

Abstract
We showed recently that chronic administration of the mitochondrial inhibitor 3-nitropropionic acid (3NP) in primates produces various dyskinetic movements and dystonic postures associated with selective striatal lesions displaying many similarities with the pathological features of Huntington’s disease (HD). In the present study, we examined whether such a toxic treatment could also induce frontal-type deficits similar to those observed in HD patients. Cognitive performances of 3NP-treated and control baboons were compared using the object retrieval detour task (ORDT), a test designed to assess the functional integrity of the frontostriatal pathway in human and nonhuman primates. During the same time, the motor function of each animal was assessed under spontaneous “no drug” conditions, and time-sampled neurological observations were used after apomorphine administration. A significant impairment in the ORDT was observed in the 3NP animals after 3–6 weeks of treatment, occurring in the absence of spontaneous abnormal movements but in the presence of apomorphine-inducible dyskinesias. Prolonged 3NP treatment resulted in the progressive appearance of spontaneous abnormal movements. Histological evaluation of these animals showed selective bilateral caudate–putamen lesions with sparing of the cerebral cortex, notably the prefrontal cortex. The present study demonstrates that chronic 3NP treatment replicates in primates the basic pathophysiological triad of HD, including spontaneous abnormal movements, progressive striatal degeneration, and a frontostriatal syndrome of cognitive impairment.
Keywords: Huntington’s disease, basal ganglia, caudate nucleus, frontal cortex, baboon, 3-nitropropionic acid, object retrieval detour task, cognitive behavior, motor behavior, apomorphine testing
Huntington’s disease (HD) is a dominantly inherited neurodegenerative disorder characterized by involuntary choreiform movements and progressive cell loss in brain regions belonging to the basal ganglia, particularly the striatum (Martin and Gusella, 1986; Harper, 1991; Kremer et al., 1992). In addition, most HD patients display memory disturbances, psychiatric manifestations, frontal-type cognitive abnormalities, decreased verbal fluency, and poor recall of recently learned information (Butters et al., 1978;Brandt et al., 1988; Massman et al., 1990; Pillon et al., 1991;Randolph, 1991; Diamond et al., 1992). Most of these cognitive alterations resemble those observed in patients with lesions of the dorsolateral prefrontal cortex (Cummings, 1993).
Despite the recent discovery of the genetic mutation associated with HD (The Huntington’s Disease Collaborative Group, 1993), the cause of this disorder is still unknown. One possibility is that the genetic defect may lead to an impairment of energy metabolism resulting in slow excitotoxic neuronal death (Albin and Greenamyre, 1992; Beal, 1992). In line with this, several studies have reported decreased striatal glucose metabolism (Kuhl et al., 1982; Young et al., 1986; Mazziotta et al., 1987; Berent et al., 1988; Grafton et al., 1990; Kuwert et al., 1990; Martin et al., 1992), abnormalities in mitochondrial electron transport chain (Brennan et al., 1985; Mann et al., 1990; Parker et al., 1990), and increased concentrations of lactate in the cerebral cortex and the striatum as detected by nuclear magnetic resonance spectroscopy, providing in vivo evidence for impaired oxidative energy metabolism in HD (Jenkins et al., 1993). Furthermore, in support of the energy impairment hypothesis, chronic blockade of energy metabolism induced in rats by systemic administration of mitochondrial toxins induces selective excitotoxic-like lesions of the striatum (Beal et al., 1993; Brouillet et al., 1994). We recently extended this observation to primates by demonstrating that chronic systemic administration to baboons of 3-nitropropionic acid (3NP), an irreversible inhibitor of succinate dehydrogenase (for review, seeLudolph et al., 1991), can produce various dyskinetic movements and dystonic postures associated with selective striatal lesions (Brouillet et al., 1993, 1995; Ferrante et al., 1993). To date, however, no studies have examined the effect of such a neurotoxic treatment on cognitive aspects of behavior.
In the present study, the cognitive performances of 3NP-treated and control baboons were compared using the object retrieval detour task (ORDT), a test designed to assess the functional integrity of the frontostriatal pathway in both humans and nonhuman primates (Diamond, 1990). At the same time, the motor function in these animals was studied under spontaneous “no drug” conditions and in the apomorphine test, a test designed to unmask presymptomatic striatal dysfunction in excitotoxically lesioned baboons (Hantraye et al., 1990) and 3NP-treated baboons (Brouillet et al., 1995). This combination of tests was used to study a possible association of frontal-type deficits with presymptomatic or clinically detectable striatal dysfunction. Finally, histological analysis of brain sections of control and 3NP-treated baboons was conducted to correlate cognitive and motor deficits with anatomical lesions.
MATERIALS AND METHODS
Animals
Fourteen Papio anubis baboons (Charles River Labs, Wilmington, MA) (7–10 kg body weight) were included in this study. Four baboons received a chronic systemic 3NP treatment, whereas the remaining 10 received saline injections and served as control animals for the behavioral studies. All animals were housed individually in standard primate cages with free access to water and food, except for bananas, which were available only while the animals were performing the ORDT. Studies were carried out in accordance with the European convention for animal care and the guide for the care and use of laboratory animals adopted by the National Institutes of Health (Bethesda, MD).
3NP lesion
3NP (Sigma, St. Louis, MO) was dissolved at a concentration of 80 mg/ml in deionized water and adjusted to pH 7.4 with NaOH (1 m). The four baboons (young adolescents) received repeated intramuscular injections of 3NP (administered in daily injections at 10 A.M. and 4 P.M.) for 20 weeks. In accordance with previous studies (Brouillet et al., 1995), the dose regimen for the administration of 3NP was designed to yield a linear increase in doses until obvious spontaneous abnormal movements could be observed (mean starting dose, 14 mg·kg−1·d−1; mean final dose, 33 mg·kg−1·d−1). As shown in Figure 1, administered doses were adjusted slightly according to the physical status of each baboon, but these alterations did not diverge significantly from the linear dose schedule (r = 0.95; p < 0.001).
