

Abstract
Neurons undergo extensive changes in growth and electrophysiological properties in response to axon injury. Efforts to understand the molecular mechanisms that initiate these changes have focused almost exclusively on the role of extrinsic signals, primarily neurotrophic factors released from target and glial cells. The objective of the present investigation was to determine whether the response to axonal injury also involves intrinsic axoplasmic signals.Aplysia neurons were removed from their ganglia and placed in vitro on a substratum permissive for growth, but in the absence of glia and soluble growth factors. Under these conditions, neurites emerged and grew for ∼4 d. Once growth had ceased, the neurites were transected. In all, 46 of 50 cells regenerated, either by resorbing the remaining neurites and elaborating a new neuritic arbor or by merely replacing the neurites that had been severed. Cut cells also exhibited enhanced excitability and, paradoxically, prolonged survival, when compared with uninjured neurons. These findings indicate that axons contain intrinsic molecular signals that are directly activated by injury to trigger changes underlying regeneration and compensatory plasticity.
Keywords: axotomy, cellular stress, regeneration, sensitization, axoplasm, retrograde transport, intracellular recording
Axons, because they extend great distances from the cell body, can sustain severe damage without lethal consequences to the neuron. This permits restoration of function by regeneration of new axons. It also allows the signals activated at the site of axonal injury to be studied separately from the processes they regulate in the distant cell body, making neurons excellent models to investigate molecular mechanisms underlying cellular adaptations to stress. Nerve injury in vertebrates and invertebrates triggers long-term alterations that involve not only regenerative growth but also altered excitability and selective cell death (Bulloch and Ridgway, 1989; Titmus and Faber, 1990; Kreutzberg, 1995). These changes commonly are assumed to be initiated by disconnection of the injured neuron from its target, thereby interrupting its supply of target-derived trophic factors (Nja and Purves, 1978; Wu et al., 1993). The idea that this deprivation can elicit regeneration has guided efforts to restore function after nerve damage (Derby et al., 1993). Far less attention has been paid to the possibility that nerve injury directly activates intrinsic signals within axons that are transported to the cell body where they induce long-term changes, including axon regeneration. Some evidence supporting this hypothesis has been obtained recently by using nociceptive sensory neurons in the marine mollusc Aplysia(Ambron et al., 1995; Walters and Ambron, 1995). Crushing the axons of these neurons induces regenerative growth and collateral sprouting, as well as hyperexcitability and synaptic facilitation, that persist for weeks (Billy and Walters, 1989; Walters et al., 1991; Clatworthy and Walters, 1994; Dulin et al., 1995; Steffensen et al., 1995). The development of long-term hyperexcitability is not caused by interrupting the retrograde transport of target-derived signals but, instead, involves “positive” molecular signals that are transported from the axon to the neuronal soma and nucleus after nerve injury (Ambron et al., 1995; Gunstream et al., 1995a; Walters and Ambron, 1995). Two central questions not answered in these earlier studies are addressed in the present paper: (1) Do intrinsic injury signals affect neuronal growth and survival as well as excitability? (2) Are soluble extrinsic factors from damaged glia, other support cells, or hemolymph (Ridgway et al., 1991; Curtis et al., 1993) necessary for axotomy-induced hyperexcitability, or are intrinsic axoplasmic signals sufficient?
MATERIALS AND METHODS
Dissociated cell culture. Polystyrene dishes coated with poly-l-lysine were exposed to hemolymph for 3 hr at room temperature. Macromolecules in the hemolymph bind to the poly-l-lysine, forming an adhesive substratum permissive for growth (Burmeister et al., 1991). The dishes were washed thoroughly to remove soluble hemolymph proteins, and isotonic L15 medium was added. Neurons from the abdominal, pedal, and pleural ganglia were dissociated by using protease, which also destroys the glial cells (Schacher and Proshansky, 1983). Individual neurons were added to the dish and were maintained at 15°C. The L15 was changed every 4–5 d.
Growth and survival. The cells were photographed with a Leitz inverted microscope equipped with phase contrast and Nomarski optics. The image was transmitted to a Hammamatsu Argus system and was then sent to a computer for storage. Composite pictures were assembled from saved images with Adobe Photoshop. Survival time was defined as the period from plating until the soma disintegrated. Survival was evaluated on a daily basis, with each cell scored unequivocally as either surviving or not.
