

Abstract
Absolute Ca2+ levels in dissociatedDrosophila photoreceptors were measured using the ratiometric indicator dye INDO-1 loaded via patch pipettes, which simultaneously recorded whole-cell currents. In wild-type photoreceptors, the ultraviolet (UV) excitation light used to measure fluorescence elicited a massive Ca2+ influx that saturated the dye (>10 μmCa2+), but lagged the electrical response by 2.8 msec. Resting Ca2+ levels in the dark, measured during the latent period before the response, averaged 160 nm in normal Ringer’s (1.5 mm Ca2+). Ca2+ increases in response to weak illumination were estimated (1) by using a weak adapting stimulus before the UV excitation light and measuring Ca2+ during the latent period; and (2) by using ninaE mutants with greatly reduced rhodopsin levels. Ca2+ rose linearly as a function of the time integral of the light-sensitive current with a slope of 2.7 nm/pC. In the transient receptor potential (trp) mutant, which lacks a putative light-sensitive channel subunit, the slope was only 1.1 nm/pC, indicating a 2.5-fold reduction in the fractional Ca2+ current. From these data, it can also be estimated that >99% of the Ca2+ influx is effectively buffered by the cell. In Ca2+-free Ringer’s, resting cytosolic Ca2+ was reduced (to 30–70 nm), but contrary to previous reports, significant light-induced increases (∼250 nm) could be elicited. This rise was reduced to <20 nm when extracellular Na+was replaced withN-methyl-d-glucamine, suggesting that it could be attributed to Na+ influx altering the Na/Ca exchanger equilibrium. It is concluded that any light-induced release from internal stores amounts to <20 nm.
Keywords: calcium entry, inositol phosphates, phototransduction, calcium fluorimetry, INDO-1, Drosophila, photoreceptor, vision
Phototransduction in invertebrate photoreceptors is believed to be mediated by the phosphoinositide (PI) signaling cascade (for review, see Payne et al., 1988; Minke and Selinger, 1991;Nagy, 1991; Hardie and Minke, 1993, 1995; Ranganathan et al., 1995), a ubiquitous G-protein-coupled signaling system characterized by the production of the second messengers inositol 1,4,5-trisphosphate (InsP3) and diacylglycerol (for review, seeBerridge, 1993). Some of the most compelling evidence for this conclusion comes from analysis of Drosophilaphototransduction mutants (for review, see Hardie and Minke, 1995;Ranganathan et al., 1995). For example, null mutants of the no-receptor potential A (norpA) gene, which encodes a light-activatable phospholipase C (PLC) (Bloomquist et al., 1988; Toyoshima et al., 1990;McKay et al., 1995), are completely unresponsive to light (Pak et al., 1969; Minke and Selinger, 1992), whereas a near null mutation in the PLC-specific G-protein (Gq) α subunit also almost completely abolishes the response to light (Scott et al., 1995).
The ability to genetically manipulate the PI cascade has been complemented by a number of experimental assays, including the ability to measure the light-activated current under whole-cell voltage clamp using a preparation of dissociated ommatidia (Hardie, 1991; Ranganathan et al., 1991). More recently, the same preparation has been used for making simultaneous measurements of intracellular Ca2+ using fluorescent Ca2+indicators. To date, only two studies have investigated the light-induced changes in cytosolic Ca2+ inDrosophila (Peretz et al., 1994a; Ranganathan et al., 1994). Both reported large light-induced Ca2+ influx signals, but failed to detect Ca2+ rises that might have been attributed to Ca2+ release in Ca2+-free solutions. Because both studies used uncalibrated single-wavelength dyes, no information is currently available on absolute Ca2+ concentration. In addition, temporal resolution was limited so that measurements, e.g., of latency, were not possible. Both studies recognized an obvious difficulty with Ca2+ fluorimetry in photoreceptors; namely, that the measuring light itself induces saturating light responses, often resulting in irreversible damage. An elegant solution to this problem was provided by Ranganathan et al. (1994) by using long-wavelength dyes combined with an ultraviolet (UV)-absorbing rhodopsin substituted transgenically into the photoreceptors. Nevertheless, the measuring light still induced sizeable responses, so that behavior near threshold was not measured. In the present study, cytosolic Ca2+ was measured using the ratiometric indicator INDO-1. The data provide the first information on absolute Ca2+ levels inDrosophila photoreceptors, both in the dark and over a range of adapting intensities. They also demonstrate that there can be substantial light-induced Ca2+ increases in the absence of extracellular Ca2+; however, these are most likely attributable to the effects of Na+influx on the Na/Ca exchange equilibrium rather than release from intracellular stores. The data also allow an estimation of the effective buffering capacity of the cells and a quantitative comparison of the fractional Ca2+ current carried by the light-sensitive channels in wild type (WT) and the transient receptor potential (trp) mutant, which has been proposed to lack a subunit of the light-sensitive channels responsible for high Ca2+ permeability (Hardie and Minke, 1992).
MATERIALS AND METHODS
Animals. The wild-type strain was white-eyed (w) Oregon Drosophila melanogaster; in addition, the following mutants were used (on a w background):trp301, a null mutant of thetrp gene that encodes a putative light-sensitive channel subunit (Montell and Rubin, 1989; Hardie and Minke, 1992; Phillips et al., 1992; Pollock et al., 1995); and two strains lacking theninaE gene, which encodes the rhodopsin of the principal photoreceptor class R1–R6 (O’Tousa et al., 1985; Zuker et al., 1985). Although both of these strains [ninaEoraes(referred to as ora, kindly supplied by W. Pak, Lafayette, IN) and ry ninaEI17es(subsequently referred to as ninaE, kindly supplied by J. O’Tousa, Notre Dame, IN)] are reported to be null mutants, R1–R6 photoreceptors in both stocks often had small amounts of functional rhodopsin, possibly attributable to low-level expression of other opsin genes. These mutants were also found to be useful for measurements of light-induced Ca2+ influx at low effective intensities. Adult WT and trp flies were used up to 4 hr posteclosion; because of age-dependent degeneration, ora andninaE flies were taken as late stage (>90 hr) pupae. Flies were reared at 25°C in the dark.
Solutions. The electrode solution contained (in mm): 140 Kgluconate, 4 MgATP, 0.4 Na2GTP, 2 MgCl2, and 100 μm of either INDO-1, Mag-INDO-1, or Fluo-3. (tetrasodium salts; Molecular Probes, Eugene, OR). The bath contained (in mm): 120 NaCl, 5 KCl, 4 MgSO4, 10 N-Tris-(hydroxymethyl)-methyl-2-amino-ethanesulphonic acid, 25 proline, 5 alanine, and either 1.5 mmCaCl2 (normal Ringer’s) or no added Ca2+ and 1 or 2 mm EGTA (Ca2+-free Ringer’s). For some experiments (see Fig. 10), all permeant ions (Na, K, Ca, and Mg) were replaced with 150 mmN-methyl-d-glucamine (NMDG) chloride. All solutions were buffered at pH 7.15; experiments were performed at room temperature (20° ± 1°C).
Fig. 10.