Fig. 1.

Schematic representation of the experimental design used in this study. The average daily dose of 3NP administered to the four baboons is indicated as a function of time. Dose regimen was adjusted every week according to the physical status of each animal to avoid any acute intoxication (Brouillet et al., 1995). The various behavioral tests used to assess the presence of motor deficits (apomorphine tests, APO1–3) or frontostriatal cognitive dysfunction (ORDT) in the 3NP-treated baboons, are represented as open boxes. Note that APO1 and APO2 were performed immediately before and after the ORDT testing to assess the functional status of the striatum at that particular time period and possibly correlate cognitive deficits with presymptomatic striatal dysfunction.
Behavioral assessment
Clinical examinations were performed before each injection to detect any signs of acute intoxication and the presence of spontaneous abnormal movements. In addition, two behavioral methods were used: time-sampled neurological observations after intramuscular administration of apomorphine (Hantraye et al., 1990, 1992b; Brouillet et al., 1995) and the ORDT (Diamond and Goldman-Rakic, 1985; Diamond et al., 1989, 1990; Taylor et al., 1990a,b; Schneider, 1992).
The apomorphine test. We previously used apomorphine administration in excitotoxically lesioned baboons to reveal preclinical dysfunction after partial striatal lesions. In these initial studies, the apomorphine test results were shown to be highly reproducible for all symptoms observed: individual animals responding with a similar set and incidence of dyskinesias over several successive test sessions with no change in symptoms for at least 1 year (Isacson et al., 1991; Hantraye et al., 1992b). In the present study, time-sampled neurological observations were obtained after intramuscular administration of 0.5 mg/kg apomorphine, as described previously (Hantraye et al., 1990, 1992b). Briefly, four different categories of abnormal movements (orofacial dyskinesia, dyskinesia of extremities, dystonia, and choreiform movements) were monitored after video-recording from front-view images and were rated as being present (= 1) or absent (= 0) during each 5 min time period of a 40 min test session. A dyskinesia index (sum of incidences) was computed by adding together the incidence of each symptom during the 40 min test period (maximum score = 32, minimum score = 0). Five normal baboons selected randomly among the 10 controls at the beginning of the study and the four 3NP-treated baboons received apomorphine testing. As shown in Figure 1, the first apomorphine test (APO 1) was performed the week before the first ORDT test session (corresponding to 3 weeks of treatment). The second apomorphine test (APO 2) was performed immediately after the last ORDT test session (corresponding to 7 weeks of treatment). Finally, the last apomorphine test (APO 3) was performed 6–10 weeks after the last ORDT test-session, i.e., after 13–17 weeks of treatment (Fig. 1).
The ORDT. The ORDT was used to assess the ability of the animals to retrieve an object (a slice of banana) from inside a transparent box open on only one side. We selected this task because it requires complex sequential motor planning and is particularly sensitive for detecting frontal cortex or striatal dysfunction (Diamond and Goldman-Rakic, 1985; Diamond et al., 1989, 1990; Taylor et al., 1990a,b; Schneider, 1992). In addition, the task involves features implicated in motor skills; however, in the majority of the trials in which the opening of the box is not facing the animal, the primates must inhibit their natural tendency to reach straight for what they want (prepotent response tendencies) and must make a detour around the transparent side of the box into the open side of the box. Inhibition of this natural tendency specifically requires the functional integrity of the frontostriatal system (Diamond, 1990) and is not affected by lesions of other brain regions such as the hippocampal formation or the parietal cortex (Diamond and Goldman-Rakic, 1985; Diamond et al., 1989).
The Plexiglas box (8 × 8 × 9 cm) was fixed on a tray adaptable to the home cage of the animal. The cognitive level and motor skills required to solve the task and retrieve the banana slice were modified by the experimenter by varying the location of the box relative to the subject, the location of the reward into the box, and finally the orientation of the open side of the box relative to the subject. As shown in Figure 2, each test session consisted of 15 trials presented randomly to the animals. The reward was visible only after an opaque screen placed between the animal and the box was raised. Subjects were then allowed 1 min to retrieve the reward, a period after which the screen was put back in place and the box set up for the next trial. The movements and responses of the animal involving the tray or the box were not restrained in any way.
Fig. 2.

ORDT experimental setup. Diagrammatic representation showing the position of the box on the tray, reward on the box, and open side of the box on the 15 trials that constituted each test session. In six of these configurations (1–6), the open side of the box is facing the subject so that no detour is required to reach the reward (“easy” trials). In nine of these configurations (7–15), a closed (transparent) side of the box is facing the subject, so that a detour is required to retrieve the reward (“difficult” trials).
Responses of the subject were recorded on videotape. Measures of performance included number of “success” responses (retrieval of the reward on the first reach of the trial), and number of “correct” responses (retrieval of the reward within the 1 min time period; the correct responses also include the success responses), number of “barrier hits” responses (hitting the closed transparent side of the box instead of making a detour), and number of “motor problems” responses [reaching the correct (open) side of the box but failing to retrieve the reward]. The number of correct and success responses are expressed as a percentage of the total number of trials. Barrier hits and motor problems are expressed as percentage of responses observed. Animal performances were also analyzed as a function of the level of cognitive difficulty (“easy” and “difficult”). The easy trials were defined as the trials in which the opening of the box was facing the baboon (no detour to make to reach the reward; configurations 1–6 in Fig. 2), whereas difficult trials were defined as the configurations in which the baboon had to make a detour around a closed side of the box to reach the reward (configurations 7–15 in Fig. 2). Animal testing consisted of four consecutive test sessions separated by a 1 week interval.
