

Abstract
Although hyperkinesis is expressed in several neurological disorders, the biological basis of this phenotype is unknown. The mouse mutant coloboma (Cm/+) exhibits profound spontaneous locomotor hyperactivity resulting from a deletion mutation. This deletion encompasses several genes including Snap, which encodes SNAP-25, a nerve terminal protein involved in neurotransmitter release. Administration of amphetamine, a drug that acts presynaptically, markedly reduced the locomotor activity in coloboma mice but increased the activity of control mice implicating presynaptic function in the behavioral abnormality. In contrast, the psychostimulant methylphenidate increased locomotor activity in both coloboma and control mice. When a transgene encoding SNAP-25 was bred into the coloboma strain to complement the Snap deletion, the hyperactivity expressed by these mice was rescued, returning these corrected mice to normal levels of locomotor activity. These results demonstrate that the hyperactivity exhibited by these mice is the result of abnormalities in presynaptic function specifically attributable to deficits in SNAP-25 expression.
Keywords: hyperactivity, ADHD, locomotor activity, amphetamine, methylphenidate, transgenic, SNAP-25, mouse mutant, psychostimulant, coloboma
Pathological hyperactivity is a common problem in pediatric neuropsychiatry. Hyperactivity syndromes are thought to account for a large proportion of children diagnosed with learning disabilities, and are extremely disruptive to families and school environments. Therefore, a great deal of attention has been focused on the causes and treatment of hyperactivity in children. It is almost certain that multiple factors, both genetic and environmental, contribute to the development of hyperactivity. This observation is supported by the fact that pathological hyperactivity exists as a major feature of several distinct neuropsychiatric syndromes. For example, attention deficit-hyperactivity disorder (ADHD) is described by poor attention span, impulsivity, hyperactive behavior, and it is the most common pediatric neuropsychiatric disorder, affecting ∼2–4% of school age children (Whalen and Henker, 1976; Shaywitz and Shaywitz, 1984; Anastopoulos and Barkley, 1988; Safer and Krager, 1988). Tourette syndrome is another childhood onset disorder, characterized by vocal and motor tics, in which hyperactivity is a comorbid condition in ∼50% of affected individuals (Pauls et al., 1986). Although hyperactivity is a cardinal sign in many neurological disorders, the lack of a genetically defined animal model of hyperkinesis has hampered efforts to identify the biological deficits underlying this behavioral phenotype.
We have recently identified the mouse mutant coloboma as a novel animal model for examining the neurobiological basis of hyperactivity (Hess et al., 1992). Mice heterozygous for the semidominant mutation coloboma (Cm) exhibit a triad of overt abnormalities consisting of profound spontaneous hyperactivity, head bobbing, and ocular dysmorphology (Searle, 1966; Hess et al., 1992). The hyperactivity exhibited by coloboma mice averages 3 times the activity of the control littermates. In fact, the activity of some mutants exceeds 10 times that of their control littermates. Although the spontaneous hyperactivity is extreme, the coloboma mouse CNS appears normal with no obvious neuropathology.
We have previously defined the genetic defect in these mice as a deletion mutation on mouse chromosome 2 comprised of 1–2 cM encompassing genes encoding phospholipase C isoform β-1 and the nerve terminal protein SNAP-25 (Hess et al., 1992, 1994). Recent evidence suggests that SNAP-25, which is associated with the plasma membrane of axon terminals (Oyler et al., 1989), plays a critical role in neurotransmitter release in the nervous system. SNAP-25 has been identified as a component of the machinery essential for docking and holding synaptic vesicles at the presynaptic membrane in readiness for Ca2+-triggered neurotransmitter exocytosis (Horikawa et al., 1993; O’Connor et al., 1993; Söllner et al., 1993a,b). That neurotransmission is blocked by botulinum neurotoxins A and E, which specifically recognize and cleave SNAP-25, provides additional evidence for the involvement of SNAP-25 in vesicle fusion (Blasi et al., 1993; Schiavo et al., 1993). In coloboma (Cm/+) mice, deletion of the Snap gene results in 50% lower amounts of SNAP-25 mRNA and protein expression than wild-type (+/+) mice (Hess et al., 1992). Because the intactSnap allele in Cm/+ mice does not compensate for the deleted Snap gene with increased transcription, it is likely that deficiencies in SNAP-25 expression, resulting in anomalous presynaptic function, are involved in the neurological abnormalities exhibited by these mutant mice. Here we demonstrate that the hyperactivity expressed by coloboma mice is clearly the result of a deletion of the Snap gene.
MATERIALS AND METHODS
Coloboma mouse mutants. Eight- to ten-week-oldCm/+ mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and subsequently bred at both The Scripps Research Institute and The Pennsylvania State University College of Medicine. The coloboma mutation originally identified on a C3H/HeH×101/H F1 male was backcrossed for 32 generations onto a C57BL/6By strain. The mutation has since been backcrossed to the C3H/HeSnJ strain for at least 10 generations at The Jackson Laboratory. The mutation has been maintained by matings of male Cm/+ with +/+ C3H/HeSnJ females, because Cm/+ females generally make poor mothers. Mutant (Cm/+) mice are identified at weaning primarily by head bobbing, but also by the presence of sunken, squinty eyes. The coloboma mutants can also be distinguished at this time by their hyperactivity and, consequently, their smaller size. In matings of Cm/+ mice, embryos homozygous for theCm allele have not been observed (Theiler and Varnum, 1981), and this has been attributed to early lethality before embryonic day 6.
Behavioral testing. Mice were maintained in group cages onad libitum food and water with a reverse 12 hr light/dark cycle. To quantitate locomotor activity, Cm/+ mice and control littermates were placed in individual, automated photocell activity cages (29.2 × 50.5 cm) with twelve 2-cm-high infrared beam detectors arranged in a 4 × 8 grid. A computer recorded beam breaks, which were accumulated every 10 min for the duration of the test with changes in beam status assessed 18 times/sec (San Diego Instruments, San Diego, CA). Before the start of the test, mice were first habituated to the testing room for at least 1 hr. The records are presented either as actual beam interruptions or as movements defined as changes in the x,y position that required breaking of two adjacent photocell beams as described by Geyer and Paulus (1992). For the amphetamine and methylphenidate dose–response analyses, coloboma mice on the C3H/HeSnJ strain were tested with wild-type age- and sex-matched littermates as controls. When transgenic mice were tested, littermates and siblings from the same founder line were tested.
For drug tests, mice were habituated to the photobeam cages for 4 hr before drug injection; injections occurred 1 hr after the start of the dark cycle. Equal numbers of male and female mice were tested in both the coloboma and control groups. Mice (7–9 weeks of age) were first treated subcutaneously with saline to obtain baseline activity before similar injections of d-amphetamine sulfate (Sigma, St. Louis, MO) or methylphenidate hydrochloride (Sigma, St. Louis, MO) in 0.9% NaCl. Because it is difficult to breed large numbers of age- and sex-matched animals, a repeated dosing paradigm was used to evaluate drug effects. Mice were tested with the lowest dose first, followed by progressively increasing doses with a 7 d interval between doses. Activity measures commenced immediately after injection.