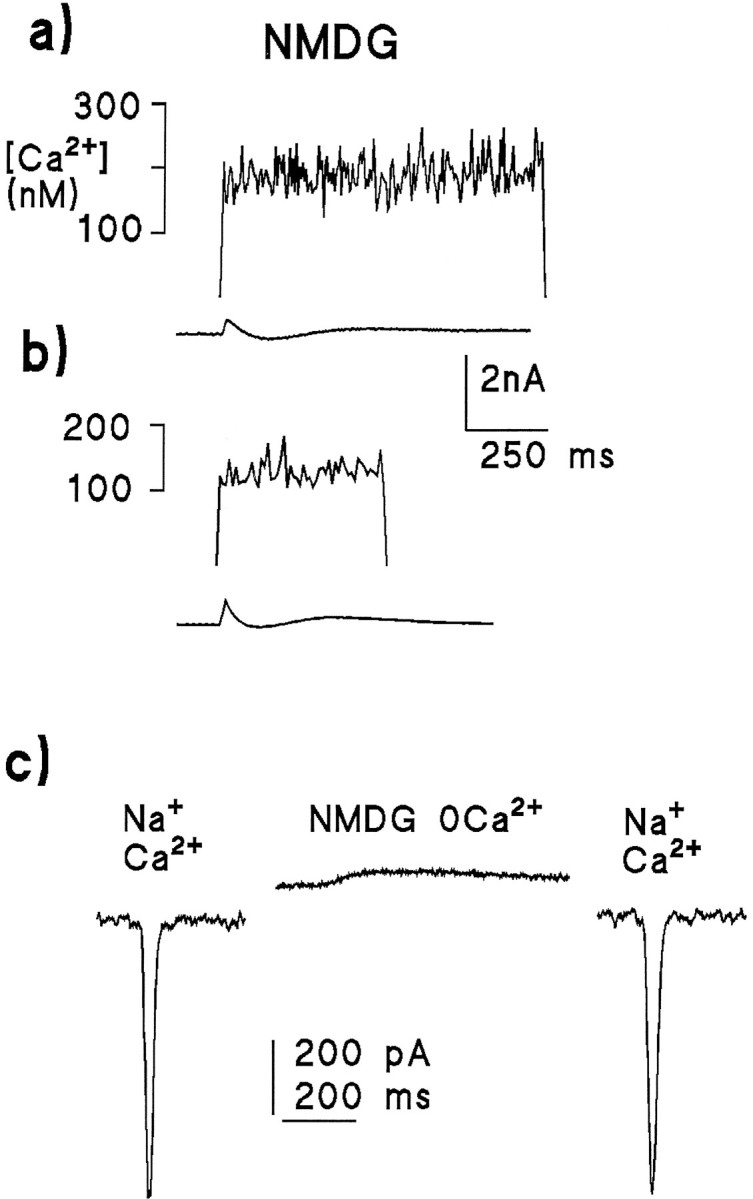
Light-induced Ca2+ signals and simultaneously recorded whole-cell currents (lower traces) measured in the absence of both extracellular Ca2+ and Na+. In contrast to Figure 9, the Ca2+ rise was virtually abolished when Na+ was substituted withN-methyl-d-glucamine (NMDG) with 0 Ca2+ and 2 mm EGTA applied by rapid perfusion from a puffer pipette. a, Response to a 1 sec saturating illumination in a cell perfused with NMDG solution, originally bathed in Ca2+-free, Na+-containing bath. b, Response from a photoreceptor perfused with NMDG, but this time after being initially bathed in normal Ringer’s (1.5 mmCa2+); again there was little or no increase in Ca2+. Both cells, clamped at −70 mV, produced small outward currents, as NMDG does not permeate the light-sensitive channels; c, Light-induced responses from the same cell as in Figure 11b to a weak 20 msec LED flash before (left), after (right), and during (middle) rapid perfusion with the Ca2+-free NMDG solution. The cell was clamped at −70 mV. After perfusion with NMDG, the response reversed and became slower (because of the absence of Ca2+-dependent feedback effects), but recovered completely after returning to normal Ringer’s. The Ca2+ measurement was made ∼60 sec after these responses were recorded, after the cell had been perfused again with NMDG for ∼30 sec.
Electrophysiology and Ca2+ measurement.Whole-cell recordings from photoreceptors were made from dissociated Drosophila ommatidia, as described previously (Hardie, 1991, 1995). Although each ommatidium contains eight photoreceptors, there is no indication of electrical or dye coupling between neighboring photoreceptors; all recordings were made from the single R1–R6 photoreceptors, identified by the whole-cell capacitance, which is at least twice as large as for two minor classes of photoreceptors, R7 and R8.
For ratiometric Ca2+ measurements, the dual emission dyes INDO-1 and Mag-INDO-1 were chosen because they allow high sampling rates without the requirement for mechanical chopping of the excitation beam (as with Fura-2). It was recently reported that Ca2+ measurements using INDO-1 in vertebrate photoreceptors were contaminated by a change in the binding properties of the dye induced by the measuring light (Gray-Keller and Detwiler, 1994). This problem has not been reported in any other preparation and, in the present experiments, the absence of all but a tiny change in fluorescence in Ca2+-free solutions strongly suggests that there is no significant Ca2+-independent light-induced change in fluorescent properties of the dye. Fluorescence of both INDO-1 and Mag-INDO-1 was excited using 360 nm light delivered via the fluorescent port of a Nikon Diaphot inverted microscope using a DM380 (Nikon, UK) dichroic mirror. For Fluo-3, light at 480 nm was delivered via a DM510 dichroic mirror. Illumination was from a 75 W Xe lamp via a monochromator (Photon Technology Instruments, Brunswick, NJ) and a Uniblitz shutter (Vincent Associates, Rochester, NY) with rise time 1.8 msec. Intensity was controlled by apertures placed at the exit of the monochromator. Fluorescence was collected via a rectangular diaphragm that just covered the recorded cell but excluded the microelectrode, and was measured simultaneously at 405 and 480 nm (INDO-1, Mag-INDO-1) or at wavelengths >520 nm (Fluo-3) via photomultipliers in photon-counting mode.
Background correction. Because of the relatively high UV autofluorescence of the photoreceptors (typically ∼30% of total signal), accurate background correction was critical for these measurements. When using ora or ninaEphotoreceptors, the background fluorescence was determined routinely for every cell after giga-seal formation, but before establishing the whole-cell configuration. However, this procedure was considered impractical for WT and trp photoreceptors, because the measuring light would have resulted in severe light adaptation before the experiment started. Consequently, background was determined separately on at least two ommatidia at the beginning and end of each preparation. The measuring light induced a small change in the autofluorescence of the photoreceptors over the first 200 msec of illumination, probably because of photoconversion of visual pigment and/or changes in mitochondrial pigments. This change (an ∼6% reduction in fluorescence measured at 480 nm) would have manifested itself as a slight (∼20–40 nm) increase in Ca2+ and was corrected for in measurements of light-induced Ca2+ changes in Ca2+-free solutions by subtracting an averaged background template (appropriately scaled) from the raw fluorescence traces before determining the fluorescence ratio. As additional controls, background fluorescence was measured from ommatidia after forming a seal with a dye-filled electrode and also from cells during whole-cell voltage clamp using otherwise identical electrode solutions, but without indicator dyes. Background fluorescence values under these conditions differed by <5% from values determined from intact ommatidia in the absence of electrodes. Background values varied little, apart from a slow reduction over the lifetime of the lamp: from the SD of the background fluorescence (∼4% of mean), it was estimated that an error of less than ± 20 nm(SD) was introduced by this indirect method of background correction.
Calibration. Free [Cai] was calculated from the ratio (R) of fluorescence at 405 and 480 nm, using the equation:
| Equation 1 |
(Grynkiewicz et al., 1985). Values forRmax, Rmin, andKd were all determined in situ both in ninaE photoreceptors and also using a cell line (Drosophila S2 cells). Cells were loaded, as for the experiments, via the patch pipette using solutions similar to the normal electrode solution but containing either no Ca2+ (20 EGTA) or 10 mmCa2+ for Rmin andRmax, respectively. To obtain estimates of the effective Kd, three different EGTA–Ca solutions were used with calculated free Ca of 111 nm (10 EGTA:3.5 Ca), 207 nm(10 EGTA:5 Ca), and 483 nm (10 EGTA:7 Ca). At least 5 min was allowed for equilibration before measuring the fluorescence ratio. The ratios obtained for all five solutions (Rmax, Rmin, and three different R values) were then fitted to Equation 1with Kd as free parameter to obtain a value for Kd (1.16 μm). Calibrations were repeated at regular intervals and not found to change significantly over the period of the experiments. Note, however, that small differences inRmin can have a large effect on the estimated Ca2+ at the lower end of the measured ranges (<100 nm).