Anatomical studies
Anatomical studies were carried out in the four 3NP-treated primates, which were killed 4 d after termination of the 3NP treatment, and in two control animals. Under chemical restraint (ketamine 10 mg/kg), animals were killed by intravenous administration of pentobarbital (120 mg/kg). Their brains were removed rapidly from the skull, cut in 2- to 3-cm-thick frontal blocks, postfixed for 48 hr in paraformaldehyde 2% sodium-m-periodate lysine, cryoprotected in phosphate buffer glycerol 20%, and kept frozen at −70°C. Histochemical and immunocytochemical procedures [calbindin immunodetection and nicotinamide adenine dinucleotide phosphate (NADPH) histochemistry] were performed on 50 μm frozen sections as described previously (Ferrante et al., 1993; Brouillet et al., 1995). Detailed immunocytochemical and anatomical descriptions of the lesions observed in these animals will also appear in a separate paper (R.J. Ferrante, P. Hantraye, E. Brouillet, M.F. Beal, unpublished observations).
Magnetic resonance imaging (MRI)
Animals were anesthetized with ketamine/xylazine, and MRI was performed on a 0.5 T MR magnet (General Electric) using a homemade Helmotz receive-only probe as described previously (Hantraye et al., 1992a).
Statistical analyses
Tests of variance homogeneity, normality, and distribution were performed to ensure that the assumptions required for a standard parametric ANOVA were satisfied. Statistical analysis of the ORDT test results was carried out using ANOVA (two factors repeated-measures ANOVA) to study interactions between groups (3NP-treated vs control subjects) or interactions between groups and session for each of the measures examined. Factorial ANOVA was used to study possible interactions between groups in the apomorphine testing as well as a possible effect of repetition of the test on the ORDT performances. All significant interactions were reanalyzed post hoc using Scheffe’sF test.
RESULTS
Histological observations
Chronic treatment with 3NP produced a marked, bilateral, neuronal cell loss restricted to the caudate and putamen nuclei in the four 3NP-treated baboons, confirming previous findings (Brouillet et al., 1995). As shown in Figure 3A,B on representative frozen cut frontal sections, there was a bilateral dorsoventral gradient of pathological effect within the striatum in each of the animals. Lesions were restricted to the dorsal aspect of the caudate nucleus and the putamen; the ventral striatum and the nucleus accumbens were relatively spared (Fig. 3A). There was also a rostrocaudal gradient of pathological damage; the rostral part of the striatum (caudate and putamen) was more affected than its most caudal part (Fig. 3A,B). MRI showed increased signal on T2-weighted images in both the caudate and putamen, at completion of the 3NP-treatment (Fig. 4A). Immunocytochemically, the striatal lesions were characterized by a severe loss in calbindin-positive neurons, a subset of medium-size spiny projection neurons, and a relative sparing of the NADPH-diaphorase-positive interneurons (Fig. 4C), as compared with control specimen (Fig. 4B). No additional damage was identified in any other area of the brain, including the prefrontal cortex (Fig. 3 and data not shown).
Fig. 3.

Photographs of fixed frozen-cut frontal section of the brain in a systemically treated primate using 3NP at the level of the caudate nucleus, putamen, and nucleus accumbens (A) and at the level of the anterior commissure and globus pallidus (B). At each of the levels considered, lesions were restricted to the dorsal aspect of the caudate nucleus and putamen; the ventral striatum and the nucleus accumbens were relatively spared. No other damage was identified in any other area of the brain. The lesion loci appear well delineated as dark areas on these frozen specimens. Scale bar, 1 cm.
Fig. 4.

Striatal pathology in 3NP-treated primate.A, MR T2-weighted image obtained after completion of the 3NP in the same animal as in Figure 3. High signal intensity within the caudate nucleus and putamen corresponds to tissue damage seen inC. B and C, Double-stained sections reacted to reveal the presence of calbindin, which labels a subpopulation of medium-size spiny neurons, and NADPH-diaphorase enzyme, which labels somatostatin/neuropeptide Y-interneurons in the caudate nucleus of the 3NP-treated animal (C) in comparison with a normal (control) specimen (B). Note the severe loss in calbindin-positive neurons and the relative sparing of the NADPH-diaphorase neurons (dark neurons with dendritic arbors) in the treated animal as compared with the control. [Same magnification inB and C; scale bar (shown in C): 100 μm.]
Behavioral observations
Motor function under spontaneous “no drug” conditions
As previously noted in baboons with partial excitotoxic striatal lesions (Hantraye et al., 1990), no spontaneous dyskinesias were observed in the 3NP-treated animals during the initial part of the neurotoxic treatment (weeks 1–11); however, in the final part of the neurotoxic treatment, spontaneous abnormal movements, primarily foot and leg dystonias, and some dyskinesias of the hindlimb were observed, starting at ∼11 weeks of treatment, which then persisted unchanged until the end of the study (i.e., for at least 9 weeks).
Apomorphine-induced dyskinesias
As early as 3 weeks after treatment, apomorphine administration elicited abnormal movements in the 3NP-treated animals. These abnormal movements consisted of irregular and partial movements of the extremities and the trunk resembling choreiform movements, orofacial dyskinesias, and dystonias. Orofacial dyskinesias consisted of repetitive jaw movements accompanied in some instances by protrusion of the tongue. The most common forms of dystonia observed in these baboons were twisting of the trunk and unilateral (occasionally bilateral) foot dystonia. In control animals, only slow jaw movements and repetitive licking behavior were elicited by apomorphine, which were taken as a baseline for comparison with the orofacial dyskinesias observed in the 3NP-treated animals. The dyskinesia index for abnormal movements was significantly higher in the treated animals as compared with the five control animals (Fig. 5) (p < 0.02) tested under the same conditions at the two latest time points (APO2 andAPO3, in Fig. 5). Although a clear tendency toward an increased dyskinesia index was already visible at APO1between controls and the 3NP-treated baboons, the difference between the two groups of animals was not statistically significant at this time point (p > 0.05).
