

Abstract
The anxiogenic benzodiazepine inverse agonist FG7142 increases dopamine turnover in rodent prefrontal cortex but not in other dopamine terminal field areas. FG7142-induced increases in prefrontal cortical dopamine receptor stimulation impair prefrontal-dependent, but not nonprefrontal-dependent, cognitive tasks in rats and monkeys. The degree of impairment correlates with levels of prefrontal cortical dopamine turnover in rats and can be blocked in rats and monkeys with dopamine receptor antagonists, suggesting that increased dopamine turnover is directly related to the cognitive deficits.
The current study examined nondopaminergic drug effects on FG7142-perturbed biochemistry and cognition. Both the noradrenergic α-2 agonist clonidine and the glycine/NMDA antagonist (+)HA966 prevented the FG7142-induced increase in dopamine turnover in rodent prefrontal cortex. Infusion of (+)HA966 into the ventral tegmental area (VTA) also blocked this increase in dopamine turnover, indicating that critical modulatory effects of (+)HA966 on FG7142-induced changes in dopamine turnover are occurring at the level of mesoprefrontal dopamine neuron cell bodies. Systemic (+)HA966 and clonidine, but not propranolol or d-cycloserine, prevented FG7142-associated spatial working memory deficits in rats and monkeys. These results support the idea of a critical range of dopamine turnover for optimal prefrontal cortical cognitive functioning, with excessive dopamine turnover leading to cognitive impairment. These studies also provide evidence for the regulation of prefrontal cortical dopamine turnover and cognition by multiple neurotransmitter systems and suggest that the VTA is an important regulatory site for these effects.
Keywords: rat, primate, FG7142, dopamine, clonidine, propranolol, norepinephrine, (+)HA-966, d-cycloserine, NMDA, prefrontal cortex, delayed alternation, delayed response, memory, cognition
The unique sensitivity of the prefrontal cortical dopamine system to stress has long intrigued researchers (Roth and Tam, 1987). Rodent studies have shown that mild stress increases dopamine turnover in the prefrontal cortex but not in other dopamine terminal field regions (Thierry et al., 1976; Herman et al., 1982; Roth et al., 1988; Deutch and Roth, 1990). The prefrontal cortical dopamine response to stress is blocked by anxiolytic benzodiazepine receptor agonists (Tam and Roth, 1990) and mimicked by anxiogenic benzodiazepine inverse agonists, such as FG7142 (Tam and Roth, 1985, 1990; Roth and Tam, 1987; Deutch and Roth, 1990). Like mild stress, FG7142 produces a prefrontal cortical-selective increase in dopamine turnover and release, which can be blocked by pretreatment with benzodiazepine receptor agonists or antagonists (Tam and Roth, 1990; Bradberry et al., 1991) and with noradrenergic α-2 receptor agonists (Tam, 1986).
In addition to a selective increase in prefrontal cortical dopamine turnover in rats, FG7142 impairs prefrontal cortical-dependent, but not nonprefrontal cortical-dependent, tasks in rats and monkeys (Murphy et al., 1994, 1996). The degree of FG7142-induced increase in prefrontal cortical dopamine turnover in rats correlates with impairment of performance on a delayed alternation task, and this impairment of spatial working memory is ameliorated in rats and monkeys with dopamine receptor antagonists (Murphy et al., 1994, 1996). These findings suggest that increased dopamine turnover in the prefrontal cortex produces deficits on spatial working memory tasks. In conjunction with research on the cognitive effects of dopamine depletion (Brozoski et al., 1979; Bubser and Schmidt, 1990), these studies indicate that there is a limited range of dopamine turnover for optimal cognitive functions in the prefrontal cortex.
The prefrontal cortex receives elaborate dopaminergic inputs from the ventral tegmental area (VTA) (Domesick, 1988; Fallon, 1988;Goldman-Rakic et al., 1992; Lewis, 1992). A variety of neurotransmitters modulates the function of cells within the VTA (Kalivas, 1993; White, 1996). Both the glutamatergic-NMDA and noradrenergic neurotransmitter systems influence the firing pattern of cells within the VTA (Grenhoff and Svensson, 1989; McMillen et al., 1992) and affect the prefrontal dopaminergic response to stress (Tam, 1986; Morrow et al., 1993). The current study examines the hypothesis that excessive dopamine release in the prefrontal cortex impairs cognitive functions by evaluating whether drugs that prevent FG7142-induced increases in prefrontal cortical dopamine turnover affect FG7142-impaired performance on spatial working memory tasks in rats and monkeys. (+)HA966, a glycine/NMDA receptor antagonist, and clonidine, an α-2 noradrenergic receptor agonist, were selected for this study, because these agents have been shown to attenuate the prefrontal cortical dopamine response to stress (Tam, 1986; Morrow et al., 1993; Goldstein et al., 1994) and to decrease the FG7142-induced increase in prefrontal cortical dopamine metabolism (Tam, 1986). In the current study, the effects of (+)HA966 and clonidine on FG7142-induced increases in prefrontal cortical dopamine turnover were determined in rats. In addition, the site of action of (+)HA966 was also investigated by direct infusion into the VTA. The cognitive effects of (+)HA966 and clonidine, as well as d-cycloserine (a glycine/NMDA agonist) and propranolol (a β-adrenergic antagonist), were examined by using spatial working memory tasks in rats and monkeys.
Portions of this data were presented originally in abstract form (Murphy et al., 1994).
MATERIALS AND METHODS
Subjects
Male Sprague Dawley rats were purchased from CAMM (Charles River, Wilmington, MA). The animals were maintained on a 12 hr light–dark cycle; experiments were conducted during the animals’ light phase. For cognitive testing, rats were pair-housed in filter-frame cages and fed a diet of autoclaved Purina rat chow immediately after behavioral testing or ad libitum on nontest days. Water was available ad libitum. Rats were ∼100 gm at the beginning of the experiment. Weights were monitored weekly; all rats continued to gain 10–30 gm/week until achieving adult weights of ∼500–550 gm. Highly palatable food rewards (miniature chocolate chips) were used during cognitive testing to minimize the need for food regulation. Rats weighing 200–300 gm were used in biochemical studies and were group-housed with food and water availablead libitum.
Four female young adult (aged 3–12 yr) rhesus monkeys (Macaca mulatta) were used in the present study. All monkeys had been used in previous behavioral pharmacological studies. Monkeys were housed individually under standard laboratory conditions and maintained on a 12 hr light–dark cycle. Animals were always tested at the same time of day immediately before feeding. Highly palatable food items (e.g., peanuts, raisins, or chocolate chips) were used as rewards during testing to minimize the need for dietary restriction.
Cognitive testing
Delayed alternation testing (rats). Animals were assigned to one trained investigator and tested 5 d a week at the same time in a room devoted to these studies. White noise was broadcast in the room to minimize auditory distractions. Rats were habituated to a T-maze (total dimensions 90 cm × 65 cm) for 2 consecutive days. During habituation, animals were allowed to explore the maze freely; food rewards were available in both maze arms. On the third day, animals began training on the delayed alternation task. On the first trial of delayed alternation, animals were rewarded for entering either arm. Thereafter, for a total of 10 trials per session, rats were rewarded only if they entered the maze arm which was not chosen previously. Between trials, the choice point was wiped with alcohol to remove potential olfactory cues. The intertrial delay was adjusted until a rat’s performance stabilized at ∼80% correct responses. This level of baseline performance allowed for detection of either improvement or impairment during drug administration.
