolaescu D, Yamazaki F, Hayasaka T, Ikegami K, Kondo T, Shibata K, Hiraide T, Taki Y, Ogura H, Shiiya N, Sanada N, Setou M, Single-cell time-of-flight secondary ion mass spectrometry reveals that human breast cancer stem cells have significantly lower content of palmitoleic acid compared to their counterpart non-stem cancer cells, Biochimie, 107 (2014) 73–77. [DOI] [PubMed] [Google Scholar]
- [20].Xie WY, Gao D, Jin F, Jiang YY, Liu HX, Study of Phospholipids in Single Cells Using an Integrated Microfluidic Device Combined with Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry, Anal Chem, 87 (2015) 7052–7059. [DOI] [PubMed] [Google Scholar]
- [21].Schober Y, Guenther S, Spengler B, Römpp A, Single Cell Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry Imaging, Anal Chem, 84 (2012) 6293–6297. [DOI] [PubMed] [Google Scholar]
- [22].Shrestha B, Vertes A, In Situ Metabolic Profiling of Single Cells by Laser Ablation Electrospray Ionization Mass Spectrometry, Anal Chem, 81 (2009) 8265–8271. [DOI] [PubMed] [Google Scholar]
- [23].Ibáñez AJ, Fagerer SR, Schmidt AM, Urban PL, Jefimovs K, Geiger P, Dechant R, Heinemann M, Zenobi R, Mass spectrometry-based metabolomics of single yeast cells, Proc Natl Acad Sci U S A, 110 (2013) 8790–8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mizuno H, Tsuyama N, Harada T, Masujima T, Live single-cell video-mass spectrometry for cellular and subcellular molecular detection and cell classification, J Mass Spectrom, 43 (2008) 1692–1700. [DOI] [PubMed] [Google Scholar]
- [25].Gong X, Zhao Y, Cai S, Fu S, Yang C, Zhang S, Zhang X, Single Cell Analysis with Probe ESI-Mass Spectrometry: Detection of Metabolites at Cellular and Subcellular Levels, Anal Chem, 86 (2014) 3809–3816. [DOI] [PubMed] [Google Scholar]
- [26].Zhu HY, Zou GC, Wang N, Zhuang MH, Xiong W, Huang GM, Single-neuron identification of chemical constituents, physiological changes, and metabolism using mass spectrometry, Proc Natl Acad Sci U S A, 114 (2017) 2586–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pan N, Rao W, Kothapalli NR, Liu RM, Burgett AWG, Yang ZB, The Single-Probe: A Miniaturized Multifunctional Device for Single Cell Mass Spectrometry Analysis, Anal Chem, 86 (2014) 9376–9380. [DOI] [PubMed] [Google Scholar]
- [28].Liu R, Pan N, Zhu Y, Yang Z, T-Probe: An Integrated Microscale Device for Online In Situ Single Cell Analysis and Metabolic Profiling Using Mass Spectrometry, Anal Chem, 90 (2018) 11078–11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Onjiko RM, Portero EP, Moody SA, Nemes P, In Situ Microprobe Single-Cell Capillary Electrophoresis Mass Spectrometry: Metabolic Reorganization in Single Differentiating Cells in the Live Vertebrate (Xenopus laevis) Embryo, Anal Chem, 89 (2017) 7069–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhang LW, Foreman DP, Grant PA, Shrestha B, Moody SA, Villiers F, Kwake JM, Vertes A, In Situ metabolic analysis of single plant cells by capillary microsampling and electrospray ionization mass spectrometry with ion mobility separation, Analyst, 139 (2014) 5079–5085. [DOI] [PubMed] [Google Scholar]
- [31].Comi TJ, Makurath MA, Philip MC, Rubakhin SS, Sweedler JV, MALDI MS Guided Liquid Microjunction Extraction for Capillary Electrophoresis–Electrospray Ionization MS Analysis of Single Pancreatic Islet Cells, Anal Chem, 89 (2017) 7765–7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Herrmann AJ, Techritz S, Jakubowski N, Haase A, Luch A, Panne U, Mueller L, A simple metal staining procedure for identification and visualization of single cells by LA-ICP-MS, Analyst, 142 (2017) 1703–1710. [DOI] [PubMed] [Google Scholar]
- [33].Fujii T, Matsuda S, Tejedor ML, Esaki T, Sakane I, Mizuno H, Tsuyama N, Masujima T, Direct metabolomics for plant cells by live single-cell mass spectrometry, Nat Protoc, 10 (2015) 1445. [DOI] [PubMed] [Google Scholar]
- [34].Zhang L, Vertes A, Single-Cell Mass Spectrometry Approaches to Explore Cellular Heterogeneity, Angew Chem Int Ed, 57 (2018) 4466–4477. [DOI] [PubMed] [Google Scholar]