Fig. 5.

Apomorphine test in control and 3NP-treated baboons. The dyskinesia index in controls and the 3NP-treated animals was determined at various time points during the chronic treatment with 3NP. APO1: 3 weeks of treatment; APO2: 7 weeks of treatment; APO3: 13–17 weeks of treatment. Values are mean ± SEM. Black bars represent control animals (CTRL, n = 5). Cross-hatched barsrepresent 3NP-treated animals (n = 4). *p < 0.02; **p < 0.0002; ns (nonsignificant), ANOVA analyzed post hoc using Scheffe’s F test.
Cognitive function
The ORDT testing was performed between APO 1 and APO 2 (see Fig.1), i.e., when the animals were essentially nonsymptomatic under “no drug” conditions but starting to show abnormal movements in the apomorphine test (Fig. 5). At this time point, ANOVA repeated-measures analysis indicated that 3NP-treated subjects were significantly less successful in obtaining the reward on the first reach during testing (F = 7.53; p < 0.02) and made significantly more barrier hits when performing the task (F = 16.3;p < 0.002) as compared with controls (Fig.6A,C). These differences could not be attributed to a physical disability of the 3NP-treated subjects, because motor function (Fig. 6D) did not differ significantly between the two groups (ANOVA, F = 0.492;p > 0.49). In addition, 3NP-treated baboons were not impaired significantly (F = 0.749; p > 0.40) in the acquisition of the task, because their correct responses did not differ significantly from controls (Fig. 6B).
Fig. 6.

ORDT performances in 4 3NP-treated baboons and 10 age-matched control animals. The percentage of “SUCCESS” (A), “CORRECT” (B), “BARRIER HITS” (C), and “MOTOR PROBLEMS” (D) responses are represented asblack bars for control animals (n = 10) andcross-hatched bars for the 3NP-treated animals (n = 4). Values are mean ± SEM. Statistical analysis was carried out using ANOVA repeated-measures and then reanalyzed post hoc using Scheffe’s F test to determine significant interactions between groups (3NP-treated vs control subjects). Success and correct responses are expressed as a percentage of the total number of trials. Correct responses include success responses. Barrier hits and motor problems are expressed as a percentage of the total responses registered. As compared with controls, 3NP animals seemed significantly impaired in their ability to reach the reward in the first attempt (success responses), which resulted in a higher number of barrier hits that reflect a lack of inhibition of their natural prepotent tendency and a dysfunction of the frontostriatal pathway.
Statistical analysis of the session effect (Fig. 7) showed a significant improvement between the first and last session in the control group for success and correct responses (success:F = 8.66, p < 0.001; correct: F = 3.23, p < 0.04) but not in the 3NP-treated group (success:F = 0.09, p > 0.96; correct: F = 0.02, p > 0.99). A similar pattern was observed for barrier hits responses (controls: F = 8.2, p < 0.001; 3NP-treated: F = 0.4, p > 0.75), indicating that 3NP-treated animals persisted in making errors in the ORDT test, whereas control animals were learning to make a detour around the closed transparent side of the box.
Fig. 7.
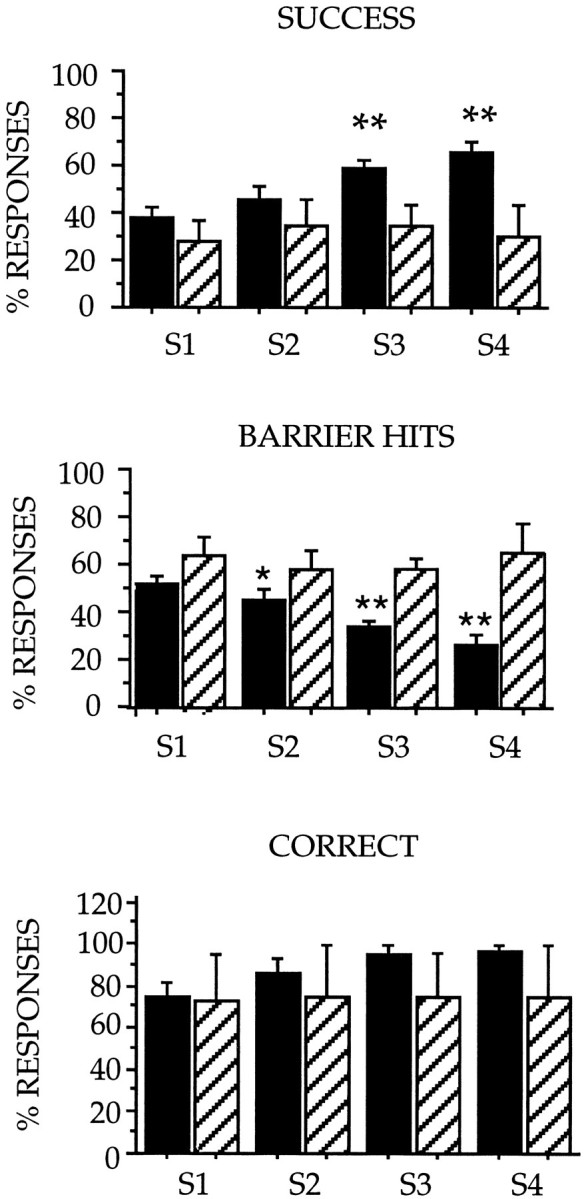
Session effect in the performances of 3NP-treated baboons and control animals in the ORDT. The percentage of “SUCCESS,” “BARRIER HITS,” and “CORRECT” responses is represented for each consecutive test session (S1, S2, S3,S4). Black bars represent control animals (n = 10), and cross-hatched bars represent 3NP-treated animals (n = 4). Values are mean ± SEM. Statistical analysis was carried out using ANOVA factorial measures and then reanalyzed post hoc using Scheffe’s F test to determine significant interactions between sessions (S1,S2, S3, S4) in 3NP-treated animals and controls. As compared with control animals whose performances in success, barrier hits, and correct responses improved significantly with time, the deficits in the ORDT observed in the 3NP animals remained constant from one session to another. Significant differences between groups were also analyzed for each test session by factorial ANOVA: **p < 0.01; *p < 0.05 (3NP-treated animals vs controls).