Electrophysiology. See Gunstream et al. (1995a) for details. Intracellular recordings were made from sensory neuron somata dissociated from the ventrocaudal cluster in each pleural ganglia and from presumptive motor neurons dissociated from the region in each pedal ganglion containing identified tail motor neurons as well as unidentified parapodial and body wall motor neurons (Walters et al., 1983; E.T. Walters, unpublished observations). Motor neurons were identified by their size (100–200 μm soma diameter), relatively brief action potentials, and patterns of spontaneous synaptic input (Walters et al., 1983). Although we did not confirm that they innervated the tail, midbody, or parapodia, all of the cells satisfying these criteria in this region of the ganglion have peripheral axons and produce movements of the posterior body when stimulated intracellularly (Walters, unpublished observations). The neurites of dissociated sensory and motor neurons were transected 2 d after plating. The next day, the cells were impaled with single-barreled glass microelectrodes filled with 3 m potassium acetate (electrode resistance 5–20 MΩ). Recordings were made after the L15 solution was replaced with buffered artificial seawater, pH 7.6. Test stimuli used to characterize soma excitability were applied in a standard test sequence (Walters et al., 1991; Gunstream et al., 1995a). Briefly, a rapid, ascending series of 20 msec depolarizing pulses was used to determine spike threshold. Then the capacity for repetitive firing was assessed by counting the number of spikes evoked by a 1 sec pulse set at 2.5 times the threshold current determined with the 20 msec pulses.
Statistics. Comparisons were made with a two-tailedt test for independent groups or, when data were not normally distributed, with Mann–Whitney U tests. A probability (p) of <0.05 was considered significant.
RESULTS
Responses of dissociated neurons to axonal injuryin vitro
We first examined the behavior of Aplysia neurons cultured in the absence of supporting cells or soluble growth factors (Fig. 1). Under these conditions, neurites emerge within hours and typically elongate until the fourth day when the growth cones become bulbous and growth ceases (Fig. 1A). After a quiescent period that lasts as long as 12 d, the neurites become varicose (Fig. 1B,C), the nucleus condenses and becomes eccentric, and within 2 d the cell soma disintegrates (Fig. 1D–F, G–I). The latter events are characteristic of programmed cell death in vitro(Deckworth and Johnson, 1993). A study of 28 cells growing in vitro yielded a mean survival of 16.5 d.
Fig. 1.
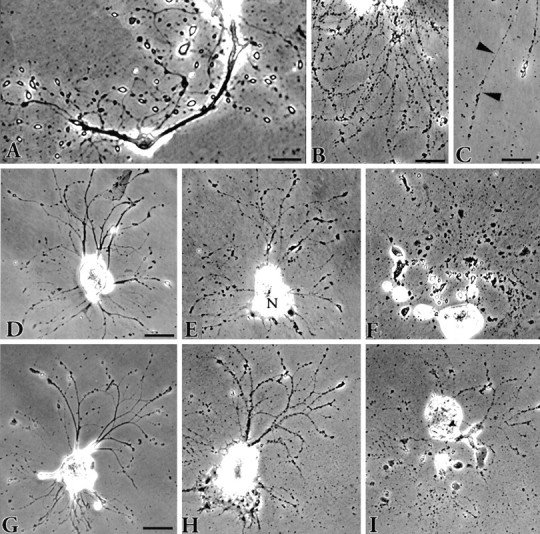
Phase-contrast views depicting the fate ofAplysia neurons in vitro.A, Neuritic processes 5 d after plating showing the bulbous, birefringent endings that have replaced the growth cones on the newly formed neurites. The appearance of these structures marks the end of the growth period. Scale bar, 50 μm. B, Cell death typically ensues when the neurites become varicose. Scale bar, 50 μm. C, The varicosities are connected via fine strands (arrowheads). These eventually break, resulting in fragmentation of the neurite. Scale bar, 30 μm. The progression of two cells toward death is shown in D–Fand G–I. The cells were photographed 1 d after plating (D, G) and then 4 d later (E, H) when the neurites were becoming varicose. The nucleus (N) in E has moved to one pole of the cell. After an additional 9 d (F) and 8 d (I), each cell had disintegrated. The entire process from plating to death takes 17 d on average. Scale bar for both cells, 100 μm.
To determine whether any axoplasmic injury signals are truly intrinsic, we grew the cells as above, waited until growth had ceased, and then injured the cells by pruning the newly formed neurites with a microneedle (Fig. 2). The extent of the pruning varied from removing only the distal-most neurites to completely severing all of the new growth. Regardless, the cells were extraordinarily resilient in that 92% (46 of 50) survived the pruning and regenerated neuritic processes. The four cells that did not (including one that was minimally trimmed), detached from the substratum within 24 hr. Two modes of regeneration were observed. Most of the cells merely elongated the severed processes (Fig. 2A), whereas the others underwent a dramatic reorganization of the entire arbor (Figs.2B, 3). The latter changes typically began with a gradual resorption of the remaining neurites and, sometimes, of the neurites that were not cut (Fig. 3). This was followed by the emergence and growth of new processes. Control cells, which were in the same dishes as the cut cells, stopped growing and died at a time when the cut cells were regenerating (Fig.2E; also see below). The factors that govern the particular mode of regeneration are not known, but neither the extent of the injury nor the origin of the cut neurites (whether from the cell body or axon stump; Figs. 2B, 3) seems to be important. Pruning the processes is a powerful signal to the cell. We have cut some cells twice and found that they were capable of regenerating another arbor. How many times a cell can recover presently is being investigated.
Fig. 2.