Values for Mag-INDO-1 were obtained in an analogous manner except that it was not necessary to obtain an Rminvalue, as resting cytosolic Ca2+ is negligible for this low-affinity dye so that Rmincould be taken from the resting value in any given cell, andKd was determined using a solution based on the low-affinity Ca2+ buffer nitrilotriacetic acid (NTA): 10 mm NTA: 3 mm Ca (yielding 50 μmfree Ca using published Kd values). When using Mag-INDO-1, Mg2+ was omitted from all solutions (bath and intracellular).
Intensities are expressed in effectively absorbed photons, calibrated, as described previously, with respect to WT photoreceptors by counting quantum bump rates in response to the dimmest stimuli and measuring the relative intensities of all other stimuli (Hardie, 1995).
RESULTS
Controls
A number of control experiments were performed to establish that the indicator dye did not affect the light response, that the resting cytosolic Ca2+ concentration was stable over the time course of the experiments, and that the measuring flash did not necessarily permanently damage the cells. Ca2+indicator dyes are themselves Ca2+ buffers and may, in principle, interfere with the kinetics of transduction, which are known to be very sensitive to cytosolic Ca2+levels (Hardie, 1991). Figure 1 shows light responses made with control solutions containing no Ca2+buffer and the solutions used for loading Ca2+dyes into the cell. The responses were essentially indistinguishable, confirming that the concentration of dye used (100 μm) did not significantly affect the response to light under these conditions. Figure 1b also shows responses recorded before and after a measurement of cytosolic Ca2+ using a 200 msec measuring flash of submaximal intensity (containing 1.5 × 107effective photons). Both the physiological response and the resting Ca2+ level (not shown) completely recovered within ∼2 min. If the brightest intensities (∼10× brighter) were used, WT photoreceptors almost invariably failed to recover sensitivity after a single measuring flash (see also Peretz et al., 1994a). However, because responses to maximal and submaximal intensities were otherwise essentially similar, it was often preferred to make single measurements on any given cell using maximum intensity to obtain superior signal-to-noise ratios.
Fig. 1.
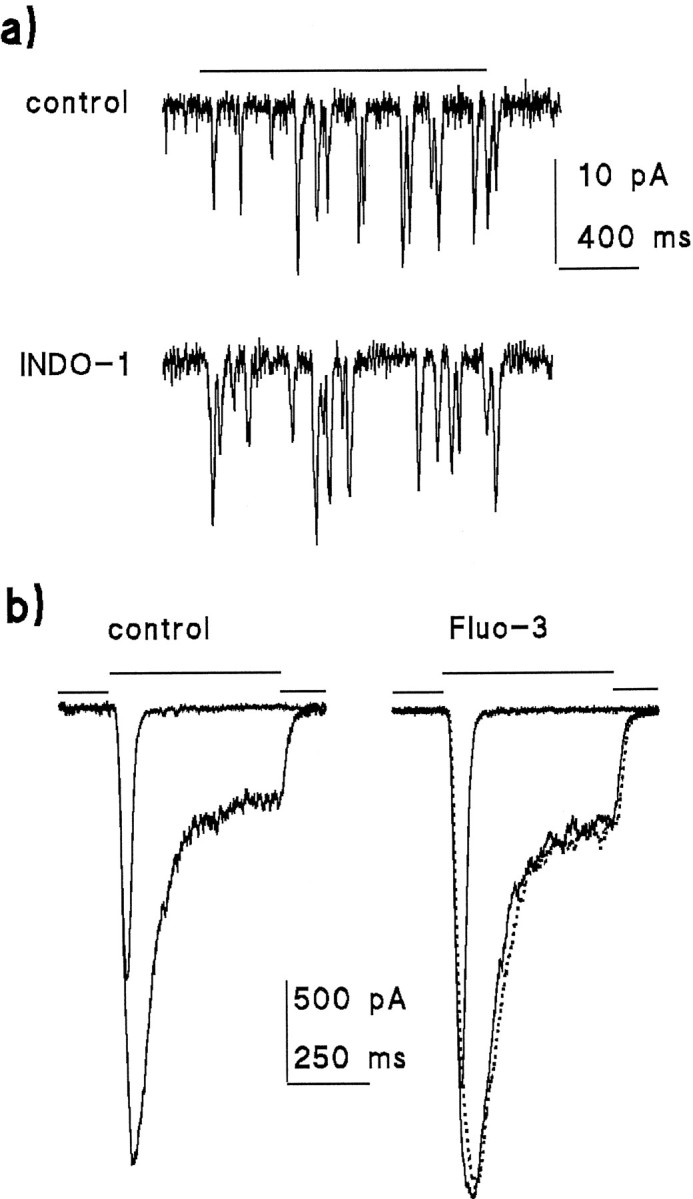
Indicator dyes do not significantly influence the light response. a, Quantum bumps recorded in response to a 1.5 sec stimulus of identical intensity in a cell loaded with 100 μmINDO-1 are similar to those recorded from another cell with control intracellular solutions containing no Ca2+ buffers.b, Similarly, Fluo-3 (100 μm) has little or no effect on responses to 20 msec flash or 500 msec step of light (the difference in absolute amplitudes is well within the experimental variability). Thedotted trace shows the response to a second 500 msec stimulus recorded 2 min after making a Ca2+measurement using a submaximal 100 msec flash containing ∼107 effective photons. All responses recorded in standard Ringer’s solution (1.5 mmCa2+) at a holding potential of −70 mV. Stimulus intensity in b was 3.8 × 104effective photons · sec−1 (calibrated from bump counts as in a).
Figure 2 shows records of fluorescence measured from a photoreceptor of an ora mutant that completely lacked rhodopsin. Fluorescence was measured continuously, starting shortly after making the giga-seal. Immediately after establishing the whole-cell configuration (break-in), the fluorescence started to increase, indicating loading of the cell with dye. The time course of loading was usually well approximated by a single exponential with a time constant in the range of 30–80 sec. The Ca2+ concentration, determined from the ratio of fluorescence at 405 and 480 nm after subtraction of the background immediately before break-in, remained effectively constant during this period, showing only an improvement in the signal-to-noise ratio as the total dye concentration increases. However, immediately after break-in there was an indication that Ca2+ may actually decline over a period of ∼10 sec; in addition, in some cells there was a slow rise in Ca2+, typically starting after 3 or 4 min of whole-cell recording. To minimize variation attributable to the increases sometimes observed after longer periods of whole-cell recording, measurements in WT flies were routinely made between 2 and 3 min after break-in.
Fig. 2.

Time course of dye loading in an oraphotoreceptor. Raw traces (dotted) show the INDO-1 fluorescence measured at 405 and 480 nm starting shortly after making a giga-seal. After establishing the whole-cell configuration (at time 0), fluorescence rises with a single exponential time course (smooth curves, single exponential fits to the traces with time constant of 68 sec). After subtraction of the background fluorescence, the ratioR between fluorescence at 405 and 480 nm was used to calculate cytosolic [Ca2+] according to Equation 1. Cytosolic [Ca2+] remained stable throughout the 200 sec recording, except for an apparent reduction during the first few seconds of recording. Recording made in normal (1.5 mm Ca2+) Ringer’s at −70 mV, briefly interrupted at ∼80 sec, for visual inspection of cell.