Mice were rated for stereotypy for 2 hr after amphetamine injection. Behavior was scored in dim light every 10 min for a 30 sec period on a behavioral scale (Creese and Iversen, 1975) modified for mice in which: 0 = sleeping; 1 = awake, inactive; 2 = active or exploring; 3 = hyperactive; 4 = hyperactive with bursts of stereotypic behavior (sniffing in one spot, licking the cage wall, repetitive grooming or gnawing); 5 = continuous persistent stereotypy; and 6 = dyskinetic behavior (reverse locomotion, body contortions, or bouncing). The first stereotypy rating was made 10 min after drug injection. Behavioral ratings were analyzed by the Mann–Whitney U test.
Transgenic mice. The mini-Snap transgene was constructed using standard recombinant DNA techniques (Sambrook et al., 1989). The nearly full-length SNAP-25 cDNA clone p8.71 (Oyler et al., 1989), which contains the majority of the 5′-untranslated region, the entire open reading encoding the major b isoform expressed in adult brain, and most of the 3′-untranslated region cloned in the plasmid vector Bluescript II KS (Stratagene, San Diego, CA), was modified by PCR to generate a BspEI site in the 5′-untranslated region corresponding to the unique but polymorphic BspEI site of the genomic DNA clone pG1.1. pG1.1 is a plasmid subclone isolated from a lambda recombinant library constructed of partial MboI genomic DNA fragments of strain 129/By mice. Primer extension and S1 mapping experiments were used to identify theSnap gene transcriptional initiation in pG1.1 to two sites separated by 29 base pairs (bp) 119 and 148 bp upstream of the 3′ boundary exon 1, and ∼1500 bp surrounding this first exon has been sequenced (Ryabinin et al., 1996). A fragment extending 10 kb upstream and including the BspEI site in exon 1 of pG1.1 was ligated to the regenerated BspEI site of p8.71 to insert 1511 bp of the cDNA sequence downstream of this genomic promoter sequence. To provide for the polyadenylation of transcripts, a 220 bpBamHI fragment containing the SV40 polyadenylation site of pCMVβ (MacGregor and Caskey, 1989) was inserted in the uniqueBamHI site of the polylinker sequence 3′ of the SNAP-25 cDNA p8.71 before ligation to the genomic promoter fragment. Finally, a rat insulin II gene fragment containing 120 bp of intron 1 sequence (Bell et al., 1980) was generated using PCR to contain flankingBspEI sites and was inserted into the uniqueBspEI in the cDNA 5′-untranslated region to complete the mini-Snap transgene. Transgenic animals were generated at TSRI Transgenic Animal Facility by standard techniques (Hogan et al., 1986) injecting linearized plasmid DNA into fertilized C57BL/6×C3H/HeJ hybrid oocytes followed by implantation into pseudopregnant mothers. Initially transgenic mice were identified by PCR using primers to amplify a unique fragment of 3′-untranslated region fused to the SV40 3′ polyadenylation sequence. The founder transgenics C3H/HeJ×C57BL/6 were first crossed with C3H/HeJ mice, and mini-Snaptransgenic lines were then established through brother–sister matings. Genotypes were determined by quantitative hybridization to slots blots of genomic DNAs isolated from tail clippings. Mice bearing theCm deletion were scored by 50% levels of hybridization with a 417 bp probe to rat Plcb-1, encoding PLCβ-1, previously shown to be codeleted with the Snap gene (Hess et al., 1994), and the number of alleles of integrated transgene was similarly distinguished using the BamHI fragment probe against the SV40 polyadenylation sequence. All blots were normalized with a probe to the c-fos gene (generously provided by T. Curran, Roche Institute), which is unlinked to the Cm deletion, to establish diploid gene copy hybridization levels for each mouse.
In situ hybridization. The in situ hybridization procedure has been described in detail (Wilson and Higgins, 1989). A35S-labeled single-stranded antisense RNA probe was generated from the BamHI fragment containing the SV40 polyadenylation sequence (220 bp) derived from the vector pCMV (Invitrogen, San Diego, CA) subcloned into pE2, an intermediate plasmid used in construction of the Snapminigene. The antisense RNA probe to SNAP-25 was synthesized from the murine SNAP-25 cDNA, subcloned into Bluescript, and linearized withMscI to produce a 250 base transcript. After in vitro transcription, the DNA template was removed by digestion with RNase-free DNase (Promega, Madison, WI). Pretreatment of the slide-mounted sections included fixation in buffered 4% formaldehyde, followed by treatment with 25 μg/ml proteinase K in 5× TE (50 mm Tris, pH 8.0, and 5 mmEDTA), treatment in 0.5N HCl, and a final post-fixation in buffered 4% formaldehyde. Sections were then dehydrated in graded ethanols and air-dried. Slides were prehybridized at 56°C for 2–3 hr in ∼750 μl of a solution containing 50% formamide, 0.75 m NaCl, 20 mm1,4-piperazinediethanesulfonic acid, pH 6.8, 10 mm EDTA, 10% dextran sulfate, 5× Denhardt’s solution (0.02% bovine serum albumin, 0.02% Ficoll, and 0.02% polyvinylpyrolidone), 50 mm dithiothreitol, 0.2% SDS, and 100 μg/ml each salmon sperm DNA and yeast tRNA. Prehybridization buffer was removed from the slides, and 75 μl of hybridization solution consisting of prehybridization buffer plus 10 ng of probe was applied to each slide. Slides were coverslipped and hybridized for 16 hr at 56°C. After hybridization, coverslips were removed in 4× SSC (0.15 m NaCl and 0.015 m sodium citrate) plus 300 mm 2-mercaptoethanol and rinsed in 4× SSC without 2-mercaptoethanol. Sections were treated with 50 μg/ml pancreatic RNase A in 0.5 m NaCl, 50 mm Tris, pH 8.0, 5 mm EDTA, rinsed in this same buffer, washed in 2× SSC at 56°C, and air-dried. Sections were exposed to x-ray film (DuPont Cronex, Boston, MA) for ∼3 d.
Protein slot blots. The level of SNAP-25 protein expressed from the Snap minitransgene was assayed using slot blots of increasing amounts of protein isolated from 1% Nonidet P-40-solubilized extracts of brain regions dissected from adult male homozygous transgenic Sp/Sp Cm/+, and nontransgenic −/−Cm/+ and −/− +/+ mice, as described previously (Hess et al., 1992). The blots were first blocked and then probed with the SNAP-25 monoclonal antibody SMI-81 (1:2000 dilution; Sternberger Monoclonals) in 5% nonfat dry milk, Tris-buffered saline, 1% Tween-20 at room temperature. After washes in the same buffer, the filters were reacted with 0.5 μCi/ml I125-labeled sheep anti-mouse secondary antibody. The radioactive secondary antibody was quantitated using a PhosphorImager (Molecular Dynamics, Sunnyvale, CA), and the slope of the relative radioactivity versus the amount of protein was used to determine the relative amount of SNAP-25 protein expressed by each genotype. Only protein concentrations that gave a linear regression to the origin (r > 0.9, generally obtained loading 0.5–2.5 μg protein/slot) were used to determine protein levels. At least three determinations of protein extract prepared from the indicated brain regions were used to compare each genotype.