Delayed response testing (monkeys). Animals were assigned to one trained investigator and were tested twice weekly in a Wisconsin General Test Apparatus situated in a sound-attenuating room. Background masking noise was used to minimize auditory distractions. The monkeys had been trained previously on a two well delayed response task. During delayed response, the animal watches as the experimenter baits one of two food wells. The food wells then are covered with identical cardboard plaques, and an opaque screen is lowered between the animal and the test tray for a specified delay period. At the end of this period, the screen is raised and the animal is allowed to choose a plaque. The reward is distributed evenly between the left and right wells over the 30 trials that make up a daily test session. To observe the effects of drugs on working memory capacity, animals were trained on a variable delayed response task. Delays varied between <1 sec (“0” sec) and the delay period that yielded chance (50% correct) performance for each animal. Five different delay intervals were distributed over the 30 trials that made up a single test session. Delays were adjusted until an individual monkey performed at ∼83% correct responses to allow for the detection of either improvement or impairment.
Pharmacological treatments
The person conducting cognitive testing in both rats and monkeys was always blind to drug conditions. For rodents, all injections were given intraperitoneally. Initially, small amounts of saline were injected to facilitate habituation to intraperitoneal injections and thereby minimize the stress associated with injections. All drugs were given to monkeys intramuscularly. Monkeys had been habituated previously to intramuscular injections and were injected before every session with either drug or vehicle.
FG7142 was purchased from RBI (Natick, MA). As FG7142 is virtually insoluble in saline, great care was taken to maintain the compound in suspension. For rats, FG7142 was suspended in a vehicle consisting of 0.05 ml of ethanol, 0.15 ml of low pH saline (pH ∼3.4), and 0.8 ml of an HBC–Tween solution [1.5 gm hydroxy-β-cyclodextrin (RBI), 1 ml of Tween-80 (Sigma, St. Louis, MO), and 7.5 ml of sterile, buffered saline]. FG7142 was sonicated in the vehicle, and the syringe was shaken immediately before injection. A difference in color, crystal size, solubility, and biochemical potency was noted for different lots of FG7142. For this reason, when a change in drug lots of FG7142 was necessary, a dose was chosen that produced an increase in prefrontal cortical dopamine turnover of approximately the same magnitude as the original lot. For rats, a dose of 20 mg/kg (Lot AL-III-47) or 30 mg/kg (lot QNL-295A) was administered intraperitoneally in a volume of 2 ml/kg 25 min before testing. FG7142 treatments were repeated throughout the study to ensure that animals did not sensitize or habituate to the effects of repeated FG7142 treatment. Although no changes were seen in cognitive or motor performance with repeated FG7142 injections, a small number of rats developed seizures (4 of 48). Rats experiencing seizures were removed immediately from the maze, and any data from that session were discarded.
For monkey treatments, FG7142 (lot AL-III-47) was suspended in a vehicle consisting of 0.2 ml of ethanol and 0.8 ml of HBC-Tween solution. FG7142 was sonicated in the vehicle, and the syringe was shaken immediately before injection. A dose of 0.2 mg/kg was administered at 0.2 ml/kg. Testing sessions were scheduled so that testing began 30–37 min after FG7142 injection.
Clonidine (RBI) for both monkeys and rats was dissolved in sterile saline, and 0.1 mg/kg (rats) or 0.04 mg/kg (monkeys) was injected 45 min before testing. (+)HA-966 was provided through the National Institute of Mental Health synthesis program (courtesy of RBI). For monkeys, (+)HA-966 (0.2 mg/kg) was dissolved in sterile saline, and 0.1 ml/kg was given 15 min before FG7142 or vehicle injections. For rat systemic injections, (+)HA-966 was dissolved in sterile saline and administered in a dose of 20 mg/kg in 1 ml/kg 15 min before FG7142 or vehicle administration. For direct infusion studies in rodents, (+)HA-966 was dissolved in artificial cerebrospinal fluid (CSF; 145 mm Na+, 1.0 mm Mg+, 2.7 mm K+, 150 mm Cl−, and 1.2 mm Ca+). (+)HA966 (15 μg) was dissolved in 1 μl of artificial CSF and infused into the VTA over 1 min, 15 min before systemic FG7142 or vehicle treatment. d-cycloserine (RBI) was dissolved in sterile saline (3 mg/kg) and administered to rats 15 min before injection with FG7142 or vehicle.
Direct infusion
Rats were anesthetised with a chloral hydrate and sodium pentobarbital mixture (0.0166 gm/kg chloral hydrate, 0.0316 gm/kg sodium pentobarbital, with 0.39 ml of propylene glycol and 0.10 ml of ethanol/ml) and placed into a stereotaxic apparatus. Supplemental anesthesia was provided as needed. A 23 gauge guide cannula was implanted 1 mm above the VTA (5.8 mm posterior, 2.0 mm lateral from bregma, 6.5 mm vertical from brain surface at a 5° angle toward midline). The cannula was affixed to the skull with stainless steel screws and dental cement. A 30 gauge stylette was placed in the guide cannula to maintain patency.
After surgery, rats were housed individually for 14 d. Immediately after recovery from anesthesia, rats were treated orally with Tylenol elixer, and water bottles containing Tylenol elixer (4 mg/ml) were placed on cages for 2 d after surgery (with an estimated daily consumption of 20 ml of water) Stylettes were changed on alternate days, and the surgical area was treated with an antibiotic powder daily. Rats were handled daily and habituated to brief hand-held restraint. During infusions, the stylette was replaced with a 30 gauge cannula, which extended 1 mm beyond the guide cannula. Polyethylene tubing (Clay Adams, Parsippany, NJ) ran from syringes in an infusion pump (Harvard Apparatus, South Natick, MA) to the infusion cannula.
Biochemistry
Rats were killed by decapitation 30 min after intraperitoneal FG7142 or vehicle administration. The brains were removed quickly, and brain regions were dissected out on a chilled platform according to Tam and Roth (1990). Immediately after dissection, brain samples were frozen on dry ice until stored at −70°C. Tissue samples were prepared using alumina extraction, with dihydroxybenzylamine as an internal standard based on the methods of Elsworth et al. (1989). Analysis of DOPAC (dihydroxyphenylacetic acid), dopamine, and norepinephrine levels was conducted on an HPLC system using an electrochemical transducer with a glassy carbon electrode at 0.7 V (BAS, West Lafayette, IN) and a reversed-phase column (3 μm C18 beads, 100A diameter; Rainin Instrument, Woburn, MA). The mobile phase was optimized for the system on the basis of the mobile phase ofElsworth et al. (1989). Analysis of total MHPG (3-methoxy-4-dydroxyphenyletheleneglycol) levels (free and conjugated) was conducted by GC-mass spectrometry, following the methods ofElsworth et al. (1983).
Slides/Probe Placement. Whole brains posterior to the striatum were placed in formalin for storage after regional brain dissection. Brains were frozen, and 120 μm thin slices were made on a microtome (American Optical, Buffalo, NY) with a cooling stage (Bailey Instruments, Saddlebrook, NJ) and placed on gel-coated slides. Histologically prepared sections were examined to verify probe placement (Fig. 1).
Fig. 1.

The location of the injection cannula for infusions of (+)HA966 shown in Figure 3. The locations of the tips of the injection cannula are indicated by black dots. Brain drawings are taken from Paxinos and Watson (1982) with the location posterior to bregma in mm, as indicated.
Data analysis. Within-subjects comparisons were used for rats and monkey cognitive testing [paired t test (t-dep)], because animals were used as their own controls in both delayed alternation and delayed response experiments. Analysis of biochemical experiments in rats used between-subjects comparisons [unpaired t test (t-ind)]. Statistical analyses were performed with Statworks (Version 1.2, Cricket Software, Philadelphia, PA) on a Macintosh computer.