Resting Ca2+ levels in the dark
Figure 3 shows a simultaneous recording of membrane current and INDO-1 fluorescence using 100 μmINDO-1 in a WT photoreceptor, sampled at 500 Hz. After a short latency (∼10 msec), fluorescence measured at both wavelengths changed rapidly and in opposite directions (Fig. 3a) indicating a rise in Ca2+. The time course of the calculated Ca2+ signal is shown below, along with the simultaneously recorded whole-cell current. The INDO-1 signal was rapidly saturated by the large Ca2+ increase (indicated by the increase in noise) so that, arguably, the most useful information to be gained from such records is the resting level determined during the latent period (Fig. 3b). When considering data only from “healthy” cells, using the criteria of negligible leak currents (<20 pA at a holding potential of −70 mV) and clearly discernible quantum bumps (5–10 pA) in response to weak illumination (e.g., Fig. 1), resting values showed rather little variation, averaging 161 ± 32 nm (n = 11) in the presence of normal Ringer’s (1.5 mm). Resting values determined in WT flies were indistinguishable from values determined in ora mutants (162 ± 24 nm; n = 12), where resting levels could be more accurately determined, both because it is possible to record uninterrupted for several minutes and because the background level could be determined for each cell from the fluorescence before break-in (Fig. 2). The correspondence of values in WT andora also indicate that the ora mutant may be usefully used for quantitative studies of factors affecting cytosolic Ca2+ other than light itself.
Fig. 3.

Light-induced Ca2+ rises measured with INDO-1 in normal (1.5 mmCa2+) Ringer’s. a, Raw traces (raw) of fluorescence (counts per second) measured at 405 and 480 nm, sampled at 500 Hz;[Ca2+], after background subtraction, the time course of the rise in Ca2+is calculated from the ratio of the raw traces according to Equation 1(see Materials and Methods). The simultaneously recorded light-induced current (LIC, lower trace, holding potential −70 mV) saturated the amplifier (>10 nA). b shows the [Ca2+] signal and LIC on an expanded scale. Notice that the dark level can be readily measured during the latent period of the response (arrow). The 150 msec UV stimulus (360 nm) contained 3 × 107 effective photons.
Identical measurements performed in the absence of extracellular Ca2+ (see further below) revealed a very significant drop in resting cytosolic Ca2+. In this case, however, values in ora mutants appeared somewhat lower than in WT (WT 70 ± 22 nm; n = 11; ora 28 nm ± 16; n = 36; measured at −70 mV), although, as emphasized in Materials and Methods, absolute levels in this range are prone to larger errors because of uncertainty of Rmin values.
Latency
A number of studies have addressed the question of whether the Ca2+ signal measured in invertebrate photoreceptors precedes the electrophysiological response. For example, in Limulus, the light-induced Ca2+signal, which is mainly attributable to release, was found to lag the electrical response by ∼3–20 msec, thus calling into question the hypothesis that Ca2+ release is causal for excitation (Ukhanov et al., 1995) (see also Walz et al., 1994 for drone bee). In Drosophila, it now seems clear that virtually all the light-induced Ca2+ signal is attributable to Ca2+ influx (Peretz et al., 1994a; Ranganathan et al., 1994; see below), which should, in principle, coincide with the electrophysiological response. Measurements of latency inDrosophila thus provide an empirical test of the temporal resolution of whole-cell Ca2+ measurements. In practice, it was found that the best signal-to-noise ratio was found using the single-wavelength dye, Fluo-3, and this was preferred for obtaining information on latency, although there was no obvious difference from results using INDO-1. Figure 4 shows that even with the brightest intensities, there was always a significant delay between the light-induced current (LIC) and the first detectable rise in Ca2+. Judged by eye, the average latency using the brightest intensity available was 9.1 ± 1.1 msec for the LIC and 11.9 ± 1.8 msec for the Ca2+ rise, or a lag of 2.8 ± 1.2 msec (n = 11 cells). The apparent lag increased considerably when lower intensities were used (e.g., a lag of 7.8 ± 2.3 msec;n = 10 with 10× lower intensity). Whatever the reasons for this lag (which presumably include the diffusional delay required for the Ca2+ to reach and bind to dye within a sufficient volume of the cell to overcome the measurement noise), these results indicate caution in interpreting temporal data derived from whole-cell fluorescent Ca2+ measurements. This problem appears now to have been resolved in Limulus, because recent measurements using confocal microscopy have shown that Ca2+ signals can indeed precede the electrical response (Ukhanov and Payne, 1995).
Fig. 4.

Latency of the Ca2+ response determined using Fluo-3 in normal Ringer’s solution. The time course of the Ca2+ signal measured using the single-wavelength dye Fluo-3 was similar to that measured with INDO-1 (compare Fig. 3); however, a better signal-to-noise ratio can be obtained. The traces show the average of LIC (upper trace) and Ca2+ signal from eight photoreceptors in response to a 200 msec measuring flash (480 nm) containing ∼108 effective photons. The current trace has been inverted for easier comparison of time courses. The first detectable rise in Ca2+ always lagged the LIC by ∼3 msec (inset on expanded scale). Holding potential was −70 mV, data sampled at 1 kHz.
Quantification of light-induced Ca influx
Ca2+ rises elicited by weak illumination
To determine absolute Ca2+ levels reached in response to weak-to-moderate stimulation, a two-flash strategy was used similar to that recently used in Limulus ventral photoreceptors by Ukhanov et al. (1995). A 500 msec adapting step of light from a light-emitting diode (LED) of variable intensity was delivered to the cells, eliciting responses in the range of 0–2 nA in amplitude. Synchronously with the cessation of the adapting step, a brief (25–50 msec) UV measuring flash was applied, and the resting Ca2+ level was determined, as before, during the latent period (Fig. 5a). After pooling data from several cells, there was a clear intensity-dependent increase in the Ca2+ level. Because the Ca2+ increase derives almost exclusively from Ca2+ influx via the light-sensitive channels (Peretz et al., 1994a; Ranganathan et al., 1994; see further below), it seemed most instructive to plot the Ca2+concentration reached as a function of the charge (time integral of current) carried by the response to the 500 msec adapting flash (Fig.5b). A linear regression line plotted through the data indicated an increase of 2.7 nm for Ca2+ for each picocoulomb of current.
Fig. 5.

Determination of Ca2+ levels in response to weak illumination in normal Ringer’s solution.a, A WT photoreceptor was first illuminated for 500 msec with a dim LED stimulus (∼2000 photons/sec) generating an inward current of ∼500 pA amplitude (dotted trace). The Ca2+ level reached during this period (530 nm) was then determined during the latent period of the response to a saturating UV measuring stimulus (50 msec, 3 × 107 effective photons). The Ca2+ signal (solid trace) is replotted on an expanded time base below. b, Ca2+ levels obtained from 19 cells (filled squares), as in Figure 5a, plotted against the total charge flowing during the 500 msec adapting step. Open square, “Dark” Ca2+ concentration determined identically, but without preillumination with the LED (mean ± SD of 12 cells). The data have been fitted by a regression line of slope 2.7 nm/pC with an intercept (dark resting Ca2+ level) of 161 nm.Triangles represent data determined using measurements of light-induced Ca2+ rises in ora orninaE flies with small amounts of residual rhodopsin (Fig. 6).
A second strategy for investigating the Ca2+rises associated with small responses is to use mutants with greatly reduced rhodopsin levels, which consequently generate only small responses to the bright measuring light. As reported previously (Johnson and Pak, 1986), there are a number of alleles of the rhodopsin gene ninaE in which the rhodopsin concentration in the photoreceptors is drastically reduced. The residual response to light in these mutants was apparently normal except that, as reported previously (Johnson and Pak, 1986), the quantum bumps were somewhat larger than in WT. Initially, the ninaEP334allele was tested, as photoreceptors in this mutant were reported to contain ∼500 rhodopsin molecules (Johnson and Pak, 1986). However, photoreceptors from newly eclosed ninaEP334adults were found to be virtually unresponsive to the brightest stimuli available (equivalent to ∼3 × 108 photons/sec in WT flies), indicating that, at this age at least, there can be at most only one or two rhodopsin molecules per cell. Surprisingly, it was found that photoreceptors from both the ora mutant and theninaEI17 alleles, although reported to be completely null alleles, often produced responses up to ∼200 pA in response to the brightest stimuli. Assuming a rhodopsin content of 108 molecules per WT cell (Johnson and Pak, 1986), the reduction in sensitivity in such cells would indicate a rhodopsin content of ∼25 functional molecules. Figure6 confirms that responses of ∼20 pA are sufficient to give clearly resolvable rises in Ca2+ in these mutants. When plotted in the same way as the data collected from WT (i.e., as a function of charge carried), there was reasonable agreement in the absolute levels reached (Fig. 5b).