RESULTS
Amphetamine responses
Amphetamine, a psychostimulant that acts at the presynaptic terminal to promote catecholamine release, is effective in ameliorating the hyperactivity expressed in ADHD-affected children (Barkley, 1977;Shaywitz and Shaywitz, 1984). Coloboma mice were challenged with the indirect dopamine agonist amphetamine, and the behavioral response was assessed by evaluating spontaneous locomotor activity and stereotypic behavior. As shown in Figure 1, coloboma mice were 3–4 times more active than normal littermates after saline administration throughout the 3 hr testing period. However, subcutaneous injection of d-amphetamine sulfate markedly suppressed coloboma mouse activity 20–90 min after injection. In the 20–90 min after injection, 2 mg/kg amphetamine significantly reduced the locomotor activity of coloboma mice compared with the locomotor activity exhibited by coloboma mice treated with saline (Student’st test, p < 0.05). After coloboma mice received 4 mg/kg amphetamine, a trend toward a reduction in locomotor activity below saline-treated baseline Cm/+ activity was also observed (Student’s t test, p < 0.09). These doses either produced no effect (2 mg/kg) or potentiated locomotor activity (4 mg/kg) in control mice.
Fig. 1.
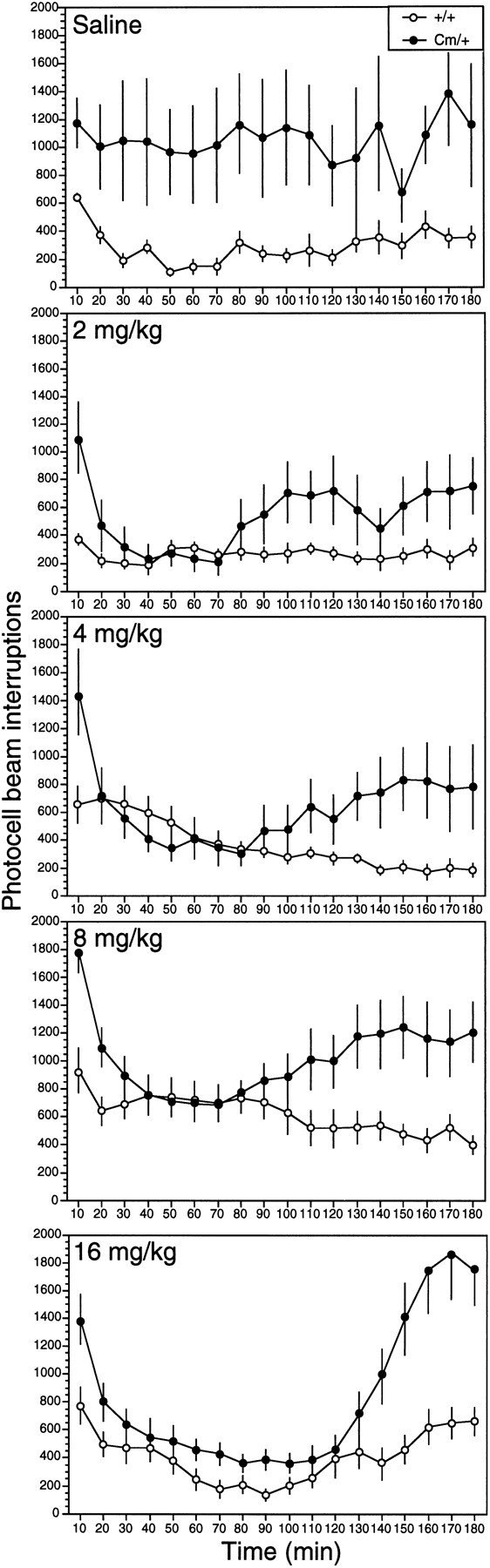
Locomotor activity exhibited by control (n = 8) and coloboma (n = 8) mice after administration of saline or d-amphetamine sulfate. Mice were habituated to the test cages for 4 hr before subcutaneous injection, and photocell beam interruptions were recorded every 10 min for 3 hr postinjection. Data represent mean ± SEM. ANOVA for repeated measures indicated a significant genotype (F(1,14) = 6.18, p < 0.05) and repeated-measures (F(1,17) = 1.689, p < 0.05) effect for the saline treatment. A significant Genotype × Time interaction effect (F(17,238) = 1.69, p < 0.05 for 2 mg/kg; F(17,238) > 5.25, p < 0.001 for all other doses) was observed for all doses of d-amphetamine tested.
Psychostimulants, such as amphetamine, can also induce repetitive, focused behaviors (stereotypy) that result in decreased locomotor activity. To distinguish between drug-induced stereotypy and a drug-induced reduction in locomotor activity without stereotypy, mice were observed and rated for their qualitative response to amphetamine for 120 min after injection. Consistent with the locomotor activity recorded by the photocell cages, the behavioral ratings demonstrated that saline-injected coloboma mice were significantly more active than their wild-type littermates (Fig. 2). Exposure to 2 or 4 mg/kg d-amphetamine decreased the activity ofCm/+ mice between 20 and 90 min postinjection without inducing stereotypy. During this time, Cm/+ mice treated with amphetamine scored in the normal mouse activity range of 1–2 (Fig. 2).
Fig. 2.
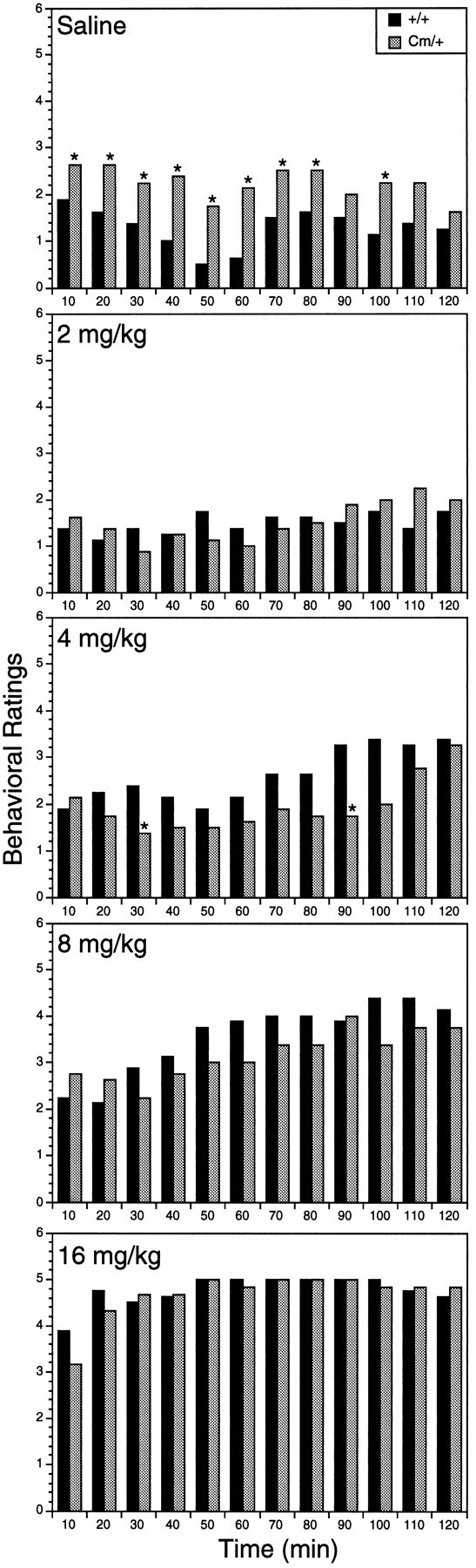
Behavioral rating scores for control (black bars; n = 8) and coloboma mice (graybars; n = 8) after administration of saline ord-amphetamine sulfate. Mice were rated every 10 min for 2 hr after injection. Data were analyzed using the Mann–WhitneyU test for nonparametric statistics; asterisksindicate significant difference between coloboma and wild-type littermates (p < 0.05). For ease of presentation, data are shown as mean scores.