Finally, when the level of cognitive difficulty was considered (Fig.8), barrier hits performances of 3NP-treated animals were significantly different from those of controls in the difficult (F = 19.5; p < 0.001) but not in the easy trials (F = 1.29; p > 0.25).
Fig. 8.

“Barrier hits” responses of 3NP-treated baboons and control animals according to the level of difficulty in the ORDT. The percentage of barrier hits responses are represented for easier front-reach trials (open side of the box directly facing the animal) and for difficult trials (transparent side of the box interposing the reward with the animal). Black barsrepresent control animals. Cross-hatched bars represent 3NP-treated animals. Values are mean ± SEM. Statistical analysis was carried out using factorial ANOVA and post hoc analysis using Scheffe’s F test to determine significant interactions between groups. 3NP-treated animals seemed significantly different from controls in the difficult but not the easy trials.
DISCUSSION
We showed recently that chronic administration of the irreversible succinate dehydrogenase inhibitor 3NP to nonhuman primates can produce selective bilateral degeneration of the caudate–putamen complex accompanied by spontaneous and apomorphine-induced abnormal movements resembling those observed in HD (Brouillet et al., 1995). The striatal lesions were strikingly reminiscent of those occurring in HD, with selective loss of medium-size spiny neurons as compared with other striatal cell types, and a dorsoventral gradient of neuronal degeneration within both caudate and putamen. Additional histological observations included a sparing of the nucleus accumbens, a preservation of the patch-matrix compartmentalization of the striatum, and the presence of growth-related neuronal alterations in spiny striatal neurons (Ferrante et al., 1993; Brouillet et al., 1995; R.J. Ferrante, P. Hantraye, E. Brouillet, M.F. Beal, unpublished observations).
In the present study we aimed to characterize further this primate model as a model of HD by studying whether such a chronic 3NP treatment would also produce HD-like cognitive deficits in primates.
As shown here, chronic 3NP produced significant deficits in the ORDT. Thus, success responses were significantly reduced, and barrier hits responses were increased significantly in the 3NP-treated animals, as compared with control baboons. When the level of cognitive difficulty was taken into account, 3NP animals seemed significantly different from controls only in the difficult trials, i.e., configurations for which a detour around a closed transparent side of the box is necessary to retrieve the reward. In contrast, the correct responses, which represent the ability of each animal to finally reach the reward, were not significantly different between the two groups, and no detectable motor problems were noted in the 3NP-treated animals as compared with controls.
Altogether these findings indicate that (1) alterations in motor control or a disinterest of the animals in the task are not responsible for the differences observed between the two groups and (2) 3NP animals differ from controls in trials specifically requiring the inhibition of a natural tendency to reach straight for a reward, which as discussed by Diamond (1990) involves the frontostriatal pathway. Therefore, a specific functional and/or anatomical dysfunction of the frontostriatal pathway had to be present in the 3NP-treated baboons. Interestingly, these observations were obtained in the first phase of the toxic treatment during which the 3NP-treated animals did not show any abnormal movements under spontaneous conditions. This indicates further that the frontal-type cognitive deficits associated with the 3NP-treatment precede the onset of spontaneous dyskinesias in this chronic model of striatal degeneration.
Similar cognitive deficits in the ORDT have been observed in monkeys with lesions of the dorsolateral prefrontal cortex (Diamond and Goldman-Rakic, 1985) or with severe striatal dopamine depletion attributable to MPTP treatment (Taylor et al., 1990a,b; Schneider, 1992). It is interesting to note that these two experimental conditions as well as the 3NP-model described here directly affect either the anatomical or the functional integrity of the frontal cortex or the striatum. The cognitive deficit observed in the 3NP-treated baboons after 5–7 weeks of treatment (ORDT sessions 3 and 4) is thus reminiscent of frontal-type deficits resulting from a dysfunction of the striatum, especially the dorsomedial caudate nucleus. This interpretation is supported by two types of observations. First, histological observations in these and other animals have demonstrated that chronic 3NP treatment in primates is associated with lesions restricted to the striatum, involving the dorsal and medial aspects of both the caudate and putamen but sparing the nucleus accumbens and the cerebral cortex, especially the prefrontal cortex. Second, by performing the apomorphine test immediately before and after the cognitive test sessions, we have shown that despite the absence of spontaneous abnormal movements, there was a presymptomatic striatal dysfunction in the 3NP-treated baboons at the time of ORDT testing. Because previous experimental and clinical studies (Battig et al., 1960; Divac et al., 1967; Cummings, 1993) have shown that striatal lesions, notably those involving the medial caudate nucleus, which receives dense projections from the orbitofrontal and anterior cingulate cortex, and those involving the dorsolateral caudate, which receives projections from the dorsolateral prefrontal cortex, can produce personality alterations and cognitive impairments indistinguishable from those accompanying frontal syndromes (Richfield et al., 1987; Mendez et al., 1989), these observations indicate that the frontal-type deficits in the 3NP-treated baboons have a striatal, probably caudate, origin.