Fig. 6.

Ca2+ influx measured in real time in ora photoreceptors containing residual levels of rhodopsin. UV measuring flashes of 1 sec duration delivered toora (or ninaE) photoreceptors sometimes elicited small responses (lower traces) attributable to residual levels of rhodopsin, thus allowing direct measurement of Ca2+ influx (upper traces) during weak effective illumination. Traces from two different cells are shown using different intensities, generating responses of ∼20–40 pA (left) and 200 pA (right). Quantum bump noise can be clearly resolved in these small responses: as reported previously (Johnson and Pak, 1986), quantum bumps in ninaE mutants with greatly reduced rhodopsin levels were in fact typically larger than in WT. Substantial Ca2+ rises were detected in each case. The data from these and three other cells are plotted on Figure5b and show reasonable agreement with measurements made in WT photoreceptors using the two-flash paradigm.
Maximum rises
The Ca2+ influx occurring during the response to the measuring stimulus clearly saturates high-affinity Ca2+indicator dyes such as INDO-1 and Fluo-3 (see also Peretz et al., 1994a; Ukhanov et al., 1995). To gain an estimate of the absolute level of Ca2+ reached during more intense illumination, lower-affinity indicator dyes must be used. Currently, the only appropriate dual-emission dye available for this purpose is Mag-INDO-1. As implied by its name, this is actually designed as a Mg2+ indicator, but has an affinity for Ca2+ of ∼100 μm and can be used for measuring Ca2+ in this range in the absence of Mg2+. Cells were loaded with 100 μm Mag-INDO-1 using an electrode solution containing no Mg2+ and stimulated in Mg2+-free external solutions. Under these conditions, Ca2+ increases to values >50 μm were observed (45 ± 23 μm; n = 4; e.g., Fig.7).
Fig. 7.

Light-induced Ca2+ rise in a WT photoreceptor loaded with the low-affinity indicator dyeMag-INDO-1. In response to a saturating UV stimulus, Ca2+ rose rapidly beyond 50 μm. Data were recorded at a holding potential of −70 mV in standard (1.5 mmCa2+) Ringer’s solution containing no Mg2+. Mg2+ was also omitted from the recording electrode solution. Similar results were obtained in four other cells.
Light-induced rise in trp
In the trp mutant, measurements of reversal potential have indicated that the Ca2+ permeability of the light-sensitive channels is severely reduced, leading to the proposal that the trp gene encodes a channel subunit responsible for the high Ca2+ selectivity of the light-sensitive channels (Hardie and Minke, 1992). To obtain independent confirmation of the reduced Ca2+ permeability of the light-sensitive channels in trp, Ca2+influx in the trp mutant was quantified, as for WT, as a function of charge carried in the presence of normal (1.5 mm) extracellular Ca2+using an identical twin-flash paradigm (as in Fig. 5). When quantified in this way, Ca2+ influx in the trpmutant was found to be reduced by a factor of ∼2.5, now showing an increase of only 1.09 nm/pC (Fig.8). By contrast, the resting Ca2+concentration in the dark (i.e., during the latent period of the response to the measuring stimulus without preillumination) was not found to differ significantly from WT (153 ± 21 nm; n = 12; see Fig. 8).
Fig. 8.

Ca2+ influx is reduced in the trp mutant. Measurements of Ca2+levels in the trp mutant were made using the two-flash paradigm (Fig. 5) and plotted against the total charge carried in response to the adapting flash. In the dark (open triangle), resting Ca2+ levels in trp were indistinguishable from WT; however, in response to the 500 msec LED-adapting flash, the Ca2+ rise was significantly less than in WT (dotted line replotted from Fig. 5). The regression line through the trp data had a slope of 1.09 nm/pC (i.e., ∼2.5× less than in WT).
Light-induced Ca2+ increase in the absence of extracellular Ca2+
Previous studies reported either that there was no detectable light-induced Ca2+ rise in the absence of extracellular Ca2+ (Ranganathan et al., 1994) or a very small rise that might have been attributable to influx of residual extracellular Ca2+ (Peretz et al., 1994a). Because of the importance of Ca2+ release for models of excitation, these experiments have been repeated and extended (see also Hardie, in press). Figure 9 shows the response of a cell exposed to Ca2+-free Ringer’s solution (2 mm EGTA, no added Ca2+, and 120 mm NaCl). In contrast to previous results (Ranganathan et al., 1994), the light response was associated with a readily detectable Ca2+ signal increasing from a resting level of ∼70 to ∼250 nm during a 1 sec saturating stimulus. It seems unlikely that this signal was attributable to influx of residual extracellular Ca2+, because similar signals were seen in each of 15 cells tested even after exposure to the Ca2+-free bath for 50 min (mean rise at a holding potential of −70 or −50 mV: 213 ± 125 nm;n = 11). In addition, the increase in Ca2+ was, if anything, increased (246 ± 146 nm; n = 4) when measurements were made at a holding potential of −10 or 0 mV, which would have substantially reduced the driving force for influx (Figs. 9, 11).
Fig. 9.

Light-induced Ca2+ signals measured in the absence of extracellular Ca2+. Substantial Ca2+ increases were detected in every cell in response to saturating UV-measuring stimuli: Ca2+ signals (upper traces), simultaneously recorded whole-cell currents at 0 and −70 mV (lower traces) (two different cells). The rise was at least as large in cells clamped at 0 mV as in those clamped at −70 mV, arguing against influx of residual Ca2+ as an explanation. Note also that the initial (dark) resting level of Ca2+ was higher in the cell clamped at 0 mV (see also Fig. 11). Bath contained 0 Ca2+, 2 mm EGTA, and 120 mmNaCl.
Fig. 11.

Summary of data collected in Ca2+-free solutions. Solid bars, Dark levels determined during the initial 20 msec of the response;open bars, levels reached after 500 msec. In the presence of external Na+, light-induced Ca2+ increases of >200 nmat both −70 mV (n = 11) and 0 mV (n = 4); note also that the depolarization alone increased the dark resting level from ∼70 nm to 150 nm. After substitution with NMDG, the dark level also rose to ∼150 nm; however, light now resulted in a barely significant rise of 16 nm. Data in NMDG, 0 Ca2+solutions (n = 14 cells) were pooled from cells bathed initially in 0 Ca or 1.5 mmCa2+ and regardless of holding potential (−70, −50 or 0 mV), since none of these conditions appeared to affect the resting Ca2+ in the absence of Na+.
Initially these results appear to indicate the light-induced release of Ca2+ from intracellular stores; however, an alternative explanation may also be suggested. Drosophilaphotoreceptors have a powerful Na/Ca exchange mechanism (Hardie, 1995), which is probably one of the major mechanisms for maintaining low levels of intracellular Ca2+. The response to light is inevitably associated with a large influx of Na+ ions through the light-sensitive channels, which should reduce the Na+ gradient available for Ca2+ extrusion. Assuming there is some internal Ca2+ source (e.g., the electrode solution, which was calculated to have a weakly buffered free Ca2+ concentration in excess of 200 nm or tonic flux from intracellular sources of Ca2+ such as the mitochondria), the resulting shift in the Na/Ca exchange equilibrium would be expected to result in a rise in cytosolic Ca2+. To test whether such a mechanism might underlie the light-induced Ca2+rises, extracellular Na+ was replaced with NMDG to block the exchanger. To minimize the possibility of any long-term effects of this substitution, the NMDG solution was applied by rapid perfusion from a puffer pipette placed close to the recorded cell; as shown in Figure 10c, control responses recovered immediately after return to the original bathing solution. Under these conditions, the light-induced Ca2+rise in Ca2+-free Ringer’s was virtually abolished, the average rise after 0.5 or 1 sec illumination now amounting to <20 nm (16 nm ± 13; n = 14; Figs. 10, 11). Although this is significantly (p < 0.005) different from zero, it is of the same order as the SD of the noise in these recordings, as well as the light-induced change in autofluorescence (which was subtracted from the raw data; see Materials and Methods), and it seems debatable whether it can be attributed to release.