Higher doses of amphetamine induced stereotypy in both control and coloboma mice. At 8 mg/kg amphetamine, occasional bursts of stereotyped behavior were observed in mutant and wild-type mice. Although +/+ locomotor activity was clearly potentiated by this dose, coloboma mouse activity was still below baseline coloboma locomotor activity. At the highest dose of amphetamine administered, persistent stereotypy was observed in both control and coloboma mice. At all doses of amphetamine, the behavioral effects of the drug diminished in parallel with control and coloboma mice returning to baseline levels of activity, reflecting comparable drug clearance in normal and mutant mice.
Because the amphetamine-induced reduction in locomotor activity in coloboma mice was remarkably similar to the response of ADHD-affected children administered psychostimulants (Barkley, 1977; Shaywitz and Shaywitz, 1984), we also tested coloboma mice in the same behavioral paradigm with methylphenidate, the drug more commonly used to treat hyperactive children. The dose-dependent response to methylphenidate in coloboma mice was markedly different from the amphetamine response. Methylphenidate (2–32 mg/kg) increased the locomotor activity inboth control and coloboma mice in a dose-dependent manner (Fig. 3). Stereotypy was also induced in parallel in control and coloboma mice (data not shown); persistent stereotypy was only observed at the highest doses of methylphenidate (16 and 32 mg/kg). Thus, unlike amphetamine, the methylphenidate response in coloboma mice appears normal.
Fig. 3.
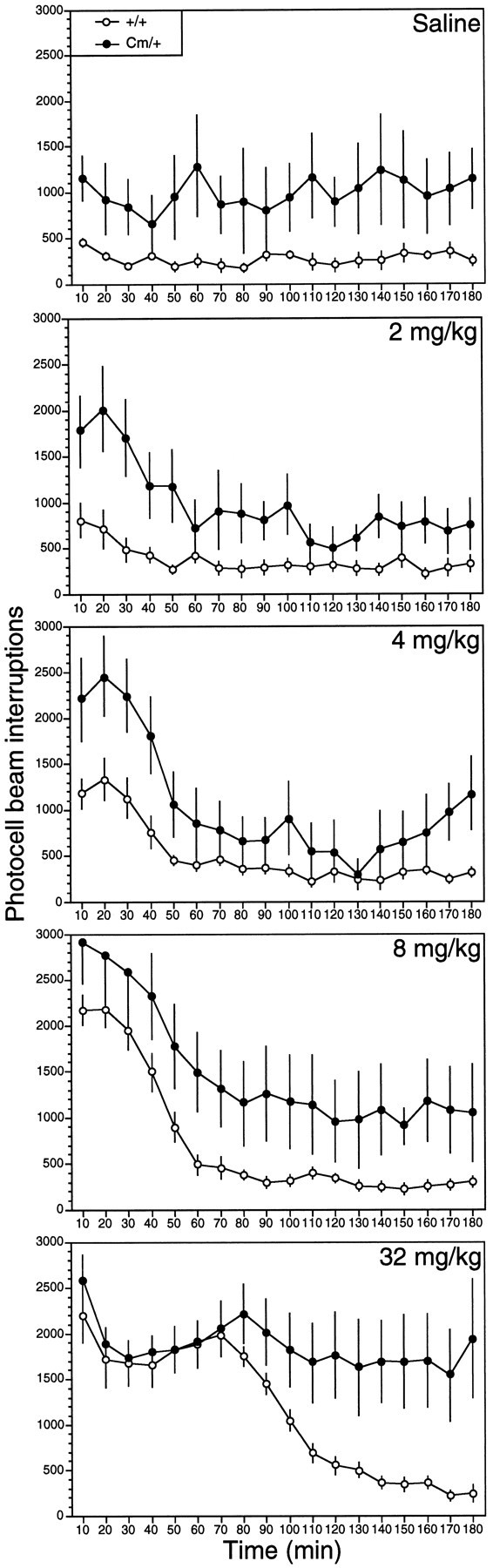
Locomotor activity exhibited by control (n = 8) and coloboma (n = 8) mice after administration of saline or methylphenidate hydrochloride. Mice were habituated to the test cages for 4 hr before subcutaneous injection, and photocell beam interruptions were recorded every 10 min for 3 hr postinjection. Data represent mean ± SEM. ANOVA for repeated measures indicated a significant repeated-measures effect for 4 mg/kg methylphenidate (F(1,17) = 18.514, p < 0.0005) and 8 mg/kg methylphenidate (F(1,17) = 38.291, p < 0.0005) with no significant effects of groups or interaction for either dose. A significant Genotype × Time interaction effect was observed for 2 mg/kg methylphenidate (F(17,238) = 2.164, p < 0.01) and 32 mg/kg methylphenidate (F(17,238) = 5.409, p < 0.0005).
Transgenic mice overexpressing SNAP-25
Because the abnormal response to amphetamine suggested a presynaptic component to the regulation of locomotor hyperactivity in coloboma mice, the contribution of reduced SNAP-25 expression to the coloboma phenotype was tested by generating transgenic mice overexpressing SNAP-25 to be crossed with Cm/+ mutants to compensate for the deleted Snap allele. For this rescue experiment, a SNAP-25 minigene (Sp) was constructed using 10 kb of genomic DNA 5′ to and including the transcriptional start site of theSnap gene (Ryabinin et al., 1996) to drive expression. Preliminary experiments showed that this region upstream of the transcriptional start site was sufficient to drive β-galactosidase gene activity in PC12 cells (E. Hess and M. Wilson, unpublished data). The Snap gene encodes two isoforms of the protein, SNAP-25a and SNAP-25b, which are generated by alternative splicing (Bark, 1993). This Snap promoter was fused to the intact coding sequence corresponding to the SNAP-25b isoform, the predominant SNAP-25 mRNA species expressed in adult brain (Bark et al., 1995). Post-transcriptional processing was provided by incorporating the short intron 1 of the rat insulin II gene into the 5′-untranslated sequences of the SNAP-25 cDNA, and the SV40 polyadenylation sequence was added to the 3′ end of the cDNA (Fig. 4). Founder lines of transgenic mice generated with C3H/HeJ×C57Bl/6 hybrid embryos were expanded and bred onto the C3H/HeJ strain, the strain on which theCm mutation has been inbred. After breeding the transgene to homozygosity, several lines were identified that expressed the SNAP-25 transgene in brain. Mice either hemizygous or homozygous for the transgene exhibited no overt behavioral defects and were phenotypically indistinguishable from nontransgenic littermates.