RESULTS
The effects of FG7142 and clonidine on dopamine and norepinephrine turnover
The effects of systemic FG7142 and clonidine administration on dopamine metabolism in dopamine terminal field areas were examined by measuring DOPAC and dopamine levels in the prefrontal cortex, nucleus accumbens, and striatum of rats (Table 1). Postmortem measures of the DOPAC/dopamine ratios were used as a measure of dopamine turnover and indicated that the administration of FG7142 significantly increased dopamine use in the prefrontal cortex to 133% of vehicle levels (Fig. 2; VEH vs FG: t= 4.07, df = 15, p = 0.001). Pretreatment with clonidine prevented the FG7142-induced increase in prefrontal cortical dopamine turnover but had no effect when given alone. In contrast to the prefrontal cortex, FG7142 had no significant effect on measures of dopamine turnover in the nucleus accumbens or striatum. The administration of clonidine had no significant effect on dopamine turnover in the nucleus accumbens, but it did produce a small but significant decrease in dopamine turnover in the striatum when given alone (VEH vs CLON: t = 4.86, df = 13,p < 0.001) or in combination with FG7142 (VEH vs CLON/FG: t = 6.95, df = 12, p < 0.001).
Table 1.
The effect of FG7142, (+)HA966, and clonidine on DOPAC/dopamine ratios in the prefrontal cortex, nucleus accumbens, and striatum and of FG7142 and clonidine on MHPG/norepinephrine ratios in the prefrontal cortex
| DOPAC/DA | ||||
|---|---|---|---|---|
| Vehicle | FG7142 | (+)HA966 | FG7142/(+)HA966 | |
| Systemic PFC | 0.26 ± 0.04 | 0.39 ± 0.031-a | 0.29 ± 0.06 | 0.28 ± 0.03 |
| (n = 13) | (n = 11) | (n = 19) | (n = 18) | |
| NAC | 0.19 ± 0.02 | 0.17 ± 0.02 | 0.18 ± 0.02 | 0.17 ± 0.01 |
| (n = 7) | (n = 7) | (n = 7) | (n = 7) | |
| STR | 0.07 ± 0.005 | 0.07 ± 0.004 | 0.06 ± 0.004 | 0.06 ± 0.003 |
| (n = 7) | (n = 7) | (n = 8) | (n = 7) | |
| Infusion PFC | 0.19 ± 0.01 | 0.32 ± 0.011-a | 0.22 ± 0.02 | 0.23 ± 0.01 |
| (n = 5) | (n = 5) | (n = 8) | (n = 7) | |
| NAC | 0.19 ± 0.03 | 0.15 ± 0.01 | 0.18 ± 0.01 | 0.15 ± 0.01 |
| (n = 5) | (n = 5) | (n = 8) | (n = 7) | |
| STR | 0.07 ± 0.003 | 0.07 ± 0.004 | 0.06 ± 0.0031-a | 0.07 ± 0.002 |
| (n = 5) | (n = 5) | (n = 8) | (n = 6) | |
| Vehicle | FG7142 | Clonidine | FG7142/Clonidine | |
|---|---|---|---|---|
| PFC | 0.30 ± 0.01 | 0.40 ± 0.021-a | 0.30 ± 0.01 | 0.27 ± 0.02 |
| (n = 7) | (n = 10) | (n = 7) | (n = 7) | |
| NAC | 0.18 ± 0.01 | 0.18 ± 0.01 | 0.15 ± 0.004 | 0.16 ± 0.02 |
| (n = 6) | (n = 9) | (n = 8) | (n = 8) | |
| STR | 0.07 ± 0.003 | 0.07 ± 0.003 | 0.05 ± 0.0041-a | 0.05 ± 0.0021-a |
| (n = 7) | (n = 9) | (n = 8) | (n = 7) |
| MHPG/NE | ||||
|---|---|---|---|---|
| Vehicle | FG7142 | Clonidine | FG7142/Clonidine | |
| PFC | 0.32 ± 0.02 | 0.35 ± 0.03 | 0.28 ± 0.03 | 0.30 ± 0.02 |
| (n = 6) | (n = 8) | (n = 8) | (n = 8) | |
The ratio of DOPAC to dopamine (DA) in the prefrontal cortex (PFC), nucleus accumbens (NAC), and striatum (STR) for the following drug combinations: FG7142 (20 mg/kg) and systemic (+)HA966 (20 mg/kg), FG7142 (20 mg/kg) and the infusion of (+)HA966 (15 μg) into the ventral tegmental area, and FG7142 (30 mg/kg) and clonidine (0.1 mg/kg). Data are expressed as the mean ± SEM with the number of animals in each category, as indicated.
Significantly different from vehicle (unpairedt test; p < 0.05).
Fig. 2.

Dopamine turnover as determined by the DOPAC-to-dopamine (DA) ratio in the prefrontal cortex, nucleus accumbens, and striatum of rats. Rats were pretreated with either clonidine (CLON; 0.1 mg/kg) or vehicle at 45 min and FG7142 (FG; 30 mg/kg) or vehicle (VEH) at 30 min before killing. Data are expressed as mean ± SEM. *p < 0.05 versus vehicle.
The effect of systemic FG7142 administration on norepinephrine metabolism in the rodent prefrontal cortex was also examined. Postmortem tissue measures demonstrated that neither the administration of FG7142 30 min before sacrifice nor the administration of clonidine 45 min before killing had a significant effect on norepinephrine turnover in the rodent prefrontal cortex as measured by the MHPG-to-norepinephrine ratio.
The effects of FG7142 and (+)HA966 on dopamine turnover
The effects of systemic FG7142 and (+)HA966 administration on dopamine metabolism in dopamine terminal field areas were examined in the prefrontal cortex, nucleus accumbens, and striatum of rats (Table1). The systemic injection of FG7142 increased dopamine turnover levels to 150% of vehicle levels in the rodent prefrontal cortex (Fig.3; VEH vs FG: t = 2.47, df = 22,p = 0.022). The systemic injection of (+)HA966 prevented the increase in the DOPAC/dopamine ratio in the prefrontal cortex but had no effect on dopamine turnover when given alone. In contrast to the prefrontal cortex, there was no significant effect of FG7142 and/or (+)HA966 in the nucleus accumbens or striatum.
Fig. 3.