- [35].Stolee JA, Shrestha B, Mengistu G, Vertes A, Observation of Subcellular Metabolite Gradients in Single Cells by Laser Ablation Electrospray Ionization Mass Spectrometry, Angew Chem Int Ed, 51 (2012) 10386–10389. [DOI] [PubMed] [Google Scholar]
- [36].Zhang L, Sevinsky CJ, Davis BM, Vertes A, Single-Cell Mass Spectrometry of Subpopulations Selected by Fluorescence Microscopy, Anal Chem, 90 (2018) 4626–4634. [DOI] [PubMed] [Google Scholar]
- [37].Onjiko RM, Moody SA, Nemes P, Single-cell mass spectrometry reveals small molecules that affect cell fates in the 16-cell embryo, Proc Natl Acad Sci U S A, 112 (2015) 6545–6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bergman H-M, Lanekoff I, Profiling and quantifying endogenous molecules in single cells using nano-DESI MS, Analyst, 142 (2017) 3639–3647. [DOI] [PubMed] [Google Scholar]
- [39].Zenobi R, Single-Cell Metabolomics: Analytical and Biological Perspectives, Science, 342 (2013) 1243259. [DOI] [PubMed] [Google Scholar]
- [40].Pan N, Rao W, Standke SJ, Yang ZB, Using Dicationic Ion-Pairing Compounds To Enhance the Single Cell Mass Spectrometry Analysis Using the Single-Probe: A Microscale Sampling and Ionization Device, Anal Chem, 88 (2016) 6812–6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rao W, Pan N, Yang Z, Applications of the Single-probe: Mass Spectrometry Imaging and Single Cell Analysis under Ambient Conditions, J Vis Exp, 112 (2016) e53911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sun M, Yang ZB, Wawrik B, Metabolomic Fingerprints of Individual Algal Cells Using the SingleProbe Mass Spectrometry Technique, Front Plant Sci, 9 (2018) 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liu R, Zhang G, Yang Z, Towards rapid prediction of drug-resistant cancer cell phenotypes: single cell mass spectrometry combined with machine learning, Chem. Commun, 55 (2019) 616–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sun M, Yang Z, Metabolomic Studies of Live Single Cancer Stem Cells Using Mass Spectrometry, Anal Chem, 91 (2018) 2384–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Standke SJ, Colby DH, Bensen RC, Burgett AWG, Yang Z, Mass Spectrometry Measurement of Single Suspended Cells Using a Combined Cell Manipulation System and a Single-Probe Device, Anal Chem, 91 (2019) 1738–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kim KS, Cho CH, Park EK, Jung M-H, Yoon K-S, Park H-K, AFM-Detected Apoptotic Changes in Morphology and Biophysical Property Caused by Paclitaxel in Ishikawa and HeLa Cells, PLOS ONE, 7 (2012) e30066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lee J-W, Park S, Kim SY, Um SH, Moon E-Y, Curcumin hampers the antitumor effect of vinblastine via the inhibition of microtubule dynamics and mitochondrial membrane potential in HeLa cervical cancer cells, Phytomedicine, 23 (2016) 705–713. [DOI] [PubMed] [Google Scholar]
- [48].Takara K, Obata Y, Yoshikawa E, Kitada N, Sakaeda T, Ohnishi N, Yokoyama T, Molecular changes to HeLa cells on continuous exposure to cisplatin or paclitaxel, Cancer Chemother Pharmacol, 58 (2006) 785–793. [DOI] [PubMed] [Google Scholar]
- [49].Heath JR, Ribas A, Mischel PS, Single-cell analysis tools for drug discovery and development, Nat Rev Drug Discov, 15 (2016) 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhang X-C, Wei Z-W, Gong X-Y, Si X-Y, Zhao Y-Y, Yang C-D, Zhang S-C, Zhang X-R, Integrated Droplet-Based Microextraction with ESI-MS for Removal of Matrix Interference in Single-Cell Analysis, Sci Rep, 6 (2016) 24730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Schuhmann K, Thomas H, Ackerman JM, Nagornov KO, Tsybin YO, Shevchenko A, Intensity-Independent Noise Filtering in FT MS and FT MS/MS Spectra for Shotgun Lipidomics, Anal Chem, 89 (2017) 7046–7052. [DOI] [PubMed] [Google Scholar]
- [52].Romano P, Profumo A, Rocco M, Mangerini R, Ferri F, Facchiano A, Geena 2, improved automated analysis of MALDI/TOF mass spectra, BMC Bioinformatics, 17 (2016) 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Xia J, Wishart DS, Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis, Curr Protoc Bioinformatics, 55 (2016) 14.10.1–14.10.91. [DOI] [PubMed] [Google Scholar]