Phase-contrast photomicrographs of neurons showing the two typical patterns of neurite regeneration. A, An example of regenerative growth in which the new growth merely extends the cut neurites 4 d after plating growth had ceased and the neurites were cut at the arrowhead. Fifteen minutes later the cell was photographed again (C). One day later (F) several prominent growth cones were visible. In the ensuing days the neurites extended, retracted, and then slowly extended again. Seventeen days later, the cell was still growing (I), and it survived >32 d. The other response (B) is much more complex. The neurites of this cell were cut after 4 d in culture (arrowheads), and the cell was photographed 15 min later (D). During the next 11 d the neurite stumps were resorbed into the soma, and new growth emerged (G). The neurites formed thick fascicles that grew extensively and by 24 d after cutting had formed an arbor very different from the original (J) (see also Fig. 3). This cell survived for 31 d. Scale bar, A, B, 100 μm. A control cell (E) in the dish with the cell in A had stopped growing after 4 d. Varicosities on the neurite began to appear 6 d later (H); by day 17 many of the neurites had disappeared, and the cell began to disintegrate (K). Scale bar, 150 μm.
Fig. 3.
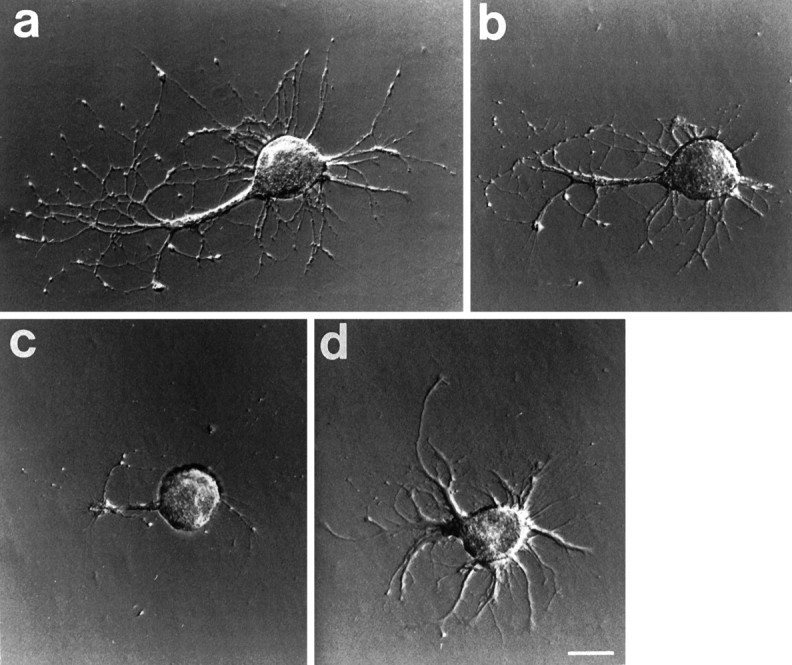
Photomicrographs of a cell after transecting neurites in vitro. a, The cell is seen 9 d after plating and just before the neurites were cut. Neurites extend from the large remnant of the original axon. b, The cell 15 min after the neurites had been severed and (c) 7 d later. The appearance is radically altered because the axon and many of the neurites were resorbed.d, After an additional 7 d, new neurites have emerged. These continued to grow for several more days, and the cell died on day 35. Scale bar, 100 μm.
Isolated axons have a limited capacity for growth
The new growth that emerges from the proximal stump almost immediately after injury is supported by the reuse of materials already present in the axon (Ashery et al., 1996). Sustained growth, however, requires the synthesis of new components in the cell body. An assessment of the relative amount of growth that can occur via the first route is made traditionally by using inhibitors of protein synthesis. This approach is not ideal, however, because some inhibitors (e.g., anisomycin) interfere with export into the axon (Ambron et al., 1975) and cause cellular stresses that can activate protein kinase cascades (Kyriakis et al., 1994). To assess directly the ability of axons to grow in the absence of the cell soma, we examined isolated axons in vitro (Fig. 4). These axons, which can survive for relatively long periods without a cell body (Benbassat and Spira, 1993), did sprout neurites, but the extent of growth did not approach nearly the levels seen after injury of neurites attached to the cell soma (compare Figs. 2, 3). Also, the neurites that emerged typically stopped growing by the second day (Fig. 4). These findings support the idea that intrinsic signals generated at a site of axonal injury regulate events in the nucleus that are necessary for effective regeneration.
Fig. 4.

Typical examples of neurite outgrowth from isolated axons. Axons, which had detached from their cell bodies during plating, sprouted neurites within the first 24 hr (A, C, E), but there was little subsequent growth. The axons were followed for 22 d (B), 9 d (D), and 4 d (F), respectively; shortly thereafter, each disintegrated. Scale bar, 100 μm.