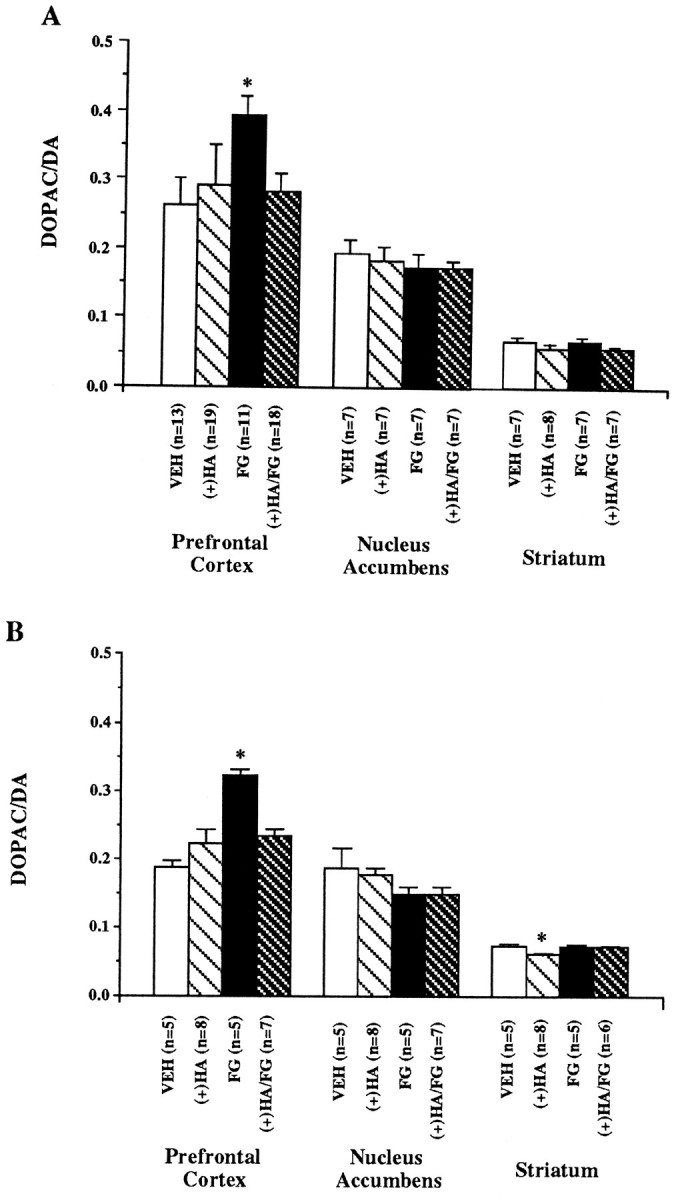
Dopamine turnover as determined by the DOPAC-to-dopamine (DA) ratio in the prefrontal cortex, nucleus accumbens, and striatum of rats. A, Rats were pretreated with systemic injections of either (+)HA966 [(+)HA; 20 mg/kg] or vehicle at 45 min and FG7142 (FG; 20 mg/kg) or vehicle (VEH) at 30 min before killing. B,Rats were pretreated with infusions into the ventral tegmental area of either (+)HA966 [(+)HA; 15 μg in 1 μl] or vehicle at 45 min and FG7142 (FG; 20 mg/kg) or vehicle (VEH) at 30 min before killing. Data are expressed as mean ± SEM. *p < 0.05 versus vehicle.
The systemic injection of FG7142 with the infusion of vehicle (artificial CSF) directly into the VTA increased the DOPAC-to-dopamine ratio to 168% of vehicle levels in the prefrontal cortex (Fig. 3; VEH vs FG: t = 9.19, df = 8, p < 0.001). The infusion of (+)HA966 into the VTA prevented the effects of a systemic FG7142-induced increase in dopamine turnover in the prefrontal cortex. In contrast, the infusion of (+)HA966 with the systemic injection of vehicle had no significant effect on the DOPAC/dopamine ratio in the prefrontal cortex.
There was no effect of treatment with FG7142 and the infusion of (+)HA966 on dopamine turnover in the nucleus accumbens. In the striatum, there was no effect of FG7142 or of FG7142 with the direct infusion of (+)HA966. When infused alone, (+)HA966 produced a small but significant decrease in the DOPAC/dopamine ratio in the striatum (VEH vs (+)HA: t = 2.38, df = 11; p= 0.036).
The effects of FG7142, clonidine, and propranolol on spatial working memory
Cognitive performance in rats performing delayed alternation after FG7142 administration was significantly impaired when compared with performance on vehicle (Fig. 4; VEH vs FG:t = 7.27, df = 8, p < 0.0001). Pretreatment with clonidine at a dose shown to prevent the FG7142 increase in dopamine turnover prevented FG7142-induced impairment of delayed alternation (FG vs CLON/FG: t = 4.35, df = 7, p = 0.003). However, the administration of clonidine alone had no significant effect on spatial working memory.
Fig. 4.

Spatial working memory performance in rats and monkeys. Data are expressed as percentage correct (mean ± SEM) on a delayed alternation task in rats and a delayed response task in monkeys. Rats were pretreated with either clonidine (CLON;0.1 mg/kg) or vehicle at 45 min and FG7142 (FG; 20 mg/kg) or vehicle (VEH) at 25 min before testing. Monkeys were pretreated with either clonidine (CLON; 0.04 mg/kg) or vehicle 45 min and FG7142 (FG; 0.2 mg/kg) or vehicle (VEH) 30–37 min before testing. *p< 0.05 versus vehicle. **p < 0.05 versus FG7142.
As in the rat, FG7142 significantly impaired delayed response performance in monkeys (Fig. 4; VEH vs FG: t = 4.40, df = 3, p = 0.02). Although clonidine had no effect on spatial working memory when given alone, pretreatment with clonidine in the monkey prevented FG7142-associated deficits of delayed response performance (FG vs CLON/FG: t = 4.71, df = 2, p = 0.04).
The administration of propranolol had no significant effect on spatial working memory performance in the rat (Table 2). When given as a pretreatment, propranolol did not improve FG7142-impaired performance. At a higher dose (10 mg/kg), propranolol produced marked sedation and reduced performance accuracy. This dose did not improve FG7142-impaired delayed alternation performance (data not shown).
Table 2.
The effect of FG7142, propranolol, andd-cycloserine on delayed alternation performance
| Vehicle | FG7142 | Propranolol | FG7142/Propranolol |
|---|---|---|---|
| 80.4 ± 2.7 | 51.6 ± 7.02-a | 70.0 ± 4.9 | 56.5 ± 7.5 |
| (n = 14) | (n = 11) | (n = 7) | (n = 7) |
| Vehicle | FG7142 | d-Cycloserine | FG7142/d-Cycloserine |
|---|---|---|---|
| 76.4 ± 4.2 | 59.9 ± 2.72-a | 88.8 ± 4.3 | 43.3 ± 13.3 |
| (n = 6) | (n = 5) | (n = 4) | (n = 3) |
Performance on delayed alternation in rats after the administration of vehicle, FG7142 (20 mg/kg), propranolol (5 mg/kg), and d-cycloserine (3 mg/kg). Data are expressed as percentage correct mean ± SEM.
Significantly different from vehicle (unpairedt test; p < 0.05).
The effects of FG7142, (+)HA966, and d-cycloserine on spatial working memory
When FG7142 was administered to rats before delayed alternation testing, cognitive performance was impaired significantly when compared with performance on vehicle (Fig. 5; VEH vs FG:t = 7.12, df = 10, p = < 0.001). Pretreatment with (+)HA966 at a dose shown to prevent FG7142-induced increases in prefrontal cortical dopamine turnover prevented the effects of FG7142 on delayed alternation performance (FG vs (+)HA/FG:t = 7.31, df = 7, p < 0.001). The administration of (+)HA966 alone had no effect on spatial working memory.
Fig. 5.

Spatial working memory performance in rats and monkeys. Data are expressed as percentage correct (mean ± SEM) on a delayed alternation task in rats and a delayed response task in monkeys. Rats were pretreated with either (+)HA966 [(+)HA;20 mg/kg] or vehicle at 45 min and FG7142 (FG; 20 mg/kg) or vehicle (VEH) 25 min before testing. Monkeys were pretreated with either (+)HA966 [(+)HA; 0.4 mg/kg] or vehicle 45 min and FG7142 (FG; 0.2 mg/kg) or vehicle (VEH) 30–37 min before testing. *p< 0.05 versus vehicle. **p < 0.05 versus FG7142.