Fig. 4.

The mini-Snap transgene. TheSnap promoter was fused to a SNAP-25 cDNA and a rat insulin intron, and SV40 polyadenylation signals were added to provide post-transcriptional processing sites. Diagonal arrowsindicate the two transcriptional start sites.
Expression of the Snap transgene
The level of transgene expression in six founder lines was compared using in situ hybridization. To distinguish the expression of the transgene from endogenous SNAP-25 expression, an RNA probe to the SV40 polyadenylation sequence was used to detect the transgene transcript. The pattern of expression of the transgene in the CNS was similar in all founder lines with some variability in relative levels of expression. Although the transgene was expressed at somewhat lower levels than the endogenous gene, the Snap minigene was expressed throughout the brain, similar to endogenous SNAP-25 expression (Fig. 5). No detectable hybridization was found in myelinated tracts or in glia-enriched regions of white matter, consistent with the neuron-specific expression of SNAP-25. For most lines, high levels of Snap transgene expression were present in granular layer of the cerebellum, and expression was also present in cortical and subcortical brain regions. Using an SV40 polyadenylation signal probe similar to that used for the in situhybridization, RNase protection assays to tissue RNAs of theSnap minigene founder 40 line confirmed the expression of the transgene in cortex as well as higher levels in cerebellum, but notably also demonstrated lack of expression in non-neuronal tissue including liver, kidney, and spleen (data not shown).
Fig. 5.

Localization of Snap transgene mRNA expression. Parasagittal sections were hybridized with a probe to the SV40 polyadenylation sequence (a) or a probe to the murine SNAP-25 cDNA (b). The SV40 probe was hybridized to a section from an Sp/Sp +/+ mouse derived from founder 40, revealing the distribution of the transgene expression (a). A similar pattern of transgene expression was observed in several other founder lines. The SNAP-25 probe was hybridized to a section obtained from a −/− +/+ littermate to illustrate the localization of endogenous SNAP-25 mRNA expression for comparison with the transgene expression. Sections were processed in parallel in the same experiment and apposed to x-ray film for ∼60 hr.
Additionally, SNAP-25 protein levels in brain were assessed in transgenic mice derived from founder line 40 using a monoclonal antibody (SMI81) to SNAP-25 in a quantitative slot blot analysis of brain extracts of coloboma mice without (genotype, −/−Cm/+) or homozygous for the transgene (genotype, Sp/SpCm/+), the genotype in which complete correction of the hyperactivity was observed (see below). For comparison, the level of SNAP-25 protein in nontransgenic, wild-type −/− +/+ mice was used as reference for “normal” levels of SNAP-25 protein expression. Although analysis of proteins extracted from whole brain demonstrated only a small (5%) increase in SNAP-25 protein expression in Sp/SpCm/+ over −/− Cm/+ mice, a regional analysis revealed a differential pattern of transgene-induced SNAP-25 protein expression in brain regions directly involved in the regulation of motor activity. In striatum and cerebellum of Cm/+ mice homozygous for the transgene SNAP-25, protein levels were increased by >30% compared with coloboma mutants (relative to normal −/− +/+ levels in striatum: Sp/Sp Cm/+ 0.812 ± 0.090, −/−Cm/+, 0.613 ± 0.007; in cerebellum: Sp/Sp Cm/+ 0.759 ± 0.053, −/− Cm/+ 0.557 ± 0.004). In contrast to the apparent restoration to ∼80% of normal mouse SNAP-25 levels in these regions, SNAP-25 expression in cerebral cortex was comparable inCm/+ mice with or without the transgene.
Transgenic rescue of hyperactivity in coloboma mice
To determine the precise relationship of SNAP-25 to the coloboma phenotype, the Snap transgene was bred onto the coloboma mouse strain. The resulting mice were evaluated visually for head bobbing and ocular dysmorphology, and their locomotor activity was quantified. As shown in Figure 6, +/+ mice without (−/−), or hemizygous (Sp/−) or homozygous (Sp/Sp) for the transgene exhibit virtually identical levels of activity, consistent with little or no obvious effect of the transgene on wild-type behavior. In contrast, the Cm/+ mice homozygous for the transgene (Sp/SpCm/+) were significantly less active than the nontransgenicCm/+ littermates; in fact, Sp/Sp Cm/+ mice exhibited activity comparable with control littermates. Additionally, there was a trend toward a reduction in activity in Cm/+ mice hemizygous for the Snap minigene (Sp/−Cm/+), consistent with a dose response of the transgene complementing the hyperactivity resulting from the Cmdeletion mutation.
Fig. 6.

Complementation of spontaneous hyperactivity in coloboma mice by the Snap transgene (Sp). Progeny (n = 92) generated from an Sp/− +/+ × Sp/−Cm/+ cross were tested for spontaneous locomotor activity. Activity was recorded for 3 hr and is expressed as average movements per 10 min. Data represent mean ± SEM. Data were analyzed by one-way ANOVA followed by a Scheffe’s post hoc test. Asteriskindicates significantly different (p < 0.05) from −/−Cm/+ mice derived from the same cross. Note that the locomotor activity expressed by Sp/Sp Cm/+ mice was not significantly different from +/+ mice either with or without the transgene. All data presented were obtained from a single transgenic founder (line 40); similar results have been obtained from the progeny of the same cross matings with two additional founder lines (lines 4 and 45) wherein homozygosity for the transgene was also effective in significantly reducing the hyperactivity of Cm/+ mice.
Although the SNAP-25 transgene bred into the coloboma line clearly resulted in mice with quantitatively normal locomotor activity, expression of the transgene was not sufficient to completely rescue all elements of the coloboma phenotype. Mice homozygous for the transgene, for example, displayed more individual variability in their level of activity than nontransgenic and transgenic normal (+/+) animals. Head bobbing, which is seen in virtually all coloboma mice, was observed in 5 of the 6 Sp/Sp Cm/+ mice. Additionally, although we have observed that the severity of the eye dysmorphology in the mutants varies considerably, and is not always observed in all Cm/+ mice, 3 of the 6 Sp/Sp Cm/+ mice had the sunken, closed-eye characteristic of the coloboma phenotype, whereas 14 of 19 Sp/−Cm/+ mice and 8 of 10 −/− Cm/+ mice bore this dysmorphology.