The dark resting level of Ca2+ was also significantly increased by the NMDG substitution (from 70 ± 22 nm to 158 ± 43 nm; Fig.11): this suggests that the Na/Ca exchange is indeed important in maintaining low Cai in Ca2+-free solutions; however, it also raises the possibility that the block of the light-induced Ca2+ rise might have been attributable to inhibition by the raised resting Ca2+. This possibility can probably be excluded because in the control (0 Ca, 120 mm NaCl) solution, depolarization caused a similar rise in the resting Ca2+ level (probably because of the voltage dependence of the Na/Ca exchange equilibrium), yet did not inhibit the subsequent light-induced increase (Figs. 9,11).
Finally, the possibility was considered that the inability to detect significant release from stores was because of the putative light-sensitive stores becoming depleted during prolonged exposure to Ca2+-free solutions (Hardie and Minke, 1992;Ranganathan et al., 1994). Therefore, measurements were also performed on photoreceptors bathed initially in normal (1.5 mm Ca2+, 120 mm Na+) Ringer’s solution, but again briefly exposed to the same Ca2+-free NMDG solution between 10 and 30 sec before making the measurements (Fig. 10b). Again, only a minimal and arguably insignificant Ca2+ rise was detected under these conditions (14 ± 8 nm; n = 8).
In summary, light-induced Ca2+ rises can indeed be detected in the absence of extracellular Ca2+; however, the most likely explanation for this rise appears to be a shift in the Na/Ca exchange equilibrium. In the absence of external Na+, the maximal rise detected is <20 nm over a time scale of 500 msec to 1 sec.
DISCUSSION
This study provides data on absolute levels of cytosolic Ca2+ in Drosophila photoreceptors and describes procedures for quantification of both dark- and light-induced Ca2+ signals in this important genetic model of phototransduction and PI signaling. The results confirm that the majority of the light-induced Ca2+ signal is attributable to Ca2+ influx; however, contrary to previous reports, significant rises can also be detected in Ca2+-free conditions, probably as a result of Na+ influx influencing the Na/Ca exchange equilibrium. With certain assumptions, the results also allow a number of useful quantitative estimates to be made, including (1) the effect of the trp mutation on the fractional Ca2+ current through the light-sensitive channels; (2) the effective buffering capacity of the photoreceptors; and (3) an upper limit on the amount of Ca2+ that might be released from internal stores—a result that has some implications for the still unresolved mechanism of excitation.
Subcellular compartments
The supposed Ca2+ stores (submicrovillar cisternae, or SMC) in Drosophila abut directly against the base of the microvilli, defining a putative “transduction compartment” consisting of the microvilli and the subrhabdomeric space, which is an ∼10–20 nm gap between the SMC and the base of the microvilli (Walz, 1982). There is some evidence to suggest that indicator dyes or other substances introduced intoDrosophila photoreceptors by patch pipettes do not permeate readily into the microvilli or the subrhabdomeric region (Ranganathan et al., 1994) (see also Hardie, 1995). The present paper adds little new information on this issue, except to reveal that even small LICs, which derive from channels believed to open into the subrhabdomeric space, give rise to readily detectable Ca2+ signals (Figs. 5, 6). However, one should be aware of the possibility that the transduction compartment might at least partially exclude the dye; the implications for some of the conclusions of this study are discussed where appropriate below.
Resting levels
By calibrating in situ, using solutions with ionic strength similar to those used for the actual measurements, many of the uncertainties associated with calibration of Ca2+indicators should have been avoided in the present study. There is, however, still some uncertainty as to whether the measured values are representative of those occurring in vivo. For example, there was an indication over the first few seconds after break-in that [Ca] actually fell from a higher level (Fig. 2). One explanation may be that the electrode solution contained virtually no Na+, whereas the photoreceptors can be expected to contain a few mm Na+, which would result in a different equilibrium of the Na/Ca exchanger. An alternative possibility is that equilibration of the contents of the cell with the electrode solution may change the effectiveKd of the dye by, for example, a change in the “viscosity factor.” The dark, resting cytosolic Ca2+ level was found to be very sensitive to extracellular Ca2+, falling from 150 to 70 nm (30 nm inora). Although not systematically explored, it seems likely that Na/Ca exchange may be one of the major mechanisms responsible, because in the absence of extracellular Ca2+, cytosolic Ca2+ rose to ∼150 nm when extracellular Na+was substituted for NMDG (Fig. 11). A similar effect was elicited by depolarization, which is also predicted to shift the Na/Ca exchange equilibrium because of its voltage dependence.
Light-induced influx and buffering capacity
Measurements with Mag-INDO-1 indicated that Ca2+ may rise globally to levels as high as 50 μm; however, some caution should be exercised in accepting this figure. Recently, the effective single-channel conductance of the light-sensitive channels was found to increase ∼10-fold in the absence of external Mg2+(Hardie and Mojet, 1995). Therefore, under the Mg2+-free conditions required to measure Ca2+ using Mag-INDO-1, the Ca2+ influx per channel is likely to be significantly greater. Conversely, this effect may be offset by the reduction in internal Mg2+ (omitted from the electrode solution), which results in an inhibition of the light response (R. Hardie, unpublished data). Nevertheless, the rapid saturation of INDO-1 signals (Fig. 2) clearly indicates that light-induced Ca2+ influx results in global concentration increases into the high micromolar range: locally, the levels are presumably even higher. These are unusually high Ca2+ loads and represent a considerable challenge for the Ca2+ homeostatic mechanisms of the photoreceptor.
For smaller currents, it was possible to quantify the Ca2+ rise more reliably (Fig. 5b). Comparison of this rise (2.7 nm/pC) with the predicted amount of Ca2+ influx via the light-sensitive channels provides an estimate of the effective buffering capacity of the photoreceptors, by which is understood here all mechanisms controlling cytosolic Ca2+, including Ca-binding proteins, sequestration, extrusion by the Na/Ca exchanger or other transporters, and exclusion from the bulk of the cell by diffusion barriers. The relative permeability of the light-sensitive channels for Ca (PCa:PNa) has been estimated at ∼40:1 on the assumptions of the Goldman–Hodgkin–Katz (GHK) theory (Hardie and Minke, 1992). Solving the GHK constant current equation for the permeant ions suggests that ∼49% of the current is carried by Ca2+ under the conditions of the experiment. Assuming the cell to be a cylinder 100 μm long and 5 μm in diameter, this should in fact raise global Ca2+ concentration by 1.29 μm/pC—suggesting that ∼99.8% of the Ca2+ influx is effectively buffered (effective buffering capacity of ∼500:1). Although the assumptions of the GHK analysis (e.g., independent mobility of ions) may be violated by the light-sensitive channels, even assuming a 10-fold lower PCa:PNa ratio, ∼16% of the current would be carried by Ca2+, yielding an effective buffering capacity of ∼150:1. Both of these figures are larger than values reported in other cells (e.g., 25–100:1 in chromaffin cells; Zhou and Neher, 1993), but similar to an estimate in the Limulus ventral photoreceptor (O’Day and Gray-Keller, 1989), suggesting that invertebrate photoreceptors may have evolved particularly powerful mechanisms for controlling cytosolic Ca2+ levels.
This calculation ignores the possibility that a component of the measured LIC is attributable to an electrogenic inward Na/Ca exchange current evoked by the Ca2+ influx. Although some contribution cannot be excluded, it is likely to be minor for the small currents used to determine the relationship between Cai and the influx current (Fig. 5b), because direct measurements of the Na/Ca exchange current using caged Ca2+ (Hardie, 1995) indicated that the exchange currents elicited by raising Ca2+ to 1 μm (the maximum level reached in Fig.5b) were barely measurable (∼4 pA).