The time course analysis of the success and barrier hits showed that 3NP-treated baboons did not improve their performances significantly from the first test day to the fourth, whereas controls significantly improved their performances from one test session to the other. Such alterations in the acquisition of the task in the 3NP-treated animals may be interpreted as a learning deficit, but as in the frontal syndrome observed in HD, which is primarily characterized by deficits in executive function and organizational strategies for learning tasks, the present results may also be interpreted as an impairment in organizational strategy in the 3NP-treated animals. Additional studies that would use more specific cognitive tasks designed to assess more specifically frontal-type deficits (Battig et al., 1960; Divac et al., 1967; Roberts et al., 1994), memory disturbances (Chavoix et al., 1994), or organizational strategies are required to characterize further the exact nature of the cognitive deficit associated with a chronic 3NP treatment.
In conclusion, this new primate model offers several advantages for the study of normal striatal function, as well as for the pathophysiology of HD and the testing of new therapeutic strategies as compared with the previously developed unilateral excitotoxic lesion model (Hantraye et al., 1990). Because 3NP readily crosses the blood–brain barrier, the toxin can be delivered by intramuscular injection. This allows the generation of a model of progressive striatal degeneration associated with selective bilateral (symmetrical) striatal lesions, which do not require repeated intrastriatal injections. In addition, the chronic nature of this lesion model enables one to study the time course of cognitive deficits associated with various degrees of striatal atrophy and their relationship with the onset of spontaneous dyskinesias. The chronic administration of the mitochondrial toxin also produces the progressive appearance of spontaneous abnormal movements, which persist over time, and frontal-type deficits, which are particularly suitable for assessment of the efficacy of neurotrophic/neuroprotective agents (Beal, 1994; Beal et al., 1994) or neural grafting procedures (Hantraye et al., 1992b) on motor and cognitive aspects of the 3NP-induced syndrome.
Footnotes
This study was supported by grants from CNRS-NSF and CNAMTS/INSERM. S.P. was supported by fellowships from Bayer-Pharma (France), Association Française contre les Myopathies (AFM), and Association Huntington France. We thank Dr. M. Mazière and Professor Y. Kéravel for continuing support and Dr. P. C. Jedynak for expert behavioral observations. Jérôme Cayla and Christophe Jouy are greatly acknowledged for the outstanding care of the primate colony and for their help in the behavioral experiments.
Correspondence should be addressed to Dr. Philippe Hantraye, URA CEA-CNRS 1285, Service Hospitalier Frédéric Joliot, DRIPP, CEA-DSV, 4 Place du Général Leclerc, 91401 Orsay Cedex, France.
REFERENCES
- 1.Albin RL, Greenamyre JT. Alternative excitotoxic hypothesis. Neurology. 1992;42:733–738. doi: 10.1212/wnl.42.4.733. [DOI] [PubMed] [Google Scholar]
- 2.Battig K, Rosvold HE, Mishkin M. Comparison of the effects of frontal and caudate lesions on delayed response and alternation in monkeys. J Comp Physiol Psychol. 1960;53:400–404. doi: 10.1037/h0047392. [DOI] [PubMed] [Google Scholar]
- 3.Beal MF. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann Neurol. 1992;31:119–130. doi: 10.1002/ana.410310202. [DOI] [PubMed] [Google Scholar]
- 4.Beal MF. Neurochemistry and toxin models in Huntington’s disease. Curr Opin Neurol. 1994;7:542–547. doi: 10.1097/00019052-199412000-00012. [DOI] [PubMed] [Google Scholar]
- 5.Beal MF, Brouillet E, Jenkins B, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:1481–1492. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beal MF, Henshaw R, Jenkins BG, Rosen BR, Schulz JB. Coenzyme Q10 and nicotinamide block striatal lesions produced by the mitochondrial toxin malonate. Ann Neurol. 1994;36:882–888. doi: 10.1002/ana.410360613. [DOI] [PubMed] [Google Scholar]
- 7.Berent S, Giordani B, Lehtinen S, Markel D, Penney JB, Buchtel HA, Starosta-Rubinstein S, Hichwa R, Young AB. Positron emission tomographic scan investigations of Huntington’s disease: cerebral metabolic correlates of cognitive function. Ann Neurol. 1988;23:541–546. doi: 10.1002/ana.410230603. [DOI] [PubMed] [Google Scholar]
- 8.Brandt J, Folstein SE, Folstein MF. Differential cognitive impairment in Alzheimer’s disease and Huntington’s disease. Ann Neurol. 1988;23:555–561. doi: 10.1002/ana.410230605. [DOI] [PubMed] [Google Scholar]
- 9.Brennan WAJ, Bird ED, Aprille JR. Regional mitochondrial respiratory activity in Huntington’s disease brain. J Neurochem. 1985;44:1948–1950. doi: 10.1111/j.1471-4159.1985.tb07192.x. [DOI] [PubMed] [Google Scholar]