Restoration of the amphetamine response
The “rescued” Cm/+ mice were tested for their response to amphetamine to determine whether the amphetamine response was corrected in parallel with the transgene-induced reduction in locomotor activity. Although amphetamine significantly reduced the locomotor activity of nontransgenic Cm/+ mice, the same dose of amphetamine increased activity of Sp/Sp Cm/+ mice (Fig.7). The amphetamine-induced increase in activity was comparable with amphetamine responses in wild-type (Fig. 1) and Sp/Sp +/+ mice (Fig. 7). Thus, the Snap transgene reversed the paradoxical suppressive effect of amphetamine attributed to theCm mutation.
Fig. 7.

Complementation of the abnormal amphetamine response in coloboma mice by the Snap transgene (Sp). Sp/Sp +/+ (n = 9) and Sp/Sp Cm/+ (n = 6) derived from line 40 were administered 4 mg/kgd-amphetamine after a 4 hr habituation to the testing room and locomotor activity was assessed. The data represent the mean ± SEM of the activity of 10 min intervals taken over a 60 min period starting 10 min after injection; a two-way ANOVA reveals a significant drug effect (F(1,26) = 25, p = 0.0001), but no significant effect of genotype (F(1,26) = 1.4, p = 0.24) or drug/genotype (F(2,26) = 0.65, p = 0.43) interaction.
DISCUSSION
Coloboma mice represent an unprecedented model in which to study the contributions of a single known gene to a complex multifactorial phenotype in humans. The expression of SNAP-25, a presynaptic protein involved in synaptic vesicle fusion and transmitter release, is reduced in coloboma mice. Replacement of the deleted Snap gene with a Snap transgene ameliorated the hyperactivity, demonstrating that the reduction in SNAP-25 expression is sufficient to cause hyperactivity. In fact, the rescue of the hyperactivity was clearly dependent on the “dose” of transgene with the minigene in the hemizygous state reducing, but not eliminating, the hyperactivity. In the homozygous state, the transgene clearly provided enough SNAP-25 to complement the hyperactivity completely, although SNAP-25 expression did not achieve wild-type mRNA or protein levels. That the SNAP-25 transgene complemented neither ophthalmic deformation nor head bobbing suggests that other genes encompassed by the Cm deletion contribute to the expression of these elements of the coloboma mutant phenotype. The correction of the hyperactivity in coloboma mice with the Snap transgene indicates that the deletion of theSnap gene clearly plays a central role in the expression of hyperactivity.
SNAP-25 is expressed as two isoforms in the CNS; in the mouse, SNAP-25a is the mRNA species associated with early CNS development and SNAP-25b is the major isoform expressed in adult brain (Bark et al., 1995). The SNAP-25b cDNA was used in the transgene and effectively complemented the hyperactivity, suggesting that the hyperactivity is a consequence of persistent SNAP-25b deprivation in the adult coloboma mouse and not a result of developmental anomalies associated with a reduction in the SNAP-25a isoform. SNAP-25b is thought to participate in docking and fusion of small, clear synaptic vesicles for neurotransmitter release (see Bark and Wilson, 1994; Bark et al., 1995). Consequently, as a structural element of regulated vesicle fusion, the reduced level of SNAP-25b expression likely results in deficits in the efficiency of neurotransmitter release that are ultimately expressed phenotypically as hyperactivity. Because hyperactivity is observed with 50% levels of SNAP-25 protein inCm/+ mice, the behavioral phenotype may represent those pathways placing a high demand on SNAP-25 function. In fact, the striatum and cerebellum, regions implicated in locomotor activity, achieved near normal levels of SNAP-25 expression in rescued mice, suggesting that SNAP-25 is integral to the normal function of these pathways.
Amphetamine dramatically reduced the locomotor activity of coloboma mice, similar to the response to psychostimulants in hyperkinetic children. In contrast, methylphenidate, another indirect acting dopamine agonist, increased the coloboma locomotor activity in a dose-dependent manner in parallel with their control littermates. The contrasting behavioral responses to the indirect acting dopamine agonists amphetamine and methylphenidate in coloboma mice likely reflect an abnormality intrinsic to presynaptic function as opposed to a more general systemic abnormality resulting from misregulation of the pathways governing locomotor activity. In normal mice, both amphetamine and methylphenidate increase synaptic catecholamine concentrations, resulting in locomotor hyperactivity through complex motor feedback loops including corticostriate, nigrostriatal, and striato–pallido–thalamic loops. If one or all of these pathways were disrupted in coloboma mice, an increase in synaptic dopamine caused by either methylphenidate or amphetamine should result in a comparable and parallel behavioral response. Instead, we have observed opposite responses to these two drugs, suggesting that the differences in the drugs’ mechanism of action are responsible for the contrasting behavioral responses to amphetamine and methylphenidate.
Because both methylphenidate and amphetamine act at the presynaptic terminal, it is likely that abnormalities in presynaptic mechanisms that involve SNAP-25 are responsible for the opposing effects of the drugs. Although methylphenidate and amphetamine produce behavioral activation by increasing the synaptic concentration of dopamine, the mechanisms of action of these two drugs are dissimilar. Psychostimulants have been categorized on the basis of whether the response is attenuated by pretreatment with reserpine (Scheel-Kruger, 1971; McMillen, 1983), a drug that disrupts vesicular release by depleting vesicular stores of catecholamines. The action of amphetamine and amphetamine-like compounds is not inhibited by reserpine pretreatment and is therefore thought to increase synaptic dopamine levels independent of vesicular release. In fact, amphetamine itself appears to disrupt vesicular stores of dopamine and causes reversal of the dopamine transporter, resulting in the efflux of dopamine via the transporter (Sulzer and Rayport, 1990; Sulzer et al., 1993, 1995). In contrast, the methylphenidate class of psychostimulants (methylphenidate, cocaine, and nomifensine) blocks reuptake (Scheel-Kruger, 1971; Butcher et al., 1991) and is inhibited by reserpine-induced vesicular depletion. In coloboma mice, methylphenidate produces classical psychostimulant effects, suggesting that reuptake mechanisms and postsynaptic responses are functional. The paradoxical effects produced by amphetamine are more difficult to interpret. That coloboma mice respond to low doses of amphetamine with a decrease in locomotor activity suggests that amphetamine may actually be reducing synaptic dopamine concentrations. In fact, it has been demonstrated recently that in rats, the response to low-dose amphetamine is significantly blunted after disruption of vesicular release by reserpine whereas vesicular disruption has little effect on the response to moderate doses of amphetamine (Florin et al., 1995), similar to the effect observed in coloboma mice in which SNAP-25, a protein involved in vesicular release, is disrupted. As yet, the mechanism underlying this effect is unknown. Having defined the abnormal molecular condition that can give rise to the paradoxical response to amphetamine, the coloboma mouse model of hyperactivity will clearly be useful in defining this unusual drug response.