Effect of the trp mutation on Ca2+ influx
Measurements of reversal potential indicated that the light-sensitive channels in the trp mutant have a reduced Ca2+ permeability (Hardie and Minke, 1992), suggesting that the trp gene encodes a light-sensitive channel subunit. Independent support for this important conclusion was provided by Peretz et al. (1994a,b), who showed that Ca2+ influx in response to bright lights intrp was substantially reduced. However, because the currents evoked in trp were also smaller, this result may have been a consequence of fewer channels being activated rather than a reduction in Ca2+ permeability per se. On the assumption that Ca2+ influx in WT and the trp has access to the same subcellular compartment(s) and is subject to the same buffering conditions, the relative fractional Ca2+ current through the light-sensitive channels in WT and trp should be given by the difference in slope of the plot of [Ca] versus charge, i.e., 2.7 nm/pC (WT): 1.09 nm/pC (trp), yielding an ∼2.5× greater fractional Ca2+ current in WT. This estimate compares favorably with the fractional current predicted via GHK analysis [49% fractional Ca2+ current in WT compared with 13% in trp, assuming permeability ratios from Hardie and Minke (1992)]. In conclusion, on the assumption that Ca2+ influx is buffered equivalently in WT and trp, the present results provide strong confirmation of a major reduction in Ca2+ permeability in the light-sensitive channels of trp photoreceptors.
The question of light-induced Ca2+ release: implications for transduction
Contrary to previous reports (Peretz et al., 1994a; Ranganathan et al., 1994), in the present study substantial rises in Ca2+ were reliably detected in Ca2+-free solutions. Previous failure to detect significant rises may have been attributable to the dyes acting as Ca2+ buffers (both studies used higher dye concentrations) or [in the case of Ranganathan et al. (1994)] less intense stimulation, or because measurements were always made from an already partially stimulated condition. Although the present results initially seemed to indicate substantial release of Ca2+ from internal stores, when Na+ was replaced with NMDG, the light-induced Ca2+ rise was virtually abolished, leaving a residual signal of <20 nm. This suggests that most of the measured rise was not attributable to release from internal stores, but was probably attributable to a shift in the Na/Ca exchange equilibrium caused by Na+ influx.
It is of some interest to inquire how much Ca2+would be required to generate an increase of 20 nm, as this is probably an upper estimate of how much Ca2+ is released by a saturating light stimulus. In turn, this could provide clues whether such release might be sufficient to account for excitation—as demanded by some hypotheses of excitation (Shin et al., 1993). If it is assumed that any Ca2+ release is buffered and detected with the same efficiency as Ca2+ influx, then the Ca2+ flux required to generate a signal of 20 nm can be taken from the slope of the regression line for Ca2+ influx (2.7 nm/pC), i.e., ∼7 pC for a 20 nm increase. Assuming 49% of the LIC is carried by Ca2+, this would represent a pure Ca2+ current of 3.5 pC, corresponding, e.g., to 7 pA flowing for 500 msec, which is the typical duration of a quantum bump in Ca2+-free Ringer’s solution. This figure can be converted into numbers of InsP3 receptors by assuming a value for their single-channel conductance. Bezprovanny and Ehrlich (1994) suggest a value of 0.5 pA under physiological conditions; however, this may be ∼10× too high because it assumes a luminal Ca2+ concentration of 2.5 mm, whereas recent measurements suggest values closer to 200 μm (Hofer et al., 1995). Using a figure of 0.04 pA, the estimated Ca2+ flux required to generate a 20 nm rise would correspond to only 175 InsP3 receptors being open on average for 500 msec.
By comparison, one can ask how many InsP3receptors might be required for excitation. As in most invertebrate photoreceptors, each effectively absorbed photon inDrosophila evokes a discrete quantum bump (Wu and Pak, 1975). The UV excitation light typically generated responses of >10 nA (underestimated because of imperfect voltage-clamp control). In Ca2+-free Ringer’s, quantum bumps are ∼2 pA in amplitude (R. Hardie and S. Henderson, unpublished data) so that at least 5000 are likely to have contributed to the response. This large mismatch in the number of elementary excitatory events contributing to the response and the estimated number of InsP3receptors that one might expect to have been recruited (175) provides little support for the hypothesis that Ca2+release via InsP3 receptors is causal for excitation in Drosophila photoreceptors.
It should be emphasized that the above arguments assume that Ca2+ influx and Ca2+release are buffered by the cell and detected by the indicator dye with similar efficiency. The possibility must also be considered that significant amounts of Ca2+ are released, but into a subcellular region distinct from that accessed by the light-sensitive channels and more remote from the indicator dye (assuming this to be at least partially excluded from the “transduction compartment”). This possibility cannot be excluded, but would contravene the widely accepted view that the putative release channels and influx channels both have access to the same compartment, i.e., the subrhabdomeric space between the SMC and the base of the microvilli.
Footnotes
This research was supported by grants from the Wellcome Trust, Biotechnology and Biological Sciences Research Council, and the Royal Society.
Correspondence should be addressed to Dr. R. C. Hardie, Cambridge University, Department of Anatomy, Cambridge CB2 3DY, UK.
REFERENCES
- 1.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 2.Bezprovanny I, Ehrlich BE. Inositol (1,4,5)-trisphosphate (InsP3)-gated Ca channel from cerebellum: conduction properties for divalent cations and regulation by intraluminal calcium. J Gen Physiol. 1994;104:821–856. doi: 10.1085/jgp.104.5.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bloomquist BT, Shortridge RD, Schneuwly S, Pedrew N, Montell C, Steller H, Rubin G, Pak WL. Isolation of a putative phospholipase C gene of Drosophila norpA and its role in phototransduction. Cell. 1988;54:723–733. doi: 10.1016/s0092-8674(88)80017-5. [DOI] [PubMed] [Google Scholar]
- 4.Gray-Keller MP, Detwiler PB. The calcium feedback signal in the phototransduction cascade of vertebrate rods. Neuron. 1994;13:849–861. doi: 10.1016/0896-6273(94)90251-8. [DOI] [PubMed] [Google Scholar]
- 5.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 6.Hardie RC. Whole-cell recordings of the light induced current in dissociated Drosophila photoreceptors: evidence for feedback by calcium permeating the light-sensitive channels. Proc R Soc Lond [Biol] 1991;245:203–210. [Google Scholar]
- 7.Hardie RC. Photolysis of caged Ca2+ facilitates and inactivates but does not directly excite light-sensitive channels in Drosophila photoreceptors. J Neurosci. 1995;15:889–902. doi: 10.1523/JNEUROSCI.15-01-00889.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hardie RC (1996) A quantitative estimate of the maximum amount of light-induced Ca2+ release inDrosophila photoreceptors. J Photochem Photobiol, in press. [DOI] [PubMed]