As in the rat, FG7142 significantly impaired delayed response performance in monkeys (Fig. 5; VEH vs FG: t = 4.40, df = 3, p = 0.02). Pretreatment with (+)HA966 in the monkey prevented FG7142-associated deficits of delayed response (Fig. 5; FG vs (+)HA/FG: t = 4.13, df = 2,p = 0.05) but had no effect when administered alone. Interestingly, performance after (+)HA966+FG7142 was significantly better than vehicle performance in the monkey (T = 5.44, df = 2, p = 0.032).
d-Cycloserine, a glycine site agonist, was also studied to determine whether it would exacerbate or ameliorate FG7142-associated impairments (Table 2). Because of concerns that agonists at the glycine/NMDA receptor might potentiate the possibility of FG7142-induced seizures (Turski et al., 1990),d-cycloserine was used as a pretreatment in only a few rats and no monkeys. Pretreatment with d-cycloserine in a small number of rats did not improve FG7142-altered performance and produced a trend toward further impairment. This dose ofd-cycloserine had no notable effects when administered alone.
DISCUSSION
FG7142 selectively increases dopamine turnover and release in the prefrontal cortex (Tam and Roth, 1985, 1990; Bradberry et al., 1991;Murphy et al., 1994, 1996). This selective increase in prefrontal cortical dopamine turnover is associated with impaired accuracy of performance of prefrontal cortical-dependent, but not nonprefrontal cortical dependent, tasks in rats and monkeys (Murphy et al., 1994,1996). Although the connection between increased dopamine receptor stimulation and impaired cognition only recently has been examined systematically, several studies support our conclusion that increased dopamine release and turnover in the prefrontal cortex are associated with impairment of spatial working memory. Drugs such as Δ9-tetrahydrocannabinol (THC; Bowers and Morton, 1994;Jentsch et al., 1996), dizocilpine (Bowers and Morton, 1994; Kashiwa et al., 1995), and ketamine (Rao et al., 1989; Verma and Moghaddam, 1996) increase measures of dopamine release and turnover in the prefrontal cortex. Systemic administration of these drugs, as well as the intracortical infusion of a dopamine D1-receptor agonist, has been shown to impair delayed alternation performance (Hauber, 1993; Maurice, 1994; Jentsch et al., 1996; Verma and Moghaddam, 1996; Zahrt et al., 1996). The idea that increased prefrontal cortical dopamine turnover is, at least in part, responsible for the FG7142-induced cognitive dysfunction is supported by findings that the degree of the deficit correlates with the increase in dopamine turnover and that the dopamine receptor antagonists clozapine, haloperidol, SCH 23390, and sulpiride (Murphy et al., 1994, 1996; unpublished results) prevent FG7142-associated cognitive deficits. These results are consistent with the effects of iontophoretically applied dopaminergic drugs on memory-related firing in prefrontal cortical pyramidal neurons (Williams and Goldman-Rakic, 1995).
To further explore the hypothesis that increased dopamine release and turnover in the prefrontal cortex is responsible for the cognitive deficits observed after FG7142 administration, we examined whether drugs that attenuate the FG7142-associated increase in prefrontal cortical dopamine turnover in rodents would ameliorate the FG7142-induced spatial working memory deficits in rats and monkeys. Previous studies have demonstrated that a strychnine-insensitive glycine site partial agonist/functional NMDA receptor antagonist (+)HA966 and an α-2 noradrenergic receptor agonist clonidine can prevent stress-induced increases in prefrontal cortical dopamine turnover (Tam, 1986; Morrow et al., 1993; Goldstein et al., 1994) and that clonidine can attenuate the FG7142-induced increase in prefrontal cortical dopamine metabolism. Thus, these two drugs were examined in the present study. The administration of (+)HA966 and clonidine blocked FG7142-induced increases in prefrontal cortical dopamine turnover. When given to rats and monkeys performing spatial working memory tasks, these drugs prevented the FG7142-associated impairments in performance accuracy. These current findings support our previous hypothesis that excessive dopamine receptor stimulation in the prefrontal cortex is associated with an impairment of prefrontal cortical-dependent cognitive functions.
Although the anatomical site of action of FG7142 in the current study is undetermined, previous studies have indicated the involvement of benzodiazepine/GABAA receptors in the VTA (Deutch and Roth, 1990; Tam and Roth, 1990). GABAergic neurons have been shown to synapse on dopaminergic and nondopaminergic neurons in the VTA, and benzodiazepine agonists delivered directly into the VTA prevent the stress-induced increase in prefrontal cortical dopamine turnover (Kalivas et al., 1990). In addition, the administration of a benzodiazepine inverse agonist DMCM directly into the VTA produces a dose-related increase in prefrontal cortical dopamine turnover (Deutch and Roth, 1990). These studies suggest that the VTA is a common site through which benzodiazepine/GABAA receptors can modulate prefrontal cortical dopamine and can mediate the FG7142-associated activation of the mesoprefrontal dopamine neurons.
Dopaminergic cells in the VTA are modulated by a number of chemically defined afferent inputs (Kalivas, 1993; White, 1996) and may represent an important regulatory site for the control of prefrontal cortical dopaminergic activity. The firing pattern of VTA dopamine cells has been demonstrated to be the major determinant of the quantity of transmitter released; for a given frequency of firing, the burst-firing pattern has been shown to result in increased transmitter release relative to the tonic discharge mode (Gonon, 1988; Grenhoff and Svensson, 1989; Bean and Roth, 1991). Interestingly, both (+)HA966 and clonidine have been shown to normalize the firing pattern in midbrain dopamine neurons and diminish the frequency of bursting (Grenhoff and Svensson, 1989; McMillen et al., 1992). By preventing the incidence of burst firing, clonidine and (+)HA966 may block the entrance of dopamine neurons into this increased release mode and thus prevent cortical hyperdopaminergic states produced by stress or FG7142 administration.
Injection of NMDA directly into the VTA produces an increase in burst firing of dopamine neurons in the VTA and an increase in dopamine release (Chergui et al., 1993; Wang et al., 1994), as well as an increase in dopamine turnover in the prefrontal cortex (Kalivas et al., 1989). Previous studies in our lab have indicated that direct infusion of (+)HA966 into the VTA prevents restraint stress-induced increases in prefrontal cortical dopamine turnover (Morrow et al., 1993). In the current study, (+)HA966 administered both systemically and through microinjection into the VTA blocked the effects of systemically administered FG7142 on prefrontal cortical dopamine turnover.
Because afferent inputs have been suggested to regulate the pattern of firing of select dopaminergic neurons in the VTA, it is possible that noradrenergic influences could differentially affect subsets of dopaminergic neurons within the VTA. Several studies have suggested this possibility. For example, lesions of the norepinephrine projections from the locus ceruleus to the VTA decrease dopamine turnover in the prefrontal cortex but not in the nucleus accumbens (Herve et al., 1982). In addition to normalizing the pattern of firing in the VTA (Grenhoff and Svensson, 1989), clonidine has been shown to block the footshock stress and conditioned fear-induced increase in prefrontal cortical dopamine turnover (Tam, 1986; Morrow et al., 1996). Consistent with what has been shown previously (Tam, 1986) and as confirmed in this study, clonidine also blocks the FG7142-induced increase in prefrontal cortical dopamine turnover, perhaps by a similar mechanism. These studies all augur an important role for the VTA in controlling mesocortical dopamine neurons.