Interestingly, a dichotomy in the response to amphetamine and methylphenidate, similar to the coloboma mouse response, has also been observed in a subgroup of ADHD-affected children. Both methylphenidate and amphetamine are efficacious in the majority of children diagnosed with ADHD. However, a subset of children responds with an amelioration of symptoms only to amphetamine ormethylphenidate whereby ∼20% are amphetamine-only responders and ∼20% respond only to methylphenidate (Arnold et al., 1978; Elia et al., 1991). From the double-blind crossover study of Arnold et al. (1978), it was not clear whether the alternative drug would produce overt hyperactivity in these children, similar to the effects of methylphenidate in coloboma mice. The heterogeneity in effective treatment strategies for ADHD-affected children likely reflects the multifactorial nature of this disorder whereby several genes or environmental influences can produce phenotypically similar syndromes. The coloboma mouse model of hyperactivity parallels ADHD amphetamine-only responders and may provide, with other animal models of hyperactivity, a starting point for biologically categorizing different forms of ADHD to enable more effective individualized treatment strategies.
It has been suggested that the sedative effect of psychostimulants in hyperactive children and animals is not truly “paradoxical” but is a rate-dependent phenomenon based on the dose–response curves for psychostimulants (Sahakian and Robbins, 1977;Glick and Milloy, 1973). The dose–response effect of amphetamine and methylphenidate on locomotor activity is an inverted U-shaped function with low-dose psychostimulant producing an increase in locomotor activity and high doses reducing locomotor activity concomitant with an increase in highly repetitive behaviors (stereotypy), thereby “focusing attention.” The rate-dependent hypothesis states that the high baseline activity in drug-naive hyperactive children essentially places them at the highest point in this U-shaped function. Therefore, treatment of hyperactive children with amphetamine, at doses that would induce hyperactivity in less active individuals, results in a decrease in locomotor activity and increased stereotyped behavior. However, our results argue against rate dependency as an explanation for the paradoxical effects of psychostimulants in ADHD. First, both methylphenidate and amphetamine produce similar U-shaped functions. Rate dependency predicts that both methylphenidate and amphetamine should reduce the locomotor activity exhibited by coloboma mice. Instead, a clear increment (up to an apparent ceiling) in coloboma mouse locomotor activity was observed in response to methylphenidate. Importantly, this response occurred in parallel with wild-type mice, suggesting that coloboma mice started at the same point in the function, but with an overall higher baseline level. Next, although low dose amphetamine clearly decreased the locomotor activity of hyperactive coloboma mice, the suppression of hyperkinesis did not result from a shift from locomotor activity to stereotypy; that is, sedation was independent of stereotypy and the normal U-shaped function in wild-type mice. Thus, the effect of amphetamine in these mice is truly paradoxical. Taken together, these results suggest that locomotor activity and stereotypy are not components of a behavioral continuum as suggested by the rate dependency hypothesis, but separate distinct and likely competing behaviors induced by psychostimulants.
Because amphetamine and methylphenidate act at the presynaptic terminal, the aberrant behavioral response exhibited by coloboma mice suggests that defects in presynaptic function, resulting from SNAP-25 deficiencies, are responsible for abnormalities in the regulation of locomotor activity in coloboma mice. In fact, correction of the amphetamine response in coloboma mice with the Snaptransgene supports this hypothesis. Thus, in addition to its central role in the expression of hyperactivity, SNAP-25 influences the behavioral response to indirect dopamine agonists. The availability of an animal model with a defined genetic defect provides a means to explore the biochemical interactions that not only mediate psychostimulant response, but may also help identify neural pathways involved in hyperactivity and facilitate the discovery of novel therapeutics for the treatment of hyperactivity syndromes.
Footnotes
This research was supported by Public Health Service Grant MH48989 (M.C.W.) and a Klingenstein Fellowship (E.J.H.). This is manuscript 8877-NP from The Scripps Research Institute. We thank Dr. Lisa Gold for advice and critical, helpful discussions and Drs. R. J. Milner, S. C. Henriksen, and G. F. Koob for their comments. We also thank T. M. Slater, J. Bergsman, I. Polis, M. Kreifeldt, and C. Slamka for excellent technical assistance.
Correspondence should be addressed to Michael C. Wilson, Department of Neuropharmacology, CVN9, The Scripps Research Institute, 10666 North Torrey Pines Road, La Jolla, CA 92037.
REFERENCES
- 1.Anastopoulos AD, Barkley RA. Biological factors in attention deficit-hyperactivity disorder. Behav Ther. 1988;11:47–53. [Google Scholar]
- 2.Arnold LE, Christopher J, Huestis R, Smeltzer DJ. Methylphenidate vs dextroamphetamine vs caffeine in minimal brain dysfunction. Arch Gen Psychiatry. 1978;35:463–473. doi: 10.1001/archpsyc.1978.01770280073008. [DOI] [PubMed] [Google Scholar]
- 3.Bark IC. Structure of the chicken gene for SNAP-25 reveals duplicated exons encoding distinct isoforms of the protein. J Mol Biol. 1993;233:67–76. doi: 10.1006/jmbi.1993.1485. [DOI] [PubMed] [Google Scholar]
- 4.Bark IC, Wilson MC. Regulated vesicular fusion in neurons: snapping together the details. Proc Natl Acad Sci USA. 1994;91:4621–4624. doi: 10.1073/pnas.91.11.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bark IC, Hahn KM, Ryabinin AE, Wilson MC. Differential expression of SNAP-25 protein isoforms during divergent vesicle fusion events of neural development. Proc Natl Acad Sci USA. 1995;92:1510–1514. doi: 10.1073/pnas.92.5.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barkley RA. A review of stimulant drug research with hyperactive children. J Child Psychol Psychiatry. 1977;18:137–165. doi: 10.1111/j.1469-7610.1977.tb00425.x. [DOI] [PubMed] [Google Scholar]
- 7.Bell GI, Pictete RL, Rutter WJ, Cordell B, Tischer E, Goodman HM. Sequence of the human insulin gene. Nature. 1980;284:26–32. doi: 10.1038/284026a0. [DOI] [PubMed] [Google Scholar]
- 8.Blasi J, Chapman ER, Link E, Binz T, Yamasaki S, De Camilli P, Sudhof TC, Niemann H, Jahn R. Botulinum neurotoxin A selectively cleaves the presynaptic protein SNAP-25. Nature. 1993;365:160–163. doi: 10.1038/365160a0. [DOI] [PubMed] [Google Scholar]