- [54].Xia J, Sinelnikov IV, Han B, Wishart DS, MetaboAnalyst 3.0—making metabolomics more meaningful, Nucleic Acids Res, 43 (2015) W251–W257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Smilde AK, van der Werf MJ, Bijlsma S, van BJC der Werff-van der Vat, R.H. Jellema, Fusion of Mass Spectrometry-Based Metabolomics Data, Anal Chem, 77 (2005) 6729–6736. [DOI] [PubMed] [Google Scholar]
- [56].Di Guida R, Engel J, Allwood JW, Weber RJM, Jones MR, Sommer U, Viant MR, Dunn WB, Non-targeted UHPLC-MS metabolomic data processing methods: a comparative investigation of normalisation, missing value imputation, transformation and scaling, Metabolomics, 12 (2016) 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ong T-H, Kissick DJ, Jansson ET, Comi TJ, Romanova EV, Rubakhin SS, Sweedler JV, Classification of Large Cellular Populations and Discovery of Rare Cells Using Single Cell Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry, Anal Chem, 87 (2015) 7036–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Morrison SJ, Shah NM, Anderson DJ, Regulatory Mechanisms in Stem Cell Biology, Cell, 88 (1997) 287–298. [DOI] [PubMed] [Google Scholar]
- [59].Do KT, Wahl S, Raffler J, Molnos S, Laimighofer M, Adamski J, Suhre K, Strauch K, Peters A, Gieger C, Langenberg C, Stewart ID, Theis FJ, Grallert H, Kastenmüller G, Krumsiek J, Characterization of missing values in untargeted MS-based metabolomics data and evaluation of missing data handling strategies, Metabolomics, 14 (2018) 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bradley W, Robert P, Multivariate Analysis in Metabolomics, Curr Metabolomics, 1 (2013) 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chai T, Cui F, Yin Z, Yang Y, Qiu J, Wang C, Chiral PCB 91 and 149 Toxicity Testing in Embryo and Larvae (Danio rerio): Application of Targeted Metabolomics via UPLC-MS/MS, Sci Rep, 6 (2016) 33481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Triba MN, Le Moyec L, Amathieu R, Goossens C, Bouchemal N, Nahon P, Rutledge DN, Savarin P, PLS/OPLS models in metabolomics: the impact of permutation of dataset rows on the K-fold cross-validation quality parameters, Mol Biosyst, 11 (2015) 13–19. [DOI] [PubMed] [Google Scholar]
- [63].Barberini L, Noto A, Saba L, Palmas F, Fanos V, Dessì A, Zavattoni M, Fattuoni C, Mussap M, Multivariate data validation for investigating primary HCMV infection in pregnancy, Data Brief, 9 (2016) 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kim J, Hu ZP, Cai L, Li KL, Choi E, Faubert B, Bezwada D, Rodriguez-Canales J, Villalobos P, Lin YF, Ni M, Huffman KE, Girard L, Byers LA, Unsal-Kacmaz K, Pena CG, Heymach JV, Wauters E, Vansteenkiste J, Castrillon DH, Chen BPC, Wistuba I, Lambrechts D, Xu J, Minna JD, DeBerardinis RJ, CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells, Nature, 546 (2017) 168–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Suarez-Garcica S, Caimari A, del Bas JM, Suarez M, Arola L, Serum lysophospholipid levels are altered in dyslipidemic hamsters, Sci Rep, 7 (2017) 10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wanichthanarak K, Fan S, Grapov D, Barupal DK, Fiehn O, Metabox: A Toolbox for Metabolomic Data Analysis, Interpretation and Integrative Exploration, PLOS ONE, 12 (2017) e0171046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Guijas C, Montenegro-Burke JR, Domingo-Almenara X, Palermo A, Warth B, Hermann G, Koellensperger G, Huan T, Uritboonthai W, Aisporna AE, Wolan DW, Spilker ME, Benton HP, Siuzdak G, METLIN: A Technology Platform for Identifying Knowns and Unknowns, Anal Chem, 90 (2018) 3156–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, Djoumbou Y, Mandal R, Aziat F, Dong E, Bouatra S, Sinelnikov I, Arndt D, Xia J, Liu P, Yallou F, Bjorndahl T, Perez-Pineiro R, Eisner R, Allen F, Neveu V, Greiner R, Scalbert A, HMDB 3.0—The Human Metabolome Database in 2013, Nucleic Acids Res, 41 (2013) D801–D807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Li S, Park Y, Duraisingham S, Strobel FH, Khan N, Soltow QA, Jones DP, Pulendran B, Predicting Network Activity from High Throughput Metabolomics, PLOS Comput Biol, 9 (2013) e1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Schrimpe-Rutledge AC, Codreanu SG, Sherrod SD, McLean JA, Untargeted Metabolomics Strategies—Challenges and Emerging Directions, J Am Soc Mass Spectrom, 27 (2016) 1897–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Huang Q, Mao S, Khan M, Zhou L, Lin J-M, Dean flow assisted cell ordering system for lipid profiling in single-cells using mass spectrometry, Chem Commun, 54 (2018) 2595–2598. [DOI] [PubMed] [Google Scholar]