Both stress and FG7142 have been shown to affect neurotransmitter systems other than dopamine. For example, FG7142 has been shown to produce significant elevations of extracellular acetylcholine in the rodent frontal cortex (Moore et al., 1995). However, recent studies from this laboratory demonstrate that the administration of scopolamine, a muscarinic cholinergic antagonist, fails to attenuate FG7142-associated cognitive impairment (our unpublished data), arguing against the possibility that increases in prefrontal cortical acetylcholine may account solely for the cognitive effects of FG714. Numerous studies have suggested that there is an important relationship between norepinephrine and stress (Bremner et al., 1996). However, in the current study it is unlikely that norepinephrine plays the major role in the cognitive impairment observed after FG7142 administration. In this study, as well as previous studies conducted in our laboratory (Ida et al., 1991), norepinephrine turnover, as measured by MHPG-to-norepinephrine ratios, was not significantly elevated in the prefrontal cortex 30 min after FG7142 administration. Additional findings from our laboratory also argue against the likelihood that a change in norepinephrine turnover is the critical component in the FG7142-induced impairment of prefrontal cortical dependent cognition. THC produces a significant increase in both dopamine and norepinephrine turnover in rodent prefrontal cortex and also impairs spatial working memory. This cognitive impairment can be ameliorated by HA966, which prevents the THC-induced increased turnover of prefrontal cortical dopamine but not that of norepinephrine (Jentsch et al., 1996). These findings are consistent with the hypothesis that a critical neurochemical component of the FG7142-induced cognitive dysfunction is an increase in dopamine transmission in the prefrontal cortex. These findings however, do not rule out a possible noradrenergic or cholinergic contribution to or modulation of this effect.
Prefrontal cortical dysfunction and dysregulation of dopaminergic systems have been suggested to underlie part of the etiology of several psychiatric disorders, including schizophrenia, attention deficit disorder, and post-traumatic stress disorder (Barkley et al., 1979;Weinberger et al., 1986, 1994; Robbins, 1990; Benson, 1991; Deutch, 1992, 1993; Goldstein and Deutch, 1992; Lewis et al., 1992;Goldman-Rakic, 1994; Deutch and Young, 1995; Southwick et al., 1995). Stress is believed to precipitate or exacerbate these disorders (Breier et al., 1991; Goldman-Rakic, 1991; Bebbington, 1993; American Psychiatric Association, 1994, Arnold and Jensen, 1995) and studies in our labs have shown that noise stress (Arnsten and Goldman-Rakic, 1990) or pharmacological stress (Murphy et al., 1994, 1996) produces impairment of prefrontal cortical dependent tasks. Clonidine ameliorates the β-carboline-induced anxiety response in monkeys (Crawley, 1985) and the noise stress-induced cognitive deficits in monkeys (Arnsten and Goldman-Rakic, 1986). In addition, the current study indicates that clonidine and (+)HA966 can attenuate the FG7142-induced cognitive deficits in both rats and monkeys. The fact that (+)HA966 and clonidine administration blocks both the stress-induced increases in prefrontal cortical dopamine turnover and the associated impairments in spatial working memory suggests that these and related drugs might be a useful therapeutic tool for the treatment of stress-exacerbated disorders.
The current findings with (+)HA966 and clonidine complement our previous study showing that the FG7142-associated cognitive impairments can be blocked by dopamine receptor antagonists. These findings support our original hypothesis that there is a critical range of dopaminergic activity for optimal prefrontal cortical-dependent cognitive functioning.
Footnotes
Support for these studies came from National Institutes of Health Grants MH14092 (R.H.R.) and AG06036 (A.F.T.A.), and from National Science Foundation Fellowship Grant GER9253954 (B.L.M.). We give special thanks to Lisa Ciavarella, Tracy White, and Jan Abele for their expert assistance. We also thank Daniel Podell, Paul Ward, and Shari Birnbaum for their reliable help.
Correspondence should be addressed to Dr. Robert H. Roth, Department of Pharmacology, P.O. Box 208066, 333 Cedar Street, New Haven, CT 06520-8066.
REFERENCES
- 1.American Psychiatric Association. DSM-IV: diagnostic and statistical manual of mental disorders, 4th Ed., pp 78–85, 424–429. American Psychiatric Association; Washington, DC: 1994. [Google Scholar]
- 2.Arnsten AFT, Goldman-Rakic PS. Reversal of stress-induced delayed response deficits in rhesus monkeys by clonidine and naloxone. Soc Neurosci Abstr. 1986;12:1464. [Google Scholar]
- 3.Arnsten AFT, Goldman-Rakic PS. Stress impairs prefrontal cortex cognitive function in monkeys: role of dopamine. Soc Neurosci Abstr. 1990;16:164. [Google Scholar]
- 4.Arnold LE, Jensen PS. Attention deficit disorders. In: Kaplan HI, Sadock BJ, editors. Comprehensive textbook of psychiatry/VI, Vol 2. Williams and Wilkins; Philadelphia: 1995. pp. 2295–2310. [Google Scholar]
- 5.Barkley RA, Grodzinsky G, DuPaul GJ. Frontal lobe functions in attention deficit disorder with and without hyperactivity: a review and research report. J Abnorm Child Psychol. 1979;20:163–188. doi: 10.1007/BF00916547. [DOI] [PubMed] [Google Scholar]
- 6.Bean AJ, Roth RH. Stimulated release of cotransmitters from rat prefrontal cortex in vivo: effects of stimulation frequency pattern and dopamine autoreceptors on dopamine and neurotensin release. J Neurosci. 1991;11:2694–2702. doi: 10.1523/JNEUROSCI.11-09-02694.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bebbington P, Wilkins S, Jones P, Foerster A, Murray R, Toone B, Shon L. Life events and psychosis. Initial results from the Camberwell collaborative psychosis study. Br J Psychol. 1993;162:72–79. doi: 10.1192/bjp.162.1.72. [DOI] [PubMed] [Google Scholar]
- 8.Benson DF. The role of frontal dysfunction in attention deficit hyperactivity disorder. J Child Neurol. 1991;6:S9–S12. doi: 10.1177/0883073891006001s02. [DOI] [PubMed] [Google Scholar]
- 9.Bowers MB, Jr, Morton JB. Regional brain catecholamines and metabolites following THC, PCP, and MK-801. Prog Neuropsychopharmacol Biol Psychiatry. 1994;18:961–964. doi: 10.1016/0278-5846(94)90111-2. [DOI] [PubMed] [Google Scholar]
- 10.Bradberry CW, Lory JS, Roth RH. The anxiogenic beta-carboline FG7142 selectively increases dopamine release in the rat prefrontal cortex as measured by microdialysis. J Neurochem. 1991;56:748–752. doi: 10.1111/j.1471-4159.1991.tb01987.x. [DOI] [PubMed] [Google Scholar]