Axotomy promotes cell survival
Both modes of regeneration after axotomy required many days to complete, suggesting the paradoxical possibility that axonal injury delays the onset of cell death. To test this hypothesis, we compared the life span of 27 cells in four dishes: 13 cells that had survived pruning and were regenerating and 14 undamaged control cells from the same dishes (Fig. 5). Using the disintegration of the soma as an obvious endpoint (Fig. 1), we found that the neurons, the neurites of which had been transected, survived 27.9 ± 1.6 d (mean ± SEM; n = 13), which was significantly longer than the 18.3 ± 2.1 d mean survival of those with unsevered axons (n = 14; t25 = 3.64; p = 0.0013).
Fig. 5.

Histogram showing the increased survival time after axotomy in vitro. Twenty-seven cells growing in four dishes in vitro were followed on a daily basis until they died. The neurites of some cells, selected at random, were severed after growth had ceased (filled bars), and their survival was compared with control cells in the same dish (open bars).
Axotomy causes electrophysiological changes in the cell body
Recently, it was reported (Salim and Glanzman, 1995) that transecting the neurites of dissociated Aplysia sensory neurons in vitro enhances the electrical excitability of the soma measured 24 hr later, indicating that hyperexcitability also is triggered directly by axon damage. The cells in this study were grown in hemolymph, however, which contains soluble factors that could contribute to this alteration (Schacher and Proshansky, 1983;Krontiris-Litowitz et al., 1989; Burmeister et al., 1991). We asked whether injury-induced hyperexcitability in vitro occurs in the absence of hemolymph and, if so, whether this intrinsic response is unique to sensory neurons. Sensory neurons from the ventrocaudal cluster of the pleural ganglion or motor neurons in the pedal ganglion that innervate the tail and parapodia (Walters et al., 1983) were placed in culture without hemolymph. Two days later their neurites were transected.
Twenty-four hours after this axotomy, sensory neurons (Fig.6A,B) and motor neurons (Fig.6C) were significantly more excitable than the corresponding neurons, the outgrowing neurites of which were not severed. As occurs after nerve crush in vivo (Walters et al., 1991; Dulin and Walters, 1993; Dulin et al., 1995; Clatworthy and Walters, 1994) and in isolated ganglia (Gunstream et al., 1995a), transection-induced hyperexcitability was expressed as an increase in repetitive firing (Fig. 6) and a tendency for action potential (spike) threshold to decrease. Repetitive firing is the electrophysiological property most sensitive to axon damage (Walters et al., 1991; Clatworthy and Walters, 1994; Gunstream et al., 1995a). Because hemolymph had little or no effect in the present study (Fig. 6B,C), we pooled the threshold data from experiments with and without hemolymph to increase statistical power. Axotomized motor neurons displayed a significant decrease in median threshold during 20 msec test pulses, as compared with unaxotomized controls: 0.12 versus 0.19 nA (p = 0.01). None of the motor neurons examined exhibited background spike activity. For axotomized and control sensory neurons, the median spike thresholds were, respectively, 0.13 and 0.19 nA (p = 0.19, Mann–Whitney U Test; see figure legend). Although not significant in this small sample, the somewhat lower threshold in transected sensory neurons shows that the significant increase in repetitive firing in these cells could not have been attributable to inadvertent delivery of higher test currents. Because the test currents were normalized to spike threshold, these cells would have received the same or lower intensity test pulses as those received by the control sensory neurons.
Fig. 6.

Long-term hyperexcitability induced by transecting neurites of isolated sensory and motor neurons in the presence or absence of hemolymph in the culture medium. A, Examples of repetitive firing during testing of a control sensory neuron and a sensory neuron, the outgrowing neurites of which had been transected 1 d earlier. The test stimulus was a 1 sec depolarizing pulse injected into the neuronal soma through the recording electrode. Injected current was set at 2.5 times that required to reach spike threshold during a previous series of 20 msec pulses. B, Repetitive firing in sensory neurons 1 d after neurite transection. The mean ± SEM number of spikes evoked by the test stimulus was enhanced by previous transection but unaffected by the presence or absence of hemolymph during the test. Hemolymph present,n = 12 control and 10 transected cells,p < 0.005; hemolymph absent, n= 14 control and 6 transected neurons, p < 0.005.C, Repetitive firing in motor neurons 1 d after neurite transection. Firing was enhanced by previous transection and unaffected by hemolymph during the test. Hemolymph present,n = 6 control and 3 transected cells,p < 0.05; hemolymph absent, n= 8 control and 8 transected neurons, p < 0.05.
Having observed that injury-induced hyperexcitability can be expressed <1 d after axotomy in a ganglion preparation (Gunstream et al., 1995a), we were surprised to find that sensory neuron somata required ∼4 d after their dissociation from the ganglion to become hyperexcitable. We do not yet know which aspects of the highly traumatic dissociation procedure (e.g., close proximity of the injury site to the soma, protease treatment, the release of heterogenous modulators by damage to surrounding tissue) is responsible for the delay in hyperexcitability triggered by dissociation. Nevertheless, the cells have recovered sufficiently within 2 d to express significant hyperexcitability in response to the severing of outgrowing neurites (Fig. 6; see also Salim and Glanzman, 1995). Detailed analyses of electrophysiological effects of axon transection produced both by cell dissociation and by severing neurites growing in culture will be described elsewhere (J.D. Gunstream, X. Liao, G.A. Castro, and E.T. Walters, unpublished data).