- 10.Brouillet E, Hantraye P, Dolan R, Leroy-Willig A, Bottlaender M, Isacson O, Mazière M, Ferrante RJ, Beal MF. Chronic administration of 3-nitropropionic acid induced selective striatal degeneration and abnormal choreiform movements in monkeys. Soc Neurosci Abstr. 1993;19:409. [Google Scholar]
- 11.Brouillet E, Hantraye P, Ferrante RJ, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal F. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci USA. 1995;92:7105–7109. doi: 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brouillet E, Hyman BT, Jenkins BG, Henshaw DR, Schulz JB, Sodhi P, Rosen BR, Beal MF. Systemic or local administration of azide produces striatal lesions by an energy impairment-induced excitotoxic mechanism. Exp Neurol. 1994;129:175–182. doi: 10.1006/exnr.1994.1159. [DOI] [PubMed] [Google Scholar]
- 13.Butters N, Sax D, Montgomery K, Tarlow S. Comparison of the neuropsychological deficits associated with early and advanced Huntington’s disease. Arch Neurol. 1978;35:585–589. doi: 10.1001/archneur.1978.00500330033006. [DOI] [PubMed] [Google Scholar]
- 14.Chavoix C, Dauvillier F, Landeau B, Le Mestric C, Huguet G, Marié RM. Comparison of mnesic performance between humans and baboons with the same automated delayed nonmatching-to-sample task. Folia Primatol (Basel) 1994;62:191–192. [Google Scholar]
- 15.Cummings JL. Frontal-subcortical circuits and human behavior. Arch Neurol. 1993;50:873–880. doi: 10.1001/archneur.1993.00540080076020. [DOI] [PubMed] [Google Scholar]
- 16.Diamond A. Developmental time course in human infants and infant monkeys, and the neural bases of inhibitory control in reaching. Ann NY Acad Sci. 1990;608:637–669. doi: 10.1111/j.1749-6632.1990.tb48913.x. [DOI] [PubMed] [Google Scholar]
- 17.Diamond A, Goldman-Rakic PS. Evidence for involvement of prefrontal cortex in cognitive changes during the first year of life: comparison of performances of human infant and rhesus monkeys on a detour task with transparent barrier. Soc Neurosci Abstr. 1985;11:832. [Google Scholar]
- 18.Diamond R, White RF, Myers R, Mastromauro C, Koroshetz WJ, Butters N, Rothstein DM, Moss MB, Varterling J. Evidence of presymptomatic cognitive decline in Huntington’s disease. J Clin Exp Neuropsychol. 1992;14:961–975. doi: 10.1080/01688639208402547. [DOI] [PubMed] [Google Scholar]
- 19.Diamond A, Zola-Morgan S, Squire LR. Successful performance by monkeys with lesions of the hippocampal formation on AB- and object retrieval, two tasks that mark developmental changes in human infants. Behav Neurosci. 1989;103:526–537. doi: 10.1037//0735-7044.103.3.526. [DOI] [PubMed] [Google Scholar]
- 20.Divac J, Rosvold HE, Szwarchart MK. Behavioral effects of selective ablation of the caudate nucleus. J Comp Physiol Psychol. 1967;63:184–190. doi: 10.1037/h0024348. [DOI] [PubMed] [Google Scholar]
- 21.Ferrante RJ, Hantraye P, Brouillet E, Kowall NW, Beal MF. Striatal pathology of impaired mitochondrial metabolism in primates profiles Huntington’s disease. Soc Neurosci Abstr. 1993;19:408. [Google Scholar]
- 22.Grafton ST, Mazziotta JC, Pahl JJ, George-Hyslop PS, Haines JL, Gusella J, Hoffman JM, Baxter LR, Phelps ME. A comparison of neurological, metabolic, structural, and genetic evaluations in persons at risk for Huntington’s disease. Ann Neurol. 1990;28:614–621. doi: 10.1002/ana.410280503. [DOI] [PubMed] [Google Scholar]
- 23.Hantraye P, Riche D, Maziere M, Isacson O. An experimental primate model of Huntington’s disease: anatomical and behavioural studies of unilateral excitotoxic lesions of the caudate-putamen in the baboon. Exp Neurol. 1990;108:91–104. doi: 10.1016/0014-4886(90)90014-j. [DOI] [PubMed] [Google Scholar]
- 24.Hantraye P, Leroy-Willig A, Denys A, Riche D, Isacson O, Syrota A. Magnetic resonance imaging to monitor pathology of caudate-putamen after excitotoxin-induced neuronal loss in the non-human primate brain. Exp Neurol. 1992a;118:18–23. doi: 10.1016/0014-4886(92)90018-l. [DOI] [PubMed] [Google Scholar]
- 25.Hantraye P, Riche D, Maziere M, Isacson O. Intrastriatal grafting of fetal striatal cross-species cells ameliorates abnormal movements in a primate model of Huntington’s disease. Proc Natl Acad Sci USA. 1992b;89:4187–4190. doi: 10.1073/pnas.89.9.4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harper PS. WB Saunders; London: 1991. Huntington’s disease (Harper PS, ed), pp 1–429. [Google Scholar]
- 27. Isacson O, Hantraye P, Riche D, Schumacher JM, Maziere M. The relationship between symptoms and functional anatomy in the chronic neurodegenerative diseases: from pharmacological to biological replacement therapy in Huntington’s disease. Lindwall O, Björklund A, Widner H. Intracerebral transplantation in movement disorders 1991. 245 258 Elsevier; Amsterdam: Science. [Google Scholar]
- 28.Jenkins BG, Koroshetz WJ, Beal MF, Rosen BR. Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized proton NMR spectroscopy. Neurology. 1993;43:2689–2695. doi: 10.1212/wnl.43.12.2689. [DOI] [PubMed] [Google Scholar]