- 11.Breier A, Wolkowitz OM, Pickar D. Stress and schizophrenia. Advances in neuropsychiatry and psychopharmacology. In: Tamminga CA, Schult SC, editors. Schizophrenia research, Vol 1. Raven; New York: 1991. [Google Scholar]
- 12.Bremner JD, Krystal JH, Southwick SM, Charney DS. Noradrenergic mechanisms in stress and anxiety. I. Preclinical studies. Synapse. 1996;23:28–38. doi: 10.1002/(SICI)1098-2396(199605)23:1<28::AID-SYN4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 13.Brozoski TJ, Brown R, Rosvold HE, Goldman PS. Cognitive deficit caused by regional depletion of dopamine in the prefrontal cortex of rhesus monkeys. Science. 1979;205:929–931. doi: 10.1126/science.112679. [DOI] [PubMed] [Google Scholar]
- 14.Bubser M, Schmidt WJ. 6-Hydroxydopamine lesions of the rat prefrontal cortex increases locomotor activity, impairs acquisition of delayed alternation tasks, but does not affect uninterrupted tasks in the radial maze. Behav Brain Res. 1990;37:157–168. doi: 10.1016/0166-4328(90)90091-r. [DOI] [PubMed] [Google Scholar]
- 15.Chergui K, Charlety PJ, Akaoka H, Saunier CH, Brunet JL, Buda M, Svensson TH, Chouvet G. Tonic activation of NMDA receptors causes spontaneous burst discharge of rat midbrain dopamine neurons in vivo. Eur J Neurosci. 1993;5:137–144. doi: 10.1111/j.1460-9568.1993.tb00479.x. [DOI] [PubMed] [Google Scholar]
- 16.Crawley JN, Ninan PT, Pickar D, Chrousos GP, Linnoila M, Skolnick P, Paul SM. Neuropharmacological antagonism of the β-carboline-induced “anxiety” response in rhesus monkeys. J Neurosci. 1985;5:477–485. doi: 10.1523/JNEUROSCI.05-02-00477.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deutch AY. The regulation of subcortical dopamine systems by the prefrontal cortex: interaction of central dopaminergic systems and the pathogenesis of schizophrenia. J Neural Transm Suppl. 1992;36:61–89. doi: 10.1007/978-3-7091-9211-5_5. [DOI] [PubMed] [Google Scholar]
- 18.Deutch AY. Prefrontal cortical dopamine systems and the elaboration of functional corticostriatal circuits: implications for schizophrenia and Parkinson’s disease. J Neural Transm Suppl. 1993;91:197–221. doi: 10.1007/BF01245232. [DOI] [PubMed] [Google Scholar]
- 19.Deutch AY, Roth RH. The determinants of stress-induced activation of the prefrontal cortical dopamine system. Prog Brain Res. 1990;85:367–403. doi: 10.1016/s0079-6123(08)62691-6. [DOI] [PubMed] [Google Scholar]
- 20. Deutch AY, Young CD. A model of the stress-induced activation of prefrontal cortical dopamine systems. Friedman MJ, Charney DS, Deutch AY. Neurobiological and clinical consequences of stress. From normal adaptation to post-traumatic stress disorder. 1995. Lippincott; New York: –Raven. [Google Scholar]
- 21.Domesick VB. Neuroanatomical organization of dopamine neurons in the ventral tegmental area. Ann NY Acad Sci. 1988;537:10–26. doi: 10.1111/j.1749-6632.1988.tb42094.x. [DOI] [PubMed] [Google Scholar]
- 22.Elsworth JD, Roth RH, Redmond DE., Jr Relative importance of 3-methoxy-4-hydroxyphenylglycol and 3,4-dihydroxyphenylglycol as norepinephrine metabolites in rat, monkey, and humans. J Neurochem. 1983;41:786–792. doi: 10.1111/j.1471-4159.1983.tb04809.x. [DOI] [PubMed] [Google Scholar]
- 23.Elsworth JD, Deutch AY, Redmond DE, Jr, Taylor JR, Sladek JR, Jr, Roth RH. Symptomatic and asymptomatic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated primates: biochemical changes in striatal regions. Neuroscience. 1989;33:323–332. doi: 10.1016/0306-4522(89)90212-1. [DOI] [PubMed] [Google Scholar]
- 24.Fallon JH. Topographic organization of ascending dopaminergic projections. Ann NY Acad Sci. 1988;537:1–9. doi: 10.1111/j.1749-6632.1988.tb42093.x. [DOI] [PubMed] [Google Scholar]
- 25.Goldman-Rakic PS. Prefrontal cortical dysfunction in schizophrenia: the relevance of working memory. In: Carroll BJ, Barrett JE, editors. Psychopathology and the brain. Raven; New York: 1991. pp. 1–23. [Google Scholar]
- 26.Goldman-Rakic PS. Working memory in schizophrenia. J Neuropsychiatry Clin Neurosci. 1994;6:348–357. doi: 10.1176/jnp.6.4.348. [DOI] [PubMed] [Google Scholar]
- 27.Goldman-Rakic PS, Lidow MS, Smiley JF, Williams MS. The anatomy of dopamine in monkey and human prefrontal cortex. J Neural Transm Suppl. 1992;36:163–177. doi: 10.1007/978-3-7091-9211-5_8. [DOI] [PubMed] [Google Scholar]
- 28.Goldstein M, Deutch AY. Dopaminergic mechanisms in the pathogenesis of schizophrenia. FASEB J. 1992;6:2413–2421. [PubMed] [Google Scholar]
- 29.Goldstein LE, Rasmussen AM, Bunney BS, Roth RH. The NMDA glycine site antagonist (+)HA966 selectively regulates conditioned stress-induced metabolic activation of the mesoprefrontal cortical dopamine but not serotonin systems: a behavioral, neuroendocrine, and neurochemical study in the rat. J Neurosci. 1994;14:4937–4950. doi: 10.1523/JNEUROSCI.14-08-04937.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonon FG. Nonlinear relationship between impulse flow and dopamine released by rat midbrain dopaminergic neurons as studied by in vivo electrochemistry. Neuroscience. 1988;24:19–28. doi: 10.1016/0306-4522(88)90307-7. [DOI] [PubMed] [Google Scholar]
- 31.Grenhoff J, Svensson TH. Clonidine modulates dopamine cell firing in rat ventral tegmental area. Eur J Pharmacol. 1989;165:11–18. doi: 10.1016/0014-2999(89)90765-6. [DOI] [PubMed] [Google Scholar]
- 32.Hauber W. Clozapine improves dizocilpine-induced delayed alternation impairment in rats. J Neural Transm Gen Sect. 1993;94:223–233. doi: 10.1007/BF01277027. [DOI] [PubMed] [Google Scholar]
- 33.Herman JP, Guillonneau D, Dantzer R, Scatton B, Semerdjan-Rouquier L, LeMoal M. Differential effects of inescapable footshocks and of stimuli previously paired with inescapable footshocks on dopamine turnover in cortical and limbic areas of the rat. Life Sci. 1982;30:2207–2214. doi: 10.1016/0024-3205(82)90295-8. [DOI] [PubMed] [Google Scholar]
- 34.Herve D, Blanc G, Glowinski J, Tassin JP. Reduction in dopamine utilization in the prefrontal cortex but not in the nucleus accumbens after selective destruction of the noradrenergic fibers innervating the ventral tegmental area in the rat. Brain Res. 1982;237:510–516. doi: 10.1016/0006-8993(82)90464-4. [DOI] [PubMed] [Google Scholar]
- 35.Ida Y, Elsworth JD, Roth RH. Anxiogenic beta-carboline FG7142 produces activation of noradrenergic neurons in specific brain regions of rats. Pharmacol Biochem Behav. 1991;39:791–793. doi: 10.1016/0091-3057(91)90166-y. [DOI] [PubMed] [Google Scholar]
- 36.Jentsch JD, Andrusiak E, Tran A, Roth RH. HA966 prevents increases in prefrontal cortical dopamine turnover and impairments of working memory induced by psychotomimetic drugs. FASEB J. 1996;20:A716. [Google Scholar]
- 37.Kalivas PW. Neurotransmitter regulation of dopamine neurons in the ventral tegmental area. Brain Res Rev. 1993;18:75–113. doi: 10.1016/0165-0173(93)90008-n. [DOI] [PubMed] [Google Scholar]