DISCUSSION
Our results show that diverse long-term neuronal changes can be triggered directly by axon injury and demonstrate that simple preparations of dissociated neurons will be useful for identifying intrinsic injury-activated signals for growth, resistance to cell death, and hyperexcitability. These alterations occur in a wide range of neuron types, including sensory and motor neurons. The neurons that were injured in our experiments were maintained in dishes that did not contain hemolymph, glial cells, or soluble factors from the CNS. Because humoral factors greatly promote growth of molluscan neurons in culture (Schacher and Proshansky, 1983; Bulloch and Ridgway, 1989), most studies of axotomy in vitro have used medium that either contained hemolymph or was conditioned with factors released from tissue (Benbassat and Spira, 1993; Williams and Cohan, 1994). The few observations of growth after injury to axons of dissociated neurons in the absence of humoral factors have been restricted to short-term effects at the level of growth cones (Williams and Cohan, 1994). Although the substratum in our experiments was pretreated with hemolymph, it is likely that the constituents of the hemolymph that bound to the poly-l-lysine merely provide an adhesive surface that is permissive for growth (Burmeister et al., 1991). Indeed, neurons from Aplysia and other invertebrates grow on lectins in the absence of hemolymph (Chiquet and Acklin, 1986; Lin and Levitan, 1987; Wilson et al., 1992). Moreover, the substrate-bound factors in our experiments do not, by themselves, initiate growth because neurite extension ceases in vitro after a few days, although the substratum still has the capacity to support growth. Interactions between bound factors and the neuritic surface may be important for growth, but our data support the idea that it is events produced within the axon by injury that initiate the growth. A critical role for signals intrinsic to injured neurites is consistent with the results of previous experiments, which showed that injection of axoplasm from injured nerves into uninjured neuronal somata induced the same electrophysiological changes that occur after nerve injury (Ambron et al., 1995).
If the responses of the cells after injury are not elicited by extrinsic growth factors, then what events associated with axonal injury activate the intrinsic signals? A good possibility would be an influx of calcium (Ziv and Spira, 1993), especially because long-term, injury-induced hyperexcitability is dependent on extracellular calcium at the axonal injury site (Gunstream et al., 1995b). The calcium could then activate protein kinase cascades in the axon. Other consequences of disrupting the axon might also activate protein kinases. Interestingly, a family of Jun kinases recently has been discovered that is responsive to cellular stress (Kyriakis et al., 1994). At activation, these kinases enter the nucleus where they phosphorylate c-Jun, a transcription factor that has been widely linked to regeneration after injury in both the CNS and peripheral nervous system (PNS) (Herdegen and Zimmermann, 1994).
The intrinsic injury signals from the axon that regulate growth, survival, and excitability in vivo would, under natural conditions, be expected to act in concert with extrinsic signals released from other cells. Different combinations of intrinsic and extrinsic signals might be used to signify the degree of injury or the stage of recovery. An important question concerns the sites of convergence of intrinsic and extrinsic injury signals and the extent of overlap of these signals with those that induce formally similar cellular alterations associated with learning (Walters and Ambron, 1995; Ambron and Walters, 1996). Thus, treatment of the sensory cells in the pleural ganglion with serotonin can produce long-term facilitation of the sensory–motor synapse (Emptage and Carew, 1993). Like injury, serotonin induces growth (Bailey and Chen, 1988; Bailey et al., 1992) and relatively long-lasting changes in excitability (Dale et al., 1987). Furthermore, both injury and serotonin induce the synthesis of some of the same proteins (Noel et al., 1993, 1995), including the transcription factor C/EBP (Alberini et al., 1994). Because long-term facilitation is also induced at distal sensory–motor synapses in response to exogenous serotonin (Clark and Kandel, 1993; Emptage and Carew, 1993), some of the signals to the nucleus from these synapses may be the same as the intrinsic injury signals that travel from a site of axonal injury. Although there are many possible sites of convergence, there also may be signals that are unique to each phenomenon. For example, the transcription factor NF-κB is strongly affected by nerve crush (M. Povelones, C. Tran, D. Thanos, and R.T. Ambron, unpublished data) but seems not to be involved in the induction of long-term facilitation (Alberini et al., 1995; Povelones, Tran, Thanos, and Ambron, unpublished data).