- 29.Kremer B, Weber B, Hayden MR. New insights into the clinical features, pathogenesis and molecular genetics of Huntington’s disease. Brain Pathol. 1992;2:321–335. doi: 10.1111/j.1750-3639.1992.tb00709.x. [DOI] [PubMed] [Google Scholar]
- 30.Kuhl DE, Phelps ME, Markham CH, Metter EJ, Riege WH, Winter J. Cerebral metabolism and atrophy in Huntington’s disease determined by 18FDG and computed tomographic scan. Ann Neurol. 1982;12:425–434. doi: 10.1002/ana.410120504. [DOI] [PubMed] [Google Scholar]
- 31.Kuwert T, Lange HW, Langen KJ, Herzog H, Aulich A, Feinendegen LE. Cortical and subcortical glucose consumption measured by PET in patients with Huntington’s disease. Brain. 1990;113:1405–1423. doi: 10.1093/brain/113.5.1405. [DOI] [PubMed] [Google Scholar]
- 32.Ludolph AC, He F, Spencer PS, Hammerstad J, Sabri M. 3-nitropropionic acid: exogenous animal neurotoxin and possible human striatal toxin. Can J Neurol Sci. 1991;18:492–498. doi: 10.1017/s0317167100032212. [DOI] [PubMed] [Google Scholar]
- 33.Mann VM, Cooper JM, Javoy-Agid F, Agid Y, Jenner P, Schapira AHV. Mitochondrial function and parental sex effect in Huntington’s disease. Lancet. 1990;336:749. doi: 10.1016/0140-6736(90)92242-a. [DOI] [PubMed] [Google Scholar]
- 34.Martin JB, Gusella J. Huntington’s disease: pathologenesis and management. N Engl J Med. 1986;315:1267–1276. doi: 10.1056/NEJM198611133152006. [DOI] [PubMed] [Google Scholar]
- 35.Martin WRW, Clark C, Ammann W, Stoessl AJ, Shtyel W, Hayden MR. Cortical glucose metabolism in Huntington’s disease. Neurology. 1992;42:223–229. doi: 10.1212/wnl.42.1.223. [DOI] [PubMed] [Google Scholar]
- 36.Massman PJ, Delis DC, Butters N, Levin BE, Salmon DP. Are all subcortical dementias alike? Verbal learning and memory in Parkinson’s and Huntington’s disease patients. J Clin Exp Neuropsychol. 1990;12:729–744. doi: 10.1080/01688639008401015. [DOI] [PubMed] [Google Scholar]
- 37.Mazziotta JC, Phelps ME, Pahl JJ, Huang SC, Baxter LR, Riege WH, Hoffmann JM, Kuhl DE, Lanto AB, Wapenski JA, Markham CH. Reduced cerebral glucose metabolism in asymptomatic subjects at risk for Huntington’s disease. N Engl J Med. 1987;316:357–362. doi: 10.1056/NEJM198702123160701. [DOI] [PubMed] [Google Scholar]
- 38.Mendez MF, Astorna NE, Skoog-Lewandowski K. Neurobehavioral changes associated with caudate lesions. Neurology. 1989;39:49–355. doi: 10.1212/wnl.39.3.349. [DOI] [PubMed] [Google Scholar]
- 39.Parker WD, Jr, Boyson SJ, Luder AS, Parks JK. Evidence for a defect in NADH: ubiquinone oxidoreductase (complex I) in Huntington’s disease. Neurology. 1990;40:1231–1234. doi: 10.1212/wnl.40.8.1231. [DOI] [PubMed] [Google Scholar]
- 40.Pillon B, Dubois B, Ploska A, Agid Y. Severity and specificity of cognitive impairment in Alzheimer’s, Huntington’s, and Parkinson’s diseases and progressive supranuclear palsy. Neurology. 1991;41:634–643. doi: 10.1212/wnl.41.5.634. [DOI] [PubMed] [Google Scholar]
- 41.Randolph C. Implicit, explicit, and semantic memory functions in Alzheimer’s disease and Huntington’s disease. J Clin Exp Neuropsychol. 1991;13:479–494. doi: 10.1080/01688639108401065. [DOI] [PubMed] [Google Scholar]
- 42.Richfield EK, Twyman R, Berent S. Neurological syndrome following bilateral damage to the head of the caudate nuclei. Ann Neurol. 1987;22:768–771. doi: 10.1002/ana.410220615. [DOI] [PubMed] [Google Scholar]
- 43.Roberts AC, De Salvia MA, Wilkinson LS, Collin P, Muir JL, Everitt BJ, Robbins TW. 6-hydroxydopamine lesions of the prefrontal cortex in monkeys enhance performance on an analog of the Wisconsin Card Sort Test: possible interactions with subcortical dopamine. J Neurosci. 1994;14:2531–2544. doi: 10.1523/JNEUROSCI.14-05-02531.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schneider JS. Behavioral and neuropathological consequences of chronic exposure to low doses of the dopaminergic neurotoxin MPTP. In: Isaacson RL, Jensen KF, editors. The vulnerable brain and environmental risks. Plenum; New York: 1992. pp. 293–308. [Google Scholar]
- 45.Taylor JR, Elsworth JD, Roth RH, Collier TJ, Sladek JR, Redmond DE. Improvements in MPTP-induced object retrieval deficits and behavioral deficits after fetal nigral grafting in monkeys. Prog Brain Res. 1990a;62:543–559. doi: 10.1016/s0079-6123(08)62645-x. [DOI] [PubMed] [Google Scholar]
- 46.Taylor JR, Redmond JD, Roth RH, Sladek JR, Redmond JR. Cognitive and motor deficits in the acquisition of an object retrieval/detour task in MPTP-treated monkeys. Brain. 1990b;113:617–637. doi: 10.1093/brain/113.3.617. [DOI] [PubMed] [Google Scholar]
- 47.The Huntington’s Disease Collaborative Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 48.Young AB, Penney JB, Starosta-Rubenstein S, Markel DS, Berent S, Giordani B, Ehrenkaufer R, Jewett D, Hichwa R. PET scan investigations of Huntington’s disease: cerebral metabolic correlates of neurologic features and functional decline. Ann Neurol. 1986;20:296–303. doi: 10.1002/ana.410200305. [DOI] [PubMed] [Google Scholar]