- 38.Kalivas PW, Duffy P, Barrow J. Regulation of the mesocorticolimbic dopamine system by glutamic acid receptor subtypes. J Pharmacol Exp Ther. 1989;251:378–387. [PubMed] [Google Scholar]
- 39.Kalivas PW, Duffy P, Eberhardt H. Modulation of A10 dopamine neurons by γ-aminobutyric acid agonists. J Pharmacol Exp Ther. 1990;253:858–866. [PubMed] [Google Scholar]
- 40.Kashiwa A, Nishikawa T, Nishijima K, Umino A, Takahashi K. Dizocilpine (MK-801) elicits a tetrodotoxin-sensitive increase in extracellular release of dopamine in rat medial frontal cortex. Neurochem Int. 1995;26:269–279. doi: 10.1016/0197-0186(94)00125-e. [DOI] [PubMed] [Google Scholar]
- 41.Lewis DA. The catecholaminergic innervation of primate prefrontal cortex. J Neural Transm Suppl. 1992;36:179–200. doi: 10.1007/978-3-7091-9211-5_9. [DOI] [PubMed] [Google Scholar]
- 42.Lewis DA, Hayes TL, Lund JS, Oeth KM. Dopamine and the neural circuitry of primate prefrontal cortex: implications for schizophrenia research. Neuropsychopharmacology. 1992;6:127–134. [PubMed] [Google Scholar]
- 43.Maurice T, Hiramatsu M, Itoh J, Kameyama T, Hasegawa T, Nabeshima T. Behavioral evidence for a modulating role of sigma ligands in memory processes. I. Attenuation of dizocilpine (MK-801)-induced amnesia. Brain Res. 1994;647:44–56. doi: 10.1016/0006-8993(94)91397-8. [DOI] [PubMed] [Google Scholar]
- 44.McMillen BA, Williams HL, Lehmann H, Shepard PD. On central muscle relaxants, strychnine-insensitive glycine receptors, and two old drugs: zoxazolamine and (+)HA966. J Neural Transm Gen Sect. 1992;89:11–25. doi: 10.1007/BF01245348. [DOI] [PubMed] [Google Scholar]
- 45.Moore H, Stuckman S, Sarter M, Bruno JP. Stimulation of cortical acetylcholine efflux by FG7142 measured with repeated microdialysis sampling. Synapse. 1995;21:324–331. doi: 10.1002/syn.890210407. [DOI] [PubMed] [Google Scholar]
- 46.Morrow BA, Clark WA, Roth RH. Stress activation of mesocorticolimbic dopamine neurons: effects of a glycine/NMDA receptor antagonist. Eur J Pharmacol. 1993;238:255–262. doi: 10.1016/0014-2999(93)90855-c. [DOI] [PubMed] [Google Scholar]
- 47.Morrow BA, George TP, Lee EJK, Roth RH. Noradrenergic alpha-2 agonists diminish behavioral and dopaminergic responses to conditioned fear. Soc Neurosci Abstr. 1996;22:810.9. [Google Scholar]
- 48.Murphy BL, Roth RH, Arnsten AFT. The effects of FG7142 on prefrontal cortical dopamine and spatial working memory in rat and monkey. Soc Neurosci Abstr. 1994;20:1010. [Google Scholar]
- 49.Murphy BL, Arnsten AFT, Goldman-Rakic PS, Roth RH. Increased dopamine turnover in the prefrontal cortex impairs spatial working memory performance in rats and monkeys. Proc Natl Acad Sci USA. 1996;93:1325–1329. doi: 10.1073/pnas.93.3.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; New York: 1982. [DOI] [PubMed] [Google Scholar]
- 51.Rao TS, Kim HS, Lehmann J, Martin LL, Wood PL. Differential effects of phencyclidine (PCP) and ketamine on mesocritical and mesostriatal dopamine release in vivo. Life Sci. 1989;45:1065–1072. doi: 10.1016/0024-3205(89)90163-x. [DOI] [PubMed] [Google Scholar]
- 52.Robbins TW. The case of frontostriatal dysfunction in schizophrenia. Schizophr Bull. 1990;16:391–402. doi: 10.1093/schbul/16.3.391. [DOI] [PubMed] [Google Scholar]
- 53.Roth RH, Tam S-Y. Regulatory control of midbrain dopamine neurons. In: Kaufman S, editor. Amino acids in health and disease: new perspectives, Vol 55. Liss; New York: 1987. pp. 159–178. [Google Scholar]
- 54.Roth RH, Tam S-Y, Ida Y, Yang J-X, Deutch AY. Stress and the mesocorticolimbic dopamine systems. Ann NY Acad Sci. 1988;537:138–147. doi: 10.1111/j.1749-6632.1988.tb42102.x. [DOI] [PubMed] [Google Scholar]
- 55. Southwick SM, Yehuda R, Morgan CA., III Clinical studies of neurotransmitter alterations in post-traumatic stress disorder. Friedman MJ, Charney DS, Deutch AY. Neurobiological and clinical consequences of stress. From normal adaptation to post-traumatic stress disorder 1995. Lippincott; New York: –Raven. [Google Scholar]
- 56.Tam S-Y (1986) Mesoprefrontal dopamine neurons: studies on their regulatory control. Doctoral thesis, Yale University.
- 57.Tam S-Y, Roth RH. Selective increase in dopamine metabolism in the prefrontal cortex by the anxiogenic beta-carboline FG7142. Biochem Pharmacol. 1985;34:1595–1598. doi: 10.1016/0006-2952(85)90708-7. [DOI] [PubMed] [Google Scholar]
- 58.Tam S-Y, Roth RH. Modulation of mesoprefrontal dopamine neurons by central benzodiazepine receptors. I. Pharmacological characterization. J Pharmacol Exp Ther. 1990;252:989–996. [PubMed] [Google Scholar]
- 59.Thierry AM, Tassin JP, Glanc G, Glowinski J. Selective activation of mesocortical DA system by stress. Nature. 1976;263:242–244. doi: 10.1038/263242a0. [DOI] [PubMed] [Google Scholar]
- 60.Turski L, Niemann W, Stephens DN. Differential effects of antiepileptic drugs and beta-carbolines on seizures induced by excitatory amino acids. Neuroscience. 1990;39:799–807. doi: 10.1016/0306-4522(90)90262-3. [DOI] [PubMed] [Google Scholar]
- 61.Verma A, Moghaddam B. NMDA receptor antagonists impair prefrontal cortex function as assessed via spatial delayed alternation performance in rats—modulation by dopamine. J Neurosci. 1996;16:373–379. doi: 10.1523/JNEUROSCI.16-01-00373.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang T, O’Connor, Ungerstedt U, French ED. N-Methyl-d-aspartic acid biphasically regulates the biochemical and electrophysiological response of A10 dopamine neurons in the ventral tegmental area: in vivo microdialysis and in vitro electrophysiological studies. Brain Res. 1994;666:255–262. doi: 10.1016/0006-8993(94)90780-3. [DOI] [PubMed] [Google Scholar]
- 63.Weinberger DR, Berman ZF, Zed RF. Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow evidence. Arch Gen Psychiatry. 1986;43:114–124. doi: 10.1001/archpsyc.1986.01800020020004. [DOI] [PubMed] [Google Scholar]
- 64.Weinberger DR, Aloia MS, Goldberg TE, Berman KF. The frontal lobes and schizophrenia. J Neuropsychiatry Clin Neurosci. 1994;6:419–427. doi: 10.1176/jnp.6.4.419. [DOI] [PubMed] [Google Scholar]
- 65.White F. Synaptic regulation of mesocorticolimbic dopamine neurons. Annu Rev Neurosci. 1996;19:405–436. doi: 10.1146/annurev.ne.19.030196.002201. [DOI] [PubMed] [Google Scholar]
- 66.Williams GV, Goldman-Rakic PS. Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature. 1995;376:572–575. doi: 10.1038/376572a0. [DOI] [PubMed] [Google Scholar]
- 67.Zahrt J, Taylor JR, Arnsten AFT. Supranormal stimulation of dopamine D1 receptors in the prefrontal cortex impairs spatial working memory performance in rats. Soc Neurosci Abstr. 1996;22:446.10. doi: 10.1523/JNEUROSCI.17-21-08528.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]