An interesting question is whether the trauma associated with excising neurons from the nervous system enhances the response of the cells to subsequent axotomy (produced by severing outgrowing neurites). A testable hypothesis is that aspects of the dissociation procedure act like the conditioning lesions that have been shown in many studies to enhance subsequently triggered axonal regeneration (McQuarrie and Grafstein, 1973; Carlsen, 1983; Jacob and McQuarrie, 1991). Our findings that intrinsic axonal injury signals promote the survival as well as growth and excitability of isolated neurons suggest that the enhanced growth seen in conditioning lesion studies and the enhanced excitability often seen after axotomy (Devor, 1994) may both be components of a common, compensatory response to neuronal stress. Indeed, it has been hypothesized that neural regeneration and memory involve very primitive mechanisms that first evolved for detecting and adapting to cellular stress (Walters, 1991, 1994). These considerations suggest that regulatory molecules prominently involved in diverse cellular stress responses, such as the Jun kinases (Kyriakis et al., 1994), are also important for initiating axonal regeneration and some forms of memory.
Footnotes
This work was supported by Grant NS12250 from National Institutes of Health (R.T.A.) and Grant MH38726 from the National Institute of Mental Health (E.T.W.). We thank D. Glanzman for helpful comments.
Correspondence should be addressed to Dr. Richard T. Ambron, Department of Anatomy and Cell Biology, Black Building 1204, Columbia University Medical Center, West 168th Street, New York, NY 10032.
REFERENCES
- 1.Alberini CM, Ghirardi M, Metz R, Kandel ER. C/EBP is an immediate-early gene required for the consolidation of long-term facilitation in Aplysia. Neuron. 1994;76:1099–1114. doi: 10.1016/0092-8674(94)90386-7. [DOI] [PubMed] [Google Scholar]
- 2.Alberini CM, Ghirardi M, Huang Y-Y, Nguyen PV, Kandel ER. A molecular switch for the consolidation of long-term memory: cAMP-inducible gene expression. Ann NY Acad Sci. 1995;256:261–286. doi: 10.1111/j.1749-6632.1995.tb24833.x. [DOI] [PubMed] [Google Scholar]
- 3.Ambron RT, Walters ET. Priming events and retrograde injury signals: a new perspective on the cellular and molecular biology of nerve regeneration. Mol Neurobiol. 1996;13:61–79. doi: 10.1007/BF02740752. [DOI] [PubMed] [Google Scholar]
- 4.Ambron RT, Goldman JE, Schwartz JH. Effect of inhibiting protein synthesis on axonal transport of membrane glycoproteins in an identified neuron of Aplysia. Brain Res. 1975;94:307–323. doi: 10.1016/0006-8993(75)90064-5. [DOI] [PubMed] [Google Scholar]
- 5.Ambron RT, Dulin MF, Zhang X-P, Schmied R, Walters ET. Axoplasm enriched in a protein mobilized by nerve injury induces memory-like alterations in Aplysia neurons. J Neurosci. 1995;15:3440–3446. doi: 10.1523/JNEUROSCI.15-05-03440.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ashery B, Penner R, Spira ME. Acceleration of membrane recycling by axotomy of cultured Aplysia neurons. Neuron. 1996;16:641–651. doi: 10.1016/s0896-6273(00)80083-5. [DOI] [PubMed] [Google Scholar]
- 7.Bailey CH, Chen M. Long-term sensitization in Aplysia increases the number of presynaptic contacts onto the identified gill motor neuron L7. Proc Natl Acad Sci USA. 1988;85:9356–9359. doi: 10.1073/pnas.85.23.9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bailey CH, Montarolo P, Chen M, Kandel ER, Schacher S. Inhibitors of protein and RNA synthesis block structural changes that accompany long-term heterosynaptic plasticity in Aplysia. Neuron. 1992;9:749–758. doi: 10.1016/0896-6273(92)90037-e. [DOI] [PubMed] [Google Scholar]
- 9.Benbassat D, Spira ME. Survival of isolated axonal segments in culture: morphological, ultrastructural, and physiological analysis. Exp Neurol. 1993;122:295–310. doi: 10.1006/exnr.1993.1129. [DOI] [PubMed] [Google Scholar]
- 10.Billy AJ, Walters ET. Long-term expansion and sensitization of mechanosensory receptive fields in Aplysia support an activity-dependent model of whole-cell sensory plasticity. J Neurosci. 1989;9:1254–1262. doi: 10.1523/JNEUROSCI.09-04-01254.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bulloch AGM, Ridgway RL. Neuronal plasticity in the adult invertebrate nervous system. J Neurobiol. 1989;20:295–311. doi: 10.1002/neu.480200504. [DOI] [PubMed] [Google Scholar]