- 9.Butcher SP, Leptrot J, Aburthnott GW. Characterisation of methylphenidate and nomifensine induced dopamine release in rat striatum using in vivo brain microdialysis. Neurosci Lett. 1991;122:245–248. doi: 10.1016/0304-3940(91)90869-u. [DOI] [PubMed] [Google Scholar]
- 10.Creese I, Iversen SD. The pharmacological and anatomical substrates of the amphetamine response in the rat. Brain Res. 1975;83:419–436. doi: 10.1016/0006-8993(75)90834-3. [DOI] [PubMed] [Google Scholar]
- 11.Elia J, Borcherdung BG, Rapoport JL, Keysor CS. Methylphenidate and dextroamphetamine treatments of hyperactivity: are there true nonresponders? Psychiatry Res. 1991;36:141–155. doi: 10.1016/0165-1781(91)90126-a. [DOI] [PubMed] [Google Scholar]
- 12.Florin SM, Kuczenski R, Segal DS. Effects of reserpine on extracellular caudate dopamine and hippocampus norepinephrine responses to amphetamine and cocaine: mechanistic and behavioral considerations. J Pharmacol Exp Ther. 1995;274:231–241. [PubMed] [Google Scholar]
- 13.Geyer MA, Paulus MP. Multivariate and nonlinear approaches to characterizing drug effects on the locomotor and investigatory behavior of rats. NIDA Res Monogr. 1992;124:203–235. [PubMed] [Google Scholar]
- 14.Glick SD, Milloy S. Rate-dependent effects of d -amphetamine on locomotor activity in mice: possible relationship to paradoxical amphetamine sedation in minimal brain dysfunction. Eur J Pharmacol. 1973;24:266–268. doi: 10.1016/0014-2999(73)90082-4. [DOI] [PubMed] [Google Scholar]
- 15.Hess EJ, Jinnah HA, Kozak CA, Wilson MC. Spontaneous locomotor hyperactivity in a mouse mutant with a deletion including the Snap gene on chromosome 2. J Neurosci. 1992;12:2865–2874. doi: 10.1523/JNEUROSCI.12-07-02865.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hess EJ, Collins KA, Copeland NG, Jenkins NA, Wilson MC. Deletion map of the coloboma (Cm ) locus on mouse chromosome 2. Genomics. 1994;21:257–261. doi: 10.1006/geno.1994.1254. [DOI] [PubMed] [Google Scholar]
- 17.Hogan B, Beddington R, Constantini F, Lacy E. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1986. In manipulating the mouse embryo: a laboratory manual, pp 217–252. . [Google Scholar]
- 18.Horikawa HPM, Saisu H, Ishizuka T, Sekine Y, Tsugita A, Odani S, Abe T. A complex of rab3A, SNAP-25, VAMP/synaptobrevin-2 and syntaxins in brain presynaptic terminals. FEBS Lett. 1993;330:236–240. doi: 10.1016/0014-5793(93)80281-x. [DOI] [PubMed] [Google Scholar]
- 19.MacGregor GR, Caskey CT. Construction of plasmids that express E . coli beta-galactosidase in mammalian cells. Nucleic Acids Res. 1989;17:2365. doi: 10.1093/nar/17.6.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McMillen BA. CNS stimulants: two distinct mechanisms of action for amphetamine-like drugs. Trends Pharmacol. 1983;10:429–432. [Google Scholar]
- 21.O’Connor VM, Shamotienko O, Grishin E, Betz H. On the structure of the “synaptosecretosome.” Evidence for a neurexin/synaptotagmin/syntaxin/Ca2+ channel complex. FEBS Lett. 1993;326:255–260. doi: 10.1016/0014-5793(93)81802-7. [DOI] [PubMed] [Google Scholar]
- 22.Oyler GA, Higgins GA, Hart RA, Battenberg E, Billingsley M, Bloom FE, Wilson MC. The identification of a novel synaptosomal-associated protein, SNAP-25, differentially expressed by neuronal subpopulations. J Cell Biol. 1989;109:3039–3052. doi: 10.1083/jcb.109.6.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pauls DL, Hurst CR, Kruger SD, Leckman JF, Kidd KK, Cohen DJ. Gilles del la Tourette syndrome and attention deficit disorder with hyperactivity: evidence against a genetic relationship. Arch Gen Psychiatry. 1986;43:1177–1179. doi: 10.1001/archpsyc.1986.01800120063012. [DOI] [PubMed] [Google Scholar]
- 24.Ryabinin AE, Sato TN, Morris PJ, Latchman DS, Wilson MC (1996) Immediate upstream promoter regions required for neurospecific expression of SNAP-25. J Mol Neurosci, in press. [DOI] [PubMed]
- 25.Safer DJ, Krager JM. A survey of medication treatment for hyperactive/inattentive students. JAMA. 1988;260:2256–2258. [PubMed] [Google Scholar]
- 26.Sahakian BJ, Robbins TW. Are the effects of psychomotor stimulant drugs on hyperactive children really paradoxical? Med Hypother. 1977;3:154–158. doi: 10.1016/0306-9877(77)90065-2. [DOI] [PubMed] [Google Scholar]
- 27.Sambrook J, Fritsch EF, Maniatis T. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. Molecular cloning: a laboratory manual. . [Google Scholar]
- 28.Scheel-Kruger J. Comparative studies of various amphetamine analogues demonstrating different interactions with the metabolism of the catecholamines in the brain. Eur J Pharmacol. 1971;14:47–59. doi: 10.1016/0014-2999(71)90121-x. [DOI] [PubMed] [Google Scholar]
- 29.Schiavo G, Rossetto O, Catsicas S, Poliverino de Laurento P, DasGupta BR, Benifenati F, Montecucco C. Identification of the nerve terminal targets of botulinum neurotoxin serotypes A, D, and E. J Biol Chem. 1993;268:23784–23787. [PubMed] [Google Scholar]
- 30.Searle AG. New mutants. II. Coloboma. Mouse News Lett. 1966;35:27. [Google Scholar]
- 31.Shaywitz SE, Shaywitz BA. Diagnosis and management of attention deficit disorder: a pediatric perspective. Ped Clin N Amer. 1984;31:429–457. doi: 10.1016/s0031-3955(16)34576-x. [DOI] [PubMed] [Google Scholar]
- 32.Söllner T, Bennett T, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation and fusion. Cell. 1993a;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 33.Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Germanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993b;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 34.Sulzer D, Rayport S. Amphetamine and other psychostimulants reduce pH gradients in midbrain dopaminergic neurons and chromaffin granules: a mechanism of action. Neuron. 1990;5:797–808. doi: 10.1016/0896-6273(90)90339-h. [DOI] [PubMed] [Google Scholar]
- 35.Sulzer D, Maidment JT, Rayport S. Amphetamine and other weak bases act to promote reverse transport of dopamine in ventral midbrain neurons. J Neurochem. 1993;60:527–535. doi: 10.1111/j.1471-4159.1993.tb03181.x. [DOI] [PubMed] [Google Scholar]
- 36.Sulzer D, Chen R-K, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Theiler K, Varnum DS. Development of coloboma (Cm /+), a mutation with anterior lens adhesion. Anat Embryol. 1981;161:121–126. doi: 10.1007/BF00318098. [DOI] [PubMed] [Google Scholar]
- 38.Whalen CK, Henker B. Psychostimulants and children: a review and analysis. Psychol Bull. 1976;83:1113–1130. [PubMed] [Google Scholar]
- 39.Wilson MC, Higgins GA. In situ hybridization. In: Boulton AA, Baker GB, Campagnoni AT, editors. Neuromethods; molecular neurobiological techniques, Vol. 16. Humana; Clifton, NJ: 1989. pp. 239–284. [Google Scholar]