- [72].Zhang Y, Jin L, Xu J, Yu Y, Shen L, Gao J, Ye A, Dynamic characterization of drug resistance and heterogeneity of the gastric cancer cell BGC823 using single-cell Raman spectroscopy, Analyst, 143 (2018) 164–174. [DOI] [PubMed] [Google Scholar]
- [73].Wang T-H, Wang H-S, Ichijo H, Giannakakou P, Foster JS, Fojo T, Wimalasena J, Microtubule-interfering Agents Activate c-Jun N-terminal Kinase/Stress-activated Protein Kinase through Both Ras and Apoptosis Signal-regulating Kinase Pathways, J Biol Chem, 273 (1998) 4928–4936. [DOI] [PubMed] [Google Scholar]
- [74].Balázsi G, van Oudenaarden A, James J Collins, Cellular Decision Making and Biological Noise: From Microbes to Mammals, Cell, 144 (2011) 910–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Touw WG, Bayjanov JR, Overmars L, Backus L, Boekhorst J, Wels M, van Hijum SAFT, Data mining in the Life Sciences with Random Forest: a walk in the park or lost in the jungle?, Brief Bioinform, 14 (2013) 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Vinaixa M, Samino S, Saez I, Duran J, Guinovart JJ, Yanes O, A Guideline to Univariate Statistical Analysis for LC/MS-Based Untargeted Metabolomics-Derived Data, Metabolites, 2 (2012) 775–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Simons K, Toomre D, Lipid rafts and signal transduction, Nat Rev Mol Cell Biol, 1 (2000) 31. [DOI] [PubMed] [Google Scholar]
- [78].Sun H, Wang B, Wang J, Liu H, Liu J, Biomarker and pathway analyses of urine metabolomics in dairy cows when corn stover replaces alfalfa hay, J Anim Sci Biotechnol, 7 (2016) 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kapatos G, The neurobiology of tetrahydrobiopterin biosynthesis: A model for regulation of GTP cyclohydrolase I gene transcription within nigrostriatal dopamine neurons, IUBMB Life, 65 (2013) 323–333. [DOI] [PubMed] [Google Scholar]
- [80].Pickert G, Lim H-Y, Weigert A, Häussler A, Myrczek T, Waldner M, Labocha S, Ferreirós N, Geisslinger G, Lötsch J, Becker C, Brüne B, Tegeder I, Inhibition of GTP cyclohydrolase attenuates tumor growth by reducing angiogenesis and M2-like polarization of tumor associated macrophages, Int J Cancer, 132 (2013) 591–604. [DOI] [PubMed] [Google Scholar]
- [81].Rowinsky EK, Signal events: Cell signal transduction and its inhibition in cancer, Oncologist, 8 (2003) 5–17. [DOI] [PubMed] [Google Scholar]
- [82].Qi YP, Jiang CT, Cheng J, Krausz KW, Li TG, Ferrell JM, Gonzalez FJ, Chiang JYL, Bile acid signaling in lipid metabolism: Metabolomic and lipidomic analysis of lipid and bile acid markers linked to anti-obesity and anti-diabetes in mice, Bba-Mol Cell Biol L, 1851 (2015) 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Gregory DH, Vlahcevic ZR, Prugh MF, Swfll L, Mechanism of secretion of biliary lipids: Role of a microtubular system in hepatocellular transport of biliary lipids in the rat, Gastroenterology, 74 (1978) 93–100. [PubMed] [Google Scholar]
- [84].Mashima T, Seimiya H, Tsuruo T, De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy, Br J Cancer, 100 (2009) 1369–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Onjiko RM, Morris SE, Moody SA, Nemes P, Single-cell mass spectrometry with multi-solvent extraction identifies metabolic differences between left and right blastomeres in the 8-cell frog (Xenopus) embryo, Analyst, 141 (2016) 3648–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Portero EP, Nemes P, Dual cationic–anionic profiling of metabolites in a single identified cell in a live Xenopus laevis embryo by microprobe CE-ESI-MS, Analyst, 144 (2019) 892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.