- 12.Burmeister DW, Rivas RJ, Goldberg DJ. Substrate-bound factors stimulate engorgement of growth cone lamellipodia during neurite elongation. Cell Motil Cytoskeleton. 1991;19:255–268. doi: 10.1002/cm.970190404. [DOI] [PubMed] [Google Scholar]
- 13.Carlsen RC. Delayed induction of the cell body response and enhancement of regeneration following a condition/test lesion of frog peripheral nerve at 15°C. Brain Res. 1983;279:9–18. doi: 10.1016/0006-8993(83)90158-0. [DOI] [PubMed] [Google Scholar]
- 14.Chiquet M, Acklin SE. Attachment to Con A or extracellular matrix initiates rapid sprouting by cultured leech neurons. Proc Natl Acad Sci USA. 1986;83:6188–6192. doi: 10.1073/pnas.83.16.6188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clark GA, Kandel ER. Branch-specific heterosynaptic facilitation in Aplysia siphon sensory cells. Proc Natl Acad Sci USA. 1993;81:2577–2581. doi: 10.1073/pnas.81.8.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clatworthy A, Walters ET. Comparative analysis of hyperexcitability and synaptic facilitation induced by nerve injury in two populations of mechanosensory neurones of Aplysia californica. J Exp Biol. 1994;190:217–238. doi: 10.1242/jeb.190.1.217. [DOI] [PubMed] [Google Scholar]
- 17.Curtis R, Adryan KM, Zhu Y, Harkness P, Lindsay RM, Distefano PS. Retrograde axonal transport of ciliary neurotrophic factor is increased by peripheral nerve injury. Nature. 1993;365:253–255. doi: 10.1038/365253a0. [DOI] [PubMed] [Google Scholar]
- 18.Dale N, Kandel ER, Schacher S. Serotonin produces long-term changes in the excitability of Aplysia sensory neurons in culture that depend on new protein synthesis. J Neurosci. 1987;7:2232–2238. doi: 10.1523/JNEUROSCI.07-07-02232.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deckworth TL, Johnson EM. Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Derby A, Engleman VW, Frierdich GE, Neises G, Rapp SR, Roufa DG. Nerve growth factor facilitates regeneration across nerve gaps: morphological and behavioral studies in rat sciatic nerve. Exp Neurol. 1993;119:176–191. doi: 10.1006/exnr.1993.1019. [DOI] [PubMed] [Google Scholar]
- 21.Devor M. The pathophysiology of damaged peripheral nerves. In: Wall PD, Melzack R, editors. Textbook of pain. Churchill Livingstone; Edinburgh: 1994. pp. 79–100. [Google Scholar]
- 22.Dulin MF, Walters ET. Similar alterations of sensory and motor neurons in Aplysia persist after regeneration. Soc Neurosci Abstr. 1993;19:578. [Google Scholar]
- 23.Dulin MF, Steffensen I, Morris C, Walters ET. Recovery of function, peripheral sensitization, and sensory neurone activation by novel pathways following axonal injury in Aplysia californica. J Exp Biol. 1995;198:2055–2066. doi: 10.1242/jeb.198.10.2055. [DOI] [PubMed] [Google Scholar]
- 24.Emptage NJ, Carew TJ. Long-term synaptic facilitation in the absence of short-term facilitation in Aplysia neurons. Science. 1993;262:253–256. doi: 10.1126/science.8211146. [DOI] [PubMed] [Google Scholar]
- 25.Gunstream J, Castro GA, Walters ET. Retrograde transport of plasticity signals in Aplysia sensory neurons following axonal injury. J Neurosci. 1995a;15:439–448. doi: 10.1523/JNEUROSCI.15-01-00439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gunstream JD, Castro GA, Walters ET. Axotomy-induced hyperexcitability of Aplysia sensory neurons requires peripheral calcium. Soc Neurosci Abstr. 1995b;21:1681. [Google Scholar]
- 27.Herdegen T, Zimmermann M. Expression of c-Jun and JunD transcription factors represent specific changes in neuronal gene expression following axotomy. Prog Brain Res. 1994;103:153–171. doi: 10.1016/s0079-6123(08)61135-8. [DOI] [PubMed] [Google Scholar]
- 28.Jacob JM, McQuarrie IG. Axotomy accelerates slow component B of axonal transport. J Neurobiol. 1991;22:570–582. doi: 10.1002/neu.480220603. [DOI] [PubMed] [Google Scholar]
- 29.Kreutzberg GW. Reaction of the neuronal cell body to axonal damage. In: Waxman SG, Kocsis JD, Stys PK, editors. The axon: structure, function, and pathology. Oxford UP; New York: 1995. pp. 355–374. [Google Scholar]
- 30.Krontiris-Litowitz JK, Cooper BF, Walters ET. Humoral factors released during trauma of Aplysia. I. Body wall contraction, cardiac modulation, and central reflex suppression. J Comp Physiol [B] 1989;159:211–223. doi: 10.1007/BF00691742. [DOI] [PubMed] [Google Scholar]
- 31.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Ruble EA, Ahmad MF, Avruch J, Woodgett JR. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 32.Lin SS, Levitan IB. Concanavalin A alters synaptic specificity between cultured Aplysia neurons. Science. 1987;237:648–650. doi: 10.1126/science.3603045. [DOI] [PubMed] [Google Scholar]