- 9.Hardie RC, Minke B. The trp gene is essential for a light-activated Ca2+ channel in Drosophila photoreceptors. Neuron. 1992;8:643–651. doi: 10.1016/0896-6273(92)90086-s. [DOI] [PubMed] [Google Scholar]
- 10.Hardie RC, Minke B. Novel Ca2+channels underlying transduction in Drosophila photoreceptors. Implications for phosphoinositide-mediated Ca2+ mobilization. Trends Neurosci. 1993;16:371–376. doi: 10.1016/0166-2236(93)90095-4. [DOI] [PubMed] [Google Scholar]
- 11.Hardie RC, Minke B. Phosphoinositide-mediated phototransduction in fly photoreceptors: the role of Ca2+ and TRP. Cell Calcium. 1995;16:256–274. doi: 10.1016/0143-4160(95)90023-3. [DOI] [PubMed] [Google Scholar]
- 12.Hardie RC, Mojet MH. Mg2+dependent block of the light-activated and trp dependent conductance in Drosophila photoreceptors. J Neurophysiol. 1995;74:2590–2599. doi: 10.1152/jn.1995.74.6.2590. [DOI] [PubMed] [Google Scholar]
- 13.Hofer AM, Schlue W-R, Curci S, Machen TE. Spatial distribution and quantitation of free luminal [Ca] within the InsP3 sensitive internal store of individual BHK-21 cell: ion dependence of InsP3 induced Ca release and reloading. FASEB J. 1995;9:788–798. doi: 10.1096/fasebj.9.9.7601343. [DOI] [PubMed] [Google Scholar]
- 14.Johnson EC, Pak WL. Electrophysiological study of Drosophila rhodopsin mutants. J Gen Physiol. 1986;88:651–673. doi: 10.1085/jgp.88.5.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKay RR, Chen D-M, Miller K, Kim S, Stark WS, Shortridge RD. Phospholipase C rescues visual defect in norpA mutant of Drosophila melanogaster. J Biol Chem. 1995;270:13271–13276. doi: 10.1074/jbc.270.22.13271. [DOI] [PubMed] [Google Scholar]
- 16.Minke B, Selinger Z. Inositol lipid pathway in fly photoreceptors, excitation, calcium mobilization and retinal degeneration. In: Osborne NN, Chader GJ, editors. Progress in retinal research, Vol. 11. Pergamon; Oxford: 1991. pp. 99–124. [Google Scholar]
- 17.Minke B, Selinger Z. The inositol-lipid pathway is necessary for light excitation in fly photoreceptors. In: Corey D, Roper SD, editors. Sensory transduction. Rockefeller UP; 1992. pp. 201–217. [PubMed] [Google Scholar]
- 18.Montell C, Rubin GM. Molecular characterization of Drosophila trp locus, a putative integral membrane protein required for phototransduction. Neuron. 1989;2:1313–1323. doi: 10.1016/0896-6273(89)90069-x. [DOI] [PubMed] [Google Scholar]
- 19.Nagy K. Biophysical processes in invertebrate photoreceptors: recent progress and a critical overview based on Limulus photoreceptors. Q Rev Biophys. 1991;24:165–226. doi: 10.1017/s0033583500003401. [DOI] [PubMed] [Google Scholar]
- 20.O’Day PM, Gray-Keller MP. Evidence for electrogenic Na+/Ca2+ exchange in Limulus ventral photoreceptors. J Gen Physiol. 1989;93:473–492. doi: 10.1085/jgp.93.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Tousa JE, Baehr W, Martin RL, Hirsh J, Pak WL, Applebury ML. The Drosophila ninaE gene encodes an opsin. Cell. 1985;40:839–850. doi: 10.1016/0092-8674(85)90343-5. [DOI] [PubMed] [Google Scholar]
- 22.Pak WL, Grossfield J, White NV. Non-phototactic mutants in a study of vision in Drosophila . Nature. 1969;222:351–354. doi: 10.1038/222351a0. [DOI] [PubMed] [Google Scholar]
- 23.Payne R, Walz B, Levy S, Fein A. The localization of calcium release by inositol trisphosphate in Limulus photoreceptors and its control by negative feedback. Philos Trans R Soc Lond Biol. 1988;320:359–379. doi: 10.1098/rstb.1988.0082. [DOI] [PubMed] [Google Scholar]
- 24.Peretz A, Suss-Toby E, Rom-Glas A, Arnon A, Payne R, Minke B. The light response of Drosophila photoreceptors is accompanied by an increase in cellular calcium: effects of specific mutations. Neuron. 1994a;12:1257–1267. doi: 10.1016/0896-6273(94)90442-1. [DOI] [PubMed] [Google Scholar]
- 25.Peretz A, Sandler C, Kirschfeld K, Hardie RC, Minke B. Genetic dissection of light-induced Ca2+ influx into Drosophila photoreceptors. J Gen Physiol. 1994b;104:1057–1077. doi: 10.1085/jgp.104.6.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phillips AM, Bull A, Kelly L. Identification of a Drosophila gene encoding a calmodulin binding protein with homology to the trp phototransduction gene. Neuron. 1992;8:631–642. doi: 10.1016/0896-6273(92)90085-r. [DOI] [PubMed] [Google Scholar]
- 27.Pollock JA, Assaf A, Peretz A, Nichols CD, Mojet MH, Hardie RC, Minke B. TRP, a protein essential for inositide-mediated Ca2+ influx, is localized adjacent to the calcium stores in Drosophila photoreceptors. J Neurosci. 1995;15:3747–3760. doi: 10.1523/JNEUROSCI.15-05-03747.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ranganathan R, Harris GL, Stevens CF, Zuker CS. A Drosophila mutant defective in extracellular calcium-dependent photoreceptor deactivation and rapid desensitization. Nature. 1991;354:230–232. doi: 10.1038/354230a0. [DOI] [PubMed] [Google Scholar]
- 29.Ranganathan R, Bacskai BJ, Tsien RY, Zuker CS. Cytosolic calcium transients: spatial localization and role in Drosophila photoreceptor cell function. Neuron. 1994;13:837–848. doi: 10.1016/0896-6273(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 30.Ranganathan R, Malicki DM, Zuker CS. Signal transduction in Drosophila photoreceptors. Annu Rev Neurosci. 1995;18:283–317. doi: 10.1146/annurev.ne.18.030195.001435. [DOI] [PubMed] [Google Scholar]
- 31.Scott K, Becker A, Sun Y, Hardy R, Zuker C. Gqα protein function in vivo: genetic dissection of its role in photoreceptor cell physiology. Neuron. 1995;15:919–927. doi: 10.1016/0896-6273(95)90182-5. [DOI] [PubMed] [Google Scholar]
- 32.Shin J, Richard EA, Lisman JE. Ca2+ is an obligatory intermediate in the excitation cascade of Limulus photoreceptors. Neuron. 1993;11:845–855. doi: 10.1016/0896-6273(93)90114-7. [DOI] [PubMed] [Google Scholar]
- 33.Toyoshima S, Matsumoto N, Wang P, Inoue H, Yoshioka T, Hotta Y, Osawa T. Purification and partial amino-acid sequences of phosphoinositide-specific phospholipase C of Drosophila eye. J Biol Chem. 1990;265:14842–14848. [PubMed] [Google Scholar]
- 34.Ukhanov KY, Payne R. Light-activated Ca2+ release in Limulus ventral photoreceptors as revealed by laser confocal microscopy. Cell Calcium. 1995;18:300–312. doi: 10.1016/0143-4160(95)90026-8. [DOI] [PubMed] [Google Scholar]
- 35.Ukhanov KY, Flores TM, Hsiao HS, Mohaptra P, Pitts CH, Payne R. Measurements of cytosolic Ca2+concentration in Limulus ventral photoreceptors using fluorescent dyes. J Gen Physiol. 1995;105:95–116. doi: 10.1085/jgp.105.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walz B. Calcium-sequestering smooth endoplasmic reticulum in retinula cells of the blowfly. J Ultrastruct Res. 1982;81:240–248. doi: 10.1016/s0022-5320(82)90079-x. [DOI] [PubMed] [Google Scholar]
- 37.Walz B, Zimmermann B, Seidl S. Intracellular Ca2+ concentration and latency of light-induced changes in photoreceptors of honey bee drone. J Comp Physiol [A] 1994;174:421–431. [Google Scholar]
- 38.Wu C-F, Pak WL. Quantal basis of photoreceptor spectral sensitivity of Drosophila melanogaster . J Gen Physiol. 1975;66:149–168. doi: 10.1085/jgp.66.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou Z, Neher E. Mobile and immobile calcium buffers in bovine adrenal chromaffin cells. J Physiol (Lond) 1993;469:245–273. doi: 10.1113/jphysiol.1993.sp019813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zuker CS, Cowman AF, Rubin GM. Isolation and structure of a rhodopsin gene from D. melanogaster . Cell. 1985;40:851–858. doi: 10.1016/0092-8674(85)90344-7. [DOI] [PubMed] [Google Scholar]