- 33.McQuarrie IG, Grafstein B. Axon outgrowth enhanced by a previous nerve injury. Arch Neurol. 1973;29:53–55. doi: 10.1001/archneur.1973.00490250071008. [DOI] [PubMed] [Google Scholar]
- 34.Nja A, Purves D. The effects of nerve growth factor and its antiserum on synapses in the superior cervical ganglion of the guinea-pig. J Physiol (Lond) 1978;277:55–75. [PMC free article] [PubMed] [Google Scholar]
- 35.Noel F, Nunez-Regueiro M, Cook R, Byrne JH, Eskin A. Long-term changes in synthesis of intermediate filament protein, actin, and other proteins in pleural sensory neurons of Aplysia produced by an in vitro analogue of sensitization training. Mol Brain Res. 1993;19:203–210. doi: 10.1016/0169-328x(93)90027-m. [DOI] [PubMed] [Google Scholar]
- 36.Noel F, Frost WN, Tian L-M, Colicos MA, Dash PK. Recovery of tail-elicited siphon-withdrawal reflex following unilateral axonal injury is associated with ipsi- and contralateral changes in gene expression in Aplysia californica. J Neurosci. 1995;15:6926–6938. doi: 10.1523/JNEUROSCI.15-10-06926.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ridgway RL, Syed NI, Lukowiak K, Bulloch AG. Nerve growth factor (NGF) induces sprouting of specific neurons of the snail, Lymnaea stagnalis. J Neurobiol. 1991;22:377–390. doi: 10.1002/neu.480220406. [DOI] [PubMed] [Google Scholar]
- 38.Salim A, Glanzman DL. Axotomy causes long-term hyperexcitability of single Aplysia sensory neurons in cell culture. Soc Neurosci Abstr. 1995;21:1267. [Google Scholar]
- 39.Schacher S, Proshansky E. Neurite regeneration by Aplysia neurons in dissociated cell culture: modulation by Aplysia hemolymph and the presence of the initial axonal segment. J Neurosci. 1983;3:2403–2413. doi: 10.1523/JNEUROSCI.03-12-02403.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steffensen I, Dulin MF, Walters ET, Morris CE. Peripheral regeneration and central sprouting of sensory neurone axons in Aplysia californica following nerve injury. J Exp Biol. 1995;198:2067–2078. doi: 10.1242/jeb.198.10.2067. [DOI] [PubMed] [Google Scholar]
- 41.Titmus M, Faber D. Axotomy-induced alterations in the electrophysiological characteristics of neurons. Prog Neurobiol. 1990;35:1–51. doi: 10.1016/0301-0082(90)90039-j. [DOI] [PubMed] [Google Scholar]
- 42.Walters ET. A functional, cellular, and evolutionary model of nociceptive plasticity in Aplysia. Biol Bull. 1991;180:241–251. doi: 10.2307/1542394. [DOI] [PubMed] [Google Scholar]
- 43.Walters ET. Injury-related behavior and neuronal plasticity: an evolutionary perspective on sensitization, hyperalgesia, and analgesia. Int Rev Neurobiol. 1994;36:325–427. doi: 10.1016/s0074-7742(08)60307-4. [DOI] [PubMed] [Google Scholar]
- 44.Walters ET, Ambron RT. Long-term alterations induced by injury and by 5-HT in Aplysia sensory neurons: convergent pathways and common signals? Trends Neurosci. 1995;18:137–142. doi: 10.1016/0166-2236(95)93891-z. [DOI] [PubMed] [Google Scholar]
- 45.Walters ET, Byrne JH, Carew TJ, Kandel ER. Mechanoafferent neurons innervating the tail of Aplysia. I. Response properties and synaptic connections. J Neurophysiol. 1983;50:1543–1559. doi: 10.1152/jn.1983.50.6.1522. [DOI] [PubMed] [Google Scholar]
- 46.Walters ET, Alizadeh H, Castro EA. Similar neuronal alterations induced by axonal injury and learning in Aplysia. Science. 1991;253:797–799. doi: 10.1126/science.1652154. [DOI] [PubMed] [Google Scholar]
- 47.Williams DK, Cohan CS. The role of conditioning factors in the formation of growth cones and neurites from the axon stump after axotomy. Brain Res Dev Brain Res. 1994;81:89–104. doi: 10.1016/0165-3806(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 48.Wilson MP, Carrow GM, Levitan IB. Modulation of growth of Aplysia neurons by an endogenous lectin. J Neurobiol. 1992;23:739–750. doi: 10.1002/neu.480230611. [DOI] [PubMed] [Google Scholar]
- 49.Wu W, Mathew TC, Miller FD. Evidence that the loss of homeostatic signals induces regeneration-associated alteration in neuronal gene expression. Dev Biol. 1993;158:456–466. doi: 10.1006/dbio.1993.1203. [DOI] [PubMed] [Google Scholar]
- 50.Ziv NE, Spira ME. Spatiotemporal distribution of Ca2+ following axotomy and throughout the recovery process of cultured Aplysia neurons. Eur J Neurosci. 1993;5:657–668. doi: 10.1111/j.1460-9568.1993.tb00531.x. [DOI] [PubMed] [Google Scholar]